

Abstract
The mechanism that controls the selective vulnerability of striatal neurons in Huntington's disease is unclear. Brain-derived neurotrophic factor (BDNF) protects striatal neurons and is regulated by Huntingtin through the interaction with the neuron-restrictive silencer factor. Here, we demonstrate that the downregulation of BDNF by mutant Huntingtin depends on the length and levels of expression of the CAG repeats in cell cultures. To analyze the functional effects of these changes in BDNF in Huntington's disease, we disrupted the expression of bdnf in a transgenic mouse model by cross-mating bdnf+/ - mice with R6/1 mice. Thus, we compared transgenic mice for mutant Huntingtin with different levels of BDNF. Using this double mutant mouse line, we show that the deficit of endogenous BDNF modulates the pathology of Huntington's disease. The decreased levels of this neurotrophin advance the onset of motor dysfunctions and produce more severe uncoordinated movements. This behavioral pathology correlates with the loss of striatal dopamine and cAMP-regulated phosphoprotein-32-positive projection neurons. In particular, the insufficient levels of BDNF cause specific degeneration of the enkephalinergic striatal projection neurons, which are the most affected cells in Huntington's disease. This neuronal dysfunction can specifically be restored by administration of exogenous BDNF.
Therefore, the decrease in BDNF levels plays a key role in the specific pathology observed in Huntington's disease by inducing dysfunction of striatal enkephalinergic neurons that produce severe motor dysfunctions. Hence, administration of exogenous BDNF may delay or stop illness progression.
Keywords: neurotrophins, cell death, striatum, knock-out, movement disorders, polyQ
Introduction
Neurodegenerative disorders such as Parkinson's, Huntington's, or Alzheimer's diseases are characterized by the loss of specific neuronal populations. The mechanism responsible for the neurodegenerative pattern of each disease is not known. In Huntington's disease (HD), the CAG triplet expansion in the huntingtin gene (htt) causes a primary loss of striatal projection neurons that, in turn, leads to progressive motor impairment (Hickey and Chesselet, 2003). Although htt is widely expressed in all animal cells and its mutation is not tissue specific, the medium-sized spiny striatal neurons, which express GABA, and dopamine and cAMP-regulated phosphoprotein (DARPP)-32, undergo selective degeneration. Within the striatum, two separate subpopulations of GABAergic neurons project to the external globus pallidus or to the substantia nigra pars reticulata and the internal globus pallidus, originating the “indirect” and “direct” pathways, respectively. The first of these subpopulations expresses enkephalin and is enriched in D2 dopamine receptors, whereas the neurons that project to the substantia nigra and the internal globus pallidus express substance P and D1 dopamine receptors (Gerfen, 1992). The primary affected striatal neurons are the enkephalinergic and those neurons that project to the substantia nigra; however, striatal output to the internal globus pallidus is relatively preserved (Richfield et al., 1995). Striatal interneurons are also spared in this disorder (Sieradzan and Mann, 2001). This specific degeneration correlates with the appearance of choreic movements (Reiner et al., 1988). However, neuronal degeneration extends to other brain regions such as the cerebral cortex, particularly in more advanced cases (Rubinsztein, 2002).
It has been suggested that mutant Htt initiates a cascade of different events in the disease that converge in the specific cell death of striatal neurons. A large number of abnormalities have been reported in HD, including transcription deficits, energy impairment, excitotoxicity, and lack of trophic support (Sugars and Rubinsztein, 2003; Alberch et al., 2004). In this context, it has been shown recently that Htt directly modulates the expression of neuron-restrictive silencer factor (NRSF)-controlled neuronal genes, including the brain-derived neurotrophic factor (bdnf) gene (Zuccato et al., 2003). Thus, wild-type (wt) Htt stimulates the production of BDNF, a neurotrophic factor for striatal neurons (Perez-Navarro et al., 2000), whereas mutant Htt reduces it (Zuccato et al., 2001). In keeping with these findings, BDNF is decreased in brain tissue from human HD patients (Ferrer et al., 2000; Zuccato et al., 2001) and in some mice transgenic for mutant htt (Zuccato et al., 2001; Duan et al., 2003; Zhang et al., 2003).
Besides this evidence that mutant Htt regulates BDNF expression, it remains to be established whether downregulation of endogenous BDNF participates in the specific motor dysfunctions observed in HD. To study the function of endogenous BDNF in HD, we generated a double-mutant animal by crossing bdnf heterozygous mice with a model of HD. Our present data show that reduction of BDNF levels advances the age of onset and exacerbates the lack of motor control. This enlarged neurological pathology correlates with morphological alterations, which shows that BDNF plays a role in the specific degeneration of the striatal enkephalinergic population.
Materials and Methods
Cell procedures. We used the conditionally immortalized striatal derived neural stem cells, M213 (a generous gift from Dr. W. Freed, National Institute on Drug Abuse, Bethesda, MD) (Giordano et al., 1993). For all experiments, cells were grown and passaged at the permissive temperature of 33°C in DMEM (Invitrogen, Renfrewshire, Scotland) supplemented with 10% fetal calf serum (Invitrogen). Cells were transfected using the rat neural stem cells nucleofector kit as described by the manufacturer (Amaxa, Cologne, Germany). M213 cells (5 × 106) were resuspended in 200 μl of nucleofector solution (Amaxa) and mixed with 5 μg of the required cDNA for each sample. Electroporation was performed using the A-33 nucleofector program (Amaxa). We transfected three different constructs, which allowed us to express the mutant exon 1 of the htt gene with different CAG/CAA repeats: 47 (qp47), 72 (qp72), and 103 (qp103) (Kazantsev et al., 1999) (generously provided by Dr. George M. Lawless, Cure HD Initiative, Reagent Resource Bank of the Hereditary Disease Foundation, New York, NY).
For the study of BDNF secretion, cells were cultured in 24-well plates for 24 hr after transfection and, thereafter, positively expressing cells were purified by cell sorting. Cell sorting was performed on a fluorescence-activated cell sorting Vantage SE (Becton Dickinson, Mountain View, CA) using a single laser set at 488 nm. Cells were sorted according to the endogenous enhanced green fluorescent protein (EGFP) fluorescence intensity, and fluorescent channel 1 threshold was set to collect only cells with high levels of expression except for qp103 clones, for which the gates were adjusted to collect separately the cells with high and low levels of expression (see Fig. 1 B, inset). Sorting conditions were as follows: drop drive of 23,000 drops per second; three drops of sorting envelope; sample rate of 500-1000 cells per second; drop delay of 12.0. Sorted cells were collected in the cell culture medium described above and seeded at a density of 10,000 cells/cm2 onto 24-well plates. One day later, the expression of EGFP was assessed in an inverted fluorescent microscope. More than 80% of cells were positive, showing larger and more abundant aggregates in cells transfected with longer constructs. The levels of mutant htt directly correlate to the levels of EGFP, because both are expressed as a fusion protein. Six days after plating, the cell culture was removed and processed for the quantification of the BDNF levels. Proteins from cell lysates were quantified to normalize the levels of BDNF with respect to the microgram of protein from cells for each well at the end of the experiment.
Figure 1.
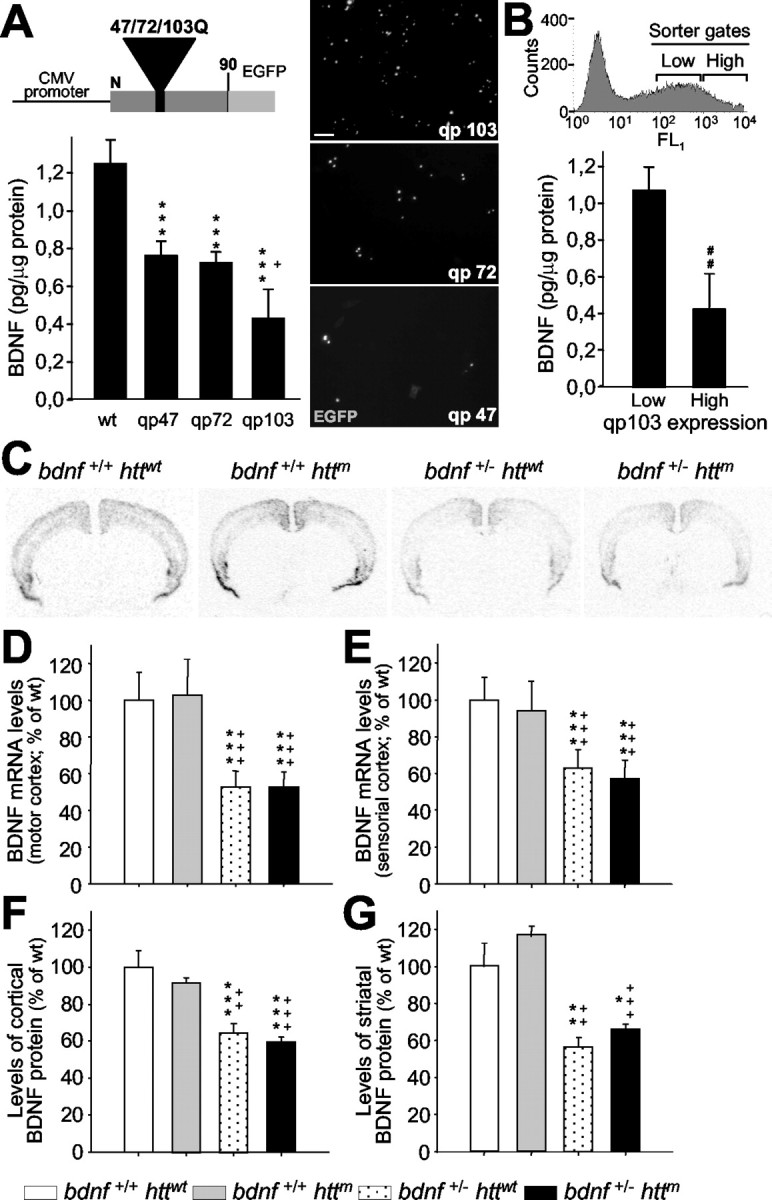
The number of CAG repeats and the levels of mutant htt expression modulate the expression of BDNF. A, ELISA for BDNF was performed on the culture media from wt M213 cells and subclones expressing exon 1 of mutant htt with 47 (qp47), 72 (qp72), and 103 CAG/CAA repeats (qp103). The expression of this neurotrophin decreases as the number of repeats is longer. The scheme at the top shows the structure of qp constructs, which expresses a fusion protein of the first 90 amino acids of the mutant htt and the EGFP. The right panels are photomicrographs of transfected cells with the qp47, 72, and 103 constructs, which show the mutant htt inclusions. CMV, Cytomegalovirus. Scale bar, 50 μm. B, Transfected cells that express different levels of qp103 were collected by cell sorting (inset shows sorting gates), and BDNF levels were assessed. The amount of BDNF in culture media is inversely proportional to the levels of expression of qp103. ***p<0.001 compared with wt cells; +p<0.05 compared with qp47 or qp72; ##p < 0.005 compared with low levels of qp103 expression. C-G, BDNF levels are not affected by the mutant exon 1 of Htt in R6/1 (bdnf+/+ httm). C-E, In situ hybridization demonstrates that mutant htt does not change the levels of bdnf expression either in the motor cortex (D) or in the sensorial cortex (E). C, A Representative coronal section (bregma, +1.1 mm) of bdnf in situ hybridization of the four genotypes analyzed. F, G, In addition, the levels of BDNF protein detected by ELISA are not modified by mutant htt in the cortex (F) or in the striatum (G). The only changes in mRNA or protein are detected in bdnf+/- mice with or without mutant htt (C-G). *p < 0.05, **p < 0.005, and ***p < 0.001 compared with wt mice (bdnf+/+ httwt); ++p < 0.005 and +++p < 0.001 compared with R6/1 mice (bdnf+/+ httm).
Mouse strains. To obtain double-mutant mice with mutant Htt and low levels of BDNF (bDM; bdnf+/ - httm) or mice with mutant Htt and low levels of neurotrophin (NT)-3 (n3DM; nt-3+/ - httm), we cross-mated R6/1 mice (Mangiarini et al., 1996) with bdnf heterozygous (Ernfors et al., 1994a) or nt-3 heterozygous mice (Ernfors et al., 1994b), respectively. F2 bdnf+/+ httm × bdnf+/ - httwt or F2 nt-3+/+ httm × nt-3+/ - httwt inbred mice were used in this study. All experiments were performed on male littermates from the F2 population to avoid strain and sex differences. Mice were housed together in numerical birth order in groups of mixed genotypes until they were 30 weeks of age with access to food and water ad libitum in a colony room kept at a constant temperature (19-22°C) and humidity (40-50%) on a 12 hr light/dark cycle. All experiments were conducted in a blind-coded manner with respect to genotype, and data were recorded for analysis by microchip mouse number. For developmental studies, some animals were killed 15 d after birth (n = 4-6 per genotype). All animal-related procedures were in accordance with the National Institutes of Health guide for the care and use of laboratory animals and approved by the local animal care committee of the Universitat de Barcelona (99/01) and by the Generalitat de Catalunya (99/1094). Animals were killed at 30 weeks of age, just before they drastically lost weight and died. Body weight was measured twice each week.
For genotyping, DNA was obtained from tail biopsy and processed for PCR. The primers used for DNA amplifications have been described previously (Ernfors et al., 1994b; Mangiarini et al., 1996; Agerman et al., 2003). PCR fragments were resolved in agarose gels, 2% for bdnf or nt-3 amplification and 1.5% for mutant htt analysis.
In vivo BDNF treatment. Administration of exogenous BDNF was performed in 20-week-old R6/1 mice (n = 5) and in 14-week-old bDM (n = 5) at the disease onset in each genotype. BDNF (4.5 μg per day) was continuously infused using an osmotic minipump at a rate of 1 μl per hour (Alzet model 2001; Alza, Palo Alto, CA). A 28-gauge stainless steel cannula was implanted into the left striatum (coordinates: anteroposterior, +0.5; lateral, +2.0 from bregma and dorsoventral, -2.7 from dura). The cannula was fixed on the skull with anchor screws and dental cement. The metal inlet of the cannula was fitted to a tube connected to the osmotic pump filled with either BDNF (187.5 ng/μl in PBS) or PBS alone (sham controls). The Alzet minipump was previously equilibrated for 4hr at 37°C in 0.9% NaCl. The pump was implanted subcutaneously in the back of the animal. After 1 week, animals were killed and brains were analyzed for in situ hybridization for neuropeptides.
Behavior analysis. Behavioral testing began at 3 weeks of age, when mice were weaned. Tests were conducted regularly until week 30 to follow the progression of each phenotype (n = 9-16 mice per group). Motor coordination and balance were evaluated on the rotarod apparatus at several revolutions per minute as described previously (Carter et al., 1999). In brief, animals were trained at a constant speed (24 rpm) for 60 sec. We performed four trials per day on three consecutive days, and the latency to fall was recorded. No differences between groups were detected during this period. After training, mice were evaluated once every 2 weeks at 16, 24, and 32 rpm starting at 6 weeks of age, and the number of falls in a total of 60 sec was recorded. The animals were put on the rotarod several consecutive times until the addition of the latency to fall off reached the total time of 60 sec. We compared the curves of the behavior pattern and calculated the percentage of rotarod impairment as described previously (Ferrante et al., 2002). The curves have been estimated through generalized linear mixed models assuming a residual distributed under a Poisson distribution. The estimation approach used was the Penalized Quasi-Likelihood, and the effects have been tested using the F-Wald test. To analyze the individual differences in each time point, we performed a one-way ANOVA followed by least significant difference (LSD; t test) post hoc test.
The walking footprint pattern, equilibrium, vision, and muscular strength were evaluated at 30 weeks of age (n = 9-16 mice per group). The footprint test was performed as described previously (Carter et al., 1999). Mice were trained three consecutive times to walk in a corridor that was 50 cm long and 7 cm wide. To register the experiments, the forefeet and hindfeet of the mice were painted with nontoxic black and red ink, respectively, and then given one run. The footprint pattern was analyzed for the number of steps on the white paper, the stride length was measured as the average distance of forward movement between each stride, and the forebase and hindbase widths were measured as the perpendicular distance between the left and right footprints of a given step.
Equilibrium was analyzed by placing the mice at the center of a raised (60 cm) horizontal rod (50 cm long), and their latency to fall off the rod was scored with a maximum of 60 sec. A flat wooden rod (12 mm wide) was used in trials 1 and 2, and a cylindrical wire rod (14 mm diameter) was used in trials 3 and 4. The distance covered on the cylindrical rod was also registered and analyzed. The latency to fall off the wooden rod was recorded with no differences between groups found. The visual cliff avoidance and the wire hang tests were performed as described previously (Lione et al., 1999; Gimenez-Llort et al., 2002).
For analyses of circling behavior, bDM were injected intraperitoneally with amphetamine (5 mg/kg) 1 week after unilateral BDNF infusion. Mice were placed in circular cages and connected to an automated rotometer. A computer recorded the number of complete (360°) turns made during a 5 min period. Mice were allowed 15 min to habituate to the rotometer before the administration of amphetamine. The values were expressed as net total numbers of full turns in 1 hr.
Results were expressed as the mean of several animals, and error bars represent the SEM. Statistical analysis was performed using one-way ANOVA followed by LSD post hoc test.
ELISA. BDNF or NT-3 contents were determined in duplicate by the Emax ImmunoAssay system (Promega, Madison, WI), as described previously (Perez-Navarro et al., 2000). For cell cultures, the cell culture media were collected after 6 d of plating cell after cell sorting. We analyzed 50 μl of supernatants from cell cultures diluted 1:1 in block and sample buffer. Values were normalized by the total protein content of the cells in each well at the end of the experiment and expressed as picogram of neurotrophin per microgram of protein. For total protein measurements, wells were washed twice with PBS and then cells were homogenized in lysis buffer (137 mm NaCl, 20 mm Tris-HCl, pH 8.0, 1% Igepal, 10% glycerol, 1 mm PMSF, 10 μg/ml Aprotinin, 1 μg/ml Leupeptin). Quantifications were performed using the Detergent Compatible Protein Assay (Bio-Rad, Hercules, CA) following the manufacturer instructions.
For analysis of BDNF levels in brain tissue, mice were deeply anesthetized in a CO2 chamber at 30 weeks of age (n = 4 per genotype). Their cortex and striatum were dissected out on ice and rapidly frozen using CO2 pellets. Samples were then homogenized in lysis buffer, sonicated, and centrifuged (10 min at 14 rpm at 4°C). Supernatants were collected and the protein contents were measured as above. Total protein (300 μg) was analyzed for each point diluted 1:1.5 in block and sample buffer. Values were calculated as picogram of BDNF per milligram of tissue protein and standardized to the average of wt animals.
Western blot analysis. We analyzed the levels of wt Htt by Western blot analysis using the same monoclonal antibody against the wt Htt fragment described above (Mab2166; Chemicon, Temecula, CA). Samples (n = 3 per genotype and time point) were prepared and processed by Western blot as described previously (Dyer and McMurray, 2001). In brief, tissue from the cerebral cortex or striatum was homogenate by sonication in 10 μl of radioimmunoprecipitation assay buffer (150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, 1 mm PMSF, 10 μg/ml Aprotinin, 1 μg/ml Leupeptin) per milligram of tissue. After homogenation, samples were centrifuged twice at 12,000 × g for 10 min. Supernatant proteins (30 μg) without head denaturation were loaded in a 7.5% SDS-PAGE and transferred to Immobilon-P membranes (Millipore, Bedford, MA). Blots were blocked in 3% nonfat dry milk in TBS-T (150 mm NaCl, 20 mm Tris-HCl, pH 7.5, 0.05% Tween 20) and then incubated with 1:5000 of primary antibody in 0.5% nonfat dry milk in TBS-T. After several washes in TBS-T, blots were incubated with 1:1000 of anti-mouse IgG HRP-conjugated (Promega) and developed by ECL Western blotting analysis system (Bioscience Europe, Freiburg, Germany). As loading controls, we reincubated the membranes with a monoclonal anti-α-tubulin antibody (Sigma, St. Louis, MO).
Immunocytochemistry. To analyze the localization of wt Htt, we performed immunocytochemistry on M213 transfected with qp72. For these experiments, expressing cells were not selected by cell sorting to have a heterogenous population in each well. In addition, we analyzed qp72 cells because their smaller and less abundant aggregates allowed us to perform more accurate localization at different levels of aggregate formation. To detect wt Htt, we used a monoclonal antibody against the Htt fragment from aa 181-810 (Mab2166; Chemicon). This antibody only detects wt Htt because this sequence is not included in the qp-mutant Htt constructs used in the present study (Kazantsev et al., 1999). Cells were fixed with 4% paraformaldehyde solution in PBS. After three washes, cells were incubated with the primary antibody (1:1000 in PBT buffer: PBS with 1% bovine serum albumin and 0.3% Triton X-100) at 4°C for 16 hr. The excess of antibody was washed out with PBS and thereafter incubated with the secondary antibody (donkey anti-mouse-Texas Red conjugated 1:300; Jackson Immuno Research, West Grove, PA), washed, and mounted with Mowiol (Calbiochem, La Jolla, CA). Control avoiding the primary antibody was performed and resulted in no signal. Photomicrographs were taken using a confocal microscope.
Histology. For neurotrophin in situ hybridization studies, mice were killed by decapitation at 30 weeks of age, and brains were dissected out and frozen in dry-ice cooled isopentane (n = 4). Serial coronal cryostat sections (14 μm) were processed for hybridization with radioactive riboprobes (Ibanez et al., 1992; Laurenzi et al., 1994; Lindefors et al., 1995) as described previously (Canals et al., 2001). Antisense cRNA probe to detect the transcripts of neurotrophins was prepared by in vitro transcription using T3 or T7 RNA polymerase (Promega) and [33P]-UTP (Bioscience Europe). For control experiments, sense cRNA probes were obtained by in vitro transcription using the opposite RNA polymerase. After deproteination and acetylation slices were incubated for 16 hr in a humidified chamber at 53°C with 3 × 105 cpm of antisense probes in 150 μl of hybridization mixture (50% formamide, 20 mm Tris-HCl, pH 7.6, 1 mm EDTA, pH 8.0, 0.3 m NaCl, 0.1 m dithiothreitol, 0.5 mg/ml yeast tRNA, 0.1 mg/ml polyA RNA, 1× Denhardt's solution, and 10% dextran sulfate). After hybridization, slices were washed at high stringency, dried, and exposed to Biomax MR (Kodak, Rochester, NY) for 10 d.
Cresyl violet staining and immunohistochemistry assays were performed on paraformaldehyde fixed material as described previously (Canals et al., 2001). Animals were transcardially perfused with 4% paraformaldehyde solution in 0.1 m sodium phosphate, pH 7.2, at 15 d and 30 weeks of age. The brains were postfixed for 2 hr in the same solution, cryo-protected in incrementing 10-30% sucrose/PBS, and frozen in dry-ice cooled isopentane. Serial horizontal cryostat sections (30 μm) separately (0.3 mm) were processed for cresyl violet staining or immunohistochemistry.
Sections were processed as free-floating for immunohistochemistry. To block endogenous peroxidases, sections were incubated for 30 min with PBS containing 10% methanol and 3% H2O2. Sections were then washed three times in PBS and blocked for 1 hr with 2-10% normal serum in PBS. Tissue was then incubated with the appropriated primary antibody in PBS containing 2% normal goat serum for 16 hr at 4°C, except for DARPP-32, which was incubated at room temperature. The following antibodies were used: anti-neuronal-specific nuclear protein (NeuN) 1:100 (Chemicon); anti-DARPP-32, 1:10,000 (a generous gift from Dr. H. C. Hemming, Rockefeller University, New York, NY); anti-parvalbumin, 1:1250 (Sigma); anti-ubiquitin, 1:800 (Dako, High Wycombe, UK), and anti-mutant Htt, 1:50 (EM48; Chemicon). Sections were washed three times and incubated with a biotinylated secondary antibody (1/220; Pierce, Rockford, IL) for 1-2 hr at room temperature in the same buffer as the primary antibody. The immunohistochemical reaction was developed using the ABC kit (Pierce). No signal was detected in controls avoiding the primary antibodies.
For double-labeling immunohistochemistry, slides were coincubated with the primary antibodies anti-ubiquitin and anti-NeuN overnight at 4°C. After three washes in PBS, the sections were coincubated with both secondary antibodies (anti-rabbit FITC conjugated, 1:100, Vector Laboratories, Burlingame, CA; and anti-mouse Texas Red conjugated, 1:200, Jackson ImmunoResearch), washed, and mounted with Mowiol (Calbiochem). Double-labeling analysis was performed using a confocal microscope.
For enkephalin and substance P analyses, sections were processed by nonisotopic in situ hybridization as described previously (Serrats et al., 2003). Frozen tissue sections were air dried, fixed in 4% paraformaldehyde in PBS for 20 min at 4°C, washed once in 3× PBS and twice in 1× PBS for 5 min each, and incubated for 2 min at 21°C in a freshly prepared solution of predigested Pronase (Calbiochem) at a final concentration of 24 U/ml in 50 mm Tris-HCl, pH 7.5, 5 mm EDTA. Enzyme activity was stopped by immersion for 30 sec in 2 mg/ml glycine in PBS. Tissues were finally rinsed in PBS and dehydrated through a graded series of ethanol. Tissue sections were then covered with 100 μl of hybridization buffer containing the probes, overlaid with Nescofilm coverslips (Bando Chemical, Kobe, Japan), and incubated overnight in humidified boxes at 42°C. Digoxigenin (DIG)-labeled probes were diluted in hybridization solution: 50% formamide, 4× SSC (1× SSC: 150 mm NaCl, 15 mm sodium citrate), 1× Denhardt's solution, 10% dextran sulfate, 1% Sarkosyl, 20 mm phosphate buffer, pH 7.0, 250 μg/ml yeast tRNA and 500 μg/ml salmon sperm DNA to a final concentration of 1.5 nm. The oligodeoxyribonucleotide probes used were as follows: the enkephalin probe was complementary to bases 513-542 of the enkephalin sequence (GenBank accession number K02807) and was synthesized on a 380 Applied Biosystem DNA synthesizer (Foster City Biosystem, Foster City, CA), and the substance P probe was complementary to bases 223-270 from the preprotachykinin A sequence (GenBank accession number M34183) and synthesized by Isogen Bioscience BV (Maarssen, The Netherlands). They were 3′-end labeled with terminal deoxynucleotidyltransferase and DIG-11-dUTP (Roche, Mannheim, Germany) as described previously (Schmitz et al., 1991). DIG-labeled oligonucleotides were purified by ethanol precipitation and resuspended in 200 μl of TE (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, pH 8.0). After hybridization, sections were washed four times (45 min each) in a buffer containing 0.6 m NaCl and 10 mm Tris-HCl, pH 7.5, at 60°C.
Nonradioactive hybridization signal was developed as described previously (Landry et al., 2000). Briefly, after washing, the slides were immersed for 30 min in a buffer containing 0.1 m Tris-HCl, pH 7.5, 1 m NaCl, 2 mm MgCl2, and 0.5% bovine serum albumin and incubated overnight at 4°C in the same solution with alkaline phosphate-conjugated anti-digoxigenin-F (ab) fragments (1:5000; Roche). They were then washed three times in the same buffer and twice in an alkaline buffer (0.1 m Tris-HCl, pH 9.5, 0.1 m NaCl, and 5 mm MgCl2). Alkaline phosphatase activity was developed by incubating the sections with 3.3 mg of nitroblue tetrazolium and 1.65 mg of bromochloroindolyl phosphate (Invitrogen) dissolved in 10 ml of alkaline buffer. The enzymatic reaction was stopped by extensive rinsing in alkaline buffer with the addition of 1 mm EDTA. The sections were then dehydrated and air-dried.
Quantification of neurotrophin expression. The expression levels of different neurotrophins [nerve growth factor (NGF), BDNF, NT-3, and NT-4/5] were quantified on in situ hybridization films (n = 4). Consecutive sections (23-29 sections per animal) were scanned, and mRNA levels were analyzed using the ImageJ program (National Institute of Mental Health, Bethesda, MD). The intensity in a squared area of 152.4 μm2 was quantified, and the background signal of the same adjacent area outside of the brain was subtracted. Results were expressed as the mean of several mice, and error bars represent the SEM. Statistical analysis was performed using one-way ANOVA followed by the LSD post hoc test.
Cell and inclusion counting. Brain, cortical, and striatal volumes were measured by using Image tool program (The University of Texas Health Center, San Antonio, TX) on a computer attached to an Olympus (Ballerup, Denmark) microscope. Consecutive sections (14-16 sections per animal) were visualized, and the borders of the anatomical landmarks were outlined. The volumes were calculated by multiplying the sum of all sectional areas (square millimeters) by the distance between successive sections (0.3 mm) as described previously (Canals et al., 2001).
All cell counts were performed blind with respect to genotype in 30-week-old mice (n = 4-7 per each group and staining). Unbiased stereological counts of striatal cells were obtained from the entire neostriatum using the computer-assisted stereology toolbox software (Olympus). The dissector counting method was used to analyze coronal sections spaced 300 μm apart. The counting frames were randomly sampled. We counted the neuron-like cells in cresyl violet staining as the cells with a large nonpyknotic nucleus with clear nucleoli. DARPP-32 neuron areas were analyzed using the program AnalySIS Auto (Soft Imaging System, Münster, Germany).
For intranuclear aggregate counting, three fields of 0.035 mm2 were analyzed in each section separated by 0.3 mm (n = 4). The number of inclusions was expressed in density. To obtain the number per cubic millimeter, we divided the counts by the area corresponding to each field examined and multiplied by the thickness of the section.
All results were expressed as the mean and the SEM of different mice. Statistical analysis was performed using ANOVA followed by the LSD post hoc test or a Student's t test for independent samples.
Results
The length and levels of CAG-repeat expression regulate BDNF in vitro
Here, we examined whether the number of repeats and the levels of mutant htt expression affect the production of BDNF (Fig. 1A, B). The levels of BDNF in supernatants of immortalized striatal neurons transfected with constructs of exon 1 of mutant htt containing 47 or 72 CAG repeats (qp47and qp72) were reduced similarly (Fig. 1A) (qp47, 63.01 ± 5.43%; qp72, 61.64 ± 3.63% of parental cells). However, supernatants from cells transfected with exon 1 of the mutant htt with 103 repeats (qp103) was significantly lower than qp47 and qp72 (Fig. 1A) (34.55 ± 12.95% of parental cells and 59.38 ± 12.25% with respect to qp72). In addition, the amount of BDNF was inversely proportional to the levels of expression of qp103 (Fig. 1B). In cells expressing low qp103 levels, BDNF production was similar to that found in supernatants from parental cells without significant differences. These findings may help to explain the differences in BDNF levels observed in distinct mouse models of HD. To evaluate the specificity of this effect, we also measured the levels of NT-3 in supernatants from qp103 transfected cells, and we did not observe modifications of this protein (104.52 ± 18.9% of parental cells).
Generation of transgenic mutant htt mice with different levels of BDNF
To analyze the role of BDNF in HD progression and to explore whether the decrease of its expression is involved in the selective vulnerability, we generated a bDM line by crossing R6/1 mice (Mangiarini et al., 1996) with bdnf heterozygous mice (Ernfors et al., 1994b) as described in Materials and Methods. We did not detect any difference in life span between wt and bdnf+/- mice. However, R6/1 mice showed shorter survival as described by Mangiarini et al. (1996). We examined the levels of BDNF, mRNA, and protein in the cerebral cortex and striatum from 30-week-old mice (n = 4 per genotype). We found that bdnf+/+ httm mice, the equivalent genotype to R6/1, have similar levels of BDNF mRNA and protein to wt mice (Fig. 1C-G). In situ hybridization for bdnf showed that the expression of this neurotrophin was only lower in bdnf heterozygous animals (bdnf+/ - httwt and bdnf+/ - httm) (Fig. 1C-E). In addition, BDNF protein levels analyzed by ELISA (Fig. 1F,G) showed the same levels in the cerebral cortex and in the striatum of the two genotypes with bdnf+/ + (in picogram per milligram of protein; striatum: wt, 46.81 ± 5.97 and R6/1 genotype, 54.70 ± 3.24; cerebral cortex: wt, 61.90 ± 5.52 and bdnf+/+ httm, 56.96 ± 1.67). To rule out a genetic background effect, we also evaluated R6/1 mice. These mice showed no changes in BDNF levels (data not shown). In addition, bDM (bdnf+/ - httm; in picogram per milligram of protein; striatum: 30.96 ± 1.41; cortex: 36.78 ± 1.70) showed the same decrease in BDNF levels as bdnf+/ - littermates with normal htt (bdnf+/ - httwt; in picogram per milligram of protein; striatum: 26.42 ± 2.98; cortex: 40.24 ± 3.49) (Fig. 1F,G). We next analyzed whether other neurotrophins compensate for the insufficient levels of BDNF by modifying their levels of expression. In situ hybridizations against NGF, NT-3, or NT-4/5 did not show changes in the mRNA levels of these neurotrophins in any genotype studied (data not shown).
BDNF regulation is downstream of the aggregate formation
It has been proposed that this wt Htt-mediated BDNF regulation can be associated to the sequestering of wt Htt into aggregates. Our results showed that in nontransfected M213 cells, wt Htt is localized in endocytic compartments, as described previously (Kegel et al., 2000) (Fig. 2A). However, in M213 cells transfected with mutant htt, wt Htt is also localized into aggregates, and in cells with large aggregates, the endocytic wt Htt labeling disappeared (Fig. 2A). No signal was observed avoiding primary antibody (Fig. 2B).
Figure 2.
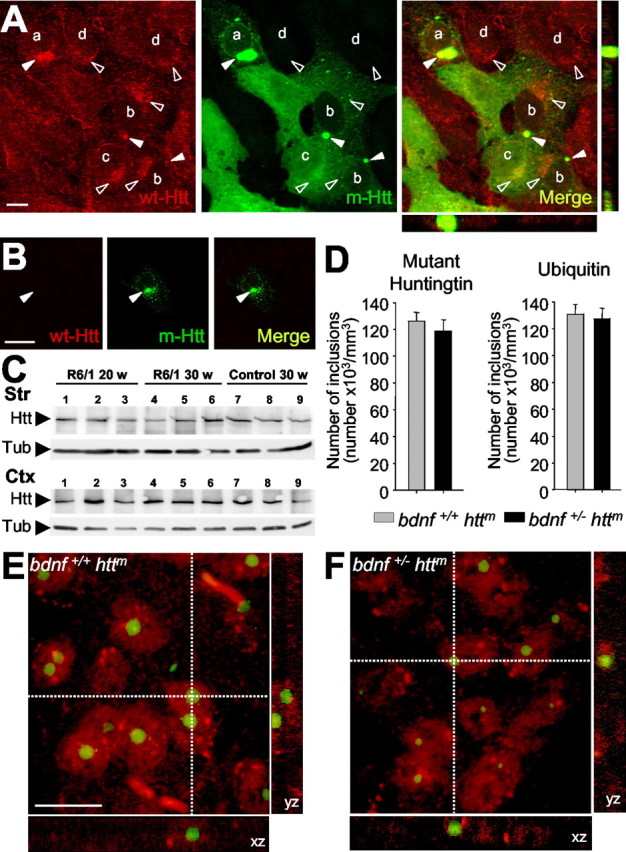
Sequestering wt Htt downregulates BDNF. A, Within nontransfected neural stem cells derived from the striatum (M213; d), wt Htt localizes in endocytic structures (open arrowheads). This localization is also detected in cells transfected with the exon 1 of htt with 72 repeats (qp72) that do not present aggregates (c) or that present small aggregates (b). However, in cells that express the qp72 construct and present large aggregates (a), wt Htt is only detected into the aggregates (filled arrowheads). We used the monoclonal antibody Mab2166 (Chemicon) to detect wt Htt (red; wt-Htt), whereas the mutant Htt was detected by the endogenous EGFP of the fusion protein (green; m-Htt). B, No signal for wt Htt was detected in the aggregates (filled arrowheads) in experiments avoiding the primary antibody. C, Western blot showing the levels of wt Htt in the cerebral cortex (Ctx) and in the striatum (Str) from R6/1 animals at 20 weeks (lanes 1-3) or at 30 weeks (lanes 4-6) or from wild-type mice at 30 weeks (lanes 7-9). Note that there are no changes in the levels of wt Htt (Htt) in R6/1. The levels of α-tubulin (Tub) are shown as a loading control. D-F, The levels of BDNF in mutant htt mice do not modify the number or morphological aspects of the intranuclear inclusions. Animals with mutant htt had intranuclear inclusions, which can be detected by mutant Htt or ubiquitin immunohistochemistry. D, The same density of aggregates was detected in both immunostaining patterns per genotype. In addition, similar results are shown in both R6/1 mice (bdnf+/+ httm) and bDM (bdnf+/ - httm), without significant differences between the two groups. E, F, Double immunohistochemistry against NeuN (red) and ubiquitin (green) demonstrated the same intranuclear location in both genotypes, containing mutant htt, with normal levels of BDNF (R6/1 mice; E) or with lower bdnf expression (bDM; F). Scale bars: A, 10 μm; B, 15 μm; (in E) E, F, 10 μm.
To test a correlation between the expression of BDNF and the levels of wt Htt in vivo, we examined the levels of the endogenous wt form in symptomatic R6/1 at 20 and 30 weeks in the cerebral cortex and the striatum (n = 3). Our results demonstrated that the levels of the protein of wt Htt were not modified in the R6/1 compared with 30-week-old wt mice (Fig. 2C).
Our results also showed that modification of BDNF levels did not alter the number and the localization of ubiquitinated aggregates (Fig. 2D-F). We counted the density of aggregates containing mutant Htt by immunohistochemistry against mutant Htt or ubiquitin in the striatum of both mutant htt genotypes, R6/1 mice and bDM at 30 weeks of age (n = 4). The number of aggregates per cubic millimeter was not modified by decreased bdnf expression [mutant Htt-positive aggregates per cubic millimeter: R6/1 (bdnf+/+ httm), 126,100 ± 6540; bDM (bdnf+/ - httm), 118,722 ± 8169] (Fig. 2D). Similarly, the ubiquitination of these aggregates showed no inter-group differences [ubiquitin-positive aggregates per cubic millimeter: R6/1 (bdnf+/+ httm), 130,751 ± 7416; bDM (bdnf+/ - httm), 127,695 ± 7600] (Fig. 2D). We also performed double immunohistochemistry to locate these inclusions. Aggregates in both genotypes had the same distribution pattern with a main intranuclear location (Fig. 2E,F). Most striatal cells had at least one intranuclear aggregate (Fig. 2E,F). Similar results were observed in the cerebral cortex (data not shown). Together, present findings suggest that BDNF regulation is downstream of the aggregate formation.
The reduction of BDNF induces advanced and more severe motor deficits in mutant htt mice
All animals that expressed mutant htt displayed a progressive neurological phenotype. However, bDM showed more severe symptoms. bDM had more tremors at rest and sudden movements that resembled chorea (supplemental material, available at www.jneurosci.org). The behavioral analyses of their motor abilities on the rotarod apparatus showed that R6/1 mice began to show significant differences from wt mice at 18 weeks at 24 and 32 rpm (Fig. 3B,C) and at 20 weeks of age at 16 rpm (Fig. 3A). However, bDM showed significant motor alterations with respect to wt or bdnf heterozygous mice from 10 weeks of age at 32 rpm (Fig. 3C) and 12 weeks at 16 and 24 rpm (Fig. 3A,B). Therefore, the onset of motor disorders advanced between 6 and 8 weeks (at 24 rpm and 16 or 32 rpm, respectively) in animals with a life span of 34 weeks (Mangiarini et al., 1996). The severity of motor dysfunction was also affected in bDM. The progression of movement deficits in bDM (bdnf+/ - httm) was more severe than in R6/1 mice (bdnf+/ + httm; p < 0.001; F-Wald test). Furthermore, the total number of falls during the entire period (30 weeks) was significantly higher in bDM (bdnf+/ - httm) than in R6/1 mice (bdnf+/+ httm) (Fig. 3D). The impairment of rotarod performance resulting from a decrease of BDNF in mutant htt mice was 154%. This effect was especially drastic in the initial period: up to 16 weeks of age, bDM showed 340% of rotarod test impairment (Fig. 3E). The progression of motor dysfunction was rapid in bDM, which were the only mice to reach the top number of falls (Fig. 3A-C). Remarkably, bdnf heterozygous mice did not show differences with respect to wt mice, indicating that the decrease of BDNF per se does not produce motor disturbances.
Figure 3.

Lower BDNF levels cause earlier onset and more severe motor dysfunctions in mutant htt mice. A-C, bDM (bdnf+/- httm) show more deficits on the rotarod than R6/1 mice (bdnf+/+ httm) tested at 16 rpm (A), 24 rpm (B), or 32 rpm (C) (p < 0.001; F-Wald test). +p < 0.05 and ++p < 0.005 compared with R6/1 mice (bdnf+/+ httm). bDM show earlier symptoms at all revolutions assayed, and the onset of motor deficits (expressed as the first significant time point with respect to control animals; A-C, arrowhead) showed advances from 6-8 weeks with respect to R6/1 mice (bdnf+/+ httm; A-C). D, In addition, bDM show more severe symptoms. The total number of accumulated falls in the period examined is significantly different between mutant htt mice with normal or low levels of BDNF. E, During early stages, the differences in the severity of motor deficits (measured as the total number of falls up to the age of 16 weeks) between R6/1 mice and bDM are very large. D, E, ***p < 0.001 compared with wt mice (bdnf+/+ httwt); +p < 0.05 and ++p < 0.005 compared with R6/1 mice (bdnf+/+ httm); ##p < 0.005 and ###p < 0.001 compared with bdnf heterozygous mice (bdnf+/ - httwt).
The instability of animals on the moving cylinder of the rotarod may be attributable to the lack of motor coordination or because of deficits in equilibrium, vision, or muscular strength. Thus, we next measured walking coordination by quantitative analysis of the footprint patterns (Fig. 4A-D). At 30 weeks of age, R6/1 mice did not show alterations in the number of steps (Fig. 4A), stride length (Fig. 4B), frontbase width (Fig. 4C), or hindbase width (Fig. 4D). However, bDM showed significant differences in all parameters studied except hindbase width (Fig. 4A-D), suggesting that the lack of coordination contributes to rotarod disturbance. We also analyzed other behavioral parameters that might affect motor dysfunction. Equilibrium was assessed for the four different genotypes (Fig. 4E). The groups did not differ in their performance on a cylindrical wire rod, although fall latency in mutant htt animals with normal or reduced BDNF levels tended to go down (Fig. 4E). Similarly, no differences in the visual cliff avoidance test or in muscular strength evaluated by the wire hang test were observed between groups (data not shown).
Figure 4.
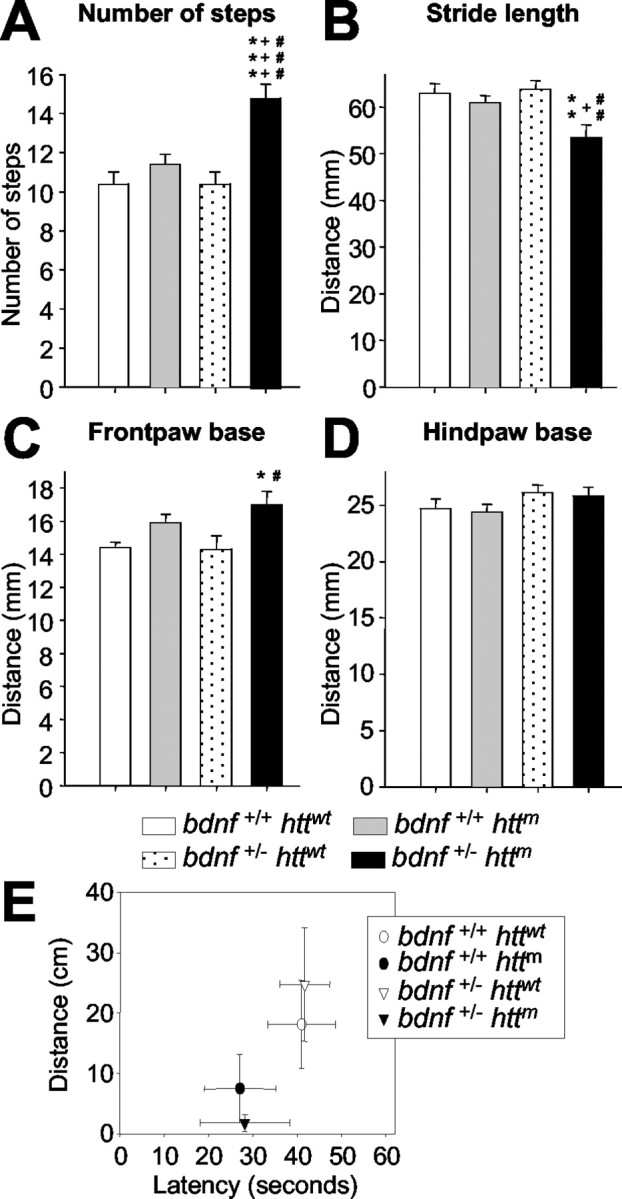
BDNF levels control motor coordination in mutant htt mice. A-D, The decrease of bdnf in mutant htt mice produces an uncoordinated walking footprint pattern. We analyzed the following four parameters of footprints: A, the number of steps; B, the distance between two consecutive steps; C, D, the distance between right and left frontbase and hindbase path, respectively. bDM shows differences in the number and distance of steps (A, B) and in the frontbase width (C). No differences were found in the hindbase pattern (D). *p < 0.05, **p < 0.005, and ***p < 0.001 compared with wt mice (bdnf+/+ httwt); +p < 0.05 and +++p < 0.001 compared with R6/1 mice (bdnf+/+ httm); #p < 0.05, ##p < 0.005, and ###p < 0.001 compared with bdnfheterozygous mice (bdnf+/ - httwt). E, The levels of BDNF do not affect the mouse's ability to walk on a wire rod. Although we did not find statistical differences between groups, animals with mutant htt and with normal or lower levels of BDNF had a higher tendency to fall off the rod. The distance covered by the animals on the rod also shows the same effect, with shorter runs in those animals expressing mutant htt.
We could not perform motor behavior analysis in n3DM because all nt-3 heterozygous mice were unable to perform the tasks. Motor deficits in these mice have been described previously (Ernfors et al., 1994b).
Neuronal cell loss in the striatum of bDM
Although all animals with mutant htt had less brain volume, modification of bdnf alleles did not affect these changes (n = 4-7; brain volume: bdnf+/+ httwt, 100 ± 8.83%; bdnf+/+ httm, 81.75 ± 6.80%; bdnf+/ - httwt, 97.29 ± 12.82%; bdnf+/ - httm, 82.43 ± 13.81% with respect to wt). Similarly, the levels of bdnf expression did not affect the volume of the cortex, which was less in both groups with mutant htt (Fig. 5A,B). In contrast, degeneration of the striatum was observed in the bDM (Fig. 5A,C). These animals showed a volume of 75.43 ± 1.12% with respect to wt animals (100 ± 5.17%), which was significantly lower than the volume in R6/1 striata (85.32 ± 0.68% with respect to wt). In addition, a decrease in BDNF levels caused neuronal loss in the striatum of mutant htt transgenic mice. The estimated number of total neurons counted by cresyl violet was down to 85.64 ± 1.29% in bDM with respect to wt mice (Fig. 5D). Similarly, the number of NeuN-positive cells was 79.60 ± 2.35% in bDM with respect to wt mice (Fig. 5E). No differences were detected in the other genotypes, either in cresyl violet or in NeuN-positive counts (cresyl violet: bdnf+/+ httwt, 100 ± 0.87%; bdnf+/+ httm, 96.93 ± 3.72%; bdnf+/ - httwt, 97.44 ± 5.54% with respect to wt; NeuN-positive cells: bdnf+/+ httwt, 100 ± 7.04%; bdnf+/+ httm, 98.31 ± 4.01%; bdnf+/ - httwt, 95.80 ± 8.90% with respect to wt).
Figure 5.
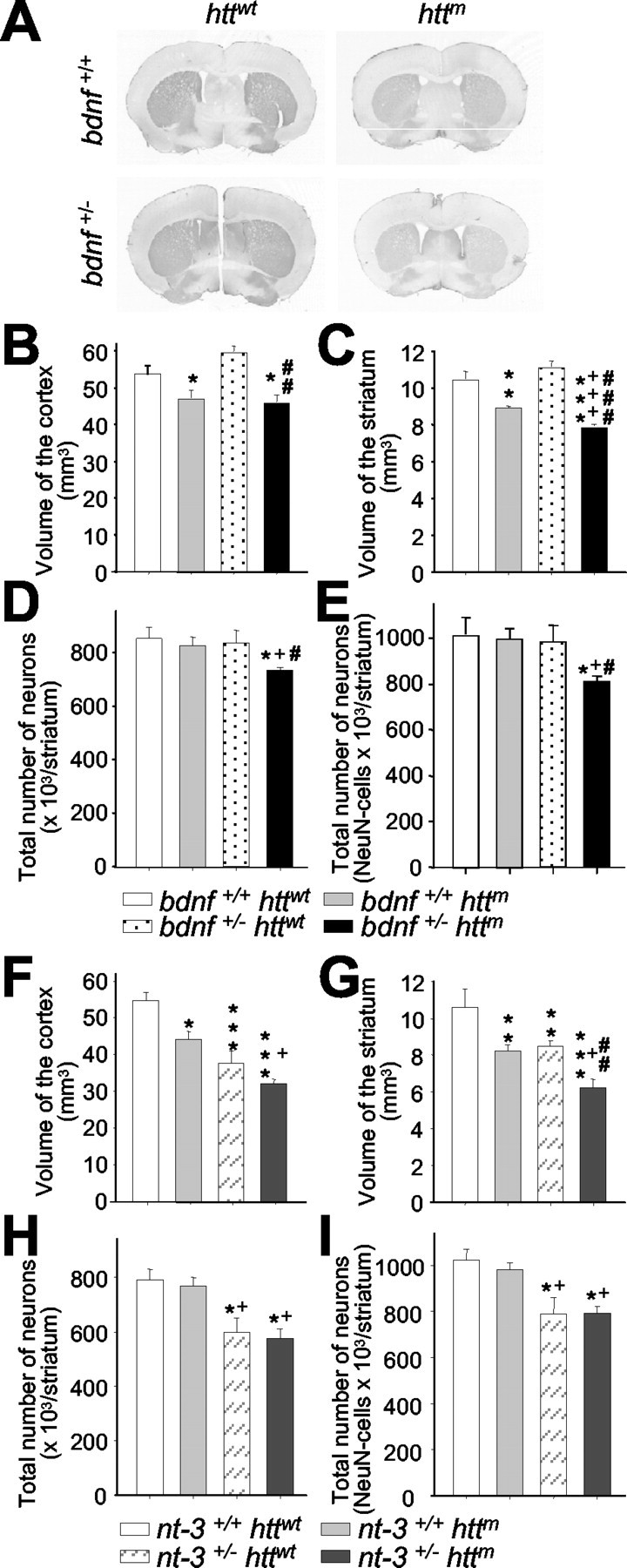
Reduction of BDNF levels in mutant htt mice produces neuronal loss in the striatum. A, Representative sections of DARPP-32 immunohistochemistry on wt mice (bdnf+/+ httwt), R6/1 mice (bdnf+/+ httm), bdnf heterozygous mice (bdnf+/ - httwt), and bDM (bdnf+/ - httm). B, C, We used DARPP-32 immunostaining to measure the volume of the cerebral cortex (B) and the striatum (C). B, The cerebral cortex shows the same atrophy in both genotypes with mutant htt. C, In contrast, the striatal volume is significantly lower in bDM with respect to R6/1 mice, indicating that the insufficient levels of this neurotrophin produce more severe striatal degeneration. D, E, At 30 weeks, bDM have striatal neuronal loss, counted by neuronal morphology in cresyl violet staining (D) or by NeuN immunohistochemistry (E). No decrease in neurons was detected in other genotypes. F, G, In n3DM (nt-3+/- httm), the effects observed result from the addition of striatal and cortical reduction observed in individual genotypes, mutant htt and nt-3+/-. H, I, No differences in cell loss were observed between nt-3 heterozygous with or without mutant htt. Both genotypes (nt-3+/- httwt and nt-3+/- httm) present a ∼25% reduction. *p < 0.05, **p < 0.005, and ***p < 0.001 compared with wt mice (bdnf+/+ httwt or nt-3+/+ httwt); +p < 0.05 and +++p < 0.001 compared with bdnf+/+ httm or nt-3+/+ httm mice; #p < 0.05, ##p < 0.005, and ###p < 0.001 compared with bdnf or nt-3 heterozygous mice, respectively.
To demonstrate the specific effect of BDNF, we generated a n3DM line in which transgenic mice present different levels of NT-3, another neurotrophin with protective effects on striatal neurons (Perez-Navarro et al., 2000). The analyses of n3DM revealed a decrease in the volume of both cortex and striatum (Fig. 5F,G) [cerebral cortex: nt-3+/ - httm, 59.02 ± 2.13% with respect to wt (nt-3+/+ httwt, 100 ± 4.18%); striatum: nt-3+/ - httm, 62.30 ± 4.50% with respect to wt (nt-3+/+ httwt, 100 ± 8.26%)]. However, this decrease in volume results from the addition of the independent effects observed in R6/1 mice and nt-3 heterozygous mice (Fig. 5F,G) [cerebral cortex: R6/1 mice (nt-3+/+ httm), 82.57 ± 4.59%; nt-3+/ - httwt, 68.90 ± 9.4% with respect to wt; striatum: R6/1 mice (nt-3+/+ httm), 78.52 ± 1.84%; nt-3+/ - httwt, 81.38 ± 2.68% with respect to wt]. Therefore, the effect of mutant htt is similar in nt-3 heterozygous mice (nt-3+/ - httm with respect to nt-3+/ - httwt: cortex, 85.65 ± 3.1%; striatum, 76.55 ± 5.5%) and in mice with normal NT-3 levels (nt-3+/+ httm with respect to wt: cortex, 82.57 ± 4.59%; striatum, 78.52 ± 1.8%). The effect of this neurotrophin on striatal neurons is similar in both NT-3 heterozygous genotypes in which the number of neurons is reduced ∼25% (Fig. 5H,I) (cresyl violet: nt-3+/+ httwt, 100 ± 4.98%; nt-3+/+ httm, 97.34 ± 3.76%; nt-3+/ - httwt, 75.59 ± 6.79%; nt-3+/ - httm, 72.80 ± 4.45% with respect to wt; NeuN-positive cells: nt-3+/+ httwt, 100 ± 4.69%; nt-3+/+ httm, 96.68 ± 3.46%; nt-3+/ - httwt, 77.03 ± 3.50%; nt-3+/ - httm, 77.76 ± 2.70% with respect to wt).
We next analyzed whether neuronal loss in the striatum is because of a deficit during development or progressive degeneration. bDM showed no neuronal alterations in the striatum at postnatal day 15 (n = 4-6 per genotype). The striatal volume of bDM was 104 ± 4.18% with respect to wt, and the number of NeuN-positive neurons was 109.93 ± 5.01% with respect to wt. In addition, we analyzed the striatal volume and neurons of animals that had complete knock-out of the bdnf gene. These animals showed a striatal volume of 98.87 ± 3.53%, and the number of NeuN-positive neurons was 105.49 ± 6.06% with respect to wt.
Specific degeneration of enkephalin striatal subpopulation is controlled by BDNF
One of the pathological characteristics of HD is its specific pattern of degeneration. Here, we show that the number of medium-sized neurons immunostained with DARPP-32 was reduced in R6/1 mice (59 ± 2% with respect to wt; 100 ± 9%) (Fig. 6A,B). However, DARPP-32-positive neurons were fewer in bDM (30 ± 16% with respect to wt) (Fig. 6A,B). We calculated the effect attributed to lower BDNF levels in animals with mutant htt by standardizing values for the R6/1 mice, showing a reduction of 50 ± 13% in bDM with respect to R6/1 mice. No differences in the number of DARPP-32-positive neurons were observed in bdnf heterozygous mice (87 ± 14% with respect to wt) (Fig. 6A,B).
Figure 6.
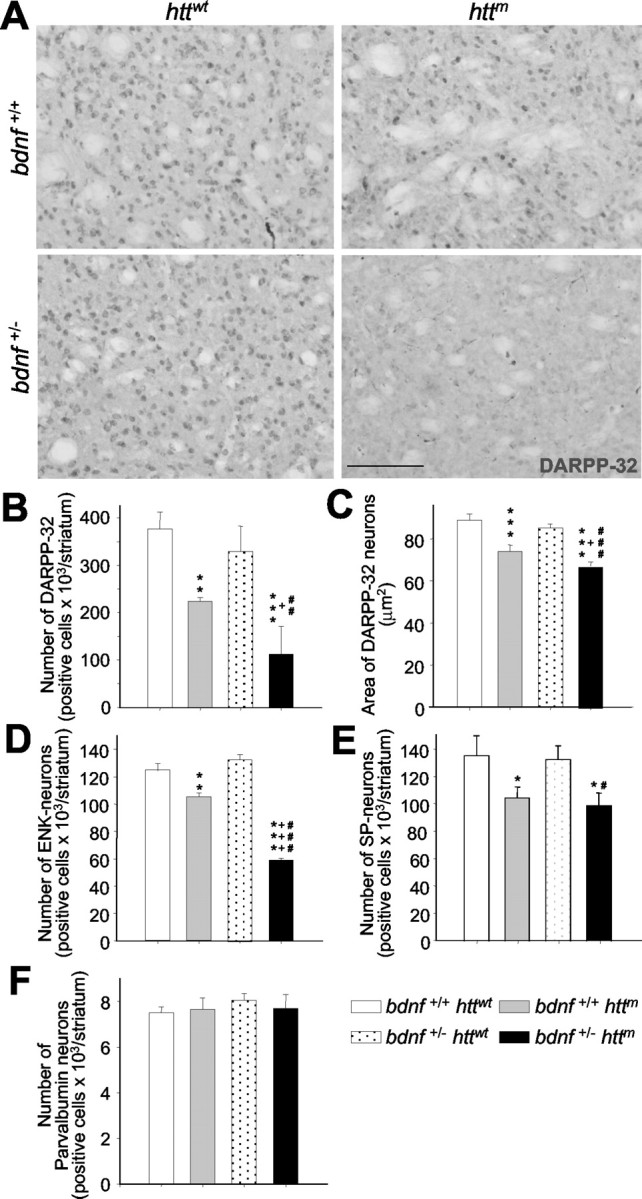
The striatal cells lost in bDM are enkephalin-positive neurons. A-C, At 30 weeks of age, bDM (bdnf+/- httm) had great atrophy of medium-sized striatal projection neurons, as demonstrated by DARPP-32 immunostaining. The number of DARPP-32-positive neurons decreased in all mutant htt mice (B). However, the lowest number of DARPP-32-positive neurons was detected in bDM. C, The cross-sectional area of DARPP-32 neurons decreased in all animals that express mutant htt, although the area is significantly smaller in bDM.D, E,The two neuronal subpopulations of medium-sized spiny neurons in the striatum were characterized by insitu hybridization. D, The number of enkephalin-positive neurons (ENK) was dramatically less in bDM. The number of ENK-positive neurons was also less in R6/1 mice (bdnf+/+ httm) than in wt mice but only by a small amount. E, The differences between the two mutant htt groups are statistically significant. In contrast, the decrease in substance P-positive neurons (SP) is similar in R6/1 mice (bdnf+/+ httm) and bDM (bdnf+/- httm),without significant differences. F, We also analyzed the number of striatal interneurons by parvalbumin immunohistochemistry. No differences were found between the four genotypes analyzed. *p < 0.05, **p < 0.005, and ***p < 0.001 compared with wt mice (bdnf+/+ httwt); +p<0.05 and +++p<0.001 compared with R6/1 mice (bdnf+/+ httm);#p<0.05,##p<0.005,and###p<0.001 compared with bdnf heterozygous mice (bdnf+/- httwt). Scale bar, 100 μm.
The cross-sectional area of DARPP-32-positive neurons showed a small reduction in R6/1 mice (bdnf+/+ httm, 85 ± 3% with respect to wt; 100 ± 3%) (Fig. 6C). However, in bDM, medium-sized cells were more atrophied with a significantly lower soma area (72 ± 1.9% with respect to wt) (Fig. 6C). In contrast, we did not find loss of parvalbumin-positive interneurons in any genotype analyzed (bdnf+/+ httwt, 100 ± 3.2%; bdnf+/+ httm, 95.6 ± 9.2%; bdnf+/ - httwt, 107.29 ± 4.0%; bdnf+/ - httm, 102.6 ± 7.9% with respect to wt) (Fig. 6 F).
We next characterized the subpopulations of striatal projection neurons by in situ hybridization (Fig. 6D,E). In bDM, the number of enkephalin-positive neurons drastically fell to 47.20 ± 0.99% with respect to wt (100 ± 4.39%), whereas in R6/1, the number of enkephalin-positive neurons decreased to only 84.32 ± 2.10% with respect to wt (Fig. 6D). In contrast, the number of substance P-positive neurons in the two genotypes expressing mutant htt was not significantly different (bdnf+/+ httwt, 100 ± 10.75%; bdnf+/+ httm, 77.10 ± 7.02%; bdnf+/- httwt, 97.91 ± 7.41%; bdnf+/- httm, 73.04 ± 6.83% with respect to wt) (Fig. 6E).
To further study the specific effect of BDNF on enkephalinergic neurons, we administered 4.5 μg of BDNF per day into the striatum of 20-week-old R6/1 mice during 1 week (n = 5) (Fig. 7). This treatment enhanced the number of enkephalin-positive neurons to 60.28 ± 24.07% above the sham-R6/1 mice (Fig. 7A). We also administered BDNF to 14-week-old bDM in which exogenous BDNF increased the number of enkephalinergic neurons 127.09 ± 31.42% above sham-bDM (Fig. 7A,B). However, BDNF treatment did not alter the number of substance P-positive neurons in R6/1 mice (12.86 ± 15.25% above sham) or in bDM (19.18 ± 10.34% above sham) (Fig. 7C,D). In addition, preliminary behavioral analysis showed that bDM, which received unilateral administration of BDNF, showed asymmetric rotations induced by amphetamine (5 mg/kg). These mice showed 65.00 ± 20.57 contralateral net rotations, whereas sham-bDM showed -40.00 ± 23.18 ipsilateral net rotations (p = 0.0137). Thus, present results suggest that BDNF restores some striatal functions related to the increase in enkephalin expression.
Figure 7.
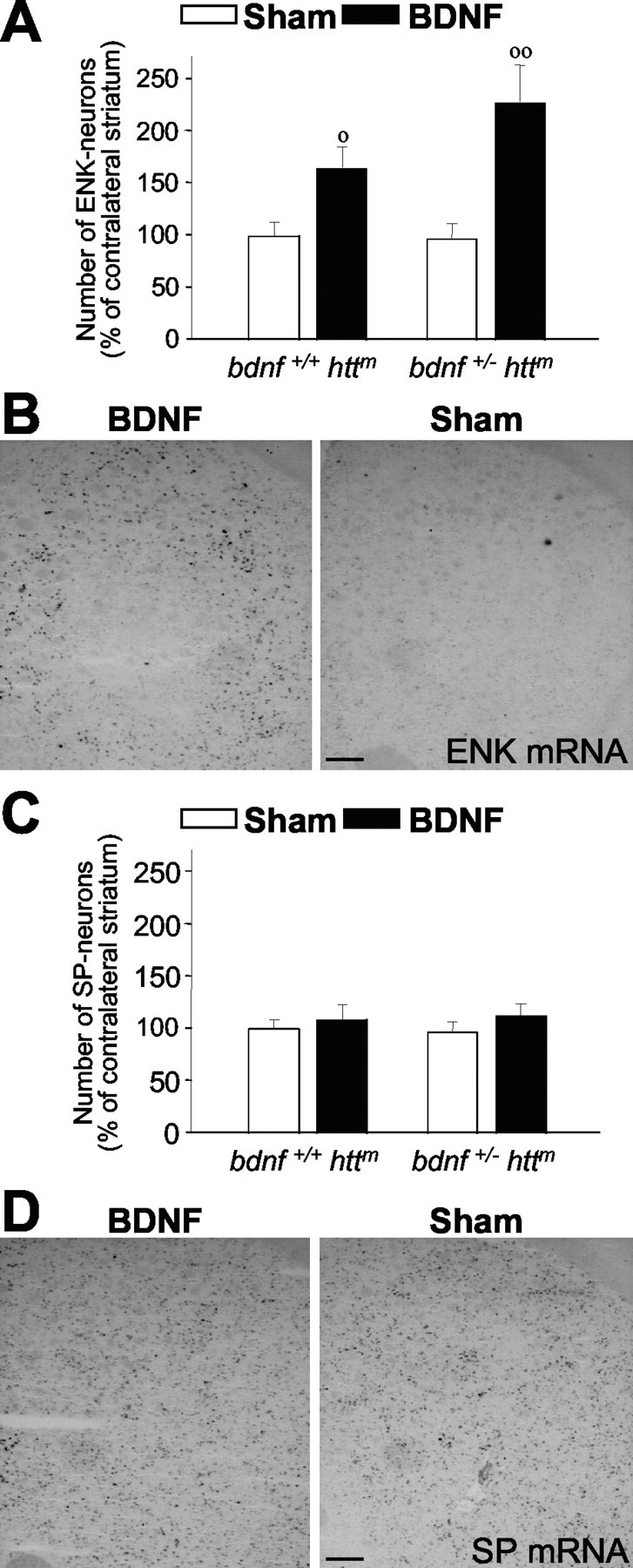
Exogenous BDNF restores the enkephalin loss in mutant htt mice. A, B, Continuous intrastriatal administration of BDNF (4.5 μg per day) during 1 week produces an increase in the number of enkephal in-positive cells (ENK). This effect may be caused by the enhancement in the enkephalin mRNA levels in both R6/1 mice (bdnf+/+ httm) and bDM (bdnf+/ - httm). C, D, However, intrastriatal administration of BDNF in the same animals does not modify the levels of substance P-positive neurons (SP). No differences were observed in PBS-infused animals (sham) with respect to untreated animals, either in enkephalin- or in substance P-positive neurons (data not shown). B, D, Representative photomicrographs of in situ hybridization for enkephalin (ENK) and substance P (SP) in bDM injected with BDNF or PBS (Sham). Note that after BDNF treatment, the enkephalin-positive neurons were detected around the injection site but not in the injection core (B). ○p < 0.05 and ○○p < 0.005 compared with sham-operated mice (Student's t test). Scale bars: B, D, 100 μm.
Discussion
In the present study, we disrupted the expression of bdnf in an HD mouse model by cross-mating bdnf+/- (Ernfors et al., 1994b) and R6/1 mice (Mangiarini et al., 1996). The characterization of these mice demonstrates that endogenous BDNF regulates the onset and the severity of motor dysfunction. These effects are caused by selective degeneration of enkephalin-positive neurons in the striatum, which can be restored by administration of exogenous BDNF.
It has been postulated that mutant Htt may act as a dominant-negative over wt protein by capturing it into aggregates (Dyer and McMurray, 2001). This effect could result in the loss of wt Htt function in NRSF inhibition (Zuccato et al., 2003), causing BDNF alterations. We observed that wt Htt colocalizes with mutant Htt into the aggregates in cells that express mutant htt gene, supporting the idea of a dominant-negative role for mutant Htt in cell cultures. In addition, a decrease in mutant Htt-mediated BDNF depends not only on the number of CAG repeats but also on its levels of expression, because low amounts of mutant Htt did not produce a decrease in BDNF levels. These data may explain the differences observed in distinct mutant htt transgenic mouse models. In fact, our findings showed that the transgenic R6/1 mice, which express only one copy of the mutant 5′-end of the human htt gene carrying (CAG)115 (Mangiarini et al., 1996), have normal levels of BDNF, which correlates with the normal levels of wt Htt detected in these mice. However, R6/2 mice, which express the transgene three times more than R6/1 and carry 150 CAG repeats (Mangiarini et al., 1996), have decreased levels of free wt Htt, which reduces BDNF concentrations (Zhang et al., 2003). Within knock-in mouse lines, a dose-dependent Htt-mediated BDNF decrease has also been established (Zuccato et al., 2001). Our results also demonstrate that decreasing the levels of BDNF in R6/1 mice does not alter the number or localization of intranuclear aggregates in vivo. Similar results have been described previously in cell culture (Saudou et al., 1998). Together, all of these findings suggest that the number of CAG repeats and its levels of expression modify the amount of wt Htt available to retain the NRSF and consequently produce changes in bdnf expression.
Besides this interaction between the expression of mutant htt and BDNF, no evidence has correlated the decrease in BDNF with the HD neuropathology. Our results show that the decrease in BDNF levels produced more severe symptoms in an HD model. bDM showed an early onset of motor dysfunction with significant motor alterations 6-8 weeks before their littermates, R6/1 mice. In addition, the progression of movement deficits in bDM was faster and more severe than in R6/1. We also found motor disorders in the analysis of footprint patterns, which demonstrated that only bDM have deficits in walking coordination. Note that although bdnf knock-outs have alterations of sensory neurons (Ernfors et al., 1994a; Jones et al., 1994), bdnf+/- mice with wt htt or mutant htt did not show deficits in the equilibrium, visual test, or muscular strength. In addition, it has been shown recently that the conditional BDNF-null mutant mice, which lack this neurotrophin in the striatum, do not have motor alterations (Baquet et al., 2004). These findings demonstrate that BDNF affects brain nuclei that control motor coordination only in the presence of mutant htt.
We next examined the neuronal populations that are affected by BDNF in HD. The lower BDNF levels of bDM reduced striatal volume without affecting the cerebral cortex. Similarly, neuronal loss in the cerebral cortex has not been observed in transgenic mutant htt mouse models (Mangiarini et al., 1996; Hodgson et al., 1999; Shelbourne et al., 1999; Yamamoto et al., 2000; Slow et al., 2003). These results indicate that downregulation of cortical bdnf mRNA found in animal models and patients with HD (Zuccato et al., 2001) primarily affects striatal neurons but not cortical structures. In fact, it has been described that BDNF protein only decreases in the striatum of HD patients and not in the cerebral cortex (Ferrer et al., 2000). In support of this view, it has been shown that cortical BDNF is transported anterogradely to the striatum (Altar et al., 1997), and it can protect striatal neurons from excitotoxicity (Martinez-Serrano and Bjorklund, 1996; Perez-Navarro et al., 1999, 2000; Canals et al., 2001; Alberch et al., 2002). Therefore, a decrease in the expression of cortical bdnf participates in selective striatal damage.
HD advances to higher grades (1-4) when specific striatal cell death occurs (Vonsattel et al., 1985). We show that the specific decrease of striatal volume in bDM is caused by a loss of striatal neurons, which was only observed in our bDM model at 30 weeks. These findings are consistent with previous reports that show that R6/1 (Mangiarini et al., 1996) and bdnf+/- mice (Jones et al., 1994; Ivkovic et al., 1997) have the same number of striatal neurons as wt mice. Thus, the lack of BDNF per se does not induce degeneration of striatal neurons, but, together with the presence of mutant htt, it produces striatal cell loss. This neuronal loss is not attributable to a deficit during development, because we did not observe alterations of the number of striatal neurons in 15-d-old bDM. In addition, the effect of BDNF on striatal neurons of HD mice is specific because no differences were detected in nt-3 heterozygous mice with or without mutant htt. Our results are in agreement with the decrease of BDNF, but not NT-3, in the transgenic yeast artificial chromosome models (Zuccato et al., 2001), which present loss of striatal neurons (Hodgson et al., 1999). Similarly, reduced levels of BDNF are also detected in R6/2 (Zhang et al., 2003), which show a rapid disease progression and striatal degeneration (Menalled and Chesselet, 2002). It has been demonstrated that mutant htt causes context-dependent neurodegeneration mediated by different N-terminal Htt fragments in transgenic mice (Yu et al., 2003), similar to what we observed for the regulation of BDNF expression.
Motor deficits in R6/1 animals are attributed to neuronal dysfunction of DARPP-32-positive neurons rather than to cell death, because no loss of striatal neurons was observed in these mice. DARPP-32 is downregulated in R6/1 mice, whereas calbindin immunolabeling, another marker for medium-sized projection neurons, is not modified (van Dellen et al., 2000). However, the drastic decrease in DARPP-32-positive cells observed in bDM is attributed to downregulation of the cellular marker and neuronal death, because this population represents 95% of striatal neurons. bDM showed a 50% reduction in DARPP-32-positive neurons without any change in the number of parvalbumin-positive interneurons. These striatal DARPP-32-positive neurons project to the globus pallidus and substantia nigra via two separate subpopulations, enkephalin- and substance P-positive neurons, respectively, which have different sensitivity to mutant htt. The analysis of each subpopulation demonstrated that the number of enkephalin-positive neurons fell drastically in bDM. In contrast, the levels of BDNF did not affect the number of substance P-positive neurons. These results are coincident with previous studies with R6/2 mice, which present a decrease of BDNF levels (Zhang et al., 2003) and a 50% reduction of enkephalin mRNA but not of the levels of substance P expression (Bibb et al., 2000).
In view of our results, which show that the deficit of endogenous BDNF specifically regulates the levels of enkephalin in HD, we next examined the effect of administration of this neurotrophin in mutant htt transgenic mice. The intrastriatal treatment with BDNF produced a reversion of the loss of the enkephalinergic phenotype in R6/1 and in bDM. This increase in the number of enkephalinergic neurons is attributable to the recovery of mRNA expression because only 20% of neurons are lost in bDM, and no cell death is observed in R6/1 mice. These findings suggest that atrophic or dysfunctional striatal neurons are recovered by BDNF administration. Consistent with these results is the observation that 1 week administration of BDNF in vivo leads to overall increased expression of enkephalin as well as an increase in the number of striatal cells expressing enkephalin (Perez-Navarro et al., 1999). In addition, our results from the circling behavior induced by amphetamine administration suggest that increasing enkephalin expression via BDNF administration may have functional implication in HD mouse models. However, additional experiments are required to improve the dose and the administration strategies to perform long-term bilateral treatments.
In conclusion, our results demonstrate that BDNF plays a pivotal role in the pathophysiology of HD. Although BDNF is not an inductor of HD and the initial steps of this disorder may occur without BDNF alterations, we show that BDNF regulates the age of onset and the severity of motor dysfunctions through the control of the survival of striatal enkephalinergic neurons. In addition, BDNF treatment restores the enkephalin levels in striatal projection neurons, one of the most affected populations in HD. Therefore, administration of BDNF or increasing endogenous levels in HD patients may stop or delay illness progression.
Footnotes
This study was supported by Ministerio de Ciencia y Tecnología Grants SAF2002-00314 (J.A.), SAF2002-00311 (J.M.C.), and SAF1999-0123 (G.M.); Redes Temáticas de Investigación Coorporativa (G03/167, G03/210; Ministerio de Sanidad y Consumo); Fundació La Caixa; and Fundación Ramón Areces. J.R.P. is a fellow of the Ministerio de Educación, Cultura y Deporte, J.F.T.-P. is a fellow of the Fundación Gran Mariscal de Ayacucho, M.B. is a fellow of the Universitat de Barcelona, and R.M.-I. is a fellow of the Ministerio de Ciencia y Tecnologia. We thank Drs. Lydia Giménez-Llort and Mara Dierssen for their help with behavior design and analyses and for critically reading this manuscript. We are grateful to Sonia Marco, Ana López, and Francesca Calderón for technical assistance, Drs. Jordi Petriz and Isabel Sanchez (Cell Separation Unit, Institut d'Investigacions Biomèdiques August Pi i Sunyer) for their support and advice in cell sorting technique, Anna Bosch (Serveis Científico-Tècnics, Universitat de Barcelona) for her support and advice in the use of confocal microscopy, Dr. Josep Lluis Carrasco (Departament de Salut Pública, Universitat de Barcelona) for statistical analyses, and Dr. Emili Martínez for the behavior set up. We are also grateful to Dr. Amèrica Jiménez and the staff of the animal facility (Facultat de Medicina, Universitat de Barcelona) for their help with mouse care. We thank Robin Rycroft for checking the English.
Correspondence should be addressed to Dr. Jordi Alberch, Departament de Biologia Cel·lular i Anatomia Patològica, Facultat de Medicina, Institut d'Investigacions Biomèdiques August Pi i Sunyer, Universitat de Barcelona, C/Casanova, 143, E-08036 Barcelona, Spain. E-mail: alberch@ub.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247727-13$15.00/0
References
- Agerman K, Hjerling-Leffler J, Blanchard MP, Scarfone E, Canlon B, Nosrat C, Ernfors P (2003) BDNF gene replacement reveals multiple mechanisms for establishing neurotrophin specificity during sensory nervous system development. Development 130: 1479-1491. [DOI] [PubMed] [Google Scholar]
- Alberch J, Perez-Navarro E, Canals JM (2002) Neuroprotection by neurotrophins and GDNF family members in the excitotoxic model of Huntington's disease. Brain Res Bull 57: 817-822. [DOI] [PubMed] [Google Scholar]
- Alberch J, Perez-Navarro E, Canals JM (2004) Neurotrophic factors in Huntington's disease. Prog Brain Res 146: 195-229. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ (1997) Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389: 856-860. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci 24: 4250-4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb JA, Yan Z, Svenningsson P, Snyder GL, Pieribone VA, Horiuchi A, Nairn AC, Messer A, Greengard P (2000) Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc Natl Acad Sci USA 97: 6809-6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals JM, Checa N, Marco S, Akerud P, Michels A, Perez-Navarro E, Tolosa E, Arenas E, Alberch J (2001) Expression of brain-derived neurotrophic factor in cortical neurons is regulated by striatal target area. J Neurosci 21: 117-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci 19: 3248-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP (2003) Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci USA 100: 2911-2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer RB, McMurray CT (2001) Mutant protein in Huntington disease is resistant to proteolysis in affected brain. Nat Genet 29: 270-278. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R (1994a) Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368: 147-150. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Kucera J, Jaenisch R (1994b) Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell 77: 503-512. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF (2002) Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci 22: 1592-1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutan E, Marin C, Rey MJ, Ribalta T (2000) Brain-derived neurotrophic factor in Huntington disease. Brain Res 866: 257-261. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1992) The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci 15: 133-139. [DOI] [PubMed] [Google Scholar]
- Gimenez-Llort L, Fernandez-Teruel A, Escorihuela RM, Fredholm BB, Tobena A, Pekny M, Johansson B (2002) Mice lacking the adenosine A1 receptor are anxious and aggressive, but are normal learners with reduced muscle strength and survival rate. Eur J Neurosci 16: 547-550. [DOI] [PubMed] [Google Scholar]
- Giordano M, Takashima H, Herranz A, Poltorak M, Geller HM, Marone M, Freed WJ (1993) Immortalized GABAergic cell lines derived from rat striatum using a temperature-sensitive allele of the SV40 large T antigen. Exp Neurol 124: 395-400. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Chesselet MF (2003) The use of transgenic and knock-in mice to study Huntington's disease. Cytogenet Genome Res 100: 276-286. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR (1999) A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 23: 181-192. [DOI] [PubMed] [Google Scholar]
- Ibanez CF, Hallbook F, Godeau F, Persson H (1992) Expression of neurotrophin-4 mRNA during oogenesis in Xenopus laevis Int J Dev Biol 36: 239-245. [PubMed] [Google Scholar]
- Ivkovic S, Polonskaia O, Farinas I, Ehrlich ME (1997) Brain-derived neurotrophic factor regulates maturation of the DARPP-32 phenotype in striatal medium spiny neurons: studies in vivo and in vitro Neuroscience 79: 509-516. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF (1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76: 989-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404-11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M (2000) Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci 20: 7268-7278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry M, Holmberg K, Zhang X, Hokfelt T (2000) Effect of axotomy on expression of NPY, galanin, and NPY Y1 and Y2 receptors in dorsal root ganglia and the superior cervical ganglion studied with double-labeling in situ hybridization and immunohistochemistry. Exp Neurol 162: 361-384. [DOI] [PubMed] [Google Scholar]
- Laurenzi MA, Barbany G, Timmusk T, Lindgren JA, Persson H (1994) Expression of mRNA encoding neurotrophins and neurotrophin receptors in rat thymus, spleen tissue and immunocompetent cells. Regulation of neurotrophin-4 mRNA expression by mitogens and leukotriene B4. Eur J Biochem 223: 733-741. [DOI] [PubMed] [Google Scholar]
- Lindefors N, Brodin E, Metsis M (1995) Spatiotemporal selective effects on brain-derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neuroscience 65: 661-670. [DOI] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB (1999) Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci 19: 10428-10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493-506. [DOI] [PubMed] [Google Scholar]
- Martinez-Serrano A, Bjorklund A (1996) Protection of the neostriatum against excitotoxic damage by neurotrophin-producing, genetically modified neural stem cells. J Neurosci 16: 4604-4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Chesselet MF (2002) Mouse models of Huntington's disease. Trends Pharmacol Sci 23: 32-39. [DOI] [PubMed] [Google Scholar]
- Perez-Navarro E, Alberch J, Neveu I, Arenas E (1999) Brain-derived neurotrophic factor, neurotrophin-3 and neurotrophin-4/5 differentially regulate the phenotype and prevent degenerative changes in striatal projection neurons after excitotoxicity in vivo Neuroscience 91: 1257-1264. [DOI] [PubMed] [Google Scholar]
- Perez-Navarro E, Canudas AM, Akerund P, Alberch J, Arenas E (2000) Brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 prevent the death of striatal projection neurons in a rodent model of Huntington's disease. J Neurochem 75: 2190-2199. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB (1988) Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85: 5733-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P (1995) Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington's disease patients. Ann Neurol 38: 852-861. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC (2002) Lessons from animal models of Huntington's disease. Trends Genet 18: 202-209. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55-66. [DOI] [PubMed] [Google Scholar]
- Schmitz GG, Walter T, Seibl R, Kessler C (1991) Nonradioactive labeling of oligonucleotides in vitro with the hapten digoxigenin by tailing with terminal transferase. Anal Biochem 192: 222-231. [DOI] [PubMed] [Google Scholar]
- Serrats J, Artigas F, Mengod G, Cortes R (2003) GABAB receptor mRNA in the raphe nuclei: co-expression with serotonin transporter and glutamic acid decarboxylase. J Neurochem 84: 743-752. [DOI] [PubMed] [Google Scholar]
- Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RM (1999) A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet 8: 763-774. [DOI] [PubMed] [Google Scholar]
- Sieradzan KA, Mann DM (2001) The selective vulnerability of nerve cells in Huntington's disease. Neuropathol Appl Neurobiol 27: 1-21. [DOI] [PubMed] [Google Scholar]
- Slow EJ, Van Raamsdonk J, Rogers D, Coleman SH, Graham RK, Deng Y, Oh R, Bissada N, Hossain SM, Yang YZ, Li XJ, Simpson EM, Gutekunst CA, Leavitt BR, Hayden MR (2003) Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet 12: 1555-1567. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC (2003) Transcriptional abnormalities in Huntington disease. Trends Genet 19: 233-238. [DOI] [PubMed] [Google Scholar]
- van Dellen A, Welch J, Dixon RM, Cordery P, York D, Styles P, Blakemore C, Hannan AJ (2000) N-Acetylaspartate and DARPP-32 levels decrease in the corpus striatum of Huntington's disease mice. NeuroReport 11: 3751-3757. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson Jr EP (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559-577. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57-66. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ (2003) Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci 23: 2193-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li M, Drozda M, Chen M, Ren S, Mejia Sanchez RO, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM (2003) Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem 87: 101-106. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493-498. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35: 76-83. [DOI] [PubMed] [Google Scholar]