

Abstract
Neuroadapted Sindbis virus (NSV) is a neurotropic virus capable of inducing the death of spinal motor neurons in mice and rats. In this study we investigated the mechanisms that underlie NSV-induced motor neuron death. We found that many degenerating spinal motor neurons were not infected directly with NSV, suggesting that bystander cell death occurs. An excitotoxic mechanism was confirmed when blockade of calcium-permeable AMPA receptors attenuated motor neuron death both in vitro and in vivo. Blockade of astroglial glutamate reuptake potentiated NSV-induced motor neuron loss in vivo, suggesting that astrocyte-mediated removal of perisynaptic glutamate is important in limiting NSV-induced excitotoxic injury. Astroglial glutamate transport was reduced markedly in the spinal cord during NSV infection, in advance of motor neuron injury in susceptible mice. In contrast, we found 5.6-fold elevated glutamate uptake in the spinal cords of mice resistant to NSV-induced paralysis. Likewise, minocycline markedly increased spinal cord glutamate transport and protected mice from NSV-induced motor neuron death. These studies suggest that NSV infection triggers a cascade of events in the spinal cord resulting in impaired astrocytic glutamate transport and excitotoxic injury of motor neurons mediated via calciumpermeable AMPA receptors. Similar changes may occur in other motor neuron disorders such as amyotrophic lateral sclerosis or West Nile Virus-induced poliomyelitis, suggesting a common tissue injury pathway.
Keywords: encephalomyelitis, motor neuron, paralysis, spinal, transport, virus, glutamate
Introduction
Excess glutamate in the motor neuron synapse produces hyper-excitation of both NMDA and AMPA-type glutamate receptors. Motor neurons are particularly sensitive to AMPA-mediated glutamate toxicity (Rothstein et al., 1993; Carriedo et al., 2000). Some studies suggest that AMPA receptors present on motor neurons mainly exclude the GluR2 subunit, thus rendering them more Ca2+-permeable (Van den Bosch and Robberecht, 2000; Krampfl et al., 2001; Van den Bosch et al., 2002a). Insufficient clearance of glutamate by astrocytes via their excitatory amino acid transporters contributes to increased susceptibility of postsynaptic neurons even to normal levels of glutamate (Rothstein et al., 1993, 1996; Ginsberg et al., 1995, 1996; Rao et al., 2001b; Heath and Shaw, 2002; Howland et al., 2002; Dunlop et al., 2003).
Glutamate transporter-1 (GLT-1) is the dominant glutamate transporter in most regions of the CNS, accounting for up to 95% of glutamate transport (Rothstein et al., 1996; Tanaka et al., 1997). The primary role of astroglial glutamate transporters is to maintain a homeostatic level of extracellular glutamate (<1 μm) by binding and reabsorbing glutamate from the synaptic cleft (Ventura and Harris, 1999; Anderson and Swanson, 2000; Chretien et al., 2002; Heath and Shaw, 2002; Nedergaard et al., 2002). In several disease states decreased astrocyte-mediated glutamate transport contributes to neural injury (Rothstein et al., 1995; Masliah et al., 1996; Belin et al., 1997; Dawson et al., 2000; Pitt et al., 2000; Rao et al., 2001a,b; Howland et al., 2002; Ikematsu et al., 2002; Namura et al., 2002; Warita et al., 2002; Dunlop et al., 2003).
Sindbis virus (SV) is a prototype member of the alphavirus family that replicates in neurons of infected animals. Intracranial infection of susceptible adult mice with a neuroadapted strain (NSV) of Sindbis results in high rates of motor neuron death as well as flaccid hindlimb paralysis or death (Jackson et al., 1987, 1988). Dying motor neurons assume a morphology characterized by pale swollen cytoplasm and swollen organelles, and they do not exhibit caspase-3 activation (Havert et al., 2000; Nargi-Aizenman and Griffin, 2001). We recently demonstrated that NSV-induced neuronal death in adult animals may occur via bystander mechanisms and may be mediated via the activation of AMPA receptors (Nargi-Aizenman et al., 2004).
There are likely two mechanisms by which motor neurons die in response to Sindbis virus: an intrinsic pathway in which infected neurons die via direct infection and an extrinsic pathway in which neurons die as a result of changes in the local environment. In the current study we extend previous observations that propose the presence of bystander neuronal death (Nargi-Aizenman et al., 2004), and we find that excitotoxic motor neuron injury is a critical determinant of NSV pathogenesis. It is interesting that related RNA viruses, most notably West Nile virus, have been shown to induce a flaccid paralysis characterized by spinal motor neuron death in animals (Kelley et al., 2003) and in a proportion of human cases (Leis et al., 2002). It is possible, therefore, that non-cell-autonomous excitotoxic death of motor neurons may be triggered by a variety of infectious agents and that strategies that target this process may be of therapeutic value.
Materials and Methods
Mice. C57BL/6 and BALB/cBy mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Male animals between 5 and 6 weeks of age were used in all of these studies. Mice were injected intracranially with 1000 plaque-forming units (PFUs) of NSV. Hindlimb grip strength was measured with a Chatillon CE digital force gauge (model DFIS 2). Readings were taken in T-peak mode and measured in pounds of force. Animals were supported by the examiner under their forelimbs, and their hindlimbs were lowered to the grip bar until the animals attempted to grasp the device. Animals then were moved away from the bar slowly, and the force was measured as each animal exerted active force against this movement. Each animal was given three trials per examining period and was trained for 3 d before infection to minimize a learning effect. Readings are expressed as an average of the percentage of the day 0 score for each animal. Animals in the minocycline study were given daily intraperitoneal injections of either 50 mg/kg minocycline (M-9511; Sigma, St. Louis, MO) or saline, beginning at the start of infection and continuing through the time course.
Rats. Male Lewis and Sprague Dawley rats (Charles River, Wilmington, MA) were used in these studies at 5-7 weeks of age. Cannulated Sprague Dawley rats were obtained from Zivic Miller (SCA04.00; Zelienople, PA). NSV was passaged into 3- to 4-week-old Lewis rats via serial intracranial inoculation to generate a Sindbis strain (rat-adapted NSV, raNSV) that causes paralysis in rats (Kerr et al., 2003). With the use of raNSV virtually all rats develop some degree of paralysis, with <5% mortality from the encephalomyelitis. Viral titer in the spinal cord reached a peak at 3-4 d after infection (average titers, 1-2 × 106 PFU/gm tissue) and was cleared in large part from these animals within 8-9 d.
Surgery. Cannulated Sprague Dawley rats were anesthetized with Avertin, an incision was made behind the head, and the subarachnoid cannula was connected to an Alzet pump (1007D; 0.5 μl/hr for 7 d; Alza Scientific Products, Mountain View, CA). Pumps were filled with 100 μl of dl-threo-β-hydroxy-aspartate (THA; 1.5 μg/μl; H-2775; Sigma), 1-naphthyl acetyl spermine (NASPM; 10 μg/μl; N-193; Sigma), or saline. Animals were coded and housed individually. Then each rat was scored for hindlimb grip strength by a blinded examiner on a daily basis.
Tissue preparation. At indicated time points the animals were anesthetized and perfused transcardially with chilled PBS, followed by cold 4% paraformaldehyde (PFA). Each lumbar spinal cord and associated nerve roots were dissected en bloc. The sciatic nerve then was traced back to the spinal cord to identify the L3-L5 spinal cord segments. Spinal columns were postfixed in 4% PFA overnight; next the spinal cords were removed and placed in either 1× PBS or 30% sucrose before being frozen. Paraffin and frozen sections were cut as 10-μm-thick sections and mounted onto glass slides. For protein assays the animals were perfused with chilled PBS, and spinal cords were snap frozen on dry ice until further use.
Motor neuron culture. Motor neurons were cultured as previously described (Vandenberghe et al., 1998). In brief, ventral spinal cords were dissected from 14-d-old Wistar rat embryos in HBSS, cut into 1 mm pieces, and digested in 0.05% trypsin in HBSS for 15 min at 37°C. A motor neuron-enriched neuronal population was purified from ventral spinal cord by centrifugation on a 6.5% metrizamide cushion, followed by immunopanning, using the 192 mouse IgG attached to an anti-mouse antibody-coated dish. These motor neuron-enriched suspensions were cultured on a glial feeder layer that had been preestablished on 18-mm-round glass coverslips coated with poly-l-ornithine and laminin. L15 culture medium was supplemented with 0.2% sodium bicarbonate, 3.6 mg/ml glucose, 20 nm progesterone, 5 μg/ml insulin, 0.1 mm putrescine, 0.1 mg/ml conalbumin, 30 nm sodium selenite, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2% horse serum. Cultures were kept in a 7% CO2-humidified incubator at 37°C. Live-Dead kits (Molecular Probes, Eugene, OR) were purchased and used per the manufacturer's instructions.
Glia culture. Primary rat astrocytes were isolated from 2-4 d postnatal rat pups. Cells were derived from the cortical region between bregma and the occipital lobe, and tissues were treated similarly to the spinal tissue described above. Glia were suspended in DMEM supplemented with 10% heat-inactivated horse serum, 4% glucose, 100 IU/ml penicillin, 100 μg/ml streptomycin. Cells were plated as follows: 200,000 cells on a 60 mm dish, 50,000 cells on a six-well plate, and 20,000 cells on a 12-well plate. All plates were coated with collagen before being seeded with cells.
Infection of cultures. All cultures in this study were infected with either NSV or NSV-green fluorescent protein (NSV-GFP) as indicated [at a multiplicity of infection (MOI) of 1 (1 PFU/cell)]. When treated with glutamate receptor-blocking agents, media were replenished daily to maintain a steady drug concentration. The following drugs were used in this study: 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) used at a concentration of 25 μm (C-127; Sigma), Joro spider toxin (JST) used at 3 μm (J-100; Sigma), and 1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazapine (GYKI-52466) used at 25 μm (G-119; Sigma).
Glutamate uptake assay. Glutamate uptake assays were performed with slight modification to a previously published protocol (Sepkuty et al., 2002). Briefly, lumbar spinal cord tissues were homogenized in buffer (10 mm Tris, pH 7.4, 5 mm EDTA, 1 mini-protease inhibitor tab from Roche Diagnostics, Mannheim, Germany), clarified twice, resuspended in tissue buffer (0.05 m Tris, 0.32 m sucrose, pH 7.4), and spun down; the resulting pellet was resuspended in 250 μl of Na+-Krebs [containing (in mm) 120 NaCl, 25 NaHCO3, 5 KCl, 2 CaCl2, 1 KH2PO4, 1 MgSO4, pH 7.4] with and without 500 μm dihydrokainate (DHK). Duplicate samples were generated in which Na+-free Krebs [containing (in mm) 120 choline-Cl, 25 Tris, 5 KCl, 2 CaCl2, 1 KH2PO4, 1 MgSO4, pH 7.4] was used as the blanking control. Samples were incubated with 3H-glutamate for 4 min at 37°C and immediately transferred to ice. Samples were adsorbed to filter paper and incubated in scintillation fluid overnight; total counts per minute (cpm) were measured. The amount of labeled glutamate bound by each sample (pmol/mg protein/min) was calculated by using the total incorporated cpm and concentration of protein in each sample. Tissue samples were processed in duplicate, and at least three independent experiments were performed for each experimental condition. The amount of labeled glutamate present at each time point during infection was divided by the glutamate levels present in uninfected samples to determine normalized glutamate transport at baseline.
Histology. Cryosectioned tissues were used for immunohistochemical staining. Sections were blocked and stained in a solution of 1% normal goat serum and 5% BSA in 1× PBS. The following primary antibodies and stains were used at the following concentrations: Nissl red (1:100; N-21482; Molecular Probes), glial fibrillary acidic protein (GFAP; 1:400 for MAB360 or 1:2000 for AB5804; Chemicon, Temecula, CA), GLT-1 (1:100) (Rothstein et al., 1994), propidium iodide (1 μg/μl; Sigma), SMI-31 (1:10,000; Sternberger Monoclonal, Lutherville, MD), choline acetyltransferase (1:2000; ChAT; Ab5042P; Chemicon), and nitrotyrosine (1:1000; 05-233; Upstate Cell Signaling, Charlottesville, VA). Secondary antibodies were conjugated to Alexa Fluor 488 (green) or Alexa Fluor 594 (red) and were used at a 1:100 dilution (Molecular Probes).
Western blot analysis. Tissues were sonicated in lysis buffer (10 mm Tris, 1% SDS, 1 mm sodium orthovanadate, pH 7.6), and 5 μg of each sample was run out in 12% acrylamide gels (BioExpress, Kaysville, UT). Proteins were transferred to Immobilon-P transfer membranes (Millipore, Bedford, MA) in 10% methanol transfer buffer. Freshly transferred membranes were stained with Ponceau S solution (P-7170; Sigma) and imaged to ensure equal loading. Membranes were rinsed in 1× PBS to remove Ponceau stain and then blocked in 5% milk buffer and stained in 1% milk buffer by using primary antibodies. Blots were developed with the SuperSignal West Femto Maximum Sensitivity Substrate (35095; Pierce, Rockford, IL) and visualized with a Fuji Luminescent Image Analyzer (LAS-1000plus camera; Fuji, Tokyo, Japan). The intensity of each band was determined by Image Gauge software (version 3.4). Protein expression in infected tissue was normalized to levels in uninfected controls by dividing measured band intensities. Because viral infection variably alters the expression of several housekeeping genes, making control immunoblots [glyceraldehyde phosphate dehydrogenase (GAPDH) or actin] unreliable, Ponceau S staining of immunoblots was performed to ensure equal loading of lanes.
Quantitation of neuronal death in tissue sections. Serial sections were generated by collection of every 20th section through spinal cord onto glass slides. One section in each series was used for histologic analysis after staining with hematoxylin and eosin. Motor neurons were defined as cells present within the gray matter, ventral to the central canal, and having a cell body >25 μm in diameter. Data are presented as the mean ± SE of the number of motor neurons per section of spinal cord. Adjacent sections were examined for GFP fluorescence in ventral gray neurons (indicative of infection with NSV-GFP), and colocalization to motor neurons was achieved by staining with ChAT antibodies.
For histologic correlation of various drug-treated animals (see Figs. 3, 4, 5), sections were prepared at day 7 after infection as above. Paraffin sections then were generated through the L3-L5 segments and stained with hematoxylin and eosin. Motor neurons again were counted if they were in the gray matter, ventral to the central canal, and had a cell body >25 μm in diameter. Data again are presented as the mean ± SE of the number of motor neurons per section. In total, three to five animals were analyzed per group, and motor neurons were counted by a blinded examiner.
Figure 3.

In vivo blockade of glutamate transporters and glutamate receptors alters NSV pathogenesis in rats. Sprague Dawley rats with cannulas surgically implanted into their spinal subarachnoid spaces were given NASPM, THA, or PBS at a rate of 0.5 μl/hr for 7 d by an Alzet mini-osmotic pump. A, Hindlimb grip strength of cannulated rats given THA, a specific inhibitor of the astrocyte glutamate transporter GLT-1, is not altered by the drug alone but contributes to a more drastic reduction of grip strength after NSV challenge. B, THA infusion also resulted in significantly increased motor neuron loss in NSV-infected rats when compared with saline-infused controls. C, Hindlimb grip strength of NSV-infected rats administered NASPM, a synthetic analog of JST (inhibitor of GluR2, excluding AMPA receptors); when compared with saline-infused controls, the NSV-infected rats did not develop any hindlimb weakness in response to NSV infection. D, NASPM infusion also resulted in significant preservation of motor neurons after NSV infection.
Figure 4.
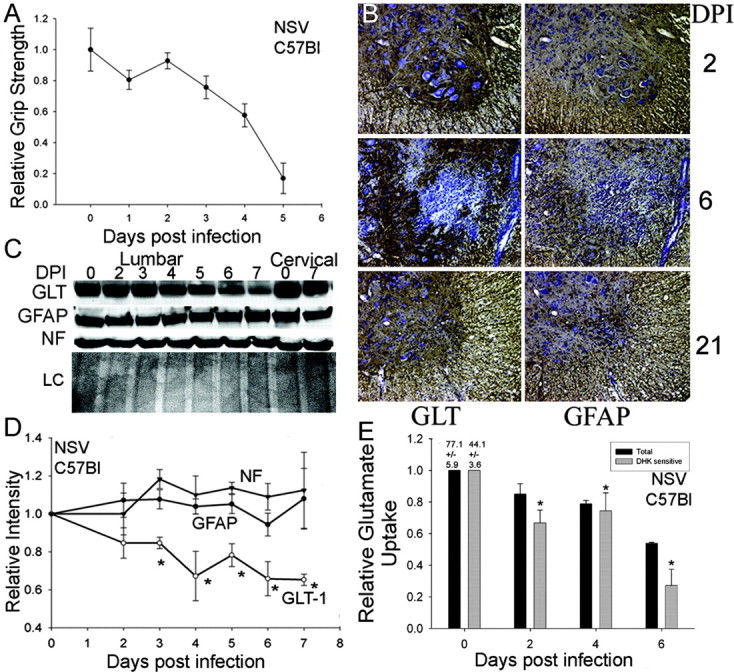
Expression of the astrocyte glutamate transporter GLT-1 in the spinal cords of NSV-infected mice. A, C57BL/6 mice develop paralysis in response to NSV. The decline in hindlimb grip strength correlates with the classic paralysis observed in the NSV model of rodent infection after NSV infection. B, Immunohistochemical detection of astrocyte glutamate transporter GLT-1 (brown staining, left panels) as well as the astrocyte marker GFAP (brown staining, right panels) in the lumbar enlargement of NSV-infected mice. The transient decrease in GLT-1 staining at day 6 after infection (DPI) is not correlated with a loss of GFAP immunoreactivity, implying a selective downregulation of GLT-1 expression. C, GLT-1 expression in lysates from lumbar and cervical spinal cords of NSV-infected mice by Western blot analysis. The intensity of the GLT-1 band decreases over the time course in the lumbar and, to a lesser degree, in cervical spinal cordlysates. GFAP and neurofilament (NF) protein immunoreactivity (SMI-31) was relatively unchanged over this time course while Ponceau S staining of membranes confirmed equivalent loading of lanes (LC). D, Quantitation of GLT-1 immunoreactivity by Western blots in spinal cordlysates of NSV-infected mice. As early as 3 d after infection, the GLT-1 expression was reduced significantly compared with either GFAP or SMI-31 expression. Asterisks indicate that intensity of GLT-2 is significantly different from GFAP at indicated time points (p < 0.05). E, Functional loss of GLT-1-mediated glutamate uptake involved in NSV infection. Spinal cord homogenates were generated at various times after infection from the lumbar region. Total and DHK-sensitive (GLT-1-mediated) glutamate transport was assessed with data normalized to uptake by uninfected, control samples. DHK-sensitive transport was decreased at days 2-6 after infection (*p < 0.05) relative to day 0.
Figure 5.

GLT-1 expression in a strain of mouse (BALB/cBy) resistant to NSV-induced paralysis. A, Hindlimb grip strength measurements in BALB/cBy mice infected with NSV show that these animals do not develop hindlimb weakness over the time course of NSV infection. B, Motor neuron counts from the lumbar spinal cords of NSV-infected BALB/cBy mice at baseline and day 6 after infection. C, Immunoblot analysis of GLT-1 expression and Ponceau S staining in the BALB/c mouse. D, Quantitative analysis of triplicate immunoblots revealed preservation of GLT-1 expression after NSV infection. E, Baseline levels of functional glutamate uptake in the BALB/cBy mouse are significantly higher than in the C57BL/6 mouse. F, Functional glutamate uptake declines by 6 d after infection in the BALB/cBy mice, although levels remain consistently above those observed in the C57BL/6 mouse. Error bars: A, B, D, F, not significant (p > 0.05); E, p < 0.05.
Statistical analysis. SPSS 11.0 (SPSS Science, Chicago, IL) was used for all of the statistical analyses. Because of the nonparametric nature of the data (as determined by tests of normality), nonparametric equivalent tests of ANOVA and repeated measures ANOVA were used to increase the robustness of the results. The Kruskal-Wallis test was performed to analyze differences between groups at each time point, and Friedman's nonparametric repeated measures comparison was used to analyze differences across time within a group. The Mann-Whitney U test was used for the comparison of two independent samples. Significance was assessed at the 0.05 level. These tests were used because they make no assumptions about the distribution of the data, such as normality.
Results
To investigate mechanisms of spinal motor neuron death after NSV infection, we used a recombinant NSV virus encoding a GFP construct (NSV-GFP), as described previously (Kerr et al., 2002, 2003). At 2 d after intracranial infection of C57BL/6 mice, virus was detected by GFP fluorescence in large ventral neurons (costained with Nissl red) in the lumbosacral enlargement of the spinal cord (Fig. 1A,B). These infected cells exhibited a normal morphology. By 4 d after infection, however, most neurons within the ventral region of the spinal cord exhibited abnormal morphology, with swollen cytoplasmic and nuclear compartments and intracellular vacuoles (Fig. 1C,D). Electron micrographs of these motor neurons (Fig. 1E, long arrow denotes synapse onto cell body) exhibited pale cytoplasm and enlarged, disrupted mitochondria (small arrows). This morphology resembled excitotoxic injury in other studies (Ginsberg et al., 1999; Krampfl et al., 2001; Van den Bosch et al., 2002b). Furthermore, relatively few motor neurons in the lumbar spinal cord exhibited GFP fluorescence (Fig. 1C,D) or SV immunoreactivity (data not shown), suggesting either that these morphologically abnormal neurons were not infected directly or that the cell death process had catabolized proteins indicative of direct infection. Because NSV capsid immunoreactivity is maintained even in cellular remnants of infected cells (Jackson et al., 1987, 1988; Lewis et al., 1999), the failure to detect NSV immunoreactivity suggests that many of the dying motor neurons were not infected directly.
Figure 1.
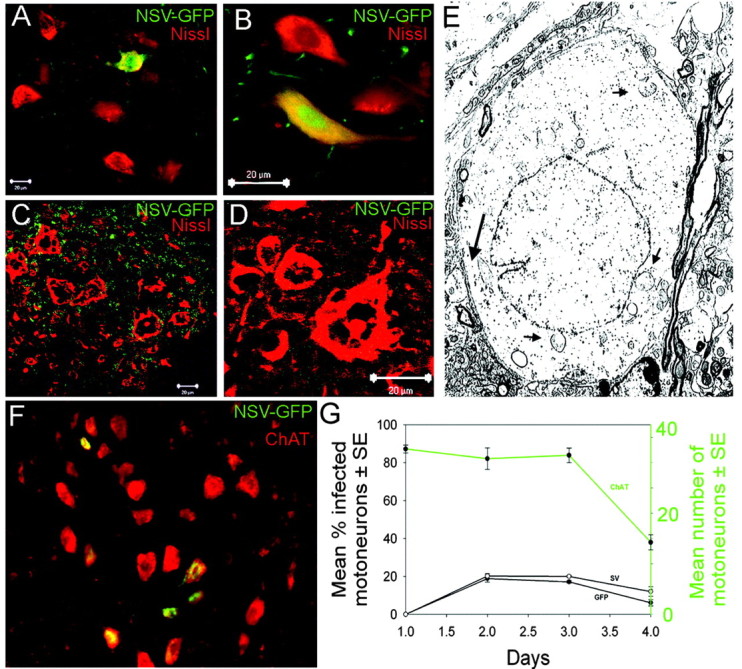
Evidence for bystander destruction of noninfected motor neuron cells during neuroadapted Sindbis virus infection. C57BL/6 mice were infected with an NSV construct expressing GFP. A, B, Lumbar enlargement of an NSV-infected C57BL/6 mouse at 2 d after infection. Infected cells are visible by the expression of GFP (green), while neurons are visualized with the Nissl counterstain (red). C, D, Lumbar enlargement of an NSV-infected C57BL/6 mouse at 4 d after infection showing many abnormally appearing neurons with swollen nuclei and intracellular vacuoles. None of these motor neurons exhibited immunoreactivity to GFP or to NSV capsid proteins (data not shown), suggesting that these neurons have not been infected directly. E, Electron micrograph of an infected lumbar enlargement at 4 d after infection. Long arrow denotes a synapse onto the cell body, confirming its neuronal identity. Swollen cytoplasm and nucleoplasm are observed, along with dilated, disrupted mitochondria (short arrows). F, ChAT staining (red) in an NSV-infected (green) lumbar enlargement at 4 d after infection. G, Quantitation of the percentage of motor neurons infected by GFP-expressing NSV (left axis) compared with the total number of motor neurons per section (right axis in green). Infection status was determined both by GFP expression and immunoreactivity to NSV glycoproteins. Error bars, p < 0.05.
Quantitative examination of motor neuron infection within the lumbar enlargement further supports the conclusion that bystander motor neuron death is occurring in vivo. When sections simultaneously were examined histologically for the presence of ChAT+ immunoreactivity (red) and NSV infection (green), total and infected motor neurons could be measured over time (Fig. 1F, 2 d after infection). The total number of motor neurons remained constant at ∼30 cells per section through day 3 of infection but then began to decline thereafter (Fig. 1G, top line, right-sided y-axis). When the percentage of infected motor neurons was determined, the highest levels were found at days 2 and 3 after infection, followed by a decline thereafter. At no point were >20% of motor neurons directly infected (Fig. 1G), yet >75-90% of motor neurons are lost during disease (Kerr et al., 2003). To exclude the possibility that ChAT staining was lost within infected motor neurons, we performed a parallel study that used a fluorescent Nissl compound (Nissl red, Molecular Probes) and again found that only 20% of Nissl-stained cells also exhibited GFP fluorescence (data not shown). Therefore, the infrequency of direct infection of motor neurons and the morphologic appearance of noninfected motor neurons suggested that NSV causes bystander motor neuron death in the spinal cord.
We next examined whether NSV could induce excitotoxic death of motor neurons in vitro, using motor neuron-enriched cocultures on astrocytes. In this system 60-70% of neurons exhibited ChAT+ immunoreactivity (data not shown). We found that this coculture system recapitulated the in vivo tropism of NSV for neurons, with no infection of the underlying astrocytes (Fig. 2A). Although these infected motor neurons initially exhibited a normal morphology (Fig. 2A), all of these cells died over time (Fig. 2B-D). Furthermore, we could detect death of uninfected motor neurons, often adjacent to infected motor neurons, as defined by propidium iodide uptake (Fig. 2B). We did not find evidence of caspase-3 activation or cytochrome c translocation within these dying neurons (data not shown). We did, however, find that dying neurons as well as adjacent, noninfected astrocytes accumulated reactive oxygen species (ROS) as defined by nitrotyrosine immunoreactivity (Fig. 2C). Therefore, motor neuron death in our culture model occurred among both infected and noninfected cells and potentiated injury of cocultured astrocytes.
Figure 2.

Bystander destruction of cultured spinal neurons by neuroadapted Sindbis virus. A, Infection of mixed spinal neurons cultured on a monolayer of astrocytes with NSV-GFP (green) at 24 hr after infection, counter stained with GFAP (red; 20×). Images were deconvolved by Simple PCI software. B, At 24 hr after treatment with NSV-GFP, the cultures were treated with propidium iodide (PI; red). Several cells are PI-positive, yet only one is infected with NSV (green). C, Nitrotyrosine reactivity (red) indicates the presence of reactive oxygen species. At 24 hr after NSV-GFP infection, the cultures were fixed with 4% paraformaldehyde and stained with a nitrotyrosine-specific antibody. D, Mixed cultures were treated with NSV-GFP in the presence of AMPA-type glutamate receptor blocking agents. At 48 hr after infection, those cultures treated with NSV-GFP alone revealed widespread death of spinal neurons, whereas parallel cultures treated with CNQX or JST showed preservation of spinal neurons. E, Quantitation of spinal motor neuron protection mediated by blockage of AMPA-type glutamate receptors. Survival of motor neurons treated with NSV and any of the three AMPA blockers is greater than with NSV alone (*p < 0.01 at 48 and 72 hr after infection).
To clarify the role of glutamate in NSV-induced motor neuron death, we added glutamate receptor antagonists to motor neuron cultures. Infection of cultured motor neurons in the presence of NMDA receptor blockers did not alter cell survival substantially, whereas AMPA receptor blockade caused significant protection from NSV-induced cell death (Fig. 2D,E). Both CNQX and JST, a specific blocker of GluR2-negative (Ca2+-permeable) AMPA receptors, produced enhanced motor neuron survival in our system. Photomicrographs taken at 36 hr after infection showed preservation of normal neuronal morphology in the presence of AMPA receptor blockers (Fig. 2D). Quantitation of neuronal survival after NSV infection revealed significant protection by CNQX, JST, or GYKI-52466 (Fig. 2E). With each of the three AMPA receptor blockers the neuron survival was ∼50% at 72 hr after infection, compared with 21% in infected control cultures (p < 0.01 at 48 and 72 hr after infection). Virus titration assays confirmed that AMPA blockade did not alter viral titers in culture supernatants (data not shown). We conclude that NSV-induced motor neuron death in vitro occurs in part by glutamate excitotoxicity mediated via calcium-permeable AMPA receptors.
To confirm whether NSV-induced motor neuron death involves glutamate excitotoxicity in vivo, we modulated extracellular glutamate levels in infected animals by blocking astrocyte-mediated glutamate transport. We used 5- to 7-week-old rats surgically implanted with cannulas extending into the lumbar CSF space at approximately the T9 vertebral level. A pump delivery system was used to continually administer THA, a nonspecific inhibitor of glutamate transporters, or a vehicle control at the time of viral challenge. In each group of 7-10 animals the hindlimb grip strength was measured daily in each animal by a blinded examiner. Consistent with previous studies (Hirata et al., 1997), we confirmed that THA administration alone did not induce paralysis or motor neuron loss (Fig. 3A), probably because it is transported like glutamate and acts in part to exchange with intracellular glutamate. However, THA administration potentiated NSV-induced hindlimb weakness (Fig. 3A) (p < 0.05 on days 6-9), and histologic analyses confirmed that this effect was associated with enhanced death of spinal motor neurons (Fig. 3B). Quantitation of motor neurons in the lumbar enlargement at 1 week after infection showed ∼50% motor neuron loss, consistent with our previous studies (Kerr et al., 2003). There were significantly fewer motor neurons present in NSV/THA-treated animals, compared with NSV/vehicle-treated animals (12.34 ± 1.92 vs 21.30 ± 0.97 motor neurons per section; p < 0.05). We then determined whether glutamate activation of calcium-permeable AMPA receptors is important in NSV-induced motor neuron death. Animals were treated with NASPM, an inhibitor of calcium-permeable AMPA receptors, given intrathecally as in previous experiments. As before, 7-10 animals were studied per group in a blinded manner. The administration of NASPM virtually eliminated NSV-induced paralysis (Fig. 3C) (p < 0.05 at day 6) without altering viral replication or spread within the spinal cord (data not shown). Histologic examination confirmed that NASPM prevented motor neuron death that was induced by NSV (Fig. 3D) (20.93 ± 1.84 vs 28.37 ± 0.79; p < 0.05). Together, we conclude that NSV-induced motor neuron death is potentiated by glutamate excitotoxicity and is mediated via calcium-permeable AMPA receptors in vivo.
Because blockage of glutamate transporter with the nonspecific inhibitor THA exacerbated the outcome of NSV infection, we investigated expression of the dominant astroglial glutamate transporter GLT-1 during NSV infection. Infected C57BL/6 adult mice developed a rapid and complete loss of hindlimb grip strength beginning at 4 d after viral challenge (Fig. 4A). Immunohistochemical analysis of spinal cord tissue at 2, 6, and 21 d after infection revealed patchy loss of GLT-1 expression within the lumbar gray matter by 4 d after infection (brown staining corresponds to GLT-1 or GFAP immunoreactivity with blue hematoxylin counterstain) (Fig. 4B). This focal reduction often was associated with inflammatory infiltrates (Fig. 4B, middle) or areas of prominent microglial activation (data not shown). Glial fibrillary acidic protein (GFAP) expression was altered minimally in response to NSV infection (Fig. 4B, right), suggesting that GLT-1 decline was a selective process and was not attributable to astrocyte death. Indeed, by day 21 after infection no areas of focal GLT-1 loss could be detected, suggesting that the earlier loss of GLT-1 staining was attributable to a transient downregulation. Immunoblots of spinal cord lysates confirmed the rapid decline in GLT-1 expression within the lumbar enlargement (Fig. 4C). Quantitation of triplicate immunoblots revealed a significant loss of expression at 3 d after challenge and a continued decline over the 7 d course of the experiment compared with either neurofilament or GFAP as a control (Fig. 4D) (p ≤ 0.05 at days 3-7). Ponceau S staining of the membranes confirmed equal loading of sample in all lanes (loading control, LC).
To determine whether reduced GLT-1 expression correlated with reduced functional glial glutamate transport, we generated membrane preparations from freshly isolated lumbar spinal cords of mice at various points after NSV infection. We then assessed the ability of these membrane preparations to transport radioactive glutamate, using an assay that principally reflects GLT-1 activity (Sepkuty et al., 2002). We confirmed the contribution of GLT-1 in these assays by incubating parallel samples with DHK, a specific antagonist of GLT-1. Relative glutamate transport over the time course of the experiment is shown relative to transport levels in uninfected control samples. In Figure 4E and all subsequent figures, relative total glutamate transport is depicted with black bars, whereas GLT-1-mediated transport (transport sensitive to DHK) is depicted in gray bars. Absolute values for total and DHK-sensitive transport are indicated above the day 0 bars. Consistent with previous studies, ∼65% of glutamate transport was eliminated in the presence of DHK (Rothstein et al., 1994). GLT-1-mediated glutamate transport was reduced significantly by 2 d after NSV infection and continued to decline thereafter (p < 0.05). By day 6 after infection GLT-1-mediated glutamate transport was only 25% that of baseline (Fig. 4E). This reduction was even more pronounced than the downregulation of GLT-1 protein levels, suggesting that mechanisms such as blockade of GLT-1 trafficking to the membrane surface might contribute to the reduction. Interestingly, because non-GLT-1-mediated transport remains constant during infection (data not shown), it is likely that other glutamate transporters present in the membrane fraction (such as GLAST) are not altered significantly in response to NSV infection. Therefore, GLT-1 protein levels and GLT-1-mediated glutamate transport are reduced in the spinal cord during NSV infection, and this reduction immediately precedes the onset of motor neuron death.
BALB/cBy mice are uniquely resistant to NSV-induced hindlimb paralysis (Thach et al., 2000). We investigated whether this resistance might be related either to increased basal glutamate transport or to sustained glutamate transport in response to NSV infection. We found that hindlimb grip strength did not decline over the time course of infection in these animals (Fig. 5A) and that there was no detectable loss of motor neurons in the spinal cord after infection (Fig. 5B). When GLT-1 expression in lumbar spinal cord lysates of NSV-infected BALB/c mice was examined, protein levels did not decline over the course of infection (5C,D, NSV). Additionally, baseline glutamate transport function was approximately fivefold greater in uninfected BALB/cBy mice compared with C57BL/6 mice (Fig. 5E)(p < 0.05). Interestingly, we did find that functional glutamate transport declined by day 6 d after infection in NSV-infected BALB/cBy mice (Fig. 5F), suggesting that the elevated baseline transport provided a buffer against excitotoxic motor neuron death and is one factor that renders these mice resistant to NSV.
Finally, because several studies have demonstrated the ability of minocycline to protect motor neurons in rodent models of amyotrophic lateral sclerosis (ALS) (Van Den Bosch et al., 2002c; Zhu et al., 2002; Kriz et al., 2003; Zhang et al., 2003), we examined whether minocycline could prevent NSV-induced motor neuron death and whether protection correlated with preserved GLT-1 expression. Minocycline has been demonstrated to have neuroprotective capacities that may relate to its inhibition of microglial activation (Yrjanheikki et al., 1998; Tikka and Koistinaho, 2001), immune effector cell activation (Brundula et al., 2002), or intracellular mitochondrial cytochrome c release (Zhu et al., 2002). When it was administered to C57BL/6 mice at the time of NSV challenge, minocycline potently protected animals from the development of hindlimb weakness (Fig. 6A) (p < 0.05 at days 2-10). Viral titration assays on spinal cord homogenates showed that minocycline had no effect on the ability of NSV to replicate or spread. Motor neurons were preserved in minocycline-treated, NSV-infected C57BL/6 mice, compared with NSV-infected mice treated with a vehicle control (Fig. 6B) (p < 0.05).
Figure 6.

GLT-1 expression in minocycline-treated, NSV-infected C65BL/6 mice. A, Hindlimb grip strength in NSV-infected mice is preserved with minocycline treatment. B, Motor neurons are protected in the lumbar spinal cord in NSV-infected minocycline-treated, NSV-infected mice. C, Immunoblot analysis of GLT-1 expression in minocycline-treated, NSV-infected mice reveals sustained GLT-1 expression with Ponceau S staining as a loading control (LC). D, Quantitative analysis of triplicate immunoblots shows a preservation of GLT-1 expression after NSV infection in minocycline-treated mice. Asterisks indicate that the intensity of GLT-1 from minocycline-treated, NSV-infected mice is significantly different from GLT-1 in untreated, NSV-infected mice at indicated time points (p < 0.05). E, Functional GLT-1-mediated glutamate uptake is increased in the lumbar spinal cord of C57BL/6 mice treated with minocycline (*p < 0.05; DHK day 6 relative to day 0). F, In NSV-infected mice treated with minocycline, functional glutamate uptake is unchanged over time, suggesting that minocycline and NSV have opposite effects that cancel each other.
Lumbar spinal cord homogenates from minocycline-treated and minocyclinetreated/NSV-infected C57BL/6 mice were examined for GLT-1 expression by Western blot, and no decline in GLT-1 immunoreactivity was found (Fig. 6C,D). Quantitation of triplicate immunoblots revealed that minocycline-treated animals had maintained a consistent GLT-1 expression pattern from days 2 to 6 after NSV infection (p ≤ 0.05 at days 2-6, comparing NSV with NSV/mino). Finally, glutamate uptake assays were performed on spinal cord lysates from animals treated with minocycline alone (Fig. 6E) or minocycline with NSV challenge (Fig. 6F). We found a significant increase in both total and GLT-1-mediated glutamate transport in minocycline-treated animals such that there was 10-fold greater transport by day 6 after treatment (p < 0.05). Interestingly, in minocycline-treated, NSV-infected animals (Fig. 6F) there was no significant change in glutamate transport over time, indicating that the viral infection and minocycline had opposite effects that canceled each other. We suggest, therefore, that the protective effects of minocycline in preventing NSV-induced paralysis may be attributable to its ability to enhance glial glutamate transport and to provide a buffer against excitotoxic motor neuron death.
Discussion
We investigated the mechanisms underlying spinal motor neuron death occurring as a result of infection with a neuronotropic alphavirus. Studies over a number of years have confirmed that Sindbis virus can induce intrinsic neuronal death (Jackson et al., 1987, 1988; Levine and Griffin, 1993; Griffin et al., 1994; Cheng et al., 1996; Levine et al., 1996; J. Lewis et al., 1996; Nava et al., 1998; Jan and Griffin, 1999; D. Lewis et al., 1999; Liang et al., 1999; Kerr et al., 2002). Most of these studies were completed in newborn or suckling mice and showed that SV induces apoptosis of infected neurons. Indeed, the use of recombinant SV constructs expressing modulators of apoptotic cell death has clarified this intrinsic neuronal apoptosis pathway (Cheng et al., 1996; Lewis et al., 1996; Nava et al., 1998; Kerr et al., 2002). However, most strains of SV are incapable of inducing neuronal apoptosis in adult animals and cause little or no morbidity. This may be attributable to the fact that postmitotic neurons are less susceptible to apoptotic triggers in adult animals after the developmental apoptotic machinery has been downregulated.
NSV, a highly neurovirulent derivative of SV, is capable of inducing significant morbidity and mortality in adult mice and rats and, unlike other SV strains, causes hindlimb paralysis with widespread spinal motor neuron destruction. Because there is evidence to suggest that excitotoxicity occurs in the Sindbis model (Nargi-Aizenman and Griffin, 2001; Nargi-Aizenman et al., 2004), we investigated the mechanisms by which NSV infection triggers excitotoxicity in motor neurons. Our findings suggest that direct NSV destruction of motor neurons may be separable from other, simultaneously active motor neuron death pathways. Specifically, we propose that NSV activates extrinsic, non-cell-autonomous bystander death in addition to its direct destruction of infected cells.
In support of such a bystander mechanism, we found that relatively few motor neurons are infected directly by NSV and that astroglial glutamate transport is a critical mediator of motor neuron death. We found that inhibition of GLT-1-mediated transport potentiated motor neuron death in response to NSV infection. Additionally, blockage of calcium-permeable AMPA receptors protected NSV-infected animals from motor neuron death. The motor neuron loss observed with NASPM-treated, NSV-infected animals is likely attributable either to persistent excitotoxic death of motor neurons via activation of AMPA or NMDA glutamate receptors or to intrinsic death of infected motor neurons. We propose, therefore, that virus-induced dysregulation of astrocyte-mediated glutamate transport mediated by GLT-1 allows excess glutamate to accumulate extracellularly in the CNS. This excess synaptic glutamate is bound by calcium-permeable glutamate receptors on postsynaptic motor neurons, causing the excitotoxic destruction of that cell whether it harbors the virus or not.
Several lines of evidence suggest that GLT-1-mediated glutamate transport is important in protecting neurons from excitotoxic death and that downregulation of its function increases extracellular glutamate and predisposes susceptible neurons to excitotoxic injury (Rosenberg and Aizenman, 1989; Bridges et al., 1991; Rosenberg et al., 1992; Robinson et al., 1993; Rothstein et al., 1993, 1996; Dugan et al., 1995; Tanaka et al., 1997). To investigate further the role of the GLT-1 transporter in the NSV model, we examined GLT-1 expression longitudinally over time. We found a transient decrease in GLT-1 expression that recovered by 3 weeks after infection, which did not correlate with a loss of GFAP staining, implying that the loss of GLT-1 was not attributable to a loss of astrocytes. Western blot analysis of spinal cord lysates from days 0 to 7 showed that GLT-1 expression began to decline by day 3 after infection and continued to decline over the study time course. The immunohistochemical staining and the Western blot analysis lead us to believe that there was a reduction in the amount of GLT-1 protein present. Furthermore, downregulation of GLT-1 protein expression correlated with loss of glutamate transport function, which diminished immediately before the development of motor neuron injury.
Because the BALB/cBy mouse is relatively resistant to NSV infection, we investigated GLT-1 expression in these animals over the course of infection. Interestingly, we found that baseline glutamate transport correlated well with host susceptibility to NSV. Specifically, C57BL/6 mice (baseline spinal glutamate transport, 77 pmol · mg-1 · min-1) were more susceptible than Lewis rats (200 pmol · mg-1 · min-1) that were more susceptible than BALB/cBy mice (434 pmol · mg-1 · min-1). We hypothesize that these marked differences prevent glutamate transport from dropping below a critical limit in more resistant hosts, reducing or preventing motor neuron injury. It is therefore possible that the maintenance of a significantly higher level of GLT-1-mediated glutamate transport in the BALB/cBy mouse contributes to the resistance of that strain to NSV-mediated motor neuron loss.
The GLT-1 promoter region recently has been found to have a nuclear factor-κB (NF-κB) binding site, suggesting that the expression of GLT-1 can be controlled at a transcriptional level by secreted factors (Su et al., 2003). Indeed, some inflammatory cytokines are known to have a downregulatory effect on glutamate transport (Huang and O'Banion, 1998; Hensley et al., 2003). Sindbis virus elicits a significant inflammatory cytokine response in the CNS (Mokhtarian et al., 1989; Wesselingh et al., 1994; Rowell and Griffin, 1999; Binder and Griffin, 2001), many of which are known to activate the NF-κB pathway. Therefore, a possible mechanism to explain the GLT-1 downregulation in the lumbar spinal cords of NSV-infected animals may be that inflammatory factors, secreted either by endogenous microglial cells or influxing leukocytes, alter astrocyte function. Currently, we are investigating whether the immune response is regionally specific and, therefore, may modulate glutamate transport differentially in the brain and spinal cord.
The finding that minocycline protects mice from NSV-induced motor neuron death and prevents the loss of GLT-1 expression and function is consistent with the hypothesis that GLT-1 transport is an important mediator of motor neuron injury. Most studies examining the neuroprotective effects of minocycline have demonstrated diminished microglial activation and often reduced leukocyte recruitment (Tikka et al., 2001; Brundula et al., 2002). Both peripheral leukocytes and endogenous microglial cells migrate to the site of injury and function in damage control and initiation of the immune response by the secretion of factors such as chemokines, cytokines, and nitric oxide (for review, see Ransohoff and Tani, 1998; Biber et al., 2002). In the CNS the effect that these molecules have on neighboring astrocytes may be to cause them to alter their glutamate-regulating capacity. Our data, using minocycline, suggest that inhibition of microglial activation may prevent this loss of astrocyte function, resulting in the enhanced protection of motor neurons from excitotoxic death.
Dysregulation of glutamate transporters is likely to play an important role in both acute and chronic neurological processes via altered synaptic transmission and via the facilitation of glutamate-mediated excitotoxicity. In ALS the loss of glutamate transport, specifically excitatory amino acid transporter-2 (EAAT2; the human homolog of GLT-1), may contribute to motor neuron degeneration (Rothstein et al., 1992, 1995; Lin et al., 1998). Antisense knockdown or functional blockade of glutamate transporters produces neurotoxicity and can model the loss of motor neurons in ALS (Rothstein et al., 1993, 1996). In other neurologic disorders, such as Alzheimer's disease, Huntington's disease, stroke, traumatic brain injury, and multiple sclerosis, loss of excitatory synapses and disruption of glutamate transport are an early common finding (Masliah et al., 1996; Pitt et al., 2000) (for review, see (Gegelashvili et al., 2001). Finally, West Nile virus causes a syndrome in humans that is pathologically identical to Sindbis in rodents. Our study of the mechanisms involved in motor neuron loss during NSV infection likely will shed light on therapeutic targets for such diseases.
Footnotes
We acknowledge the Robert Packard Center for Amyotrophic Lateral Sclerosis Research, the Muscular Dystrophy Association, and the National Institutes of Health [Grants NS34175 (J.D.), NS02130 (D.A.K.), and NS33958 (J.D.R.)] for supporting this work. We also acknowledge Diane Griffin and Marie Hardwick for scientific input and Irina Shats, James Harper, and Deepa Deshpande for technical assistance.
Correspondence should be addressed to either of the following: Dr. Douglas Kerr, Johns Hopkins Hospital, Pathology 627 C, Baltimore, MD 21287, E-mail: dkerr@jhmi.edu; or Dr. David N. Irani, Johns Hopkins Hospital, JHOPC 5073A, Baltimore, MD 21287, E-mail: dirani@jhmi.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249566-10$15.00/0
References
- Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32: 1-14. [PubMed] [Google Scholar]
- Belin MF, Didier-Bazes M, Akaoka H, Hardin-Pouzet H, Bernard A, Giraudon P (1997) Changes in astrocytic glutamate catabolism enzymes following neuronal degeneration or viral infection. Glia 21: 154-161. [DOI] [PubMed] [Google Scholar]
- Biber K, Zuurman MW, Dijkstra IM, Boddeke HW (2002) Chemokines in the brain: neuroimmunology and beyond. Curr Opin Pharmacol 2: 63-68. [DOI] [PubMed] [Google Scholar]
- Binder GK, Griffin DE (2001) Interferon-γ-mediated site-specific clearance of alphavirus from CNS neurons. Science 293: 303-306. [DOI] [PubMed] [Google Scholar]
- Bridges RJ, Stanley MS, Anderson MW, Cotman CW, Chamberlin AR (1991) Conformationally defined neurotransmitter analogues. Selective inhibition of glutamate uptake by one pyrrolidine-2,4-dicarboxylate diastereomer. J Med Chem 34: 717-725. [DOI] [PubMed] [Google Scholar]
- Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW (2002) Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain 125: 1297-1308. [DOI] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH (2000) AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro J Neurosci 20: 240-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Levine B, Boise LH, Thompson CB, Hardwick JM (1996) Bax-independent inhibition of apoptosis by Bcl-XL. Nature 379: 554-556. [DOI] [PubMed] [Google Scholar]
- Chretien F, Vallat-Decouvelaere AV, Bossuet C, Rimaniol AC, Le Grand R, Le Pavec G, Creminon C, Dormont D, Gray F, Gras G (2002) Expression of excitatory amino acid transporter-2 (EAAT-2) and glutamine synthetase (GS) in brain macrophages and microglia of SIVmac251-infected macaques. Neuropathol Appl Neurobiol 28: 410-417. [DOI] [PubMed] [Google Scholar]
- Dawson LA, Djali S, Gonzales C, Vinegra MA, Zaleska MM (2000) Characterization of transient focal ischemia-induced increases in extracellular glutamate and aspartate in spontaneously hypertensive rats. Brain Res Bull 53: 767-776. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Bruno VM, Amagasu SM, Giffard RG (1995) Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci 15: 4545-4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop J, Beal MH, She Y, Howland DS (2003) Impaired spinal cord glutamate transport capacity and reduced sensitivity to riluzole in a transgenic superoxide dismutase mutant rat model of amyotrophic lateral sclerosis. J Neurosci 23: 1688-1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegelashvili G, Robinson MB, Trotti D, Rauen T (2001) Regulation of glutamate transporters in health and disease. Prog Brain Res 132: 267-286. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Martin LJ, Rothstein JD (1995) Regional deafferentation down-regulates subtypes of glutamate transporter proteins. J Neurochem 65: 2800-2803. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Rothstein JD, Price DL, Martin LJ (1996) Fimbria-fornix transections selectively down-regulate subtypes of glutamate transporter and glutamate receptor proteins in septum and hippocampus. J Neurochem 67: 1208-1216. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Portera-Cailliau C, Martin LJ (1999) Fimbria-fornix transection and excitotoxicity produce similar neurodegeneration in the septum. Neuroscience 88: 1059-1071. [DOI] [PubMed] [Google Scholar]
- Griffin DE, Levine B, Ubol S, Hardwick JM (1994) The effects of alphavirus infection on neurons. Ann Neurol [Suppl] 35: S23-S27. [DOI] [PubMed] [Google Scholar]
- Havert MB, Schofield B, Griffin DE, Irani DN (2000) Activation of divergent neuronal cell death pathways in different target cell populations during neuroadapted Sindbis virus infection of mice. J Virol 74: 5352-5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath PR, Shaw PJ (2002) Update on the glutamatergic neurotransmitter system and the role of excitotoxicity in amyotrophic lateral sclerosis. Muscle Nerve 26: 438-458. [DOI] [PubMed] [Google Scholar]
- Hensley K, Fedynyshyn J, Ferrell S, Floyd RA, Gordon B, Grammas P, Hamdheydari L, Mhatre M, Mou S, Pye QN, Stewart C, West M, West S, Williamson KS (2003) Message and protein level elevation of tumor necrosis factor alpha (TNFα) and TNFα-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol Dis 14: 74-80. [DOI] [PubMed] [Google Scholar]
- Hirata A, Nakamura R, Kwak S, Nagata N, Kamakura K (1997) AMPA receptor-mediated slow neuronal death in the rat spinal cord induced by long-term blockade of glutamate transporters with THA. Brain Res 771: 37-44. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci USA 99: 1604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TL, O'Banion MK (1998) Interleukin-1β and tumor necrosis factor-α suppress dexamethasone induction of glutamine synthetase in primary mouse astrocytes. J Neurochem 71: 1436-1442. [DOI] [PubMed] [Google Scholar]
- Ikematsu K, Tsuda R, Kondo T, Nakasono I (2002) The expression of excitatory amino acid transporter 2 in traumatic brain injury. Forensic Sci Int 130: 83-89. [DOI] [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Griffin DE, Johnson RT (1987) The pathogenesis of spinal cord involvement in the encephalomyelitis of mice caused by neuroadapted Sindbis virus infection. Lab Invest 56: 418-423. [PubMed] [Google Scholar]
- Jackson AC, Moench TR, Trapp BD, Griffin DE (1988) Basis of neurovirulence in Sindbis virus encephalomyelitis of mice. Lab Invest 58: 503-509. [PubMed] [Google Scholar]
- Jan JT, Griffin DE (1999) Induction of apoptosis by Sindbis virus occurs at cell entry and does not require virus replication. J Virol 73: 10296-10302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley TW, Prayson RA, Isada CM (2003) Spinal cord disease in West Nile virus infection. N Engl J Med 348: 564-566. [DOI] [PubMed] [Google Scholar]
- Kerr DA, Larsen T, Cook SH, Fannjiang YR, Choi E, Griffin DE, Hardwick JM, Irani DN (2002) BCL-2 and BAX protect adult mice from lethal Sindbis virus infection but do not protect spinal cord motor neurons or prevent paralysis. J Virol 76: 10393-10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DA, Llado J, Shamblott MJ, Maragakis NJ, Irani DN, Crawford TO, Krishnan C, Dike S, Gearhart JD, Rothstein JD (2003) Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci 23: 5131-5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krampfl K, Schlesinger F, Wolfes H, Dengler R, Bufler J (2001) Functional diversity of recombinant human AMPA-type glutamate receptors: possible implications for selective vulnerability of motor neurons. J Neurol Sci 191: 19-23.11676988 [Google Scholar]
- Kriz J, Gowing G, Julien JP (2003) Efficient three-drug cocktail for disease induced by mutant superoxide dismutase. Ann Neurol 53: 429-436. [DOI] [PubMed] [Google Scholar]
- Leis AA, Stokic DS, Polk JL, Dostrow V, Winkelmann M (2002) A poliomyelitis-like syndrome from West Nile virus infection. N Engl J Med 347: 1279-1280. [DOI] [PubMed] [Google Scholar]
- Levine B, Griffin DE (1993) Molecular analysis of neurovirulent strains of Sindbis virus that evolve during persistent infection of SCID mice. J Virol 67: 6872-6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Goldman JE, Jiang HH, Griffin DE, Hardwick JM (1996) Bc1-2 protects mice against fatal alphavirus encephalitis. Proc Natl Acad Sci USA 93: 4810-4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DL, DeCamillis MA, Brunetti CR, Halder G, Kassner VA, Selegue JE, Higgs S, Carroll SB (1999) Ectopic gene expression and homeotic transformations in arthropods using recombinant Sindbis viruses. Curr Biol 9: 1279-1287. [DOI] [PubMed] [Google Scholar]
- Lewis J, Wesselingh SL, Griffin DE, Hardwick JM (1996) Alphavirus-induced apoptosis in mouse brains correlates with neurovirulence. J Virol 70: 1828-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Goldman JE, Jiang HH, Levine B (1999) Resistance of interleukin-1β-deficient mice to fatal Sindbis virus encephalitis. J Virol 73: 2563-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD (1998) Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20: 589-602. [DOI] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L (1996) Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol 40: 759-766. [DOI] [PubMed] [Google Scholar]
- Mokhtarian F, Grob D, Griffin DE (1989) Role of the immune response in Sindbis virus-induced paralysis of SJL/J mice. J Immunol 143: 633-637. [PubMed] [Google Scholar]
- Namura S, Maeno H, Takami S, Jiang XF, Kamichi S, Wada K, Nagata I (2002) Inhibition of glial glutamate transporter GLT-1 augments brain edema after transient focal cerebral ischemia in mice. Neurosci Lett 324: 117-120. [DOI] [PubMed] [Google Scholar]
- Nargi-Aizenman JL, Griffin DE (2001) Sindbis virus-induced neuronal death is both necrotic and apoptotic and is ameliorated by N-methyl-d-aspartate receptor antagonists. J Virol 75: 7114-7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargi-Aizenman JL, Havert MB, Zhang M, Irani DN, Rothstein JD, Griffin DE (2004) Glutamate receptor antagonists protect from virus-induced neural degeneration. Ann Neurol 55: 541-549. [DOI] [PubMed] [Google Scholar]
- Nava VE, Rosen A, Veliuona MA, Clem RJ, Levine B, Hardwick JM (1998) Sindbis virus induces apoptosis through a caspase-dependent, CrmA-sensitive pathway. J Virol 72: 452-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Takano T, Hansen AJ (2002) Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci 3: 748-755. [DOI] [PubMed] [Google Scholar]
- Pitt D, Werner P, Raine CS (2000) Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med 6: 67-70. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Tani M (1998) Do chemokines mediate leukocyte recruitment in post-traumatic CNS inflammation? Trends Neurosci 21: 154-159. [DOI] [PubMed] [Google Scholar]
- Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, Rothstein JD, Dempsey RJ (2001a) Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci 21: 1876-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VL, Bowen KK, Dempsey RJ (2001b) Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem Res 26: 497-502. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Djali S, Buchhalter JR (1993) Inhibition of glutamate uptake with l-trans-pyrrolidine-2,4-dicarboxylate potentiates glutamate toxicity in primary hippocampal cultures. J Neurochem 61: 2099-2103. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E (1989) Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett 103: 162-168. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, Leitner M (1992) Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci 12: 56-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW (1992) Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med 326: 1464-1468. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW (1993) Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA 90: 6591-6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13: 713-725. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38: 73-84. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675-686. [DOI] [PubMed] [Google Scholar]
- Rowell JF, Griffin DE (1999) The inflammatory response to nonfatal Sindbis virus infection of the nervous system is more severe in SJL than in BALB/c mice and is associated with low levels of IL-4 mRNA and high levels of IL-10-producing CD4+ T-cells. J Immunol 162: 1624-1632. [PubMed] [Google Scholar]
- Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, Coulter DA, Rothstein JD (2002) A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci 22: 6372-6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Leszczyniecka M, Kang DC, Sarkar D, Chao W, Volsky DJ, Fisher PB (2003) Insights into glutamate transport regulation in human astrocytes: cloning of the promoter for excitatory amino acid transporter 2 (EAAT2). Proc Natl Acad Sci USA 100: 1955-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276: 1699-1702. [DOI] [PubMed] [Google Scholar]
- Thach DC, Kimura T, Griffin DE (2000) Differences between C57BL/6 and BALB/cBy mice in mortality and virus replication after intranasal infection with neuroadapted Sindbis virus. J Virol 74: 6156-6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE (2001) Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol 166: 7527-7533. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21: 2580-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberghe W, Van den Bosch L, Robberecht W (1998) Glial cells potentiate kainate-induced neuronal death in a motoneuron-enriched spinal coculture system. Brain Res 807: 1-10. [DOI] [PubMed] [Google Scholar]
- Van den Bosch L, Robberecht W (2000) Different receptors mediate motorneuron death induced by short and long exposures to excitotoxicity. Brain Res Bull 53: 383-388. [DOI] [PubMed] [Google Scholar]
- Van den Bosch L, Van Damme P, Vleminckx V, Van Houtte E, Lemmens G, Missiaen L, Callewaert G, Robberecht W (2002a) An α-mercaptoacrylic acid derivative (PD150606) inhibits selective motor neuron death via inhibition of kainate-induced Ca2+ influx and not via calpain inhibition. Neuropharmacology 42: 706-713. [DOI] [PubMed] [Google Scholar]
- Van den Bosch L, Schwaller B, Vleminckx V, Meijers B, Stork S, Ruehlicke T, Van Houtte E, Klaassen H, Celio MR, Missiaen L, Robberecht W, Berchtold MW (2002b) Protective effect of parvalbumin on excitotoxic motor neuron death. Exp Neurol 174: 150-161. [DOI] [PubMed] [Google Scholar]
- Van den Bosch L, Tilkin P, Lemmens G, Robberecht W (2002c) Minocycline delays disease onset and mortality in a transgenic model of ALS. NeuroReport 13: 1067-1070. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19: 6897-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warita H, Manabe Y, Murakami T, Shiote M, Shiro Y, Hayashi T, Nagano I, Shoji M, Abe K (2002) Tardive decrease of astrocytic glutamate transporter protein in transgenic mice with ALS-linked mutant SOD1. Neurol Res 24: 577-581. [DOI] [PubMed] [Google Scholar]
- Wesselingh SL, Levine B, Fox RJ, Choi S, Griffin DE (1994) Intracerebral cytokine mRNA expression during fatal and nonfatal alphavirus encephalitis suggests a predominant type 2 T-cell response. J Immunol 152: 1289-1297. [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J (1998) Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA 95: 15769-15774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Narayanan M, Friedlander RM (2003) Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann Neurol 53: 267-270. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM (2002) Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 417: 74-78. [DOI] [PubMed] [Google Scholar]