

Abstract
The initiation of the psychostimulant sensitization process depends on the mesolimbic system, which projects from the ventral tegmental area (VTA) to the nucleus accumbens. Although such initiation is primarily dependent on glutamatergic activity in VTA neurons, the exact role VTA excitatory synapses play in this process is poorly understood. Here, we examine the effects of repeated in vivo injections of cocaine on the magnitude and duration of the increase in strength at VTA excitatory synapses reported previously to be elicited by a single in vivo exposure to cocaine (Ungless et al., 2001; Saal et al., 2003). We also compare the synaptic modifications induced by cocaine with its effects on locomotor activity. Surprisingly, repeated cocaine exposure potentiated the ratio of AMPA receptor-mediated to NMDA receptor-mediated EPSCs to a similar extent and duration as a single in vivo cocaine exposure. In naive animals, the magnitude of the cocaine-induced locomotor activity after a single injection of cocaine correlated with the magnitude of the accompanying synaptic enhancement. This correlation was lost on the seventh day of repeated cocaine administration, as well as when a challenge injection was given 10 d after the cessation of repeated cocaine administration. These results suggest that the cocaine-induced synaptic plasticity at VTA excitatory synapses is transient, and its duration depends on the last exposure to cocaine. Furthermore, chronic cocaine exposure disrupts the normal, presumably adaptive relationship between synaptic enhancement in the VTA and behavior.
Keywords: cocaine, synaptic plasticity, VTA, dopamine, AMPA, NMDA, behavioral sensitization
Introduction
Repeated administration of psychostimulants, such as cocaine, produces an augmented locomotor response to a subsequent psychostimulant challenge injection. This process is referred to as behavioral sensitization (Segal and Schuckit, 1983; Robinson and Becker, 1986; Post and Weiss, 1988; Kalivas and Stewart, 1991) and is a prominent animal model for the intensification of drug craving in humans (Robinson and Berridge, 1993). Development of behavioral sensitization can be conceptualized into two distinct temporal domains termed initiation and expression (Robinson and Becker, 1986; Kalivas and Stewart, 1991). The initiation of psychostimulant-mediated behavioral sensitization is thought to involve transient cellular and molecular changes in the ventral tegmental area (VTA), which, with the nucleus accumbens (NAc), constitutes the mesolimbic dopamine (DA) system. For example, administration of DA receptor antagonists into the VTA disrupts the development of sensitization to amphetamine (Vezina and Stewart, 1989), whereas repeated administration of drugs that affect DA receptor signaling directly into the VTA elicit a sensitized motor response to systemically administered amphetamine or cocaine (Vezina, 1993; Perugini and Vezina, 1994; Bjijou et al., 1996; Pierce et al., 1996a; Cornish and Kalivas, 2001).
Although psychostimulants, such as cocaine, directly affect DA receptor-mediated signaling in a manner that is critical for behavioral sensitization, several lines of evidence suggest that excitatory synaptic transmission in the VTA also plays a critical role. First and most importantly, initiation of sensitization is prevented by intra-VTA administration of NMDA receptor (NMDAR) antagonists (Kalivas and Alesdatter, 1993; Wolf et al., 1993a; Vezina and Queen, 2000). Second, electrophysiological studies have demonstrated an increase in the responsiveness of VTA dopamine neurons to the excitatory effects of AMPA after repeated amphetamine or cocaine administration (White et al., 1995; Zhang et al., 1997). Third, shortly after discontinuation of repeated amphetamine administration, intra-VTA microinjections of AMPA increased both NAc and VTA dopamine levels, suggesting the increased ability of VTA AMPA receptors (AMPARs) to regulate dopamine and glutamate transmission in the VTA and NAc (Giorgetti et al., 2001). Thus, potentiation of glutamatergic synaptic transmission in the VTA could account for the transient increase in dopamine neuronal activity thought to be critical in transferring sensitization to forebrain regions such as the NAc that are important in its maintenance and expression (Wolf, 1998; Vandenschuren and Kalivas, 2000). Indeed, our laboratory and others have recently demonstrated that AMPAR-mediated synaptic transmission is strengthened in the VTA after even a single in vivo exposure to cocaine, amphetamine, and other drugs of abuse (Ungless et al., 2001; Faleiro et al., 2003; Saal et al., 2003). If this drug-induced enhanced synaptic transmission in the VTA is a component of the neural adaptations that lead to locomotor sensitization as well as the accompanying sensitization of the incentive-motivational system (Robinson and Berridge, 1993), it is important to further examine its detailed properties. In the present study, we sought to determine how excitatory synaptic transmission in the VTA is modulated by repeated cocaine exposure and whether the consequent changes in excitatory synaptic strength are correlated with the effects of cocaine on locomotor activity.
Materials and Methods
Treatment regimen and locomotor activity. Male Sprague Dawley rats [postnatal day 26 (P26)-P40; Charles River Laboratories, Hollister, CA] were housed two per cage in treatment groups with food and water available ad libidum. A 12 hr light/dark cycle was used, with the lights on at 7:00 A.M. All cocaine injections and behavioral testing were performed during the light cycle. Rats were given either a single injection (when aged P26-P28) or multiple injections of cocaine (initiated at P19-P21). The multiple cocaine injection treatment protocol used here has been shown to induce behavioral sensitization and alterations in glutamate neurotransmission in the nucleus accumbens (Pierce et al., 1996b). All animals were habituated to the photocell boxes (MED Associates, Inc., St. Albans, VT) for 3 hr 1 d before the start of the experiment. On the first treatment day, all animals were further habituated to the photocell boxes for 1 hr. After habituation, animals received either cocaine (15 mg/kg, i.p.) or saline (0.9% NaCl), and subsequent behavior was monitored for 1 hr. For rats receiving multiple injections, on days 2-6, rats received daily injections of cocaine (30 mg/kg, i.p.) or saline in the home cage. On day 7, all animals were habituated again to the photocell boxes for 1 hr, followed by intraperitoneal administration of 15 mg/kg cocaine or saline, and locomotor behavior was monitored for 60 min after injection. In some experiments, sensitized rats (12 of 16) were retested for locomotor activity 10 d after the chronic cocaine treatment with 15 mg/kg intraperitoneal cocaine or saline. To permit a more accurate quantification in changes in photocell counts in all experiments, a lower dose of cocaine was administered for behavioral testing because this dose is approximately the ED50 for cocaine-induced increases in horizontal photocell counts (Kalivas and Weber, 1988). Locomotor activity was measured in 17 × 17 inch chambers lined with three 16-beam infrared arrays. A 50 msec scanning rate was used for measuring beams broken. Distance traveled and stereotypic counts were measured using Open Field Activity software (MED Associates, Inc., St. Albans, VT). Distance traveled was analyzed for estimates of locomotion based on the movement of a given distance and resting delay (movement in a given period). Stereotypy was measured as consecutive breaking of the same photobeam that indiscriminately includes scratching and grooming, as well as more classic psychostimulant-induced stereotypies such as focused sniffing and head bobbing (Kalivas et al., 1988; Kalivas and Alesdatter, 1993; Cornish and Kalivas, 2001).
Electrophysiology. All the electrophysiological recordings were performed in rats ranging in age from P26 to P40. Briefly, rats were anesthetized with halothane and killed. Horizontal sections of the VTA (230 μm) were prepared with a vibratome (Leica, Nussloch, Germany). Slices were placed in a holding chamber and allowed to recover for at least 1 hr before being placed in the recording chamber and superfused with bicarbonate-buffered solution (artificial CSF) saturated with 95% O2 and 5% CO2 and containing (in mm): 119 NaCl, 1.6 KCl, 1.0 NaH2PO4, 1.3 MgCl2, 2.5 CaCl2, 26.2 NaHCO3, and 11 glucose (at 32-34°C). Picrotoxin (100 μm) was added to block GABAA receptor-mediated IPSPs. Cells were visualized using infrared differential interference contrast video microscopy. Whole-cell voltage-clamp recordings were made using an Axopatch 1D amplifier (Axon Instruments, Union City, CA). Electrodes (2.8-4.0 MΩ) contained (in mm): 120 cesium methanesulfonate, 20 HEPES, 0.4 EGTA, 2.8 NaCl, 5 TEA-Cl, 2.5 MgATP, and 0.25 NaGTP, pH 7.2-7.3 (270-285 mOsm). Series resistance (10-40 MΩ) and input resistance were monitored on-line with a 4 mV depolarizing step (50 msec) given after every afferent stimulus. Dopaminergic VTA neurons were identified by the presence of a large Ih current (Lacey et al., 1990; Johnson and North, 1992). Because Ih is present in both principal and tertiary VTA neurons (Margolis et al., 2003), we recognize that its presence does not unequivocally identify dopamine neurons in midbrain slices. However, in previous work (Ungless et al., 2001; Saal et al., 2003) and in the present study, this criterion was enough to obtain clear differences between control and experimental treatments. A bipolar stimulating electrode was placed 100-300 μm rostral to the recording electrode and was used to stimulate excitatory afferents at 0.1 Hz. Neurons were voltage-clamped at -70 mV and 40 mV to record AMPAR- and NMDAR-mediated EPSCs. EPSCs were filtered at 2 kHz, digitized at 5-10 kHz, and collected on-line using Igor Pro software (Wavemetrics, Lake Oswego, OR). To calculate the decay (τ) of NMDAR-mediated EPSCs, Igor Pro software fit exponential decay curves using the formula y = Aexp(-invτ × x).
To calculate the AMPAR/NMDAR ratio, an average of 12 EPSCs at 40 mV was computed before and after application of the NMDAR blocker AP-5 (50 μm) for 5 min. NMDAR responses were calculated by subtracting the average response in the presence of AP-5 (AMPAR only) from that seen in its absence; the peak of the AMPAR EPSC was divided by the peak of the NMDAR EPSC to yield an AMPAR/NMDAR ratio. One caveat of this technique is that AMPAR- and NMDAR-mediated EPSCs are recorded at a nonphysiological potential (40 mV), resulting in an outward current. At this potential, it is possible that a series of intracellular events might be triggered as a consequence of this robust depolarization. However, at 40 mV, both AMPAR and NMDAR have a maximal open probability, and thus it is easier to make a reliable comparison between AMPAR- and NMDAR-mediated EPSCs using the same stimulation intensity. The current-voltage relationship for AMPAR- and NMDAR-mediated currents at 40 mV is linear and superimposeable. Consequently, changes in membrane potential will affect both currents to the same extent.
Data analysis. All values are expressed as mean ± SEM. Statistical significance was assessed using two-tailed Student's t tests or a two-way ANOVA for multiple group comparisons. A Bonferroni post hoc test after an ANOVA was used to test significant differences between multiple groups. In each set of experiments, there were no differences in the results from blinded and nonblinded experiments; therefore, results were combined. Correlation analysis was performed to determine whether there was a significant association between the AMPAR/NMDAR ratio and locomotor activity. If more than one AMPAR/NMDAR ratio was recorded per rat, the mean AMPAR/NMDAR ratio was correlated to that rat's locomotor response. A regression slope and Pearson's correlation coefficient were calculated using GraphPad (San Diego, CA) Prism version 4 software.
Results
Effect of multiple injections of cocaine on the AMPAR/NMDAR ratio
In our first series of experiments, we examined whether repeated cocaine administration increased the magnitude or duration of the increase in AMPAR-mediated synaptic transmission previously found to occur after a single in vivo exposure to cocaine or other drugs of abuse (Ungless et al., 2001; Saal et al., 2003). To evaluate synaptic strength, we measured the relative contribution of AMPARs and NMDARs to the EPSCs because this measure minimizes the effects of slice-to-slice variability and has been used successfully in the past (Ungless et al., 2001; Saal et al., 2003).
The chronic treatment protocol involved injecting rats with cocaine over 7 consecutive days in a manner known to induce behavioral sensitization as well as alterations in dopamine and glutamate transmission (Kalivas and Duffy, 1993; Pierce et al., 1996b) (Fig. 1A). Rats injected with multiple doses of cocaine exhibited a significantly larger AMPAR/NMDAR ratio on day 8 (0.76 ± 0.09; n = 30) compared with controls receiving multiple injections of saline (0.38 ± 0.04; n = 12; p > 0.05) or naive rats (0.33 ± 0.08; n = 8; p < 0.05) (Fig. 1B,C). The magnitude of this increase, however, was similar to that elicited by a single injection of cocaine (0.64 ± 0.05; n = 29, p < 0.05).
Figure 1.
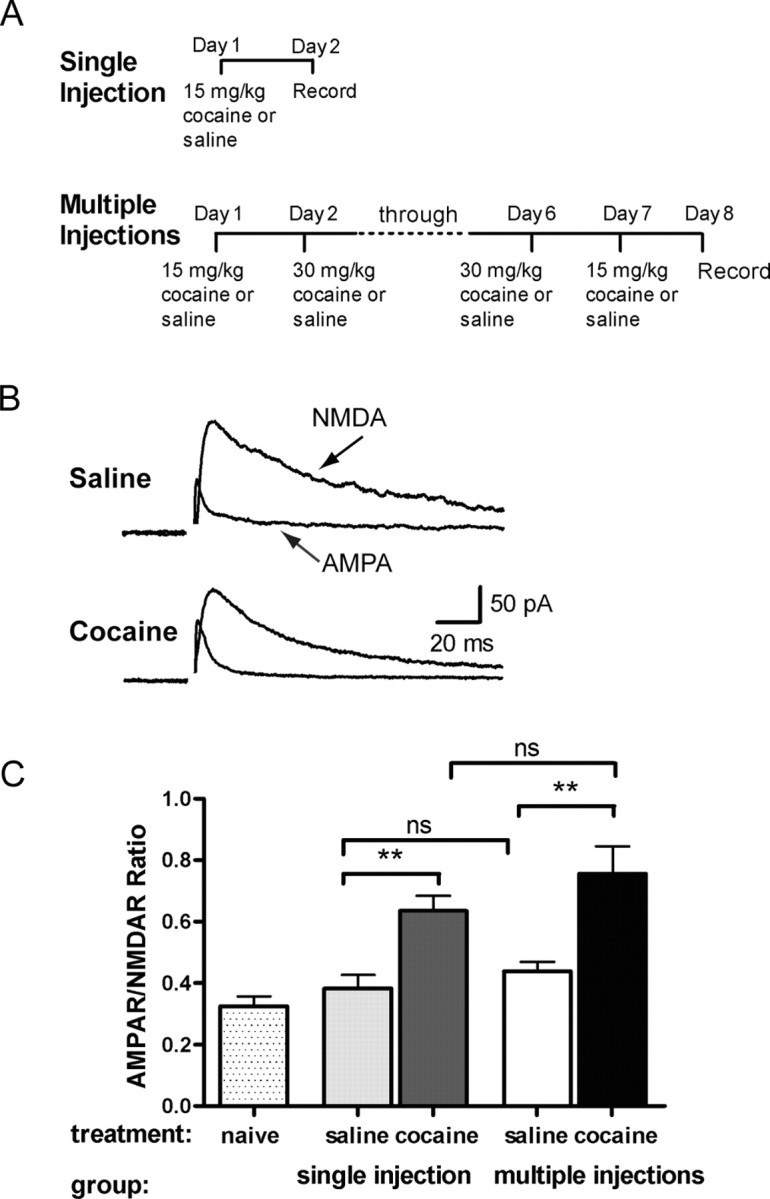
Multiple injections of cocaine potentiate AMPAR/NMDAR ratio to a similar extent as a single injection of cocaine. A, Diagram of injection protocol for single or multiple injections. B, Example recordings of evoked NMDA and AMPA EPSCs from VTA neurons of rats 24 hr after treatment with seven consecutive injections of cocaine or saline. C, Rats were given a single injection of cocaine (15 mg/kg; dark shaded bar) or saline (light shaded bar), multiple injections of cocaine (filled bars) or saline (open bars), or no treatment (dotted bars). The AMPAR/NMDAR ratio was taken as the peak amplitude of AMPAR current over peak amplitude of NMDAR current. AMPAR/NMDAR ratio of cells from rats treated with a single injection (n = 19) or multiple injections (n = 30) of cocaine was significantly larger than saline (n = 17; p < 0.05; n = 27; p < 0.01, respectively) or naive (n = 8) controls. Bars represent mean AMPAR/NMDAR ratio and SEM.
The increase in the relative ratio between AMPAR- and NMDAR-mediated currents may be attributable to various factors: an increase in AMPAR number and/or function, a decrease in NMDAR number and/or function, or a change in both. Therefore, AMPA (10 μm for 30 sec) with the AMPAR desensitization inhibitor cyclothiazide (100 μm) was superfused onto neurons held at -70 mV in both saline-treated animals and in animals chronically exposed to cocaine. Similarly to what we observed after a single injection of cocaine (Ungless et al., 2001), AMPAR-mediated currents were significantly increased in rats repeatedly exposed to cocaine (-171 ± 29 pA; n = 4) (Fig. 2A) compared with rats injected with saline (-80 ± 17 pA; n = 5; p < 0.05) (Fig. 2A). Attenuation of NMDAR function has been demonstrated in situations with increased levels of calmodulin, resulting in Ca2+-dependent inactivation (Ehlers et al., 1996) or reduced postsynaptic density-95 levels (Roche et al., 2001). Thus, to determine whether a significant change in NMDAR function had occurred as a consequence of chronic cocaine exposure, we superfused NMDA (10 μm, 30 sec) onto neurons and found no difference in current amplitude between rats chronically treated with cocaine (46 ± 8 pA; n = 5) or saline (40 ± 8 pA; n = 6; p > 0.5) (Fig. 2B). Finally, chronic cocaine exposure may have produced an alteration in NMDAR subunit composition that could be reflected by a specific change in the decay time of evoked NMDAR-mediated EPSCs (Cull-Candy et al., 2001). However, an analysis of the time constant of decay of NMDAR-mediated currents did not reveal any difference between rats treated with chronic cocaine (τ = 50 ± 7 msec; n = 15) and those treated with saline (τ = 53 ± 9 msec; n = 15; p > 0.05). Taken together, these results suggest that the increase in AMPAR/NMDAR ratio after chronic cocaine treatment is primarily attributable to an increase in AMPAR function and/or number, whereas VTA NMDAR function is not altered by the chronic cocaine exposure.
Figure 2.
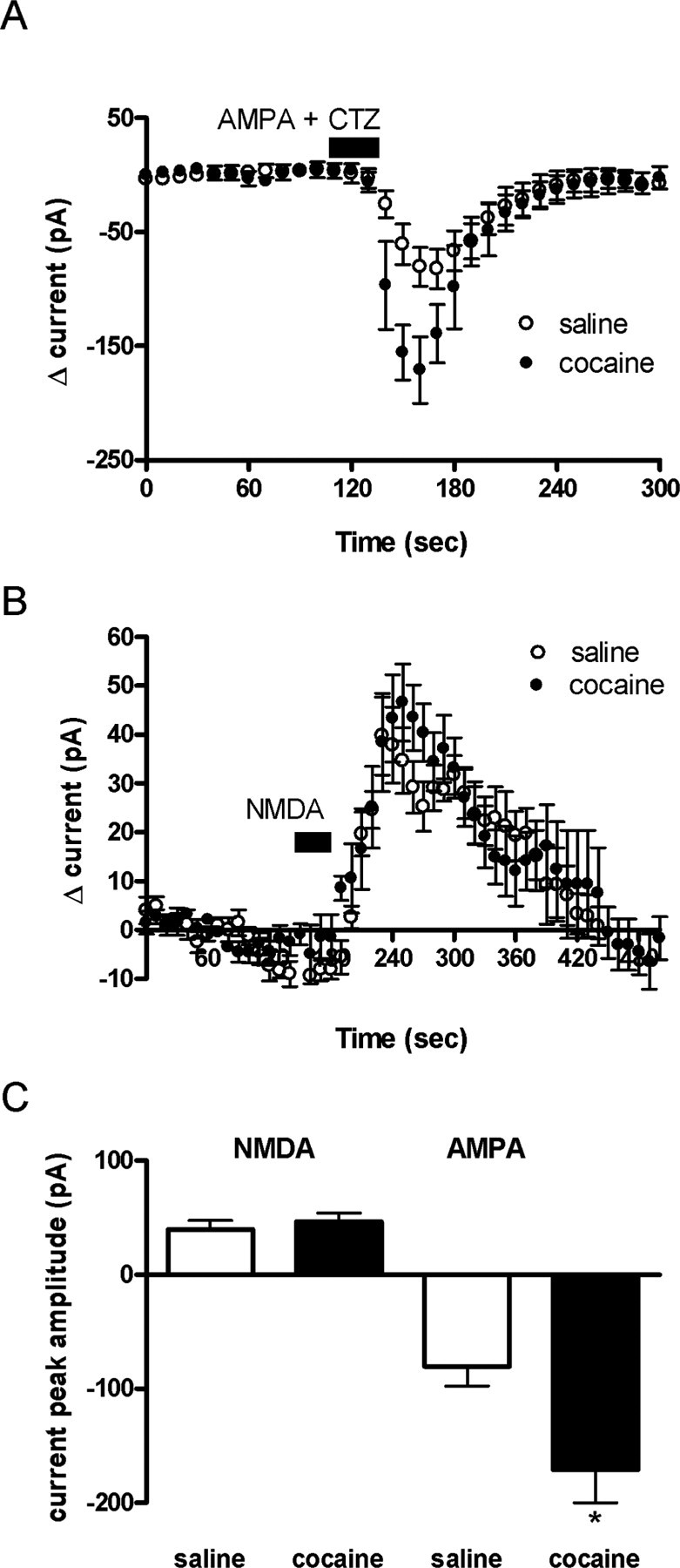
Potentiation of AMPAR/NMDAR ratio after multiple injections of cocaine is attributable to increases in AMPA current. A, Bath application of AMPA (10 μm) with cyclothiazide (100 μm) for 30 sec elicited a greater inward current (10 sec bins) in chronic cocaine-treated animals (n = 4) than in saline-treated animals (n = 5; p < 0.05). B, In neurons voltage-clamped at 40 mV, bath application of NMDA (10 μm) for 30 sec elicited similar current (10 sec bins) in both chronic cocaine- and saline-treated animals (n = 5 and 6; p < 0.05). C, Maximal current change after 30 sec AMPA or NMDA application from chronic cocaine- or saline-treated rats. Bars represent means and SEM.
Because a single cocaine exposure in mice transiently enhanced synaptic strength for 5 but not 10 d after the injection (Ungless et al., 2001), we proceeded to characterize the duration of the synaptic potentiation after repeated cocaine injections. Similar to mice, rats receiving a singleinjection of cocaine had a potentiated AMPAR/NMDAR ratio after 5 d (cocaine, 0.67 ± 0.08; n = 9; vs saline, 0.39 ± 0.07; n = 6; p < 0.05) but not 10 d after withdrawal (cocaine, 0.38 ± 0.08; n = 5; vs saline, 0.44 ± 0.03; n = 5; p > 0.05) (Fig. 3B). Repeated daily injections of cocaine for 7 d did not prolong the duration of this synaptic modification because, again, a significant increase in the AMPAR/NMDAR ratio was observed 5 d (saline, 0.39 ± 0.07; n = 6; cocaine, 0.67 ± 0.08; n = 9; p < 0.05) but not 10 d after these multiple injections (saline, 0.38 ± 0.06; n = 10; cocaine, 0.47 ± 0.05; n = 8; p > 0.05) (Fig. 3C). These results suggest that the cocaine-induced synaptic potentiation is transient, regardless of the number of injections of cocaine, and that the potentiation of VTA excitatory synaptic transmission isdependent on the last exposure to cocaine, rather than on the number of exposures.
Figure 3.

Multiple injections of cocaine potentiate AMPAR/NMDAR ratio lasting for 5 but not 10 d. A, Diagram of injection protocol for single or multiple injections. Rats were treated with single (B) or multiple (C) injections of saline (open bars) or cocaine (filled bars) as previously described. Recordings of AMPAR/NMDAR ratio made 5 d after the final cocaine injection (single injection, n = 9; multiple injections, n = 8) were significantly larger than the saline controls (*p < 0.05; single, n = 6; **p < 0.01; multiple, n = 10). However, AMPAR/NMDAR ratio 10 d after the final cocaine injection (n = 10) was not significantly different from that of the saline controls (n = 10). Bars represent mean AMPAR/NMDAR ratio and SEM.
Correlation of locomotor activity to AMPAR/NMDAR ratio after single or multiple injections of cocaine
Increases in the level of excitatory amino acids and their receptors in the VTA appear to be correlated with the initiation of behavioral sensitization (Kalivas and Alesdatter, 1993; Fitzgerald et al., 1996). We therefore investigated the relationship between increased synaptic strength in the VTA, as measured by potentiation of the AMPAR/NMDAR ratio and cocaine-induced locomotor activity, as well as behavioral sensitization. Rats treated with a single injection of cocaine had increased average ambulatory and vertical photocell counts (data not shown), as well as average distance traveled (3887 ± 707 cm; n = 11), compared with saline-treated animals (581 ± 104 cm; n = 10; p < 0.01) (Fig. 4A) when assayed immediately after injection. Additionally, acute cocaine treatment increased average stereotypic counts (1588 ± 232; n = 5; vs 7059 ± 634; n = 8; p < 0.001) (Fig. 4B). To test whether the synaptic plasticity observed after a single injection of cocaine correlated with its effect on locomotor activity in individual rats, cocaine-induced locomotor activation was compared with the AMPAR/NMDAR ratios measured the following day (Fig. 1B). Indeed, the distance traveled positively correlated with AMPAR/NMDAR ratio after a single injection of cocaine [Pearson's r = 0.85; confidence interval (CI) = 0.37-0.97; n = 8; p < 0.01] such that the greater the acute cocaine effect on locomotor activity, the greater the change in AMPAR/NMDAR ratio. Stereotypic counts did not significantly correlate with the AMPAR/NMDAR ratio for individual rats, although the data exhibited a positive trend (Fig. 4D) (Pearson's r = 0.62; CI = -0.15-0.92; n = 8; p = 0.10). In contrast to the effect of cocaine, saline-injected rats did not exhibit a correlation between distance traveled or stereotypy and the AMPAR/NMDAR ratio (distance traveled, Pearson's r = 0.35; CI = -0.77-0.94; n = 5) (Fig. 4E) (stereotypic counts, Pearson's r = -0.27; CI = -0.93-0.80; n = 5) (Fig. 4F).
Figure 4.

Locomotor activity positively correlates with potentiated AMPAR/NMDAR ratio after a single injection of cocaine. Rats were monitored for locomotor activity for 60 min immediately after a single cocaine (15 mg/kg) or saline injection. A, B, Distance traveled (A) and stereotypic counts (B) increased after cocaine (n = 11) compared with saline (n = 10; p < 0.01). Bars represent mean AMPAR/NMDAR ratio and SEM. C, D, Distance traveled (C) and stereotypic counts (D) correlated positively with the AMPAR/NMDAR ratio of cocaine-treated rats (distance traveled, r = 0.85; p < 0.01; n = 8; stereotypic counts, r = 0.62; p = 0.1; n = 8; filled squares). E, F, In saline-treated rats, distance traveled (E) and stereotypy (F) did not correlate with AMPAR/NMDAR ratio (distance traveled, r = 0.35; n = 5; stereotypy, r = -0.27; n = 5; open squares). The mean of AMPAR/NMDAR ratios per rat was compared with its locomotor activity.
We next examined the degree of behavioral sensitization resulting from seven daily cocaine or saline injections. As expected, rats treated with multiple injections of cocaine had significantly increased locomotor activity on day 7 (5920 ± 925 cm; n = 11) compared with day 1 (3603 ± 821 cm; n = 11; p < 0.05) (Fig. 5A) as well as an increase in stereotypic counts (day 7, 8523 ± 490; day 1, 6124 ± 391; p < 0.01) (Fig. 5B). Saline-injected animals, on the other hand, had no significant change in locomotor activity from day 1 (912 ± 178 cm; n = 13) to day 7 (976 ± 109 cm; n = 13; p > 0.05) nor any change in stereotypy response (Fig. 5A,B).
Figure 5.

After multiple injections of cocaine, resulting in sensitization, locomotor activity no longer correlates with AMPAR/NMDAR ratio. Rats were monitored for locomotor activity on the first (day 1) and the last (day 7) day of injection regimen. A, B, Distance traveled (A) (*p < 0.05) and stereotypic counts (B) (**p < 0.01) were greater on days 1 and 7 in cocaine-treated rats compared with saline-treated rats (cocaine, n = 11; saline, n = 12; p < 0.01). C, D, Correlation of distance traveled (C) and stereotypic counts (D) on the last injection day (day 7) and AMPAR/NMDAR ratio in rats treated with multiple injections of cocaine (n = 11). E, F, Correlation of the relative sensitization, the ratio of distance traveled (E) and stereotypic counts (F) on days 7 and 1, and AMPAR/NMDAR ratio in cocaine-treated rats (n = 11). G, H, Correlation of distance traveled (G) and stereotypic counts (H) on day 7, and AMPAR/NMDAR ratio in saline-treated rats (n = 12). Bars represent mean AMPAR/NMDAR ratio and SEM. The mean of AMPAR/NMDAR ratios per rat was compared with its locomotor activity.
To determine whether the magnitude of the potentiation of VTA synaptic strength measured after chronic cocaine treatment correlated with the degree of behavioral sensitization, we compared the AMPAR/NMDAR ratio measured on day 8 after the 7 d of repeated cocaine or saline injection treatment with the distance traveled or stereotypic counts measured on the final day of injection. Unlike the positive correlation observed with a single injection of cocaine, in rats treated with multiple injections, there was no longer a correlation between the AMPAR/NMDAR ratio and distance traveled (Pearson's r = -0.003; CI = -0.60-0.60; n = 11) (Fig. 5C) or stereotypic counts (Pearson's r = -0.47; CI = -0.47-0.17; n = 11) (Fig. 5G). After multiple saline injections, distance traveled (Pearson's r = -0.29; CI = -0.74-0.34; n = 12) (Fig. 5D) or stereotypic counts (Pearson's r = -0.16; CI = -0.67-0.45; n = 12) (Fig. 5H) also did not correlate with AMPAR/NMDAR ratios.
In contrast to the robust positive correlation between the locomotor effects of a single cocaine injection and its effects on VTA synaptic strength, there was no correlation between the AMPAR/NMDAR ratio and the degree of sensitization measured either by distance traveled (Pearson's r = -0.25; CI = -0.74-0.41; n = 11) (Fig. 5E) or stereotypic activity (Pearson's r = -0.39; CI = -0.8-0.27; n = 11) (Fig. 5F). Thus, the correlation between cocaine-induced locomotor activity and the AMPAR/NMDAR ratio was lost as the rats became sensitized to cocaine.
Effect of a cocaine challenge injection after withdrawal from chronic cocaine treatment
To determine whether VTA synaptic strength could again be potentiated after withdrawal from the chronic 7 d cocaine treatment, we tested the effect of a single injection of cocaine 10 d after the last cocaine injection. Cocaine-pretreated rats given a cocaine injection 10 d later had larger AMPAR/NMDAR ratios (0.71 ± 0.06; n = 12) than cocaine-pretreated rats receiving a saline challenge injection (0.45 ± 0.03; n = 7; p < 0.05) or saline-pretreated rats receiving a saline challenge injection (0.42 ± 0.02; n = 11; p < 0.01) (Fig. 6B). Rats pretreated with saline and injected with cocaine after 10 d of no injections also had a potentiated AMPAR/NMDAR ratio (0.62 ± 0.08; n = 9; p < 0.05) (Fig. 6B) that was not significantly different from that of the cocaine-pretreated animals. This suggests that cocaine pretreatment did not affect the potentiation of VTA synaptic strength elicited by a subsequent single injection of cocaine.
Figure 6.

After 10 d withdrawal from multiple cocaine injections, a challenge injection of cocaine reinstates potentiated AMPAR/NMDAR ratio in sensitized rats. A, Diagram of injection regimen. Distance traveled was measured on the first (day 1) and last (day 7) days of the multiple injection regimen, and then distance traveled was measured again immediately after a challenge injection of cocaine (15 mg/kg) or saline 10 d later (day 17). AMPAR/NMDAR ratios were measured 24 hr after the challenge injection (day 18). B, Rats pretreated with cocaine and given a cocaine challenge injection (filled bar; n = 12) and rats pretreated with saline and given a cocaine challenge injection (light shaded bar; n = 9) had potentiated AMPAR/NMDAR ratios compared with rats pretreated with cocaine and given a saline challenge injection (dark shaded bar; n = 7) and rats pretreated with saline and given a saline challenge injection (open bar; n = 11). C, Rats pretreated with multiple injections of cocaine and a challenge injection of cocaine after 10 d of withdrawal (filled circles; n = 6), had elevated locomotor activity compared with rats pretreated with saline and challenged with a cocaine injection (open circles; n = 8; p < 0.01). Rats pretreated with cocaine did not show enhanced locomotor response to a saline challenge (filled squares; n = 6). Rats pretreated with saline and given a challenge injection of saline (open squares; n = 8) represented baseline locomotor activity. D, Locomotor activity on day 17 in rats pretreated with cocaine receiving a cocaine (filled bar; n = 6) or saline (dark shaded bar; n = 6) challenge injection and rats pretreated with saline receiving a cocaine (light shaded bar; n = 8) or saline (open bar; n = 8) challenge injection. Bars represent mean cumulative distance traveled and SEM. p values were calculated using a two-way ANOVA.
Although the AMPAR/NMDAR ratio was no longer potentiated 10 d after the chronic cocaine pretreatment, behavioral sensitization was still present as evidenced by an enhanced locomotor response to cocaine when compared with the cocaine-induced locomotor response of saline-pretreated animals (Fig. 6C,D). This indicates that enhanced synaptic strength in the VTA is not required for the expression of behavioral sensitization.
In a final analysis, we examined whether the chronic cocaine pretreatment affected the correlation that was observed in naive animals between the AMPAR/NMDAR ratio and the acute cocaine-induced locomotor activity. Consistent with our previous results, in saline-pretreated animals there was a positive correlation between the AMPAR/NMDAR ratio and the distance traveled (Pearson's r = 0.75; CI = 0.31-0.92; p < 0.01; n = 12) (Fig. 7A) as well as stereotypic counts (Pearson's r = 0.63; CI = 0.08-0.88; p < 0.05; n = 12) (Fig. 7B) in response to the cocaine injection on day 17. In contrast, this correlation disappeared in the animals that were pretreated with cocaine for 7 d and then also received a cocaine injection on day 17 (distance traveled, Pearson's r = -0.16; CI = -0.74-0.56; n = 9) (Fig. 7C) (stereotypic counts, Pearson's r = 0.05; CI = -0.64-0.69; n = 9) (Fig. 7D). Animals receiving a saline injection 10 d after multiple saline or cocaine injections also showed no correlation between the AMPAR/NMDAR ratio and distance traveled (saline, Pearson's r = 0.10; CI = -0.65-0.75; n = 8; cocaine, Pearson's r = -0.02; CI = -0.89-0.87; n = 5) or stereotypic counts (saline, Pearson's r = 0.41; CI = -0.41-0.86; n = 8; cocaine, Pearson's r = -0.03; CI = -0.89-0.87; n = 5). Thus, it is possible that the lack of correlation between behavioral sensitization and synaptic enhancement after chronic cocaine treatment may represent a change in the functional consequences of the cocaine-induced potentiation of excitatory synaptic transmission in the VTA.
Figure 7.

AMPAR/NMDAR ratios correlate with locomotor activity measured after a cocaine challenge injection in saline-pretreated rats but not in cocaine-pretreated rats. Rats were pretreated for 7 d with saline or cocaine and given a saline or cocaine (15 mg/kg) challenge injection after 10 d of withdrawal. Circles represent the correlate of distance traveled, and squares represent the correlate of stereotypic counts measured immediately after challenge injection to AMPAR/NMDAR ratio recorded 24 hr later. A, B, Correlation of distance traveled (A) (r = 0.75; p < 0.05; n = 12) and stereotypic counts (B) (r = 0.63; p < 0.05; n = 12) and AMPAR/NMDAR ratio in saline-pretreated rats given a cocaine challenge injection. C, D, Correlation of distance traveled (C) [n = 9 (1 point is occluded); r = -0.16] or stereotypic counts (D) (n = 9; r = 0.05) and AMPAR/NMDAR ratio in cocaine-pretreated rats given a cocaine challenge injection. The mean of AMPAR/NMDAR ratios per rat was compared with its locomotor activity.
Discussion
Cocaine-induced plasticity at VTA excitatory synapses is transient
Synaptic plasticity in the VTA has been suggested to play an important role in the behavioral consequences of in vivo exposure to drugs of abuse (Robinson and Berridge, 1993; Kalivas, 1995; Overton and Clark, 1997; Ungless et al., 2001; Everitt and Wolf, 2002). However, only the effects of single injections of psychoactive drugs have been examined directly for their effects on VTA excitatory synapses (Ungless et al., 2001; Faleiro et al., 2003; Saal et al., 2003). Important questions that had not yet been addressed are what effect chronic in vivo cocaine treatment has on synaptic strength in the VTA and whether these effects differ from those elicited by a single exposure to drugs of abuse. Surprisingly, we found that after chronic cocaine treatment, excitatory synapses in the VTA are potentiated to a similar extent and for a similar duration as that observed after a single in vivo injection of cocaine. Thus, the duration or total dose of cocaine treatment has little effect on at least one of the adaptations occurring at VTA excitatory synapses. Furthermore, because the cocaine-induced synaptic enhancement in the VTA remains transient, lasting 5 but not 10 d, and is independent of the number of times the animal has been exposed to cocaine, this synaptic adaptation cannot account for the expression or maintenance of behavioral sensitization but must play some other role in the behavioral responses to cocaine and presumably other drugs of abuse.
Other studies have also found transient alterations in the VTA after chronic exposure to cocaine. These include a transient increase in the responsiveness of VTA dopamine neurons to AMPA 3 d but not 10 d, after withdrawal from chronic cocaine treatment (Zhang et al., 1997), as well as a subsensitivity of dopamine autoreceptors that lasts 1-3 but not 10-14 d after chronic cocaine (Ackerman and White, 1990; Wolf et al., 1993b). Basal extracellular dopamine levels in the VTA are also elevated in cocaine-treated animals compared with saline-treated animals when measured 1 d after discontinuing daily treatments but not after 14 d (Kalivas and Duffy, 1993). Together these transient alterations, including the synaptic potentiation observed in this study, may contribute to the increase in the basal dopamine cell-firing rate, which has been observed after short withdrawals from repeated administration of cocaine (Henry et al., 1989). Furthermore, all of these findings are consistent with the hypothesis that the VTA plays an essential but transient role in the induction of sensitization (Robinson and Berridge 1993; Kalivas 1995; Wolf, 1998; Everitt and Wolf, 2002).
Cocaine-induced locomotor activity and synaptic enhancement in the VTA
We observed a strong positive correlation between the locomotor activity induced by a single dose of cocaine and the synaptic enhancement measured 1 d later in both naive animals and those pretreated with saline and receiving a single injection of cocaine 10 d later. We also observed a weaker correlation between cocaine-induced stereotypy and the synaptic enhancement. These correlations cannot be attributed to the locomotor activity itself enhancing VTA synaptic strength because animals that only received saline injections did not exhibit similar correlations. Because external stimuli that are associated with the firing of midbrain dopamine cells are granted motivational significance (Schultz, 2002), we propose that the cocaine-induced synaptic enhancement in the VTA contributes to the neural circuit adaptations that are responsible for attaching appropriate incentive value to stimuli associated with the cocaine injection and that the magnitude of locomotor activity in naive animals reflects the degree of the motivational significance of cocaine. The transient nature of the cocaine-induced synaptic enhancement in the VTA would presumably allow this region to “reset” itself, thus facilitating its involvement in attaching appropriate incentive value to subsequent motivationally significant stimuli.
Ten days after chronic cocaine treatment, which induced behavioral sensitization, a single injection of cocaine could again increase synaptic strength in the VTA, but its magnitude no longer correlated with the cocaine-induced increase in locomotor activity. This disconnect may reflect the fact that the locus of the critical adaptations responsible for sensitization shifted from its initiation site in the VTA to its sites of expression, the most critical of which is the NAc (Wolf, 1998; Vanderschuren and Kalivas, 2000). That is, the same cocaine-induced adaptation in the VTA, which has presumably returned to its basal state after the withdrawal of cocaine, has a profoundly different behavioral effect because cocaine-induced adaptations in the NAc and other target regions are much longer-lasting, perhaps even permanent (Robinson and Berridge, 1993; Wolf, 1998; Vanderschuren and Kalivas, 2000). Long-lasting changes in neurotransmission in the motive circuit include D1 receptor supersensitivity in the NAc (Xu et al., 1994), as well as decreased dopamine transporter binding sites (Sharpe et al., 1991; Pilotte et al., 1994) and mRNA after 10 d of withdrawal from chronic cocaine treatment (Xia et al., 1992). In addition, repeated amphetamine administration in the VTA resulted in an increased sensitivity of NAc medium spiny neurons to the inhibitory effects of a D1 receptor agonist on firing rates (Hu et al., 2002). Pierce et al. (1996a) also observed that after inducing behavioral sensitization, using the same injection protocol described here, D1 dopamine receptor sensitivity in the NAc was enhanced. Consequences of D1 receptor supersensitivity include induction of immediate early genes such as Fos, FosB, and JunB (Hu et al., 2002), protein kinase A phosphorylation of AMPA receptors (Pierce et al., 1996b), and phosphorylation of voltage-dependent sodium channels (Zhang et al., 1998). These adaptations would modulate the effect of dopamine in the NAc and likely contribute to the expression of cocaine sensitization. Additionally, changes in excitatory drive from regions other than the VTA, such as the basolateral amygdala and/or the prefrontal cortex, might also underlie the enhanced motor response attributable to chronic cocaine (Pierce and Kalivas, 1997).
In summary, we have demonstrated that the cocaine-induced enhancement of synaptic strength in the VTA is transient and independent of the duration of cocaine exposure. In naive animals, VTA synaptic strength correlates with drug-induced locomotor activity, which we propose reflects the motivational effects of cocaine. However, after repeated exposure to cocaine and during the expression of behavioral sensitization, the correlation between synaptic strength and this simple behavior is lost. This is likely because of the much longer-lasting adaptations that occur in VTA target regions after chronic cocaine exposure. These results provide a potentially powerful example of how chronic drug exposure can disrupt the normal, presumably adaptive relationship between neural circuit modifications and behavior.
Footnotes
This work was supported by National Institute on Drug Abuse Grants 1R01DA15096-01 (A.B.) and 2RO1DA009264-08 (R.C.M.). We thank Drs. F. W. Hopf, P. Janak, and J. Pierce-Shimomura for critical reading of this manuscript and Lisa Guggenheim Daitch for proofreading.
Correspondence should be addressed to A. Bonci, Ernest Gallo Clinic and Research Center, Department of Neurology, University of California, San Francisco, Emeryville, CA 94608. E-mail: bonci@itsa.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249482-09$15.00/0
References
- Ackerman JM, White FJ (1990) A10 somatodendritic dopamine autoreceptor sensitivity following withdrawal from repeated cocaine treatment. Neurosci Lett 117: 181-187. [DOI] [PubMed] [Google Scholar]
- Bjijou Y, Stinus L, Moal ML, Cador M (1996) Evidence for selective involvement of dopamine D1 receptors of the ventral tegmental area in the behavioral sensitization induced by intra-ventral tegmental area injections of d-amphetamine. J Pharmacol Exp Ther 277: 1177-1187. [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW (2001) Repeated cocaine administration into the rat ventral tegmental area produces behavioral sensitization to a systemic cocaine challenge. Behav Brain Res 126: 205-209. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Brickley S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327-335. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL (1996) Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 84: 745-755. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Wolf ME (2002) Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22: 3303-3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faleiro LJ, Jones S, Kauer JA (2003) Rapid AMPAR/NMDAR response to amphetamine: a detectable increase in AMPAR/NMDAR ratios in the ventral tegmental area is detectable after amphetamine injection. Ann NY Acad Sci 1003: 391-394. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ (1996) Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-sensitizing agents. J Neurosci 16: 274-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgetti M, Hotsenpiller, Ward P, Teppen T, Wolf ME (2001) Amphetamine-induced plasticity of AMPA receptors in the ventral tegmental area: effects on extracellular levels of dopamine and glutamate in freely moving rats. J Neurosci 21: 6362-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry DJ, Greene MA, White FJ (1989) Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: repeated administration. J Pharmacol Exp Ther 251: 833-839. [PubMed] [Google Scholar]
- Hu X-T, Koeltzow TE, Cooper DC, Robertson GS, White FJ, Vezina P (2002) Repeated ventral tegmental area amphetamine administration alters dopamine D1 receptor signaling in the nucleus accumbens. Synapse 45: 159-170. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA (1992) Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 450: 455-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW (1995) Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug Alcohol Depend 37: 95-100. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Alesdatter JE (1993) Involvement of NMDA receptor stimulation in the ventral tegmental area and amygdala in behavioral sensitization to cocaine. J Pharmacol Exp Ther 267: 486-495. [PubMed] [Google Scholar]
- Kalivas PW, Duffy P (1993) Time course of extracellular dopamine and behavioral sensitization to cocaine. II. Dopamine perikarya. J Neurosci 13: 276-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Stewart J (1991) Dopamine transmission in the initiation and expression of drug- and stress-induced sensitization of motor activity. Brain Res Rev 16: 223-244. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Weber B (1988) Amphetamine injection into the A10 dopamine region sensitizes rats to peripheral amphetamine and cocaine. J Pharmacol Exp Ther 245: 1095-1102. [PubMed] [Google Scholar]
- Kalivas PW, Duffy P, DuMars LA, Skinner C (1988) Behavioural and neurochemical effects of acute and daily cocaine administration in rats. J Pharmacol Exp Ther 245: 485-592. [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA (1990) Actions of cocaine on rat dopaminergic neurons in vitro. Br J Pharmacol 99: 731-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL (2003) κ-Opioid agonists directly inhibit midbrain dopaminergic neurons. J Neurosci 23: 9981-9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton PG, Clark D (1997) Burst firing in midbrain dopaminergic neurons. Brain Res Rev 25: 312-334. [DOI] [PubMed] [Google Scholar]
- Perugini M, Vezina P (1994) Amphetamine administered to the ventral tegmental area sensitizes rats to the locomotor effects of nucleus accumbens amphetamine. J Pharmacol Exp Ther 270: 690-696. [PubMed] [Google Scholar]
- Pierce RC, Kalivas PW (1997) A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Res Rev 25: 192-216. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Born B, Adams M, Kalivas PW (1996a) Repeated intra-ventral tegmental area administration of SKF-38393 induce behavioral and neurochemical sensitization to a subsequent cocaine challenge. J Pharmacol Exp Ther 278: 384-392. [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW (1996b) Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci 16: 1550-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotte NS, Sharpe LG, Kuhar MJ (1994) Withdrawal from repeated intravenous infusions of cocaine persistently reduces binding to dopamine transporters in the nucleus accumbens of Lewis rats. J Pharmacol Exp Ther 269: 963-969. [PubMed] [Google Scholar]
- Post RM, Weiss SRB (1988) Sensitization and kindling: implications for the evolution of psychiatric symptomatology. In: Sensitization in the nervous system (Kalivas PW, Barnes CD, eds), pp 257-292. Caldwell, NJ: Telford.
- Robinson TE, Becker JB (1986) Enduring changes in brain and behavior produced by chronic amphetamine administration: a review and evaluation of animal models of amphetamine psychosis. Brain Res Rev 11: 157-198. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Rev 18: 247-291. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Ly CD, Ehlers MD, Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4: 794-802. [DOI] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577-582. [DOI] [PubMed] [Google Scholar]
- Schultz W (2002) Getting formal with dopamine reward. Neuron 36: 241-263. [DOI] [PubMed] [Google Scholar]
- Segal DS, Schuckit MA (1983) Animal models of stimulant-induced psychosis. In: Stimulants: neurochemical, behavioral, and clinical perspectives (Creese I, ed), pp 131-177. New York: Raven.
- Sharpe LG, NS Pilotte, Mitchell WM, DeSouza EB (1991) Withdrawal of repeated cocaine decreases autoradiographic [3H]mazindol-labeling of dopamine transporter in rat nucleus accumbens, Eur J Pharmacol 203: 141-144. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583-587. [DOI] [PubMed] [Google Scholar]
- Vanderschuren LJ, Kalivas PW (2000) Alterations in dopaminergic and glutamatergic transmission in the induction and expression of behavioral sensitization: a critical review of preclinical studies. Psychopharmacology (Berl) 151: 99-120. [DOI] [PubMed] [Google Scholar]
- Vezina P (1993) Amphetamine injected into the ventral tegmental area sensitizes the nucleus accumbens dopaminergic response to systemic amphetamine: an in vivo microdialysis study in the rat. Brain Res 605: 332-337. [DOI] [PubMed] [Google Scholar]
- Vezina P, Queen AL (2000) Induction of locomotor sensitization by amphetamine requires the activation of NMDA receptors in the rat ventral tegmental area. Psychopharmacology (Berl) 151: 184-191. [DOI] [PubMed] [Google Scholar]
- Vezina P, Stewart J (1989) The effect of dopamine receptor blockade on the development of sensitization to the locomotor activating effects of amphetamine and morphine. Brain Res 499: 108-120. [DOI] [PubMed] [Google Scholar]
- White FJ, Hu X-T, Zhang X-F, Wolf ME (1995) Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. J Pharmacol Exp Ther 273: 424-454. [PubMed] [Google Scholar]
- Wolf ME (1998) The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679-720. [DOI] [PubMed] [Google Scholar]
- Wolf ME, White FJ, Hu XT (1993a) Behavioral sensitization to MK-801 (dizocilpine): neurochemical and electrophysiological correlates in the mesoaccumbens dopamine system. Behav Pharmacol 4: 429-442. [PubMed] [Google Scholar]
- Wolf ME, White FJ, Nassar R, Brooderson RJ, Khansa MR (1993b) Differential development of autoreceptor subsensitivity and enhanced dopamine release during amphetamine sensitization. J Pharmacol Exp Ther 364: 249-255. [PubMed] [Google Scholar]
- Xia Y, Goebel DJ, Kapatos G, Bannon JM (1992) Quantitation of rat dopamine transporter mRNA: effects of cocaine treatment and withdrawal. J Neurochem 59: 1179-1182. [DOI] [PubMed] [Google Scholar]
- Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S (1994) Elimination of cocaine-induced hyperactivity and dopamine mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell 79: 945-955. [DOI] [PubMed] [Google Scholar]
- Zhang X-F, Hu X-T, White FJ, Wolf ME (1997) Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. J Pharmacol Exp Ther 281: 699-706. [PubMed] [Google Scholar]
- Zhang X-F, Hu X-T, White FJ (1998) Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci 18: 488-498. [DOI] [PMC free article] [PubMed] [Google Scholar]