

Abstract
Fighting off neuronal degeneration requires a well controlled T-cell response against self-antigens residing in sites of the CNS damage. The ability to evoke this response is normally suppressed by naturally occurring CD4+CD25+ regulatory T-cells (Treg). No physiological compound that controls Treg activity has yet been identified. Here, we show that dopamine, acting via type 1 dopamine receptors (found here to be preferentially expressed by Treg), reduces the suppressive activity and the adhesive and migratory abilities of Treg. Treg activity was correlated with activation of the ERK1/2 (extracellular signal-regulated kinase 1/2) signaling pathway. Systemic injection of dopamine or an agonist of its type 1 receptors significantly enhanced, via a T-cell-dependent mechanism, protection against neuronal death after CNS mechanical and biochemical injury. These findings shed light on the physiological mechanisms controlling Treg and might open the way to novel therapeutic strategies for downregulating Treg activity (e.g., in neuronal degeneration) or for strengthening it (in autoimmune diseases).
Keywords: CD4+CD25+ regulatory T-cells, neurotransmitters, dopamine, dopamine receptors, ERK1/2, autoimmune response, neuroprotection, neurodegeneration, CNS
Introduction
It is becoming increasingly clear that an autoimmune response against self-antigens residing in the site of insult (Moalem et al., 1999) can protect the body against CNS neurodegeneration. Normally, autoimmunity is suppressed by naturally occurring regulatory CD4+CD25+ T-cells (Treg) (Shevach et al., 2001). Therefore, to elicit the desired autoimmune response for protection of CNS neurons at risk of degeneration, the Treg-imposed suppression must be alleviated. Depletion of Treg promotes survival of neurons after CNS insults (Kipnis et al., 2002) as well as boosts antitumor autoimmunity (Sakaguchi et al., 2001). Treg-imposed suppression is a multifactorial process, involving cell-to-cell contacts (Nakamura et al., 2001) and the activity of soluble factors [which presumably include interleukin (IL)-10 (Sundstedt et al., 2003) and TGF-β (Piccirillo et al., 2002)]. Studies have shown that the suppressive activity of Treg can be inhibited by the addition of exogenous IL-2 (Thornton and Shevach, 1998), or blocking of the cytotoxic T-lymphocyte-associated antigen receptor 4 (CTLA-4) (Nakamura et al., 2001), or activation of the newly discovered glucocorticoid-induced TNF-α receptor (McHugh et al., 2002).
Some key adhesion molecules are more abundant on the surfaces of Treg than of effector (CD4+CD25-) T-cells (Teff) (Kohm et al., 2002). The ability of Treg to enter tissues might help prevent autoimmune disease progression. In fighting off neurodegeneration or cancer, however, the presence of Treg is a liability. Compounds capable of reducing the trafficking ability (adhesion and migration) of Treg, or their suppressive activity, or both, might therefore be promising candidates for therapy against both cancer and CNS insults. As a corollary, compounds capable of upregulating the inhibitory or trafficking activity of Treg, or both, might be potential candidates for therapy against autoimmune diseases. A fine balance would then be needed to fight off the conditions leading to neuronal degeneration without creating conditions that foster neural tissue-specific autoimmune diseases. Up to now, however, no physiological compounds have been discovered that can control the activity of Treg.
The present study was undertaken in an attempt to identify physiological compounds potentially capable of controlling the Treg activity after CNS injury. We postulated that because stress- or pain-related physiological compounds are increased after CNS injury (Rothblat and Schneider, 1998; Thiffault et al., 2000), one or more of them might transmit an early signal to Treg, with consequent reduction of the trafficking or suppressive activity, or both, of the latter. We reasoned that likely candidate compounds might be key neurotransmitters such as dopamine, norepinephrine, serotonin, and substance P, all of which have been shown to participate in interactions between the brain and the immune system (Edgar et al., 2002).
Of all the tested neurotransmitters, dopamine was the only one that reduced the activity of Treg, and it did so via an ERK (extracellular signal-regulated kinase)-dependent pathway. Dopamine affected both the suppressive and the trafficking activities of Treg, via dopamine type 1 (D1-R and D5-R) receptors, found here to be preferentially expressed by Treg. Using mouse models of neurodegenerative conditions caused by partial crush injury of the optic nerve or glutamate intoxication in the eye, we showed that systemic administration of dopamine or its D1-type agonist can induce neuroprotection after mechanical and chemical CNS injury by alleviating the suppression imposed by Treg.
Materials and Methods
Animals. Inbred adult wild-type, severe combined immunodeficient (scid), and nu/nu BALB/c and C57Bl/6 mice were supplied by the Animal Breeding Center of The Weizmann Institute of Science. All animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee.
Antibodies and reagents. The antibodies and reagents used included: mouse recombinant IL-2, anti-mouse ζ-CD3, anti-mouse CTLA-4, and purified rabbit anti-mouse ERK2 antibody (R & D Systems, Minneapolis, MN); rat anti-mouse phycoerythrin (PE)-conjugated CD25 antibody (PharMingen, Becton-Dickinson, Franklin Lakes, NJ); FITC-conjugated anti-CD4 antibody (Serotec, Oxford, UK); anti D1-R (Calbiochem, Darmstadt, Germany); 3-hydroxytyramine (dopamine), norepinephrine, SKF-38393, SCH-23390, quinpirole, clozapine, genistein, and PD98059 (Sigma-Aldrich, Rehovot, Israel); phosphatidyl serine detection kit (IQ Products, Houston, TX). Purified anti-pERK1/2 antibody was a gift from Prof. R. Seger from The Weizmann Institute of Science.
Intravitreal glutamate injection. The right eyes of anesthetized mice were punctured with a 27 gauge needle in the upper part of the sclera, and a10 μl Hamilton syringe with a 30 gauge needle was inserted as far as the vitreal body. A total volume of 1 μl of l-glutamate (400 nmol) dissolved in saline was injected into the eye (Schori et al., 2001).
Retrograde labeling of retinal ganglion cells. Mice were anesthetized and placed in a stereotactic device. The skull was exposed and kept dry and clean. The bregma was identified and marked. The designated point of injection was at a depth of 2 mm from the brain surface, 2.92 mm behind the bregma in the anteroposterior axis, and 0.5 mm lateral to the midline. The neurotracer dye FluoroGold (5% solution in saline; Fluorochrome, Denver, CO) was applied (1 μl, at a rate of 0.5 μl/min in each hemisphere) using a Hamilton syringe, and the skin over the wound was sutured.
Crush injury of the optic nerve in mice. Animals were anesthetized deeply by intraperitoneal injection of 2% Xyl-M (xylazine, 10 mg/kg; VMD, Arendonk, Belgium) and Ketaset (ketamine, 50 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA) and subjected to severe crush injury of the intraorbital portion of the optic nerve. The uninjured contralateral nerve was left undisturbed. The optic nerve was crushed 3 d after retrograde labeling of retinal ganglion cells with FluoroGold, as described above (Fisher et al., 2001).
Enzyme-linked immunosorbent assay. Treg or Teff (0.5 × 106 cells/ml) were cultured for 48 hr in the presence of anti-CD3 and anti-CD28. After 48 hr, the cells were centrifuged and their supernatants were collected and sampled. Concentrations of IL-2 in the samples were determined by the use of sandwich ELISA kits (R & D Systems). For detection of secreted IL-10, cells were centrifuged every 24 hr and replaced with a fresh medium. Supernatants obtained from cells after 24, 48, and 72 hr in culture were subjected to an ELISA kit (Diaclone Research, Fleming, France).
Purification of murine CD4+CD25+/CD4+CD25- T-cells. Lymph nodes (axillary, inguinal, superficial cervical, mandibular, and mesenteric) and spleens were harvested and mashed. T-cells were purified (enriched by negative selection) on T-cell columns (R & D Systems). The enriched T-cells were incubated with anti-CD8 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), and negatively selected CD4+ T-cells were incubated with PE-conjugated anti-CD25 (30 μg/108 cells) in PBS/2% FCS. They were then washed and incubated with anti-PE microbeads and subjected to magnetic separation with AutoMACS. The retained cells were eluted from the column as purified CD4+CD25+ cells. The negative fraction consisted of CD4+CD25- T-cells. Purified cells were cultured in 24-well plates (1 ml) with T-cell-depleted spleen cells as accessory cells (irradiated with 3000 rad) and 0.5 μg/ml anti-CD3, supplemented with 100 U of mouse recombinant IL-2.
T-cell adhesion. Adhesion of activated CD4+CD25+ and CD4+CD25- T-cells to chondroitin sulfate proteoglycans (CSPG) was analyzed as described previously (Ariel et al., 1998). Briefly, flat-bottomed microtiter (96-well) plates were precoated with CSPG (1 μg/well, 40 min, 37°C). 51Cr-labeled T-cells were left untreated or were preincubated (30 min, 37°C) with dopamine or the specified agonist or antagonist (10-5 M). The cells (105 cells in 100 μl of RPMI medium containing 0.1% BSA) were then added to the CSPG-coated wells, incubated (30 min, 37°C), and washed. Adherent cells were lysed, and the resulting supernatants were removed and counted in a gamma counter. Results were expressed as the mean percentage of the total population before adhesion of bound T-cells from quadruplicate wells for each experimental group.
Chemotaxis assay. The migration of T-cells across polycarbonate filters (pore size, 5 μm; diameter, 6.5 mm) toward stromal cell-derived factor-1 (SDF-1) and macrophage-derived chemokine (MDC) (CCL22) was assayed in 24-well Transwell chambers (Costar, Corning, Corning, NY). T-lymphocytes (1.67 × 106 cells/ml) were suspended in RPMI medium/0.1% BSA, and 150 μl of the cell suspension was added to the upper chamber after incubation with or without dopamine (90 min, 37°C). Chemokines were added to the lower chamber at concentrations of 1 μg/ml SDF-1 (CytoLab, Ness-Ziona, Israel) and 0.25 μg/ml MDC (R & D Systems). The plates were incubated for 90 min at 37°C in 9.5% CO2. T-cells that migrated to the lower chambers were collected and stained with anti-CD4 and anti-CD25 antibodies. The numbers of migrating T-cells were measured by flow cytometer acquisition for a fixed time (60 sec). To calculate specific migration, the number of cells in each subpopulation in the absence of chemokine was subtracted from the number in the corresponding cell subpopulation that migrated in the presence of chemokines. The number of migrating CD4+CD25+ T-cells was calculated as a percentage of the total T-cell population before migration. For migration of purified population, we used a similar protocol.
Activation of Treg. Purified Treg (0.5 × 106 cells/ml) were activated in RPMI medium supplemented with l-glutamine (2 mm), 2-mercaptoethanol (5 × 10-5 m), sodium pyruvate (1 mm), penicillin (100 IU/ml), streptomycin (100 μg/ml), nonessential amino acids (1 ml/100 ml), and autologous serum 2% (v/v) in the presence of mouse recombinant IL-2 (mrIL-2; 5 ng/ml) and soluble anti-CD3 antibodies (1 ng/ml). Irradiated (2500 rad) splenocytes (1.5 × 106 cells/ml) were added to the culture. Cells were activated for 24 or 96 hr. In some of the 96 hr experiments, fresh dopamine was added to the culture every 24 hr during activation.
Inhibition assay (coculturing of Teff with Treg). Naive Teff (50 × 103 cells/well) were cocultured with decreasing numbers of activated Treg for 72 hr in 96-well flat-bottomed plates in the presence of irradiated splenocytes (106/ml) supplemented with anti-CD3 antibodies. [3H]-thymidine (1 μCi) was added for the last 16 hr of culture. After the cells were harvested, their [3H]-thymidine content was analyzed by the use of a gamma counter.
Immunocytochemistry. T-cells were fixed for 10 min with a mixture (1:1) of methanol and acetone at -20°C, incubated in blocking solution (PBS containing 0.3% Triton-X100 and 1% of normal rabbit serum) for 60 min at room temperature, and then incubated overnight with a specific antibody (dilution, 1:1000) in the blocking solution. The T-cells were then washed and incubated with the secondary antibody (PE-labeled goat anti-rabbit IgG) for 30 min at room temperature, then washed, and analyzed by fluorescence and confocal microscopy.
Western blotting. Cells were stimulated for 20 min with anti-CD3 and anti-CD28 antibodies in the presence or absence of dopamine or SKF-38393. Cell lysates were prepared using radioimmunoprecipitation assay lysis buffer (50 mm Tris, pH 8, 0.1% SDS, 0.5% deoxycholate, 1% NP-40, 500 mm NaCl, and 10 mm MgCl2). Supernatants were collected, and 5× sample buffer (containing 25 mm Tris, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromophenol blue, and 0.5 m β-mercaptoethanol) was added before boiling. Activated ERK1/2 was detected by probing blots with a monoclonal antibody. Total ERK protein was detected by using a polyclonal rabbit antibody. The blots were developed by HRP-conjugated anti-mouse or anti-rabbit Fab and ECL (Amersham Biosciences, Freiburg, Germany). Signals were quantified using NIH Image version 1.62.
Polymerase chain reaction. Total RNA was purified with the RNeasy Mini kit (Qiagen, Germantown, MD). For PCR, the following primers were used (for dopamine receptors, primers were used from Lemmer et al., 2002); CCR-4: sense 5′-GTGCAGTCCTGAAGGACTTCAAGCTCCACCAG-3′, antisense 5′-GGCAAGGACCCTGACCTATGGGGTCATCAC-3′; FOXP3: sense 5′-CAGCTGCCTACAGTGCCCCTAG-3′, antisense 5′-CATTTGCCAGCAGTGGGTAG-3′.
Signals were quantified using a Gel-Pro analyzer 3.1 (Media Cybernetics, Silver Spring, MD). Real-time PCR was performed with a LightCycler instrument (Roche, Mannheim, Germany) using the FastStart DNA Master SYBR Green 1 kit (catalog #3003230; Roche) as described by the manufacturer. The following primers were used: D5-R: sense 5′-CCTTTATCCCGGTCCA-3′, antisense 5′-GATACGGCGGATCTGAA-3′; IL-10: sense 5′-ACCTGGTAGAAGTGATGCCCCAGGCA-3′, antisense 5′-CTATGCAGTTGATGAAGATGTCAAA-3′.
Results
Dopamine reduces the suppression imposed by Treg
Coculturing of Teff with Treg isolated from naive mice results in suppression of Teff proliferation. The suppressive potency depends on the Treg/Teff ratio and the state of Treg activation; the suppression is significantly increased, for example, if the Treg are activated before being added to Teff (Thornton and Shevach, 1998). Inhibition of Teff proliferation, assayed by [3H]thymidine incorporation, can therefore be taken as a measure of the suppressive effect. We examined the ability of major neurotransmitters and neuropeptides (dopamine, norepinephrine, substance P, and serotonin) to alleviate the Treg-induced suppression of Teff in vitro. Each compound was tested at several concentrations. Proliferation of Teff was significantly inhibited by cocultivation of Teff with naive Treg or with Treg that had been activated by incubation for 24 hr with anti-CD3 antibodies and IL-2 in the presence of antigen-presenting cells (APCs; lethally irradiated splenocytes) (Fig. 1). After incubating the activated Treg for 2 hr with a neurotransmitter or a neuropeptide, we washed the cells and then cocultured them with Teff. Proliferation of Teff cocultured with activated Treg that had been incubated with dopamine (10-5 M) was more than twofold higher than proliferation in coculture with activated Treg not incubated with dopamine (Fig. 1a). A significant effect on Treg-suppressive activity was also obtained with 10-7 M dopamine (Fig. 1a), whereas 10-9 M had no significant effect (data not shown). The inhibitory effect of dopamine at 10-5 and 10-7 M on Treg activity was reproduced when freshly isolated (nonactivated) Treg were used (Fig. 1b). At the dopamine concentration of 10-9 M, the obtained effect was slight and not statistically significant (Fig. 1b). It should be noted, however, that the effect of dopamine on Treg-suppressive activity was only partial and that complete blocking was not seen at any of the concentrations tested. We also examined the effect of dopamine on the activity of Treg that had been activated as described above (Fig. 1a), but for 96 hr, and to which dopamine (10-5 M) was added for 2 hr at the end of the activation period and then washed off before the activated cells were cocultured with naive Teff. Again, Teff proliferation was significantly higher in the presence of activated Treg treated with dopamine than in the presence of activated Treg without dopamine (Fig. 1c). A direct effect of dopamine on Teff proliferation was ruled out by incubation of Teff for 2 hr with 10-5 M dopamine, then washing off the dopamine and adding activated Treg without dopamine. The resulting proliferation of Teff did not differ from that seen in cultures of Teff in the absence of dopamine. Moreover, the inhibitory effects of Treg on naive Teff and on Teff exposed to dopamine were similar (Fig. 1c), indicating that dopamine did not alter the susceptibility of Teff to Treg suppression. The uptake of thymidine by Teff and the Treg-induced inhibition of such uptake varied from one experiment to another. In all experiments, however, the effect of dopamine on Treg (tested >20 times) was consistent, and in most cases, the proliferation of Teff cocultured with Treg treated with dopamine was more than twofold higher than that in the absence of dopamine treatment. The Treg used in this study were always obtained from naive animals, therefore, it is unlikely that they contained any activated effector T-cells. The purity of the Treg population used in all experiments was high (between 92 and 98% of the total CD4+ population). Moreover, the use of anti-CD25 antibodies to isolate Treg reportedly does not interfere with either the suppressive activity or the state of activation of Treg (Thornton and Shevach, 1998).
Figure 1.

Dopamine (DA) reduces the suppressive activity mediated by CD4+CD25+ regulatory T-cells. Proliferation of Teff (a CD4+CD25- population) was assayed by incorporation of [3H]-thymidine into Teff cocultured with naturally occurring Treg. Recorded values are from one of three representative experiments and are expressed as means ± SD of four replicates. a, Treg were activated by incubation for 24 hr with anti-CD3 antibodies in the presence of mrIL-2. Incubation of the activated Treg for 2 hr with dopamine (10-5 or 10-7 m) before their coculturing with Teff reduced their suppression of Teff compared with that obtained with Treg not exposed to dopamine. b, Dopamine (10-5, 10-7, or 10-9 m) added to freshly purified Treg. Dopamine (10-5 and 10-7 m) had a similar effect on activity of naive Treg to that of to activated Treg, whereas the effect of dopamine at 10-9 m on Treg-mediated suppression was not significant. c, Activation of Treg for 96 hr, followed by the addition of dopamine (10-5 m) for 2 hr at the end of activation, significantly reduced the suppressive activity of Treg on Teff. Incubation of Teff with dopamine (10-5 m) for 2 hr did not affect their susceptibility to Treg-induced suppression.
In contrast to the effect seen with dopamine, no effect on the ability of Treg to suppress Teff proliferation could be detected when Treg were preincubated with different concentrations of norepinephrine (another member of the catecholamine family) (Fig. 2a), substance P (a pain- and stress-related neurotransmitter; data not shown), or serotonin (data not shown).
Figure 2.

Dopamine effect on Treg is mediated via D1-type receptor family. Proliferation of Teff (a CD4+CD25- population) was assayed by incorporation of [3H]-thymidine into Teff cocultured with naturally occurring Treg. Recorded values are from one of three representative experiments and are expressed as means ± SD of four replicates. a, Addition of norepinephrine (NE; 10-5 or 10-7 m) to Treg for 2 hr after their activation for 24 hr did not affect the suppressive activity of Treg. Significant differences between groups were analyzed by Student's t test (p < 0.001). b, The inhibitory effect of dopamine (DA) on the suppressive activity of Treg was mimicked by SKF-38393, a specific agonist of the D1-type family. The D2-type agonist quinpirole did not alter the effect of dopamine on Treg. SCH 23390, a specific D1-type antagonist, wiped out the dopamine effect on the suppressive activity of Treg. Each experiment was performed at least five times, and representative results are shown. c, Incubation of Treg or Teff with dopamine did not cause apoptosis, as shown by propidium iodide staining for DNA content and FACS analysis of Treg and Teff, 48 hr after their incubation for 2 hr with dopamine. d, Staining for apoptosis with annexin V for phosphatidylserine on a surface membrane. No increase in annexin V-labeled cells was detected on incubation of Treg with dopamine or with the D1-type agonist SKF-38393.
To establish whether the observed effect of dopamine on Treg is exerted through a receptor-mediated pathway, we used specific agonists and antagonists of dopamine receptors. Incubation of Treg with 10-5 M SKF-38393, an agonist of the type 1 family of dopamine receptors (consisting of D1-R and D5-R), reproduced the dopamine effect (Fig. 2b). The specific D1-type antagonist SCH-23390 (10-5 M), when added together with dopamine (10-5 M), prevented the dopamine effect, further substantiating the contention that the effect of dopamine on Treg is mediated through the type 1 receptor family. Also in line with this contention was the finding that incubation of Treg with 10-5 M quinpirole, an agonist of the type 2 family of dopamine receptors (comprising D2-R, D3-R, and D4-R), had no effect on the suppressive activity of Treg.
To exclude the possibility that dopamine exerts its effect by causing the death of Treg, we examined whether dopamine at the concentrations used here cause Treg apoptosis. No signs of apoptosis were detectable in Treg, which, after being incubated with dopamine, were stained with propidium iodide and analyzed for apoptotic cells (sub-G1) by flow cytometry (Fig. 2c). To further verify the absence of apoptotic death in Treg, after incubating Treg with dopamine, we stained them for phosphatidylserine with annexin V. Again, we could not detect any signs of apoptosis in Treg beyond the background levels seen in the absence of dopamine (Fig. 2d). Thus, the reduction in Treg activity after their encounter with dopamine or a related agonist evidently results not from the death of Treg but rather from alteration of their behavior.
Because dopamine reduced the suppressive activity of Treg on Teff but did not alter the susceptibility of Teff to suppression by Treg, we examined the possibility that Teff and Treg express different subtypes or different amounts of the relevant dopamine receptors. This was done by assaying the expression of the dopamine type 1 receptors, D1-R and D5-R, in Treg and Teff. PCR assays showed that Treg expressed significantly more D1-R and D5-R transcripts (4-fold and 14-fold, respectively) than Teff (Fig. 3a,b). To further verify the differences in expression of dopamine receptors by Teff and Treg, we performed real-time PCR, which showed that the amounts of D1-R and D5-R in Treg were 5-fold and 13-fold higher, respectively, than in Teff (Fig. 3c).
Figure 3.
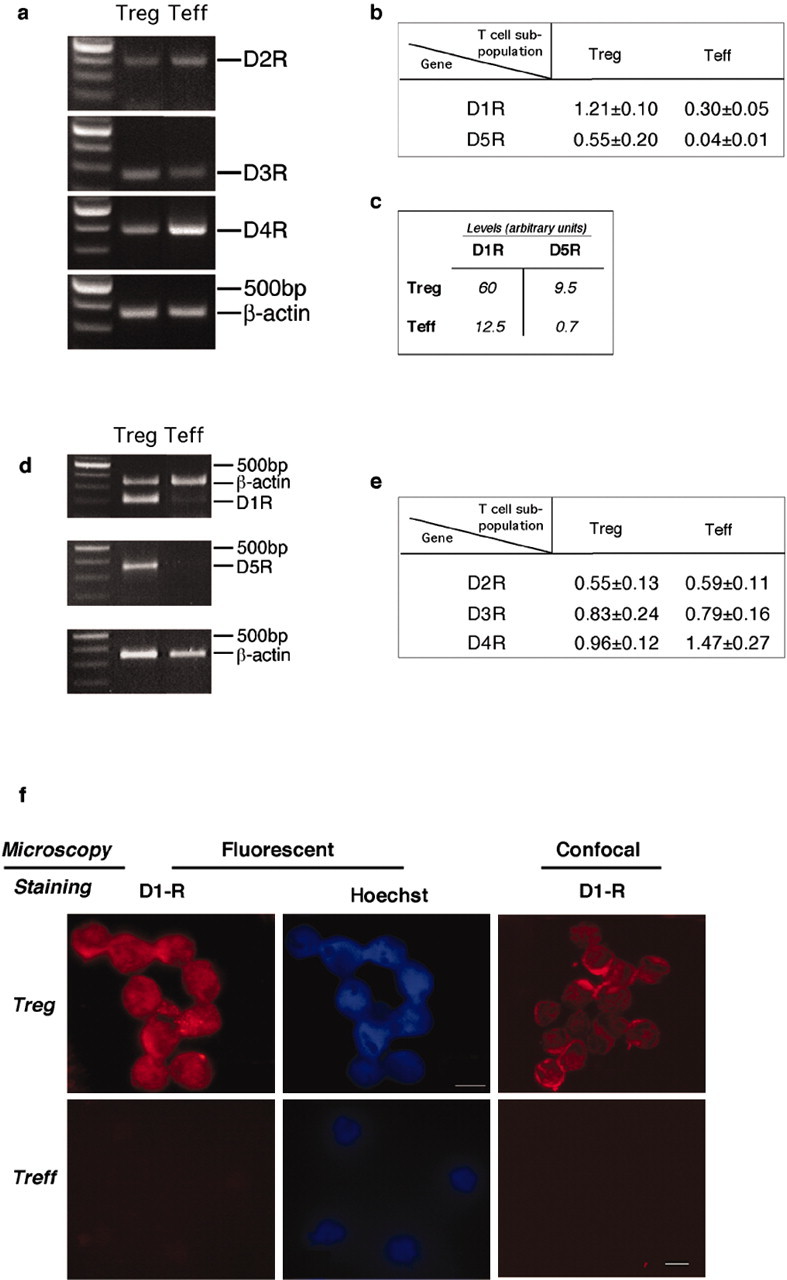
Preferential expression of D1-type receptors by Treg. Expression of dopamine receptors on Treg and Teff was investigated on mRNA and protein levels. a, b, Semiquantitative reverse transcription-PCR (RT-PCR) analysis for D1-R and D5-R expression. mRNA was extracted from freshly isolated Teff and Treg. The housekeeping gene β-actin was used for quantitative analysis. The results of one of five representative experiment are shown. c, Quantitative real-time PCR using primers for D1-R and D5-R to verify the differences in the expression of dopamine receptors on Teff and Treg. The results presented are arbitrary units and are from one of three representative experiments performed. d, e, Semiquantitative RT-PCR for D2-R, D3-R, and D4-R expression. mRNA was extracted from freshly purified Teff and Treg. The housekeeping gene β-actin was used for quantitative analysis. The results of one of five representative experiments are shown. f, Representative micrographs of D1-R-immunoreactive T-cells using fluorescence and confocal microscopy. Also shown are micrographs stained with Hoechst and visualized by fluorescence microscopy. D1-R immunoreactivity was observed in Treg but not in Teff.
We also used PCR to assay the expression of dopamine type 2 family receptors, namely D2-R, D3-R, and D4-R, in Treg and Teff. Although the expression of D4-R was somewhat more abundant in Teff than in Treg, the difference between the expression of each of these receptors in the two T-cell subpopulations was not significant (Fig. 3d,e). To verify that the difference in D1-R between Treg and Teff observed at the transcript level is manifested also at the protein level, we subjected the cells to immunocytochemical analysis. D1-R immunoreactivity was detected in naive Treg but not in naive Teff (Fig. 3f).
To gain additional insight into the mechanism whereby dopamine affects Treg activity, we examined CTLA-4, a molecule characteristic of Treg (Im et al., 2001). Expression of this molecule was slightly, but consistently, decreased on exposure of Treg to dopamine. A similar effect on CTLA-4 expression was obtained with the D1-type-specific agonist SKF-38393 (Fig. 4a). Another molecule that participates in the suppressive activity of Treg is IL-10 (Maloy et al., 2003). It was therefore of interest to measure the production of IL-10 by Treg after their exposure to dopamine. Media collected after incubation of Treg with dopamine (10-5 M) for 24, 48, and 72 hr showed a significant decrease in the amount of IL-10 at all time points examined (Fig. 4b). Dopamine did not, however, alter the anergic state of Treg; production of IL-2 was not detected in Treg that had been incubated in the presence of dopamine, as verified by ELISA for a secreted cytokine in media conditioned for 48 hr by activated Treg (Fig. 4c). Teff, as expected, secreted IL-2, the level of which was not affected by dopamine (Fig. 4c). It should be noted that activation of both T-cell populations was performed in the absence of mrIL-2.
Figure 4.

Molecular mechanism underlying the effect of dopamine on Treg. a, Expression of CTLA-4. Treg were activated for 24 hr, then incubated for 2 hr with dopamine or SKF-38393 (control cells were activated but were not incubated with either dopamine or SKF-38393; note that different cell preparations were used for each treatment, and therefore the controls used for each treatment were not the same) and were stained 24 hr later for CTLA-4 on cell surfaces. CTLA-4 expression was reduced after exposure to dopamine or to SKF-38393. Representative results of one of five independent experiments with each treatment are shown. b, Production of IL-10. Treg were activated for 24 hr with anti-CD3 and IL-2 in the presence of lethally irradiated splenocytes (APCs) and then for an additional 2 hr with dopamine. Conditioned media were collected after 24, 48, or 72 hr of culture and were assayed for IL-10 using a sandwich ELISA. At any given time, significantly less IL-10 was detected in media conditioned by dopamine-treated Treg than in media conditioned by Treg not exposed to dopamine. Statistical significance was verified using Student's t test analysis (**p < 0.01; *p < 0.05). The results shown are of one of three independent experiments, performed at each time point. c, Lack of IL-2 production by Treg. Treg and Teff were activated separately for 48 hr with anti-CD3 and anti-CD28 (without mrIL-2) with or without dopamine. Conditioned media were collected after 48 hr and subjected to ELISA. Treg with or without dopamine did not secrete detectable levels of IL-2. Production of IL-2 by Teff was not affected by dopamine. d, Foxp3 expression in Treg. Treg were activated for 24 hr with anti-CD3 and anti-CD28 in the presence of IL-2, then exposed to dopamine for 2 hr, washed, and analyzed 30 min later for Foxp3 expression. No changes in Foxp3 were detected after 30 min of dopamine treatment of naive Treg. DA, Dopamine.
A gene encoding the Foxp3 protein was recently found to be associated with Treg (Ramsdell, 2003). We therefore examined whether the dopamine-induced reduction of Treg activity alters the expression of this gene. mRNA isolated from Treg that were activated for 24 hr, exposed for 2 hr to dopamine, and maintained in culture for an additional 30 min or 24 hr was analyzed for Foxp3 expression. Foxp3, as expected, was detected in Treg, but no significant change in its expression was observed after Treg were exposed to dopamine for 30 min (Fig. 4d) or 24 hr (data not shown).
ERK1/2 is deactivated by dopamine in Treg
The finding that dopamine downregulated Treg activity via D1-type, but not D2-type, receptors, taken together with the recent report that the ERK pathway can be activated by D1-R-dependent signaling (Takeuchi and Fukunaga, 2003), led us to suspect that the downregulatory effect of dopamine on the suppressive activity of Treg might be exerted via the ERK pathway. To examine this possibility, we first treated Treg with the protein tyrosine kinase inhibitor genistein (4′,5,7-trihydroxy isoflavone), which inhibits ERK and MEK (MAP/ERK kinase) activation (Mocsai et al., 2000). This treatment blocked the suppressive activity of Treg on Teff (Fig. 5a). Genistein at the same concentration had no effect on the proliferation of Teff (Fig. 5a).
Figure 5.

ERK1/2 phosphorylation inhibitors downregulate Treg-suppressive activity. a, Treg were activated by incubation for 30 min with anti-CD3 and anti-CD28 antibodies in the presence of IL-2 and in the presence or absence of a tyrosine kinase inhibitor (genistein) and were then cocultured with Teff. The suppression of Teff by Treg was significantly reduced in the presence of genistein. b, Similarly, incubation of activated Treg with the specific MEK inhibitor PD98059, which inhibits the ERK1/2 signaling pathway, almost completely abolished their suppression of Treg.
In light of these results, we also examined whether Treg activity is affected by PD98059, a specific MEK inhibitor that blocks the ERK1/2 signaling pathway (Sharp et al., 1997). PD98059 significantly reduced the suppressive activity of Treg relative to that of control-activated Treg (Fig. 5b).
The above findings prompted us to examine the state of ERK phosphorylation in activated Treg in the presence or absence of dopamine. Treg were activated with anti-CD3 and anti-CD28 for 20 min in the presence or absence of dopamine (10-5 M), and Western blot analysis of phospho-ERK1/2 expression in lysates of Treg and Teff was performed. Significantly more phosphorylated ERK1/2 was detected in activated Treg than in activated Teff. Moreover, phospho-ERK1/2 was found to be downregulated in Treg that had been activated in the presence of dopamine (Fig. 6a). ERK1/2 phosphorylation in Treg was also reduced by the specific D1-type receptor agonist SKF (Fig. 6b). Results of the quantitative analysis of the phospho-bands are shown in Figure 6c.
Figure 6.

Correlation between Treg activity and activation state of ERK1/2. a,b, Western blot analyses of Treg lysates after activation for 20 min with anti-CD3 and anti-CD28, in the presence or absence of dopamine (a) or SKF-38393 (b). After activation, the amounts of phospho-ERK1/2 seen in Treg are larger than in Teff (a) but are reduced by dopamine (a) or by SKF-38393 (b). Dopamine did not cause a significant change in phospho-ERK1/2 levels in Teff (a, b). c, Quantitative analysis of phospho-bands using NIH Image version 1.62.
Dopamine alters the adhesive and migratory properties of Treg
One of the main features of T-cells is their ability to migrate to tissues in need of rescue or repair [such as a diseased or damaged CNS (Hickey, 1999)]. We therefore considered the possibility that dopamine reduces not only the suppressive activity but also the migratory ability of Treg. Because T-cell migration and adhesion have been linked to ERK activation (Tanimura et al., 2003), this assumption appeared even more feasible in light of the above observation that dopamine reduced ERK activation in Treg. We incubated Treg with dopamine for 2 hr and then examined their adhesion to CSPG, extracellular matrix proteins often associated with injured tissues (Jones et al., 2003). The ability of Treg to adhere to CSPG was significantly greater than that of Teff (Fig. 7a) and was significantly decreased, in a concentration-dependent manner (10-9 to 10-5 M), by dopamine (Fig. 7a). The dopamine effect on Treg could be mimicked by the D1-type-specific agonist SKF-38393 and inhibited by the D1-type antagonist SCH-23390. Dopamine had only a slight, nonsignificant effect on the adhesion of Teff to CSPG (Fig. 7a). The ability of Treg to adhere to fibronectin was greater than that of Teff (Fig. 7b). Exposure to dopamine resulted in no effect on adhesion of Treg to fibronectin and a slight increase in the adhesion of Teff (Fig. 7b).
Figure 7.

Dopamine alters the adhesive properties of Treg. a, Treg and Teff were activated for 24 hr with anti-CD3 and anti-CD28 and were then incubated, with or without dopamine (10-5 to 10-9m), for 2 hr. In the absence of dopamine, adhesion of Treg to the CSPG matrix was significantly stronger than that of Teff. Incubation with dopamine significantly reduced the adhesion of Treg in a concentration-dependent manner. The effect of dopamine on Treg adhesion could be mimicked by SKF-38393, a specific agonist of the D1-type family. The dopamine effect was blocked by SCH-23390, a D1-type antagonist. Dopamine did not significantly alter the adhesion of Teff. A Mann-Whitney nonparametric U test was used for statistical analysis. b, In the absence of dopamine, adhesion of Treg to fibronectin was only slightly (but still significantly) stronger than that of Teff. However, dopamine did not significantly alter the adhesion of either Treg or Teff. A Mann-Whitney nonparametric U test was used for statistical analysis. c, Treg were activated for 30 min in the presence or absence of the ERK1/2 signaling pathway inhibitor PD98059 and then subjected to an adhesion assay to CSPG. Adhesion of Treg incubated with PD98059 was significantly weaker than that of control Treg cells. d, CD44 expression in Treg and in Teff. FACS analysis showed that significantly larger amounts of CD44 are expressed in Treg than in Teff. After incubation with dopamine, CD44 expression was significantly decreased in Treg but was not affected in Teff.
To verify that the effect of dopamine on adhesion of Treg is exerted through the ERK1/2 pathway, we incubated Treg with the ERK1/2 signaling pathway inhibitor PD98059 before performing the adhesion assay. PD98059 significantly reduced the ability of Treg to adhere to CSPG (Fig. 7c). Because interaction of T-cells with CSPG is mediated in part by the CD44 receptor (Henke et al., 1996), and in light of the known dependence of CD44 expression on the ERK signaling pathway, it was conceivable that dopamine might affect the expression of CD44 in Treg. To examine this possibility, we assayed CD44 immunoreactivity in Treg and Teff that had been activated with anti-CD3 and anti-CD28 antibodies for 24 hr and then incubated for 2 hr with or without dopamine. In the absence of dopamine, CD44 immunoreactivity was significantly stronger in Treg than in Teff. Dopamine decreased CD44 immunoreactivity in Treg but not in Teff (Fig. 7d). Other adhesion molecule receptors that we tested, such as leuktoactic factor activity-1, intercellular adhesion molecule, and vascular cell adhesion molecule (Lee and Benveniste, 1999), did not show any dopamine-related changes in Treg (data not shown).
Migration of human Treg in humans is dependent on the chemokine receptors CCR-4 and CCR-8, which are abundantly present on Treg (Sebastiani et al., 2001). We therefore examined whether exposure to dopamine would also affect Treg migration. For this experiment, we used a normal population of CD4+ T-cells, of which Treg (CD4+CD25+) accounts for ∼11% (Fig. 8a). Of the CD4+ cells that migrated toward CCL22 (MDC; a chemokine for CCR-4),
Figure 8.

Dopamine alters the migratory properties of Treg. a, The total population of purified CD4+ T-cells was subjected to a migration assay. The percentage of CD4+CD25+ T-cells in the total population after migration toward CCL22 (MDC) was significantly higher than in the original population. Exposure of Treg to dopamine significantly decreased their migration toward MDC. Migration of Teff toward SDF-1 was significantly stronger than that of Treg, and neither Teff nor Treg migration was affected by exposure to dopamine. A Mann-Whitney nonparametric U test was used for statistical analysis. b, c, Migration of purified Treg was significantly decreased after incubation of Treg with dopamine. Treg in the lower (postmigration) chamber were collected and counted by FACS for a defined time period after staining for a membrane CD4 marker. Values are representative results of the FACS analysis (b), and the mean number of cells from triplicates of the same experiment are shown in c. d, e, Semiquantitative reverse transcription-PCR for CCR-4 expression in Treg and Teff. mRNA was isolated from Teff and Treg, incubated for 2 hr with or without dopamine. The PCR products were quantified (e) relative to a housekeeping gene (β-actin). The results of one representative experiment are shown (d). Each experiment was performed in triplicate and repeated at least three times. DA, Dopamine. ***p < 0.01; **p < 0.01.
17% were Treg (CD4+CD25+), pointing to the greater migratory ability of Treg than of Teff toward MDC. However, after exposure of the CD4+ cell population to dopamine, migrating Treg accounted for ∼10% (the same as their percentage in the total CD4+ population at the start of the experiment), suggesting that after their exposure to dopamine, Treg lost their preference for migration toward MDC.
We also examined the migration of a mixed T-cell population toward SDF-1. Migration of Teff toward SDF-1 was significantly greater than that of Treg (postmigration percentage of Treg in the total CD4+ population was <4%), and dopamine did not alter this pattern (Fig. 8a). To examine the direct effect of dopamine on the migration of Treg, we assayed the effect of dopamine on the migration of purified Treg toward MDC. The migratory Treg were stained for CD4 to ensure that cell debris and aggregates would not be counted among them. Dopamine almost completely abolished Treg migration (Fig. 8b,c) but had no effect on the migration of Teff (data not shown).
In an attempt to link the changes in migration to specific receptors, we examined the expression of mRNA for CCR-4, CCR-8, and CXCR-4. Before the cells were exposed to dopamine, their CCR-4 expression, as expected from previous findings in human Treg (Sebastiani et al., 2001), was significantly higher in Treg than in Teff, but on exposure to dopamine, the expression of CCR-4 in Treg was decreased (Fig. 8d,e). The expression of mRNA encoding for CXCR-4 and CCR-8 did not change in Treg after these cells were exposed to dopamine (data not shown).
Exogenous dopamine increases the ability to fight off neurodegeneration
A previous study by our group showed that injection of activated Treg into mice (BALB/c) immediately after CNS injury significantly inhibits the spontaneous neuroprotective response, with the result that fewer neurons survive the consequences of the insult (Kipnis et al., 2002). In the same study, we showed that depletion of Treg increases the ability to withstand the insult. The present observation that Treg and Teff respond differentially to dopamine prompted us to examine the effect of dopamine on the ability to withstand neurotoxic conditions in vivo. We reasoned that systemic injection of dopamine after a CNS insult, by weakening the Treg activity, would improve recovery after a mechanical CNS injury. We subjected two groups of mice to a severe optic nerve crush injury and immediately thereafter gave injections of dopamine (0.4 mg/kg) to the mice in one group and injections of PBS to those in the other group. Two weeks later, their retinas were excised and neuronal survival was assessed. Significantly more viable neurons (1110 ± 56/mm2; mean ± SD) were found in the retinas of dopamine-injected mice than in the retinas of vehicle-treated mice (789 ± 23) (Fig. 9a).
Figure 9.
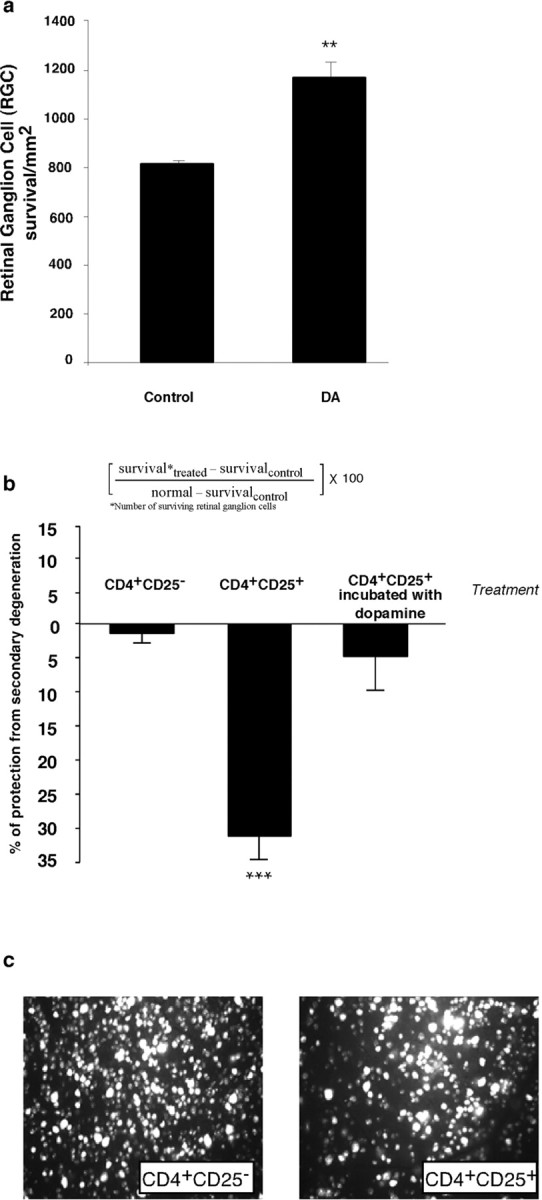
Systemic injection of dopamine, through downregulation Treg, activity increases neuronal survival after optic nerve crush injury. a, BALB/c mice were given injections of dopamine (0.4 mg/kg) immediately after being subjected to a partial crush injury of the optic nerve. Two weeks later, their retinas were excised and the numbers of surviving neurons were determined (see Materials and Methods). Significantly more neurons survived in dopamine-injected mice than in vehicle-injected controls (p < 0.01; Student's t test). Bars represent mean numbers of retinal ganglion cells per square meter of the retina. Each experiment was performed twice; n = 6-8 mice in each group. A two-tailed Student's t test was used for statistical analysis; ***p < 0.001; **p < 0.01. a, Neuronal survival was significantly worse in BALB/c mice that were inoculated (immediately after their exposure to a toxic excess of intraocular glutamate) with activated Treg than in Teff-inoculated mice. Neuronal loss is expressed as a percentage of the number of neurons in untreated glutamate-injected controls. Neuronal survival in BALB/c mice that were exposed to a toxic excess of intraocular glutamate and then treated with activated Treg that were incubated for 2 hr with 10-5 m dopamine before being administered in vivo did not differ from that in vehicle-treated glutamate-injected mice. b, Representative micrographs of retinas from mice given injections of glutamate and either Teff or Treg. Each experiment was performed twice; n = 6-8 mice in each group. A two-tailed Student's t test was used for statistical analysis; ***p < 0.001.
We tested the beneficial effect of systemic dopamine in an additional model of neuronal degeneration induced by glutamate, a common player in many neurodegenerative conditions (Katayama et al., 1990; Xiong et al., 2003). Injection of glutamate into the eyes of adult mice causes retinal ganglion cell death that is measurable 1 week after the injection (Schori et al., 2001). We gave BALB/c mice intraperitoneal injections of dopamine, or its specific D1-type agonist SKF-38393 (3.3 mg/kg), or its specific D1-type antagonist SCH-23390 (3 mg/kg), immediately after their exposure to glutamate toxicity. We also gave scid BALB/c mice injections of SKF-38393 (3.3 mg/kg) immediately after glutamate intoxication. A single systemic injection of dopamine (0.4 mg/kg) or its D1-type agonist given immediately after intraocular injection of a toxic dose of glutamate increased neuronal survival by 18 ± 2.5 or 19 ± 3.2%, respectively, relative to that in glutamate-injected controls treated with PBS (Table 1). Injection of the same agonist to scid mice resulted in no effect, thus supporting the assumption that systemic dopamine benefit CNS neurons via the peripheral immune system. The systemic injection of D1-type antagonist was done in an attempt to find out whether dopamine is involved, at least in part, in the spontaneous ability to withstand the insult. The injection of the antagonist resulted in a decrease in neuronal survival (11 ± 1.5%; p < 0.01) (Table 1) relative to that in PBS-injected mice, conceivably because it competed with the endogenous dopamine for reduction of the suppressive activity of Treg after an injury. The above results suggested that dopamine might be one of the endogenous signals initiating the cascade that leads to spontaneous T-cell-dependent neuroprotection.
Table 1.
Neuronal survival after glutamate intoxication in mice given injections of dopamine or its type 1 receptor agonist and antagonist
|
|
Treatment |
|
|
|---|---|---|---|
| Mice |
Dopamine |
SKF-38393 |
SCH-23390 |
| Wild type | 18 ± 2.5*** | 19 ± 3.2** | −11 ± 1.5** |
| SCID |
NT |
3 ± 1.8 (ns) |
NT |
Immediately after glutamate intoxication, mice were given systemic injections of the indicated drugs. Neuronal survival was determined 10 d later (see Materials and Methods). The results are expressed by changes (in percentage) in neuronal survival in treated mice relative to untreated mice. Each value represents a mean ± SEM of a group of at least five animals, and each experiment was performed at least twice, independently. Asterisks (***p < 0.001; **p < 0.01) indicate statistical significance of the presented data from a single experiment using a Student's t test statistical analysis. NT, Not tested; ns, no statistical significance.
Exposure of Treg to dopamine in vitro reduces their suppressive activity in vivo
The above results suggested that systemic dopamine can benefit injured CNS tissue in a T-cell-dependent pathway. To show this could be a consequence of a direct effect of dopamine on Treg activity in vivo, we examined whether dopamine can reduce the suppressive activity of Treg in an in vivo model of neuronal survival. Systemic injection of Treg after glutamate intoxication resulted in a 25% increase in neuronal death. Incubation of Treg with dopamine before their systemic injection into mice abolished their suppressive effect, indicated by the lack of change in the number of surviving neurons. No effect on neuronal survival after glutamate intoxication could be detected in control mice given injections of Teff (Fig. 9b). Figure 9c shows representative micrographs of fields from retinas excised from mice that were exposed to intravitreally injected glutamate and then injected with either CD4+CD25+ or CD4+CD25-.
Discussion
The results of this study show that dopamine reduces the suppressive and trafficking activities of Treg through a family of type 1 dopamine receptors (D1-R and D5-R, found here to be abundantly expressed by Treg) via the ERK signaling pathway. The physiological and pharmacological effects of dopamine, as a compound capable of downregulating Treg activity needed for fighting off neurodegeneration by a T-cell-dependent mechanism, is shown in models of CNS mechanical and biochemical (glutamate-induced) insults.
Recent studies strongly suggest that the ability to withstand CNS insults, including glutamate toxicity, is T-cell dependent and is amenable to boosting by self-antigens residing in the site of damage (Moalem et al., 1999; Yoles et al., 2001; Wekerle, 2002; Frenkel et al., 2003). An alternative way to achieve beneficial enhancement of the autoimmune response against the self-antigens needed for protection and repair after a CNS injury or for fighting off tumors is by eliminating the normally suppressive effect of Treg (Sakaguchi et al., 2001; Kipnis et al., 2002). Physiological compounds that control Treg activity on a daily basis probably underlie the mechanisms whereby the body overcomes commonly occurring adverse conditions, which in most cases resolve without development of tumors or neuronal degeneration. The results of the present study suggest that one such physiological compound is dopamine. In this context, it is important to note that transient changes in dopamine levels in mesolimbic brain areas in rats, associated with neuronal activity, can reach a concentration as high as 600 nm (Gonon, 1997).
Dopamine reduced Treg activity, and this was correlated with a decrease in ERK1/2 activation. In line with this observed correlation was the finding that adhesive and migratory abilities of Treg were reduced by dopamine via the ERK pathway.
Treg might exert their suppressive activity on Teff (autoimmune T-cells) either in the lymphoid organs or at the site of the neural tissue degeneration. Mediation of the suppressive activity of Treg has been attributed partially to IL-10 and CTLA-4, whereas their migration and adhesion have been attributed to the specific repertoire of chemokine receptors and adhesion molecules that they express (Sebastiani et al., 2001). Reduction of the suppressive activity of Treg was correlated with a decrease in their IL-10 production (Zhang et al., 2004) and CTLA-4 expression, which might participate in the cytokine-mediated and cell-cell-mediated suppression by Treg, respectively. Moreover, Treg express relatively large amounts of the CD44 receptor (needed for their adhesion to CSPG) and the chemokine receptor CCR-4 (needed for their migratory ability). The exposure of Treg to dopamine resulted in a decrease in both their adhesion to CSPG and their migration toward MDC, in correlation with their diminished expression of CD44 and CCR-4, respectively.
We found that significantly more D1-R and D5-R are expressed by Treg than by Teff. The marked difference in D1-R and D5-R expression, which is hardly detectable on Teff or any other immune cells (Ricci et al., 1997), makes the D1-type receptor family a likely candidate for the dialog of dopamine with Treg, leading, via the ERK pathway, to reduction of the suppressive activity of Treg. It is interesting to note that D2-R, which antagonizes D1-R, activates ERK (Pozzi et al., 2003).
We found that the effect of dopamine on the suppressive activity of Treg was weak compared with the effect of a protein tyrosine kinase inhibitor such as genistein (Mocsai et al., 2000) or the ERK1/2 signaling pathway inhibitor PD98059, indicating that dopamine is a suitable candidate as a physiological immunomodulator mainly in the context of autoimmune activity.
Treg exist in a state of anergy, neither proliferating in response to mitogenic stimuli nor producing IL-2. Although dopamine downregulated the suppressive activity of Treg, it did not reverse the anergic state of these T-cells with respect to proliferation or IL-2 production, supporting the contention that dopamine induces changes in the activity rather than in the phenotype of Treg.
The in vivo relevance of the effect of dopamine on the suppressive activity of Treg was demonstrated in the experimental paradigms of mouse optic nerve mechanical crush injury and glutamate intoxication in the mouse eye. Significantly more neurons survived consequences of optic nerve crush or neurotoxic insult in mice given injections of dopamine or its D1-type agonist. The observed lack of effect of the D1-type agonist SKF-38393 in mice devoid of mature T-cells substantiated our conclusion that the effect of peripheral dopamine on neuronal survival is exerted via the immune system and not directly on neural tissue. Moreover, dopamine, when injected systemically, does not cross the blood-brain barrier. That the in vivo effect of dopamine is through Treg was further demonstrated by the passive transfer of Treg after their exposure to dopamine. Treg suppressed the ability to resist neurodegeneration, as indicated by an increased loss of neurons. Incubation of Treg with dopamine before their transfer wiped out their suppressive effect on neuronal survival. The loss of Treg activity in vivo might reflect the effect of dopamine both on homing of Treg to the damaged site and on their suppression. The potential ability of endogenous dopamine to operate spontaneously in vivo was demonstrated by the decrease in neuronal survival in mice given injections of the D1-type antagonist SCH-23390 immediately after glutamate intoxication. The weak (11%) effect of SCH-23390 on neuronal survival appears to be attributable, at least in part, to the nature of the experimental model (Kipnis et al., 2002). The 11% decrease observed in the wild-type mice represents >30% of the maximal possible T-cell-dependent effect (the difference between nude and wild type). It is also possible that dopamine is a member of a family of physiological compounds capable of controlling Treg activity after a CNS insult.
Previous studies have documented the effect of dopamine on T-cell adhesion (Levite et al., 2001), on activation (Ilani et al., 2001), and on T-lymphocyte suppression of IgG production by peripheral blood mononuclear cells (Kirtland et al., 1980). No attempt was made in any of those studies to attribute the dopamine effect to subpopulations of CD4+ T-cells. Our results suggest that dopamine has a direct and preferential effect on Treg in initiating the immune response but will not circumvent the need for the two signals known to be needed for eliciting a T-cell response [antigen recognition by T-cells on class II major histocompatibility complex proteins and costimulatory molecules (Bretscher and Cohn, 1970)]. It was recently suggested that in the presence of strong immunogens, Teff, with the aid of APCs, can overcome the suppression imposed by Treg (Pasare and Medzhitov, 2003). This mechanism is not likely to operate in response to self-antigens, possibly because the self-antigens are neither present in sufficient amounts nor sufficiently potent to induce the needed response.
In light of the observed effect of dopamine on Treg in this study, the uncontrolled presence of dopamine known to occur in patients with mental disorders (such as schizophrenia) might explain the high incidence of aberrant immune activity in these patients (Muller et al., 2000). The dopamine metabolite homovanillic acid (HVA) in plasma originates mainly from central dopamine neurons or from central and peripheral noradrenergic (NA) neurons. The latter give rise to norepinephrine metabolites such as 3-methoxy-4-hydroxyphenylglycol (MHPG), in addition to HVA. It has been shown in primates that the association between HVA and MHPG in plasma or urine under varying rates of NA metabolism can be used to obtain an estimate of the central dopamine neuronal contribution of HVA to plasma or urine. This estimate is called the central dopaminergic index, which is significantly upregulated in schizophrenic patients (Chang et al., 1990; Amin et al., 1995). The present results that dopamine, via its effect on Treg, can lead to an increased ability to mount T-cell response to self-antigens, coupled with the known participation of autoimmune T-cells in fighting off cancer (Teunis et al., 2002) and the increased plasma dopamine levels in schizophrenic patients, might explain the relatively low incidence of cancer development observed in patients with schizophrenia (Dummer et al., 2002).
The observed correlation between the state of ERK activation and the activity of Treg opens the way, via dopamine or its related compounds, to novel therapeutic strategies for fine-tuning Treg activity and, hence, for fighting off conditions in which Treg activity needs to be weakened (such as neuronal degeneration and cancer) or strengthened (autoimmune diseases).
Footnotes
This work was supported by Proneuron Ltd. Industrial Park (Ness-Ziona, Israel) to M.S.M.S. holds the Maurice and Ilse Katz Professorial Chair in Neuroimmunology. We thank S. Smith for editing this manuscript, A. Shapira for animal maintenance, H. Avital for graphical assistance, and Dr. R. Eilam for histological assistance.
Correspondence should be addressed to Dr. Michal Schwartz, Department of Neurobiology, The Weizmann Institute of Science, 76100 Rehovot, Israel. E-mail: michal.schwartz@weizmann.ac.il.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246133-11$15.00/0
References
- Amin F, Davidson M, Kahn RS, Schmeidler J, Stern R, Knott PJ, Apter S (1995) Assessment of the central dopaminergic index of plasma HVA in schizophrenia. Schizophr Bull 21: 53-66. [DOI] [PubMed] [Google Scholar]
- Ariel A, Yavin EJ, Hershkoviz R, Avron A, Franitza S, Hardan I, Cahalon L, Fridkin M, Lider O (1998) IL-2 induces T cell adherence to extracellular matrix: inhibition of adherence and migration by IL-2 peptides generated by leukocyte elastase. J Immunol 161: 2465-2472. [PubMed] [Google Scholar]
- Bretscher P, Cohn M (1970) A theory of self-nonself discrimination. Science 169: 1042-1049. [DOI] [PubMed] [Google Scholar]
- Chang WH, Chen TY, Lin SK, Lung FW, Lin WL, Hu WH, Yeh EK (1990) Plasma catecholamine metabolites in schizophrenics: evidence for the two-subtype concept. Biol Psychiatry 27: 510-518. [DOI] [PubMed] [Google Scholar]
- Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA, Theofilopoulos AN (2002) T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest 110: 185-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar VA, Cremaschi GA, Sterin-Borda L, Genaro AM (2002) Altered expression of autonomic neurotransmitter receptors and proliferative responses in lymphocytes from a chronic mild stress model of depression: effects of fluoxetine. Brain Behav Immun 16: 333-350. [DOI] [PubMed] [Google Scholar]
- Fisher J, Levkovitch-Verbin H, Schori H, Yoles E, Butovsky O, Kaye JF, Ben-Nun A, Schwartz M (2001) Vaccination for neuroprotection in the mouse optic nerve: implications for optic neuropathies. J Neurosci 21: 136-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Huang Z, Maron R, Koldzic DN, Hancock WW, Moskowitz MA, Weiner HL (2003) Nasal vaccination with myelin oligodendrocyte glycoprotein reduces stroke size by inducing IL-10-producing CD4+ T cells. J Immunol 171: 6549-6555. [DOI] [PubMed] [Google Scholar]
- Gonon F (1997) Prolonged and extrasynaptic excitatory action of dopamine mediated by D1 receptors in the rat striatum in vivo J Neurosci 17: 5972-5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke CA, Roongta U, Mickelson DJ, Knutson JR, McCarthy JB (1996) CD44-related chondroitin sulfate proteoglycan, a cell surface receptor implicated with tumor cell invasion, mediates endothelial cell migration on fibrinogen and invasion into a fibrin matrix. J Clin Invest 97: 2541-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey WF (1999) Leukocyte traffic in the central nervous system: the participants and their roles. Semin Immunol 11: 125-137. [DOI] [PubMed] [Google Scholar]
- Ilani T, Ben-Shachar D, Strous RD, Mazor M, Sheinkman A, Kotler M, Fuchs S (2001) A peripheral marker for schizophrenia: Increased levels of D3 dopamine receptor mRNA in blood lymphocytes. Proc Natl Acad Sci USA 98: 625-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SH, Barchan D, Maiti PK, Fuchs S, Souroujon MC (2001) Blockade of CD40 ligand suppresses chronic experimental myasthenia gravis by down-regulation of Th1 differentiation and up-regulation of CTLA-4. J Immunol 166: 6893-6898. [DOI] [PubMed] [Google Scholar]
- Jones LL, Margolis RU, Tuszynski MH (2003) The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp Neurol 182: 399-411. [DOI] [PubMed] [Google Scholar]
- Katayama Y, Becker DP, Tamura T, Hovda DA (1990) Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J Neurosurg 73: 889-900. [DOI] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Hauben E, Shaked I, Shevach E, Schwartz M (2002) Neuroprotective autoimmunity: naturally occurring CD4+CD25+ regulatory T cells suppress the ability to withstand injury to the central nervous system. Proc Natl Acad Sci USA 99: 15620-15625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirtland 3rd HH, Mohler DN, Horwitz DA (1980) Methyldopa inhibition of suppressor-lymphocyte function: a proposed cause of autoimmune hemolytic anemia. N Engl J Med 302: 825-832. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Carpentier PA, Anger HA, Miller SD (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169: 4712-4716. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Benveniste EN (1999) Adhesion molecule expression and regulation on cells of the central nervous system. J Neuroimmunol 98: 77-88. [DOI] [PubMed] [Google Scholar]
- Lemmer K, Ahnert-Hilger G, Hopfner M, Hoegerle S, Faiss S, Grabowski P, Jockers-Scherubl M, Riecken EO, Zeitz M, Scherubl H (2002) Expression of dopamine receptors and transporter in neuroendocrine gastrointestinal tumor cells. Life Sci 71: 667-678. [DOI] [PubMed] [Google Scholar]
- Levite M, Chowers Y, Ganor Y, Besser M, Hershkovits R, Cahalon L (2001) Dopamine interacts directly with its D3 and D2 receptors on normal human T cells, and activates beta1 integrin function. Eur J Immunol 31: 3504-3512. [DOI] [PubMed] [Google Scholar]
- Maloy K, Salaun L, Cahill R, Dougan G, Saunders N, Powrie F (2003) CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med 197: 111-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC (2002) CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16: 311-323. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999) Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5: 49-55. [DOI] [PubMed] [Google Scholar]
- Mocsai A, Jakus Z, Vantus T, Berton G, Lowell CA, Ligeti E (2000) Kinase pathways in chemoattractant-induced degranulation of neutrophils: the role of p38 mitogen-activated protein kinase activated by Src family kinases. J Immunol 164: 4321-4331. [DOI] [PubMed] [Google Scholar]
- Muller N, Riedel M, Gruber R, Ackenheil M, Schwarz MJ (2000) The immune system and schizophrenia. An integrative view. Ann NY Acad Sci 917: 456-467. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kitani A, Strober W (2001) Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J Exp Med 194: 629-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R (2003) Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299: 1033-1036. [DOI] [PubMed] [Google Scholar]
- Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM (2002) CD4(+)CD25(+) regulatory T cells can mediate suppressor function in the absence of transforming growth factor beta1 production and responsiveness. J Exp Med 196: 237-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi L, Hakansson K, Usiello A, Borgkvist A, Lindskog M, Greengard P, Fisone G (2003) Opposite regulation by typical and atypical anti-psychotics of ERK1/2, CREB and Elk-1 phosphorylation in mouse dorsal striatum. J Neurochem 86: 451-459. [DOI] [PubMed] [Google Scholar]
- Ramsdell F (2003) Foxp3 and natural regulatory T cells: key to a cell lineage? Immunity 19: 165-168. [DOI] [PubMed] [Google Scholar]
- Ricci A, Mariotta S, Greco S, Bisetti A (1997) Expression of dopamine receptors in immune organs and circulating immune cells. Clin Exp Hypertens 19: 59-71. [DOI] [PubMed] [Google Scholar]
- Rothblat DS, Schneider JS (1998) Effects of GM1 ganglioside treatment on dopamine innervation of the striatum of MPTP-treated mice. Ann NY Acad Sci 845: 274-277. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Takahashi T, Yamazaki S, Kuniyasu Y, Itoh M, Sakaguchi N, Shimizu J (2001) Immunologic self tolerance maintained by T-cell-mediated control of self-reactive T cells: implications for autoimmunity and tumor immunity. Microbes Infect 3: 911-918. [DOI] [PubMed] [Google Scholar]
- Schori H, Yoles E, Schwartz M (2001) T-cell-based immunity counteracts the potential toxicity of glutamate in the central nervous system. J Neuroimmunol 119: 199-204. [DOI] [PubMed] [Google Scholar]
- Sebastiani S, Allavena P, Albanesi C, Nasorri F, Bianchi G, Traidl C, Sozzani S, Girolomoni G, Cavani A (2001) Chemokine receptor expression and function in CD4+ Tlymphocytes with regulatory activity. J Immunol 166: 996-1002. [DOI] [PubMed] [Google Scholar]
- Sharp LL, Schwarz DA, Bott CM, Marshall CJ, Hedrick SM (1997) The influence of the MAPK pathway on T cell lineage commitment. Immunity 7: 609-618. [DOI] [PubMed] [Google Scholar]
- Shevach EM, McHugh RS, Thornton AM, Piccirillo C, Natarajan K, Margulies DH (2001) Control of autoimmunity by regulatory T cells. Adv Exp Med Biol 490: 21-32. [DOI] [PubMed] [Google Scholar]
- Sundstedt A, O'Neill E, Nicolson K, Wraith D (2003) Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol 170: 1240-1248. [DOI] [PubMed] [Google Scholar]
- Takeuchi Y, Fukunaga K (2003) Differential regulation of NF-kappaB, SRE and CRE by dopamine D1 and D2 receptors in transfected NG108-15 cells. J Neurochem 85: 729-739. [DOI] [PubMed] [Google Scholar]
- Tanimura S, Asato K, Fujishiro SH, Kohno M (2003) Specific blockade of the ERK pathway inhibits the invasiveness of tumor cells: downregulation of matrix metalloproteinase-3/-9/-14 and CD44. Biochem Biophys Res Commun 304: 801-806. [DOI] [PubMed] [Google Scholar]
- Teunis MA, Kavelaars A, Voest E, Bakker JM, Ellenbroek BA, Cools AR, Heijnen CJ (2002) Reduced tumor growth, experimental metastasis formation, and angiogenesis in rats with a hyperreactive dopaminergic system. FASEB J 16: 1465-1467. [DOI] [PubMed] [Google Scholar]
- Thiffault C, Langston JW, Di Monte DA (2000) Increased striatal dopamine turnover following acute administration of rotenone to mice. Brain Res 885: 283-288. [DOI] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM (1998) CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 188: 287-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wekerle H (2002) Immune protection of the brain-efficient and delicate. J Infect Dis 186 [Suppl 2]: S140-S144. [DOI] [PubMed] [Google Scholar]
- Xiong H, McCabe L, Skifter D, Monaghan DT, Gendelman HE (2003) Activation of NR1a/NR2B receptors by monocyte-derived macrophage secretory products: implications for human immunodeficiency virus type one-associated dementia. Neurosci Lett 341: 246-250. [DOI] [PubMed] [Google Scholar]
- Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, Kuchroo V, Cohen IR, Weiner H, Schwartz M (2001) Protective autoimmunity is a physiological response to CNS trauma. J Neurosci 21: 3740-3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Koldzic DN, Izikson L, Reddy J, Nazareno RF, Sakaguchi S, Kuchroo VK, Weiner HL (2004) IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25(+)CD4(+) regulatory T cells. Int Immunol 16: 249-256. [DOI] [PubMed] [Google Scholar]