

Abstract
Fibroblast growth factor receptor 1 (Fgfr1) is expressed at high levels by progenitor cells of the ventricular zone (VZ) within the hippocampal primordium. To investigate the role of Fgfr1 in these cells, in vivo Cre recombination of “floxed” Fgfr1 alleles was directed to cells of the radial glial lineage by using the human glial fibrillary acidic protein promoter. Radial glial-like cells of the hippocampal VZ are the progenitors of pyramidal neurons and granule cells of hippocampal dentate gyrus (DG). Mice carrying null Fgfr1 alleles (Fgfr1Δflox) in cells of this lineage showed a dramatic loss of Fgfr1 gene expression throughout the embryonic dorsal telencephalon. These Fgfr1Δflox mice exhibited a ∼30% decrease in dividing radial glial progenitor cells in the hippocampal VZ and DG in the late embryonic period, progressing to a ∼50-60% loss at birth, without any changes in cell survival. In addition, no FGF2-sensitive neural stem cells could be isolated from the Fgfr1Δflox hippocampal neuroepithelium, whereas epidermal growth factor-sensitive neural stem cells were not affected. The number of hippocampal pyramidal neurons and DG granule cells was ∼30-50% decreased from the perinatal period through adulthood, and the number of parvalbumin-containing interneurons was similarly decreased in both the DG and pyramidal cell fields. We conclude that Fgfr1 is necessary for hippocampal growth, because it promotes the proliferation of hippocampal progenitors and stem cells during development.
Keywords: mouse, progenitor, neural stem cell, FGF, growth factor, tyrosine kinase, radial glia
Introduction
Fibroblast growth factors (FGFs) are extracellular molecules that regulate tissue outgrowth during organ morphogenesis (Niswander et al., 1993; Crossley et al., 1996; Zhao et al., 2001; Ohkubo et al., 2002). The Fgf8 and Fgf2 gene products are expressed in the telencephalic neuroepithelium (Ernfors et al., 1990; Crossley and Martin, 1995; Vaccarino et al., 1999a; Crossley et al., 2001). FGF2 is required for cell proliferation in the cortical ventricular zone (VZ) and for the genesis of cortical pyramidal cells (Vaccarino et al., 1999a; Raballo et al., 2000; Korada et al., 2002) as well as for hippocampal granule cell neurogenesis in postnatal mice (Cheng et al., 2002). In addition, FGF2 is required for the clonal expansion of neural stem cells isolated from cortical and hippocampal primordia (Cattaneo and McKay, 1990; Kilpatrick and Bartlett, 1993; Ray et al., 1993; Vescovi et al., 1993; Vaccarino et al., 1995; Tropepe et al., 1999). FGF8 increases the growth and survival of neuroepithelial cells of the telencephalic vesicles (Shimamura and Rubenstein, 1997) and at high levels may promote cell death (Storm et al., 2003). FGF8 is thought to control the regional specification of the neocortex (Fukuchi-Shimogori and Grove, 2001; Garel et al., 2003; Storm et al., 2003). Thus, different FGFs may play different roles in CNS development, but the receptors and downstream signaling events required for these actions are not yet clear.
FGFs bind to four distinct cell-surface receptors, FGF receptor (FGFR) 1-FGFR4 (for review, see Ornitz, 2000). Different functional effects of FGFs are likely mediated by differential binding affinity for FGFRs, which may eventually induce changes in gene expression (for review, see Vaccarino et al., 1999b). The Fgfr1, Fgfr2, and Fgfr3 genes are expressed in a partially overlapping manner in the dorsal telencephalic VZ (Orr-Urtreger et al., 1991; Peters et al., 1993; Vaccarino et al., 1999a; Ragsdale and Grove, 2001). Investigating the role of Fgfr1 and Fgfr2 during brain development has been difficult because germline null mutations for these receptors are lethal in early embryogenesis (Deng et al., 1994; Yamaguchi et al., 1994; Arman et al., 1998; Xu et al., 1998). Furthermore, the analysis of embryonic chimeras made from Fgfr1 null and wild-type cells (Deng et al., 1996; Tropepe et al., 1999) and of mice harboring region-specific deletions of the Fgfr1 gene (Pirvola et al., 2002; Hebert et al., 2003) have yielded conflicting results pertaining to the specific role of FGFR1 in neural development (Pirvola et al., 2002; Hebert et al., 2003). Among cells of the telencephalon, Fgfr1 expression is highest in the hippocampus, both during embryogenesis and throughout adulthood (Heuer et al., 1990; Wanaka et al., 1991; Asai et al., 1993; our study). Here, we directed Cre-dependent excision of the Fgfr1 gene in telencephalic radial cells of the VZ using the human glial fibrillary acidic protein (hGFAP) promoter. During the period of neurogenesis, the majority of neuroepithelial cells of the VZ express molecular markers of the astrocyte lineage, including the astrocyte-specific glutamate transporter (GLAST) and the brain lipid binding protein (BLBP) (for review, see Kriegstein and Gotz, 2003). The excision of Fgfr1 using hGFAP-Cre produced a near complete absence of Fgfr1 gene expression in the embryonic telencephalon. FGFR1 loss of function decreases the proliferation of cells expressing radial glial markers in the developing hippocampus and precludes the isolation of FGF2-sensitive neural stem cells from the hippocampal primordium. This results in defective hippocampal growth that progressively worsens during postnatal development.
Materials and Methods
Cell culture and immunocytochemistry. The hippocampal primordium was isolated from embryonic day 14.5 (E14.5) embryo and mechanically dissociated into single cells. Dissociated cells were cultured for 3 d within eight-well chamber slides (Nunc, Rochester, NY). Primary cell culture and immunostaining was performed according to standard protocols (Vaccarino et al., 1995). The primary antibodies for FGFR1 were as follows: (1) the rabbit antiserum Yu302 (a gift from Dr. J. Schlessinger, Yale University, New Haven, CT), and (2) the 19B2 mouse monoclonal antibody (Upstate Biotechnology, Charlottesville, VA). Other primary antibodies were anti-nestin (Chemicon, Temecula, CA), anti-GLAST (guinea pig polyclonal antibodies; kind gift from Dr. M. Watanabe, Hokkaido University, Sapporo, Hokkaido, Japan), anti-BLBP (kind gift from Dr. N. Heintz, Rockefeller University, New York, NY), RC2 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), anti-βIII tubulin (Promega, Madison, WI), and GFAP (Sigma, St. Louis, MO; and Zymed, San Francisco, CA). Biotinylated anti-rabbit or mouse antibodies, followed by fluorescein avidin DCS (Vector Laboratories, Burlingame, CA) or anti-mouse Alexa 594 (Molecular Probes, Eugene, OR) were used as secondary reagents.
Immunohistochemistry. Brains older than postnatal day 7 (P7) were perfused intracardially with 4% paraformaldehyde. Embryonic brains were immersed in the same fixative overnight. After cryoprotection in 20% sucrose, brains were frozen and then cryosectioned at either 10 μm for embryonic brains or 50 μm for postnatal brains. For the analyses of cell proliferation, 2-bromodeoxyuridine (BrdU) was injected (50 μg/body weight, i.p.) 30 min or 2 hr before the animals were killed. Sections were reacted with mouse anti-BrdU (Becton Dickinson, Mountain View, CA) and with the ABC reagent (Vectastain ABC kit; Vector Laboratories) as described previously (Ganat et al., 2002) and were counterstained with cresyl violet.
Other primary antibodies used were calbindin (Chemicon), parvalbumin (PV) (Sigma), neurofilaments (SMI-32; Sternberger Monoclonals, Lutherville, MD), anti-β galactosidase (β-gal; Cappel, Aurora, OH), anti-Cre recombinase (Covance, Berkeley, CA), anti-GLAST, anti-GFAP, anti-BLBP, RC2, and anti-βIII tubulin (described above). The specimens were examined using Zeiss Stemi SV11 or Zeiss Axioskope equipped with AxioCam 4.1 (Zeiss, Thornwood, NY).
Transgenic mouse lines. All animal procedures described in this study were performed in accordance with Yale Animal Resources Center policies.
A transgenic mouse expressing Cre under the control of a human GFAP promoter element was generated by Zhuo et al. (2001) and maintained on a FVB mouse strain. The Cre transgene was detected by genomic PCR of tail DNA (Zhuo et al., 2001). A mouse carrying a recombinant Fgfr1 allele that contained loxP sites (Fgfr1Flox) flanking the transmembrane and kinase domains (supplemental Fig. 2 A, available at www.jneurosci.org) was generated by Pirvola et al. (2002). To investigate the efficiency of Cre-mediated recombination, genomic DNA extracted from the cerebral cortex, hippocampus, or tail was independently subjected to PCR to detect the Fgfr1Flox and the recombined allele (Fgfr1ΔFlox) using the primers shown in supplemental Figure 2 A (available at www.jneurosci.org). PCR was performed at 98, 61, and 72°C for 1 min each cycle for 27 cycles to amplify DNA in a semiquantitative manner. The Rosa 26R Cre reporter line (Soriano, 1999) was used to detect the spatial and cellular pattern of Cre-recombinase activity by assaying β-gal expression and activity.
Reverse transcriptase-PCR analysis of Fgfr1 expression. Brain tissue was homogenized with a 21 ga needle in TRIZOL reagent (Invitrogen, Paisley, UK). One microgram of total RNA was used for each reverse transcriptase (RT)-PCR reaction. Primers for the Fgfr1 extracellular domain were designed within the cDNA sequence of the first IgG-like domain of Fgfr1 (5′-TGGAAGTGCCTCCTCTTCTGG-3′ and 5′-AAGCGTAGAGGCCAGAGTCAGC-3′), which yield a 291 bp PCR product. To amplify the deleted portion of Fgfr1, a forward primer was designed within the IgG-III domain (5′-ATGGAGGTGCTTCATCTACG-3′) and a reverse primer within the intracellular domain (5′-GGAAGTCGCTCTTCTTGGTGC-3′), giving a 258 base pair PCR product. Nucleotide sequence was taken from GenBank (accession number NM 010206). Semiquantitative RT-PCR for Fgfr1 was performed using the same conditions described above.
Western blot analyses. Embryonic brain samples were dissected out, homogenized in 6× SDS sample buffer (12% SDS, 600 mm DTT, 300 mm Tris-HCl, pH 6.8), and Western blotting was performed as described previously (Ganat et al., 2002). The blotted membrane was reacted with either anti-FGFR1 rabbit polyclonal antibody for the amino terminal (Ab-15; kind gift from Dr. L. T. Williams, Five Prime Therapeutics Inc., San Francisco, CA) or anti-FGFR1 rabbit polyclonal antibody for the carboxyl terminal (antibody Yu302, described above; dilution 1:1200), together with a mouse anti-actin antibody (dilution 1:1000; Roche Diagnostics, Hertforshire, UK). Signals were detected with a chemiluminescent detection kit (SuperSignal; Pierce, Rockford, IL) and exposed to Kodak (Rochester, NY) Biomax film.
In situ hybridization. Cryostat sections (10 μm) were processed as described by Vaccarino et al. (1999a). The following cDNA were used as probes: Fgfr1 (exon 9-15); Fgfr2 [5′ untranslated region (UTR)]; Fgfr3 (5′UTR); KA-1, Foxg-1, SCIP (Tole et al., 1997); Gli3 (Grove et al., 1998); Hes 5 (Ohtsuka et al., 2001); Lef-1 (Galceran et al., 2000), Lhx2, Lhx5 (Zhao et al., 1999), L1-CAM (3′ UTR) (Moos et al., 1988), Wnt3A (Lee et al., 2000).
Neurosphere assay. The cortical hem tissue (Grove et al., 1998) was isolated from E14.5 brains and processed for neurosphere culture following previously described protocols (Tropepe et al., 1999). Dissociated cells were plated in 24-well uncoated culture wells (Corning, Corning, NY) at 1.0 × 105 cells/well and cultured in DMEM/F12 serum-free culture media supplemented with N2 (Invitrogen), 0.6% glucose, 2 mm glutamine, 3 mm sodium bicarbonate (Sigma), and 10 ng/ml FGF-2 or EGF (human recombinant; Invitrogen). After 7 d, neurospheres (diameter >150 μm) were counted under a dissection microscope. To test for multipotency, neurospheres were harvested, replated at clonal density into poly-l-ornithine/laminin (Sigma)-coated eight-well slide chambers (Nunc), and cultured with DMEM/F12 containing 2% FCS for 5 d. Neurospheres were then fixed with 4% paraformaldehyde, washed with PBS, and analyzed by immunocytochemistry using βIII tubulin and GFAP antibodies as described above.
Unbiased morphometric methods. For unbiased stereological methods, series of sections (1 every 10) were immunostained with various markers and lightly counterstained with cresyl violet. Cell number was assessed by the optical fractionator method (West et al., 1991; Raballo et al., 2000) using StereoInvestigator (MicroBrightField, Colchester, VT). The boundaries of the hippocampal ventricular zone, dentate gyrus (DG), hippocampal pyramidal cell fields (CA), and cerebral cortex were drawn using the StereoInvestigator and Neurolucida softwares. A volume estimate was obtained by the Cavalieri method using the area estimates, section thickness, and spacing of sections. Stained cells were counted within sampling frames chosen in a systematically random manner within the areas of interest. Sampling frames (size, 20 × 20 × 5 μm) were counted at the frequency one frame every 100 or 200 μm2 for the hippocampal DG and CA fields, respectively.
Analysis of apoptosis. Apoptosis was detected in embryonic tissue sections by terminal transferase-mediated dUTP nick end labeling (TUNEL) and by immunostaining with anti-Cleaved Caspase-3 antibody (Cell Signaling Technology, Beverly, MA). TUNEL was performed with the ApopTag in situ apoptosis detection kit (Chemicon) according to the instructions of the manufacturer.
Results
FGFR1 is expressed by many cell types in the hippocampus
Fgfr1 mRNA is detectable in the early embryonic telencephalon and is maintained in the hippocampus (Wanaka et al., 1991) (Fig. 1A, C); however, the pattern of expression of the FGFR1 protein at the cellular level has not been investigated. To analyze the cell types that express FGFR1, we performed immunocytochemistry in short-term dissociated primary cell cultures using double-staining methods with two different anti-FGFR1 antibodies. The FGFR1 antibodies did not yield a detectable reaction product with cells transfected with Fgfr2 or Fgfr3 gene constructs (supplemental Fig. 1, available at www.jneurosci.org) or with hippocampal tissue from our conditional Fgfr1 knock-out mice (supplemental Fig. 2D, available at www.jneurosci.org), suggesting that they are specific for FGFR1.
Figure 1.
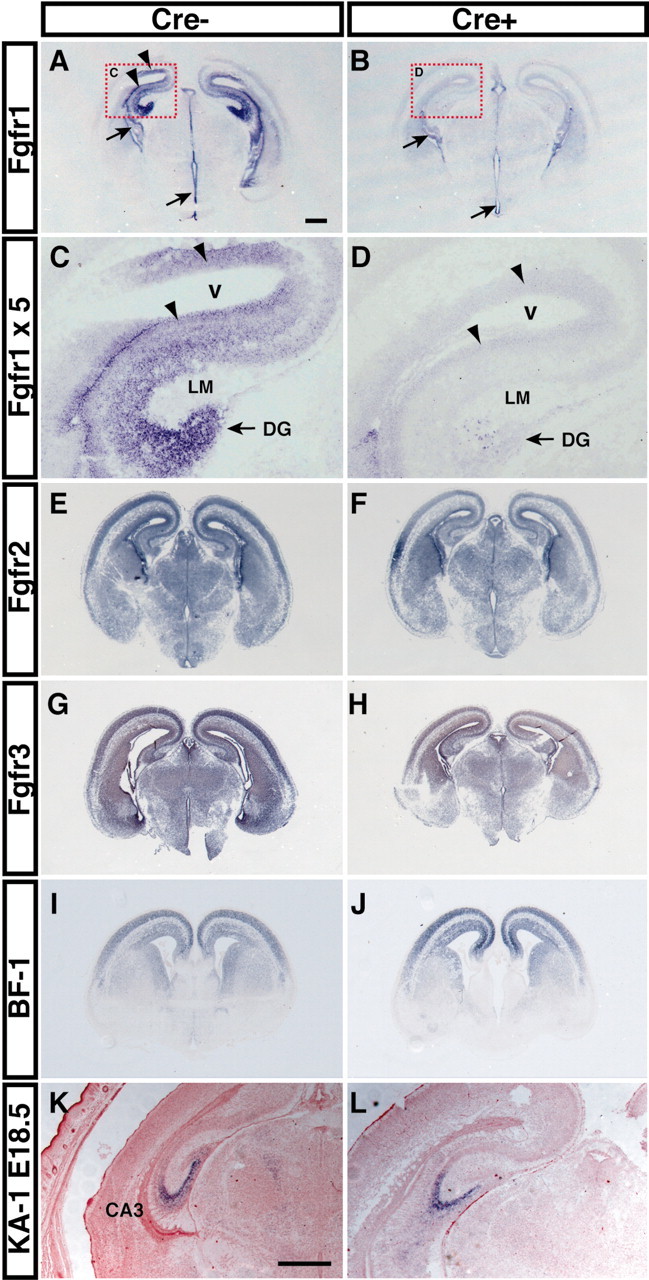
Fgfr gene expression and region-specific gene expression in Fgfr1 conditional null mice. A, In E16.5 Fgfr1flox/flox mice lacking Cre (Cre-), Fgfr1 mRNA is expressed in the cortical VZ (top arrowhead), hippocampal VZ (bottom arrowhead), and DG. B, hGFAP-Cre/+; Fgfr1flox/flox (Cre+) mutants clearly lack Fgfr1 expression in these areas, although expression is unaltered in the fimbria (top arrow) and VZ of the diencephalon (bottom arrow). Higher magnification images of red frames in A and B are presented in C and D. Very few cells expressing Fgfr1 are remaining in Cre+ mutants in the stratum LM and around the edge of the DG (arrow). E-H show that Fgfr2 and Fgfr3 expression is not changed in the absence of Fgfr1. I-L, The expression of Foxg1 (BF-1) (I, J) or KA-1 (K, L) mRNAs is not altered by the lack of a functional Fgfr1 gene. Scale bars: (in A) A, B, E-J, 500 μm; C, D, 111 μm; (in K) K, L, 500 μm.
In primary cultures prepared from the E14.5 hippocampal primordium maintained in vitro for 3 d, various cell types were identified by antibody staining for specific marker proteins. Cells of the radial glia lineage were visualized by immunostaining for GLAST, RC2, and BLBP antibodies (Shibata et al., 1997) and frequently exhibited a spindle shape with two or three main fibers (Fig. 2B,E,H). GLAST, RC2, and BLBP-positive radial glial cells were also positive for FGFR1 (100%; n = 40), as demonstrated by double immunofluorescence with either monoclonal or polyclonal anti-FGFR1 antibodies. The FGFR1 protein was accumulated in the cell body and the proximal part of the fibers (Fig. 2A,C,D,F,G,I, arrowheads). This localization was similar to that of RC2 (Fig. 2E,F), whereas GLAST was distributed in the cell body and throughout the length of the fibers (Fig. 2B,C), and BLBP was accumulated inside the soma and nucleus (Fig. 2H,I). FGFR1 was also detected in differentiating neurons and glia in these hippocampal cultures. βIII tubulin-positive neuronal cells expressed FGFR1 mainly in the cell body and initial segment of the presumptive axon (Fig. 2J,K,L) (91%; n = 45), whereas GFAP-positive cells expressed FGFR1 throughout the cell and all processes (Fig. 2M-O) (70%; n = 40). Thus, FGFR1 was expressed by most of the cells in these cultures, although with different intensity and intracellular distribution. In agreement with these results, in situ hybridization experiments (Fig. 1A,C) indicated that Fgfr1 mRNA is distributed throughout all layers of the hippocampal formation, including the hippocampal VZ, which contains dividing progenitor cells, as well as the nascent pyramidal and granule cell layers, containing differentiating neuronal cells. In aggregate, these immunocytochemical results, together with the Fgfr1 in situ hybridization analyses, suggest that FGFR1 is widely expressed by various cell types of the developing hippocampus, including cells expressing radial glial cell markers.
Figure 2.
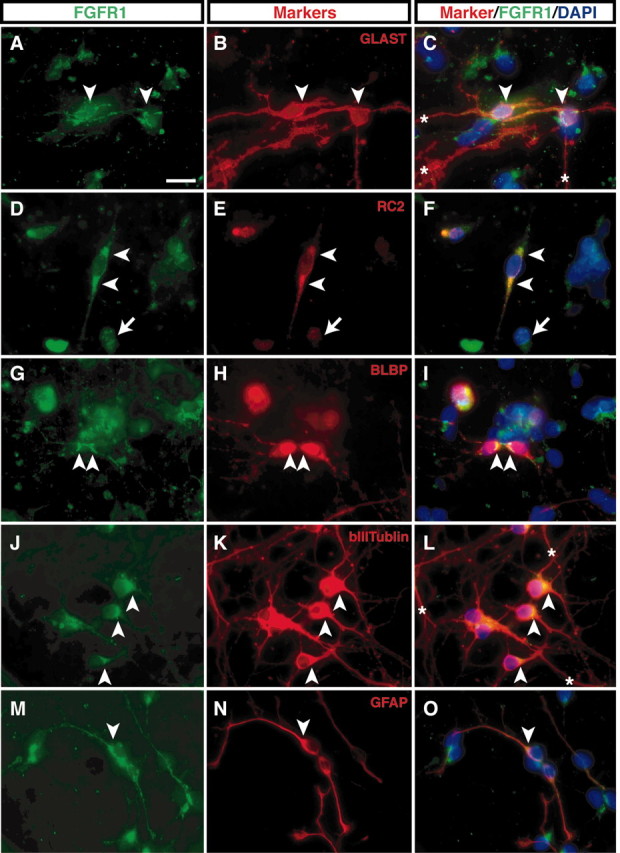
FGFR1 expression is detectable in cells of the radial glial lineage as well as immature neurons and astrocytes. Dissociated cell cultures from E14.5 hippocampal tissue were immunostained for FGFR1 using a mouse monoclonal (A, G) or a rabbit polyclonal antibody (D, J, M) and double stained with various antibodies as indicated (B, E, H, K, N). Images in each horizontal column show the same field with FGFR1 immunoreactivity at the left (green), antibody markers in the middle (red), and merged images with DAPI nuclear staining (blue) at the right. White arrow heads indicate the accumulation of FGFR1 in cell bodies. White arrows in D-F indicate a cell with weak staining for FGFR1. White asterisks in C and L indicate GLAST or β III-tubulin-positive fibers that are negative for FGFR1. However, GFAP-positive cells express FGFR1 immunoreactivity in their fibers (M, O). Scale bar, 20 μm.
Pattern of Cre activity driven by the hGFAP-Cre transgene
To target Fgfr1 recombination to telencephalic progenitor cells, we used a transgenic line in which the human GFAP regulatory element targets Cre recombination to radial glia of the mouse telencephalon (hGFAP-Cre) (Malatesta et al., 2000, 2003; Zhuo et al., 2001). We investigated the regional and cellular patterns of Cre expression using an antibody to Cre. To assess the recombinase activity driven by this hGFAP-Cre transgene, we crossed the hGFAP-Cre line with Rosa26R, a reporter line in which Cre recombination activates the transcription of an heritable β-gal marker gene (Soriano, 1999). We observed that hGFAP-driven Cre recombinase activity, as detected by lacZ staining or by β-gal immunoreactivity, started at E13.5 in the dorsal telencephalon. At this stage, Cre recombinase activity was clearly limited to the dorsal VZ, including the prospective hippocampal region (supplemental Fig. 3A,B, available at www.jneurosci.org). By E16.5, the positive area included not only the VZ but also scattered cells and processes (presumably radial glia processes) in the cortical plate (Fig. 3 A, B; supplemental Fig. 3C,D, available at www.jneurosci.org). The β-gal reporter was expressed in the entire neocortex and dorsal archicortex, including the hippocampal formation but excluding the diencephalon (Fig. 3A,B; supplemental Fig. 3C,D, available at www.jneurosci.org).
Figure 3.
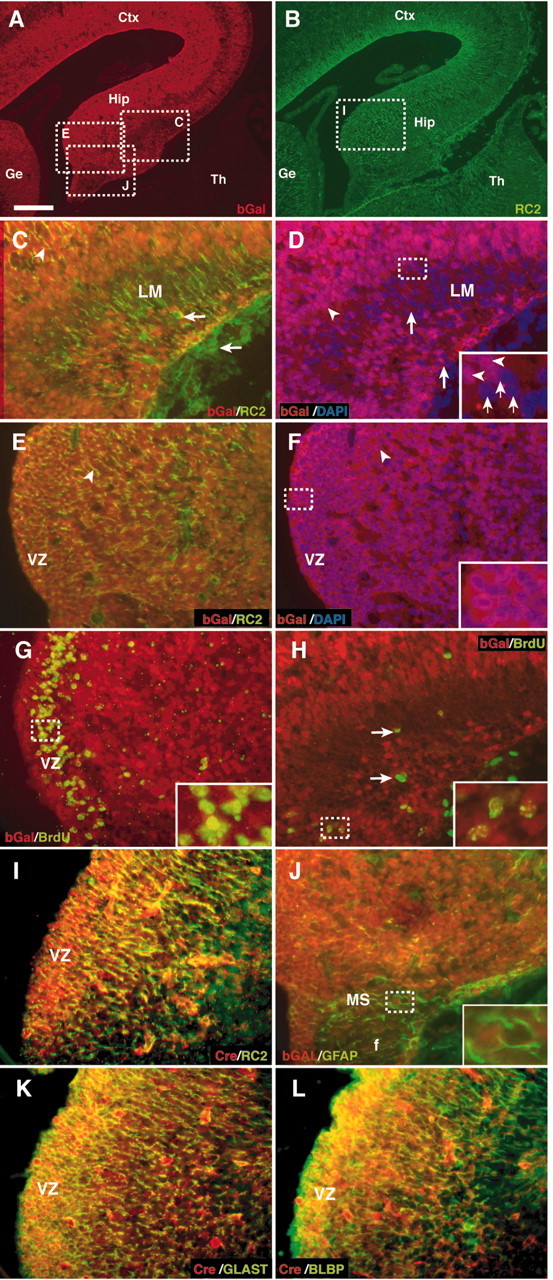
Distribution of Cre recombination driven by the hGFAP-Cre transgene. A, C-L, Brain sections from hGFAP-Cre; ROSA26R double transgenic mice were assayed for β-gal antibody staining (A, C-H, J) or Cre recombinase antibody (I, K, L) at E16.5. A, Coronal sections showing β-gal immunoreactivity (red) in cells of the cortical primordium (Ctx), the developing hippocampus (Hip), and more lightly in the ganglionic eminences (Ge) but not in the thalamus (Th). B, The same section is double stained with the neuroepithelial marker RC2. C-H and J represent higher magnifications of the corresponding white dashed squares in A. The majority of β-gal immunoreactivity colocalizes with RC2 in the hippocampal VZ (white arrowheads in C andE), except for some RC2-positive cells lacking β-gal immunoreactivity in and near the DG (C, arrows). Nuclear staining with DAPI in the same sections confirms that almost all cells are positive for β-gal in the hippocampal VZ (D, F, purple cells, arrowheads), and that some cells are not β-gal-positive in the stratum LM and adjacent DG (D, blue cells, arrows). All of the proliferating cells in hippocampal VZ, detected by BrdU, are also positive for β-gal (G); however, some BrdU-positive cells areβ-gal negative in the DG (H, arrows). Most proliferating cells in the migratory stream (MS) between the VZ and DG are β-gal positive (H, small window). J, GFAP/β-gal double immunostaining shows that GFAP-positive cells detectable in the MS and fimbria (f) can be positive or negative for β-gal; inset in J shows β-gal-positive and β-gal-negative cells (DAPI counterstaining verified the presence of nuclei). I, K, and L represent higher magnifications of the corresponding white dashed squares in B. Cre immunoreactivity is most prominent in the VZ, and in almost all cases, Cre-positive cells colocalize RC2 (I), GLAST (K), and BLBP (L). Scale bars: A, B, 200 μm; C-L, 50 μm. All inserted small windows show 3× higher magnification.
Almost all of the cells in the dorsomedial (hippocampal) VZ and developing hippocampal pyramidal layers expressed Cre recombinase activity at E16.5, as demonstrated by colocalization of β-gal immunoreactivity with DAPI-positive nuclei (Fig. 3D,F, arrowheads). Many cells in the VZ are proliferative at these stages, and virtually all express radial glial markers (Hartfuss et al., 2001). Consistently, BrdU-positive proliferating cells were all positive for β-gal in this region (Fig. 3G, inset). Radial glia markers, including RC2, GLAST, and BLBP, were colocalized with β-gal (Fig. 3C,E, arrowheads). Similar to the β-gal reporter gene, Cre immunoreactivity was colocalized with BLBP or GLAST in cell bodies of radial glia cells (Fig. 3I,K,L); in contrast to the widespread expression of the β-gal reporter gene, Cre recombinase immunoreactivity was more restricted to cells of the VZ. Some RC2 and BrdU-positive cells in the stratum lacunosum moleculare (LM), subpial layer, and adjacent DG lacked β-gal immunostaining (Fig. 3C,D,H, arrows) or Cre immunoreactivity (data not shown). We also assessed whether the cells targeted by the hGFAP-Cre transgene expressed endogenous GFAP immunoreactivity. In the E16.5 hippocampal formation, GFAP immunoreactivity was not detected in the hippocampal VZ but predominantly in a stream of elongated cells situated between the VZ, DG, and fimbria as well as in the LM and underneath the pia (Fig. 3J) (data not shown). Only some of these GFAP-positive cells expressed β-gal immunoreactivity (Fig. 3J, small window, cell on the left).
In adult mice, the recombined reporter gene was inherited by the large majority of cells within the CA fields and the DG; however, most PV-positive interneurons were β-gal negative (supplemental Fig. 3G, available at www.jneurosci.org). Hippocampal GABAergic interneurons, including PV neurons, originate from the ganglionic eminences and migrate to the hippocampus through a medial subpial route (Anderson et al., 1999; Pleasure et al., 2000; Nery et al., 2002). These data suggest that hippocampal pyramidal neurons and DG granule cells originate from the neuroepithelial astroglial progenitors within the dorsomedial VZ, whereas PV interneurons may arise from a different cell lineage. With regard to astrocytes, only some of the GFAP-positive cells in the adult hippocampus were β-gal positive. GFAP-positive cells colocalizing the reporter gene were primarily located in the DG molecular layer and within the DG itself, whereas GFAP-positive cells in the hilus or in the stratum radiatum were apparently not targeted by recombination (supplemental Fig. 3H, available at www.jneurosci.org). Hence, it is possible that some of the hippocampal GFAP-positive astrocytes, similarly to PV neurons, come from outside sources, which would be consistent with the β-gal-negative progenitor cells that we detected in the LM and DG (Fig. 3C,D,H, arrows). However, the possibility of a down-regulation of the reporter gene within mature astrocytes must be considered.
Conditional mutagenesis of Fgfr1
To investigate the role of Fgfr1 in the developing telencephalon, we generated mice that harbor a disruption of the Fgfr1 gene specifically in progenitor cells within this area. Mice carrying the Fgfr1flox allele (Pirvola et al., 2002) were crossed with the hGFAP-Cre transgenic mice to produce a truncated, recombined Fgfr1 allele, which lacks genomic regions coding for the transmembrane and the kinase domains (supplemental Fig. 2A, available at www.jneurosci.org). After intercrossing the Fgfr1flox homozygous line with the hGFAP-Cre transgenic line, we obtained hGFAP-Cre-positive, Fgfr1flox homozygous animals (indicated hereafter as hGFAP-Cre/+; Fgfr1flox/flox), which presumably harbored a tissue-specific bi-allelic deletion of Fgfr1 (indicated as Fgfr1Δflox). Littermate mice that were hGFAP-Cre negative and harbored either one or two Fgfr1flox alleles were used as controls (indicated hereafter as hGFAP-Cre/-; Fgfr1flox/flox). Both hGFAP-Cre/+; Fgfr1flox/flox mutants and control mice grew at a similar rate as wild-type mice and were fertile.
The deleted Fgfr1 allele was located in the cerebral cortex and hippocampus, the tissues that express the Cre transgene (supplemental Fig. 2B, available at www.jneurosci.org). In the hippocampus, the recombination was virtually complete, because the unrecombined Fgfr1flox was undetectable by PCR. In the cerebral cortex, there was still a small amount of unrecombined Fgfr1flox, suggesting incomplete recombination or contamination with cellular elements that did not express the Cre gene, most likely cortical cells that were generated before E13.5 (supplemental Fig. 2B, available at www.jneurosci.org).
We assessed Fgfr1 expression by in situ hybridization using a probe complementary to the recombined region or by RT-PCR using primers for either the extracellular or the transmembrane domains. In control mice, Fgfr1 mRNA was detectable in the telencephalic and diencephalic VZ and in the nascent pyramidal layer and DG, with highest concentrations in the developing DG (Fig. 1A,C). The cortical VZ and the DG contain radial glial cell bodies (Richmann et al., 1987; Gadisseux et al., 1992). In contrast, little Fgfr1 mRNA was present in the developing stratum LM, which contains afferent fibers from the entorhinal cortex and migrating cells (Fig. 1A,C, LM). In hGFAP-Cre/+; Fgfr1flox/flox brains, the Fgfr1 mRNA became undetectable in the entire hippocampal primordium, including the hippocampal and cortical VZ (Fig. 1D, arrowheads), whereas it was still detectable in the hippocampal fimbria and VZ of the diencephalon (Fig. 1B, arrows), which do not express Cre recombinase activity. Consistent with the presence of some neuroepithelial cells in and near the DG that did not express Cre recombinase activity (Fig. 3C,D,H), a few cells strongly expressing Fgfr1 mRNA were still detected in the DG primordium inhGFAP-Cre/+; Fgfr1flox/flox mice (Fig. 1D, arrow). Fgfr1 mRNA was barely detectable in the hippocampus of hGFAP-Cre/+; Fgfr1flox/flox mice using RT-PCR and primers for either the transmembrane or the extracellular domains (supplemental Fig. 2C, available at www.jneurosci.org). Finally, the FGFR1 protein was analyzed by Western blotting. Using specific FGFR1 polyclonal antibodies to either the C terminus or the extracellular domain, the FGFR1 protein product was undetectable in the mutant cortex and hippocampus (supplemental Fig. 2D, available at www.jneurosci.org; and data not shown).
In summary, our RT-PCRs in situ hybridization and Western blot analyses suggest that we obtained a disruption of the Fgfr1 gene resulting in undetectable expression of the FGFR1 message and protein in the hippocampal region. In contrast, other Fgfr family members showed no significant change in their expression level (Fig. 1E-H). Thus, we generated a mouse harboring a selective inactivation of Fgfr1 expression during hippocampal development.
Regional specification of the hippocampus in hGFAP-Cre/+; Fgfr1flox/flox mice
The expression of several genes that delimit regional identities in the hippocampal formation was not changed by the lack of Fgfr1. For example, the expression boundaries of Foxg1 (BF-1) at the dorsal limit of the hippocampal VZ were unchanged (Fig. 1I,J). Similarly, KA-1 expression in the presumptive CA3 cell field was unchanged in the mutants (Fig. 1K,L). Other genes that were not significantly altered at E13.3-E18.5 included L1-CAM, Hes5, Lef-1, Lhx-2/5, and SCIP (data not shown). These results suggest that hippocampal cell fates and regional identities identified by these markers may not be compromised by the lack of Fgfr1 gene function.
Progressive hippocampal atrophy after Fgfr1 deletion
Adult hGFAP-Cre/+;Fgfr1flox/flox mutant mice harboring the deleted Fgfr1Δflox allele showed marked hippocampal atrophy (Fig. 4). The volume of the hippocampal pyramidal layer (CA1, CA2, CA3) was decreased by ∼35% in Fgfr1 mutants compared with control littermate mice harboring nonrecombined Fgfr1flox alleles. The volume of the DG was decreased by 41% at P7 and 21% at 6 weeks (Table 1). The total number of neurons in the pyramidal cell layer of Fgfr1 recombinant mice was decreased by approximately 50% at all ages, whereas in the DG it was decreased by 65% at P7 and 29% at 6 weeks with respect to control littermates; these decreases were statistically significant (Table 2).
Figure 4.
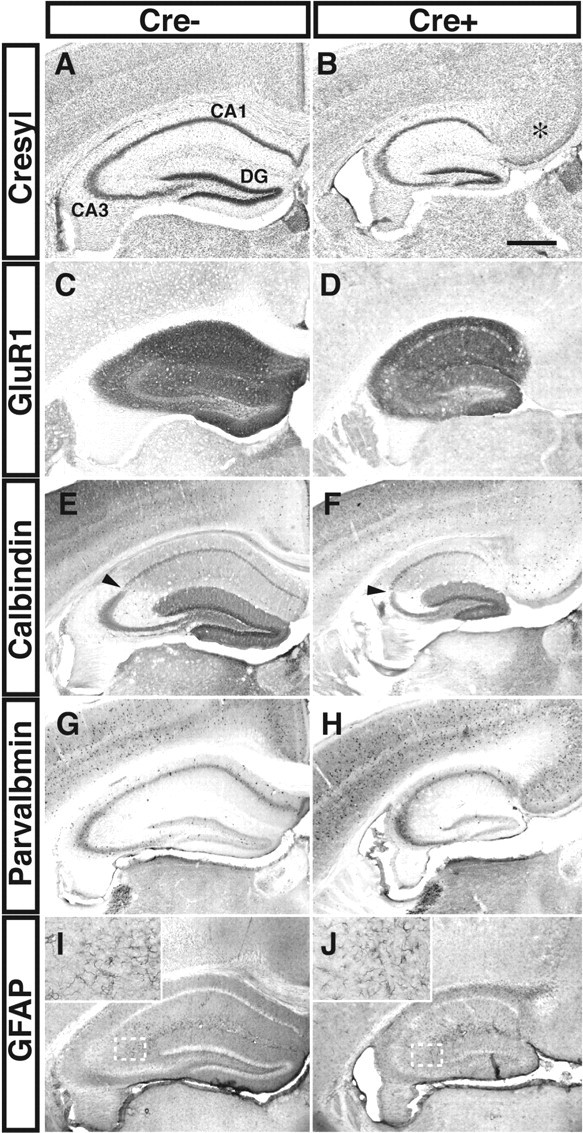
The hippocampus of Fgfr1 mutants is reduced in size but exhibits normal differentiation of neuronal or glial cell types. A, C, E, G, and I are sections from Fgfr1flox/flox mice (Cre-), and B, D, F, H, and J are from Fgfr1Δflox mutants (Cre+) at corresponding anteroposterior level. A, B, Hippocampal morphology in coronal sections stained by cresyl violet. Fgfr1Δflox mutants have decreased hippocampal size, with maintenance of pyramidal (CA1 to CA3) and granular layers (DG). Note the absence of corpus callosum and the unusual morphology of the medial cerebral cortex in the mutants (B, asterisk). C, D, Hippocampal neurons express the GluR1 in both control and mutant mice. E-J, Hippocampal granule cells express calbindin (E, F), and inhibitory interneurons express parvalbumin (G, H) in both control and mutant mice. However, the total number of these cells appears reduced in the mutants. Specific boundaries within the CA region are also distinguishable in Cre- and Cre+ hippocampi (E, F, arrowheads). The density of GFAP-positive cells is similar in control and mutant cre+ mice (I, J); insets show 4× magnification of the white boxed areas in each images. Scale bar, 500 μm.
Table 1.
Decreased volume in the hippocampal cell fields of Fgfr1 conditional null mice
|
Volume (mm3) |
|
|
|
||
|---|---|---|---|---|---|
| Perinatal period (P7) |
|||||
| Genotype |
Cerebral cortex |
Hippocampal CA fields |
Dentate gyrus |
||
| Fgfr1flox/flox | 29.8 ± 2.0 | 1.55 ± 0.15 | 0.44 ± 0.039 | ||
| hGFAP-Cre/+; Fgfr1flox/flox | 25.2 ± 0.27** | 0.99 ± 0.06 | 0.26 ± 0.028 | ||
| Percentage decrease |
15.5% |
36.1% |
40.9% |
||
| Adult (6 weeks) |
|||||
|---|---|---|---|---|---|
| Genotype |
Cerebral cortex |
Hippocampal CA fields |
Dentate gyrus |
||
| Fgfr1flox/flox | 94.2 ± 5.88 | 2.20 ± 0.29 | 0.96 ± 0.11 | ||
| hGFAP-Cre/+; Fgfr1flox/flox | 82.5 ± 3.8 | 1.44 ± 0.12** | 0.76 ± 0.07 | ||
| Percentage decrease |
12.4% |
34.6% |
20.8% |
||
Values were obtained by unbiased stereological analyses in serial sections stained with cresyl violet. Values were statistically analyzed by ANOVA, which showed an overall significant effect of genotype on volume (p < 0.01). Post hoc Sheffe tests showed that control mice significantly differed from mutants (*p < 0.05; **p < 0.01). n = 3-4 mice per genotype.
Table 2.
Decreased hippocampal cell number in Fgfr1 conditional null mice
|
Embryonic and perinatal mice |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total number of cresyl violet-stained cells (× 103) |
||||||||||||
| Hippocampal VZ |
Hippocampal DG |
|||||||||||
| Genotype |
E16.5 |
P0 |
P7 |
E16.5 |
P0 |
P7 |
||||||
| Fgfr1flox/flox | 156.5 | 377.9 | 452.9 | 152.5 | 598.5 | 637.6 | ||||||
| hGFAP-Cre/+; Fgfr1flox/flox | 189.2 | 178.2 | 304.4 | 131.1 | 381.5 | 224.0 | ||||||
| Percentage decrease |
— |
52.9 |
32.8 |
14.1 |
36.3 |
64.9 |
||||||
| Adult mice |
||||||||
|---|---|---|---|---|---|---|---|---|
| Total number of immunostained cells (× 103) |
||||||||
| Hippocampal CA fields |
Hippocampal DG |
|||||||
| Genotype |
NeuN |
PV |
NeuN |
PV |
||||
| Fgfr1flox/flox | 697 ± 74.8 | 26.0 ± 1.6 | 614.2 ± 57.6 | 5.94 ± 1.16 | ||||
| hGFAP-Cre/+; Fgfr1flox/flox | 339 ± 26.5*** | 13.0 ± 1.0*** | 437.3 ± 14.6* | 2.09 ± 0.57* | ||||
| Percentage decrease |
51.4 |
50.0 |
28.8 |
64.8 |
||||
Values were obtained by unbiased stereological analyses in serial sections stained as indicated. For cresyl violet staining at the three perinatal ages shown, a total of six mice per genotype was statistically analyzed by ANOVA, which showed an overall significant effect of genotype on cell number (p < 0.001). For the NeuN-and parvalbumin (PV)-stained sections of adult mice, three brains per genotype were analyzed. ANOVA showed an overall significant effect of genotype on NeuN cell number (F = 29.0; p < 0.001) and on PV cell number (F = 54; p < 0.0001). Post hoc Sheffe tests showed that control mice significantly differed from mutants in both regions (*p < 0.05; **p < 0.01; ***p < 0.001).
Hippocampal subregions and several hippocampal cell types appeared to be present in the smaller hippocampus of Fgfr1 conditional mutants, as shown by immunostaining with glutamate receptor 1 (GluR1), calbindin, PV, and GFAP in adult mice (Fig. 4). In addition, the CA3 region expressed the KA1 receptor during late embryonic development and SMI-32-positive neuronal fibers in adulthood (Fig. 1K,L) (data not shown), suggesting that the hippocampal fields were normally patterned and that cell differentiation was not altered in Fgfr1 conditional null mutants. However, mutant mice might have had altered number or density of some hippocampal cell types resulting from a preferential loss of particular cell lineages. We approached this question by assessing the number or density of several cell types in both control and mutant hippocampi (n = 3 mice per genotype). The number of PV interneurons was decreased as strongly as the number of total neurons in both the CA fields and DG of the mutant hippocampus (Table 2). The density of GFAP-positive astrocytes in the subgranular layer of the DG was 432,650 ± 80,369 and 494,240 ± 86,330 cells/mm3 in control and mutant mice, respectively; the density of these cells in the stratum radiatum was 167,234 ± 45,875 and 141,010 ± 19,000 cells/mm3 in control and mutant mice, respectively; the density of GFAP-positive astrocytes in the hilus of the DG was 209,210 ± 20,300 and 290,380 ± 26,430 cells/mm3 in control and mutant mice, respectively. These density values, as well as the values for total astrocyte cell number within these regions, were not significantly different between control and mutants, suggesting that the number of astrocytes was not decreased in the Fgfr1Δflox hippocampal regions.
The adult cerebral cortex of Fgfr1Δflox mice showed only a minor (∼12%) decrease in volume, and it expressed calbindin and SMI-32 immunoreactivities in a layer-appropriate manner (Fig. 4, Table 1). The differential effect of the Fgfr1 loss of function with respect to hippocampal and cortical development was already evident in the perinatal period. For example, neocortical volume was 15% decreased in Fgfr1Δflox brains at P7, whereas in the same animals, the volume of the hippocampal CA fields and that of the dentate gyrus were decreased 36 and 41%, respectively, with respect to littermate controls. However, Fgfr1Δflox mice showed dysgenesis of the corpus callosum resulting in unusual cortical morphology in the dorsomedial region (Fig. 4A,B, asterisk). Analyses of the defects in cortical connectivity of Fgfr1Δflox mice are currently in progress.
Cell proliferation is decreased in Fgfr1Δflox mice
The decrease in hippocampal cell number detected in adulthood was already present in the neonatal period (Table 2), suggesting that the removal of Fgfr1 caused either a decreased generation of cells or impaired survival. To distinguish between these possibilities, we investigated cell proliferation in Fgfr1Δflox and control littermates harboring nonrecombined Fgfr1 alleles after short-term in vivo labeling with BrdU (30 min or 2 hr). From cell counts of BrdU-labeled and -unlabeled cells in the hippocampal VZ and DG, we derived the percentage of dividing cells (BrdU-labeling index). We also estimated the total size of the proliferative population by unbiased counts of BrdU-labeled cells in the VZ and DG, a measure that does not take into account the number of “quiescent” cells (this is important because the DG neurons do not migrate away from the proliferative zone and are included in the quiescent population). Both measures of cell proliferation were strongly affected by the removal of FGFR1. In the hippocampal VZ of Fgfr1Δflox mice, the BrdU-labeling index was ∼40-45% decreased at E16.5 and P0 (Fig. 5A). In the same region, the total number of proliferative cells was decreased by 33% at E16.5 and 75% at P0 with respect to littermate controls (Figs. 5C, 6D,F). Thus, the impairment in cell proliferation resulted in a progressive decrease in dividing progenitor cell number in the VZ. In the DG, there was a ∼30-50% decrease in BrdU-labeling index (Fig. 5B), whereas the total number of proliferative cells was decreased by ∼50% at P0 and by ∼75% at P7 in Fgfr1Δflox mice with respect to control littermates (Figs. 5D, 6C,E).
Figure 5.

Decreased number and proportion of proliferative cells in the Fgfr1Δflox hippocampus. A-D, BrdU-labeling index (A, B) and number of BrdU-immunoreactive cells (C, D) in the hippocampal VZ (A, C) and DG (B, D) after 30 min BrdU labeling in vivo. The labeling index (LI) is the ratio between the number of BrdU-labeled cells and total cell number. Cross-hatched bars are hGFAP-Cre/+; Fgfr1flox/flox mutants, and black bars are Cre-negative Fgfr1flox/flox control littermates. ANOVA revealed an overall significant effect of genotype on the number of BrdU-labeled cells (F-ratio = 27.9; p < 0.0001) and on labeling index (p < 0.0005). Sheffe post hoc tests indicated significant differences in the number of BrdU labeled cells between genotypes in the hippocampal VZ (p < 0.001) and the DG (p < 0.005) and strong differences in LI in the hippocampal VZ (p < 0.001) but moderate in the DG (p < 0.05). There is no significant effect at E16.5, with significant differences between control and mutants at P0 (p < 0.01) and P7 (p < 0.001); n = 6 animals per genotype.
Figure 6.
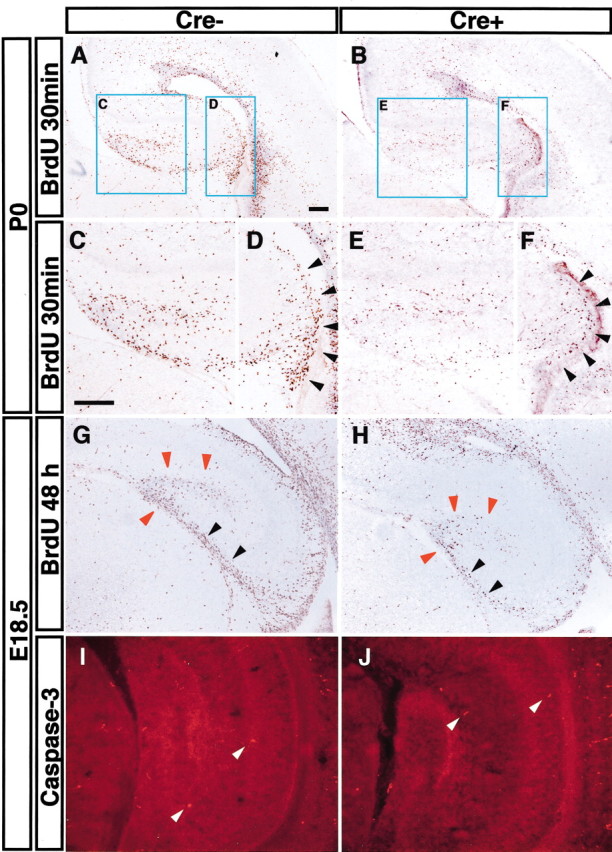
Decreased proliferation but normal survival in the Fgfr1Δflox hippocampus. A-F, Proliferating cells identified by a 2 hr BrdU incorporation at P0. C-F show a 2× higher magnification of blue squares in A and B. Proliferating cells in DG (C, E) and in hippocampal VZ (D, F, black arrowheads) are greatly decreased in hGFAP-Cre/+; Fgfr1flox/flox mutants. G, H, BrdU was injected at E16.5, and embryos were collected at E18.5. The number of cells along the migrating pathway to the DG (black arrowheads) as well as cells that are locally proliferating in the DG (red arrowheads) are clearly decreased in hGFAP-Cre/+; Fgfr1flox/flox mutants. I, J, Apoptotic cells (white arrowheads) are detected by an antibody for cleaved Caspase-3, revealing no major differences between Cre- and Cre+ mice at E18.5. Cre-, Fgfr1flox/flox; Cre+, hGFAP-Cre/+; Fgfr1flox/flox. Scale bar, 200 μm.
From E14.5 until the end of gestation, dividing cells in the dorsomedial VZ generate hippocampal pyramidal neurons, which migrate to the underlying prospective CA1-CA3 regions (Super et al., 1998). A portion of these dividing cells in the VZ is thought to migrate medially to form the dentate germinal matrices. These cells continue to divide in their subpial location and differentiate into granule cells of the dentate gyrus in the first weeks after birth (Altman and Bayer, 1990; Sievers et al., 1992). We further analyzed the effect of this conditional mutation on migrating cells en route to the DG. Proliferative cells labeled by a BrdU injection at E16.5 were visualized in the migratory pathway to the DG at E18.5 (Fig. 6G,H). We counted BrdU-positive cells along the migratory pathway from the VZ to the DG in sections from control and mutant littermate mice. The number of migrating cells was ∼53% decreased in mutant with respect to control hippocampus (average 76 cells/hippocampal section in control and 36 cells/hippocampal section in Fgfr1Δflox; n = 2). Thus, the number of progenitor cells that migrate to the DG is ∼50% decreased in mutants with respect to controls, resulting in an atrophic DG containing ∼65% fewer cells with respect to littermate controls at P7 (Table 2).
To verify whether the decrease in proliferative progenitor cells in Fgfr1 recombinant mice was attributable to a decreased survival, we stained for apoptotic cells using TUNEL. These experiments revealed only few apoptotic cells in the hippocampus at E16.5 and P0 and no detectable differences in cell death between hGFAP-Cre+ mutants and hGFAP-Cre- mice (data not shown). To confirm these data, cell death was also assayed using an anti-cleaved Caspase-3 antibody, but again, no changes were noticed in the mutant brains (Fig. 6I,J). Overall, these data indicate that the hippocampal atrophy is attributable to a decreased proliferation of VZ-derived progenitor cells.
Abnormal proliferation of radial glia-like progenitor cells in the VZ of Fgfr1Δflox mice
Cells of the hippocampal VZ and the emergent DG are positive for the neuroepithelial marker nestin as well as the radial glial markers GLAST and BLBP at E16.5-E18.5 (Fig. 7E,G,I,K,Q,S,U,W). These astroglial cellular elements densely populated the hippocampal VZ and stretched fibers to the developing pyramidal layer. However, a bipolar morphology similar to cortical radial glial cells could not be precisely defined because of the variable orientation of their fibers within the hippocampal formation; accordingly, we denominate these astroglial cells “radial glia-like” cells. To confirm whether dividing cells of the hippocampal VZ and DG are radial glia-like in vivo, we performed double immunostaining in thin frozen sections with BrdU and nestin, GLAST, or BLBP after short-term in vivo labeling with BrdU at E16.5, E18.5, P0, and P7. Cells were also immunostained with an antibody to the proliferating cell nuclear antigen (PCNA). In the hippocampal VZ and DG of control and mutant mice, all detectable BrdU-labeled or PCNA-positive cells expressed these radial glial markers at E16.5 and E18.5 (Fig. 7E-L). Importantly, the density of BrdU-labeled or PCNA-positive radial glial cells was decreased in Fgfr1Δflox hippocampal VZ and DG at E18.5 (n = 5) (Fig. 7, compare G with H and K with L).
Figure 7.
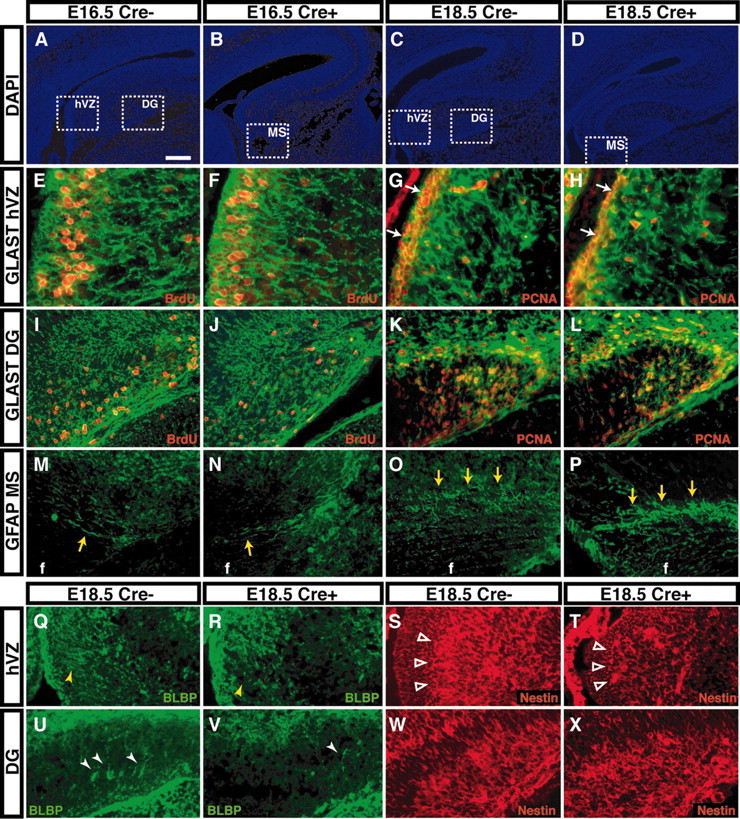
Reduction in the proliferation and density of radial glia-like cells in hGFAP-Cre/+; Fgfr1flox/flox mice. A-D, DAPI staining showing the general morphology of coronal sections in control (Cre-) and mutant (Cre+) at the indicated ages. White dashed squares in A and C indicate frame positions shown in E-H and Q-T for the hippocampal VZ (hVZ) and in I-L and U-X for the DG. White dashed squares in B and D indicate frame position for the migratory stream (MS) shown in M-P. E-H, Anti-GLAST staining in hVZ is unchanged at E16.5 (E, F) but is decreased in mutants at E18.5 (G, H). BrdU- or PCNA-positive cells colocalize GLAST immunoreactivity, and the number of PCNA-positive cells is decreased in the mutant hVZ at E18.5 (G, H, white arrows). I-L, A similar reduction in BrdU- or PCNA-positive cells double immunoreactive for GLAST is also observed in the DG at E16.5-E18.5. M-P, GFAP immunoreactivity is present in cells within the MS with no apparent change between control (Cre-) and mutant (Cre+) (yellow arrows). Q-R, U-V, BLBP-immunoreactive cells and fibers are decreased and disorganized in the mutant VZ (Q, R, yellow arrowheads) and in the DG (U, V, white arrowheads). S, T, W, X, Although nestin immunoreactivity is not substantially changed in the DG, there is an obvious reduction in hippocampal hVZ and presumptive pyramidal layer (S, T, open arrowheads). f, Fimbria. Scale bars: A-D, 200 μm; E-H, 50 μm; I-X, 100 μm.
Qualitative (Figs. 6C-F, 7E-L) and quantitative (Fig. 5C,D) analyses demonstrated that this decrease in number of proliferative cells was noticeable at E16.5, but it became more robust at E18.5 and P0, peaking at P7. Correspondingly, there was no detectable decrease in density of radial glia or in total cell number within the Fgfr1Δflox hippocampal VZ or DG at E16.5 (Fig. 7E,F,I,J; Table 2). By E18.5, however, Fgfr1 recombinant mice showed a decrease in GLAST-positive cell densities. This included cell bodies in the hippocampal VZ, processes stretching to the hippocampal pyramidal cell layer, as well as cells within the DG anlage (Fig. 7G,H,K,L). Similar results were obtained with BLBP and nestin (Fig. 7Q-X, arrowheads). There was an especially pronounced decrease in BLBP immunostaining in the Fgfr1Δflox DG (Fig. 7U,V, arrowheads). By P0, GLAST immunoreactivity was prominently decreased in the VZ (data not shown), and correspondingly there was a 53% decrease in the number of total cells within the mutant hippocampal VZ (Table 2).
To verify whether the decrease in astroglial cells labeled by the radial glial markers listed above encompassed GFAP-immunoreactive cells in the hippocampus, we performed GFAP immunostaining in adjacent sections. GFAP immunoreactivity was localized within cells and processes that were particularly abundant along the caudal border of the DG and above the fimbria (Fig. 7M-P). There was no apparent difference between the density of these cells, their location, or the abundance of their processes among control and mutant mice (Fig. 7M-P).
In aggregate, these results indicate that FGFR1 is required to promote cell proliferation within the radial glia-like neuroepithelial cell pool. The decreased proliferation of astroglial cells in the VZ of these Fgfr1 mutants is the most likely cause for the reduction in their number during the later stages of embryogenesis.
Lack of FGF2-dependent hippocampal stem cells in Fgfr1Δflox mice
It has been shown that the hippocampus contains multipotent, self-renewing stem cells that can be isolated and maintained in vitro in the presence of FGF2 (Gage et al., 1998). To determine whether FGF2-dependent hippocampal stem cells require Fgfr1, we prepared neurosphere cultures from either control or Fgfr1Δflox hippocampal primordia at E14.5. In this assay, neural stem cells in the presence of FGF2 or EGF give rise to floating neurospheres that are the clonal progeny of single neural stem cells, such that under standard conditions, the number of neurospheres should reflect the number of stem cells present in the tissue (Reynolds and Weiss, 1992; Tropepe et al., 1999). The hippocampal primordium of control mice consistently generated about 200 neurospheres per brain when cultured in the presence of FGF2 (Fig. 8A,B). These neurospheres could generate secondary neurospheres after dissociation and replating. As reported previously, primary neurospheres could give rise to neurons and glial cells when they were cultured under adherent conditions in the presence of a small amount of serum (1-3%) (data not shown). In contrast, very few neurospheres could be isolated from the hippocampus of Fgfr1Δflox mice in repeated experiments (Fig. 8A), even after adding additional amounts of FGF2 or prolonging the culture conditions to 21 d (data not shown). Repeated attempts at generating secondary neurospheres from Fgfr1Δflox hippocampi were unsuccessful, and no differentiated cells could be obtained after plating these small neurosphere-like cell clones, suggesting that the mutant cell clusters did not contain neural stem cells or that these cells were too rare to survive or yield progeny. These results suggest that there is an absolute requirement for Fgfr1 to generate neurospheres in the presence of FGF2 in vitro. In contrast, EGF retained its capability to generate neurospheres in tissue lacking a functional Fgfr1 (Fig. 8 B). However, EGF was able to generate only ∼30 neurospheres per well, ∼15% of those obtained under FGF2 treatment.
Figure 8.

Reduction of FGF2-sensitive neurospheres in hGFAP-Cre/+; Fgfr1flox/flox mice. A, The hippocampal primordium from E14.5 mice was dissociated and cultured with or without FGF2 for 10 d. Tissue from Fgfr1flox/flox mice (Cre-) produced a large number of neurospheres when treated with FGF2. Tissue of mice lacking a functional Fgfr-1 gene (Cre+) failed to produce neurospheres under identical conditions (bottom right). The cell aggregates visible in Cre+ mutant mice have a diameter much smaller than the typical neurospheres detected in Cre- control (top right). Scale bar, 2 mm. B, Hippocampal tissue (n = 5 preparations) was dissociated and cultured as above with or without FGF2 or EGF (10 ng/ml). Neurospheres with a diameter >150 μm were counted after 10 d of culture. Each histogram shows the average number of neurospheres grown per well (±SEM).
Discussion
Using mice harboring a nonfunctional Fgfr1 gene in cells of radial glial lineage, we produced a loss of Fgfr1 gene expression in the telencephalon at midneurogenesis (E16.5), and we demonstrated that the Fgfr1 gene is required for the development of the hippocampus. Fgfr1 loss-of-function mice showed decreased proliferation of radial glia-like progenitor cells in the hippocampus as well as an apparent absence of FGF2-sensitive hippocampal neural stem cells, leading to permanent hippocampal atrophy.
Radial glial-like cells of the dorsomedial VZ are progenitors for pyramidal cells and DG granule neurons
Dividing progenitors within the hippocampal VZ coexpress BLBP, GLAST, and other radial glial cell markers but no detectable GFAP immunoreactivity. Nevertheless, recombination targeted by the hGFAP-Cre line begins in the dorsomedial VZ at E13.5 and expands to the rest of the telencephalon, excluding the archicortex, by E15.5. Seemingly, all cells within the hippocampal VZ express the Cre protein and Cre recombinase activity, and Cre-positive cells coexpress radial glial markers. Although the Cre protein is restricted to radial glial cells of the VZ, the β-gal reporter gene is inherited by their progeny and is thus more widely distributed in the developing hippocampal primordium. Hence, this hGFAP-Cre transgene directs Cre recombination to radial glia-lineage cells, although we cannot exclude that few nonradial glia-like cells are also targeted. Cells with no detectable Cre recombinase activity were identified in the stratum lacunosum-moleculare and adjacent areas of the DG. These Cre-negative cells may include neuronal and glial precursors, some of which are known to migrate to the hippocampus from the basal telencephalon through a medial subpial route (Nery et al., 2002).
It has been suggested that 99% of hippocampal pyramidal neurons are the progeny of cells targeted by the hGFAP-Cre transgene (Malatesta et al., 2003). We also observed (supplemental Fig. 3, available at www.jneurosci.org) that hippocampal pyramidal cells and DG granule neurons express the β-gal heritable reporter gene in adulthood. In contrast, most PV-positive cells within these fields were β-gal negative (supplemental Fig. 3G, available at www.jneurosci.org). These results suggest that pyramidal neurons and DG granule neurons originate from the radial glia-like cell scaffold of the dorsomedial VZ, whereas hippocampal PV interneurons do not share this origin.
Astrocytes within the DG were positive for β-gal, whereas astrocytes within the hilus, stratum radiatum, and stratum pyramidalis were β-gal negative. These data suggest that although DG astrocytes descend from radial glia of the VZ, other astrocytes may migrate into the region from extrahippocampal sources. However, it is also possible that some astrocytes fail to express the R26R reporter gene. Indeed, using a different reporter line, the hGFAP-Cre line is able to confer Cre recombinase activity to GFAP-positive astrocytes within the forebrain (Theis et al., 2003).
Fgfr1 recombination in VZ cells leads to downregulation of Fgfr1 expression in the dorsomedial VZ and hippocampus
By mating the hGFAP-Cre line with mice carrying “floxed” Fgfr1 alleles, we surprisingly observed a virtually complete absence of Fgfr1 gene expression in the developing hippocampus. Our inability to detect truncated Fgfr1 mRNA and protein suggests that the deletion in Fgfr1Δflox may decrease transcriptional activity of the allele or the stability of its mRNA product. In contrast, the absence of a functional FGFR1 did not affect Fgfr2 and Fgfr3 expression, allowing us to study the specific role of the Fgfr1 gene in hippocampal development. Interestingly, the Fgfr1 gene was not expressed within the stratum lacunosum-moleculare (Fig. 1A, C), containing cells that, according to our lineage analysis, did not derive from the VZ. The enrichment of Fgfr1 mRNA within VZ radial glial cells and their progeny may explain its successful targeting by hGFAP-Cre.
The loss of FGFR1 signaling decreases hippocampal size without altering its patterning
The main phenotypic characteristic of mice carrying Fgfr1Δflox alleles is a decrease in the growth of the hippocampus, with a lower number of pyramidal and DG neurons and a corresponding decrease in intervening white matter. In addition, the number of PV-containing interneurons was dramatically reduced (Fig. 4, Table 2). Pyramidal neurons and dentate granule cells originate from radial glia-like cells of the hippocampal VZ, which are targeted by Fgfr1 recombination, as discussed above. However, hippocampal PV interneurons originate from the ganglionic eminences (Lavdas et al., 1999; Pleasure et al., 2000) and are apparently not targeted by recombination. We therefore hypothesize that PV interneurons are secondarily affected by the abnormal development of hippocampal VZ cells and that an unknown mechanism matches incoming GABA interneurons with resident glutamatergic neurons within the hippocampus. Definite proof for this hypothesis will require additional experiments. Surprisingly, the number of GFAP-immunoreactive astrocytes was not significantly decreased in the Fgfr1Δflox hippocampus, either at E16.5 or adulthood. This could be attributable to nonhomogeneous recombination of Fgfr1 in all hippocampal astrocytes or to the fact that FGFR1 is not involved in hippocampal astrocyte development.
The Fgfr1Δflox hippocampus appears to be correctly patterned, suggesting that Fgfr1 is not essential for the maintenance of hippocampal cell fates. We further explored this question by inducing Fgfr1 recombination earlier in development, using a Nestin-Cre line. We observed that Nestin-Cre/+; Fgfr1flox/flox mutant mice have decreased cell proliferation in the hippocampus and no apparent change in dorsoventral hippocampal patterning (Y. Ohkubo and F. M. Vaccarino, unpublished observations). Although it is tempting to conclude that FGFR1 signaling does not affect hippocampal patterning, it is still possible that Fgfr2 and Fgfr3 may compensate for the lack of Fgfr1.
Mechanisms of Fgfr1-dependent regulation of hippocampal cell number
We propose that FGFR1 expands the size of the primordial radial glial scaffold during late embryogenesis, secondarily regulating neuron number within the DG and CA cell fields. A first set of observations supporting this hypothesis is that Fgfr1Δflox mice exhibited a smaller number of proliferative cells in the hippocampal VZ and DG (Figs. 5, 6, 7) without any change in cell death. The decrease in labeling index in Fgfr1 conditional knock-out mice could have been attributable to an accumulation of post-mitotic cells in the VZ because of abnormal differentiation or migration. However, we also observed a decrease in the pool of proliferative cells in both the hippocampal VZ and DG, which peaked in the neonatal period. Furthermore, the density of radial glial cells in the VZ and DG was decreased, starting from the late embryonic period in the mutant mice. At least three explanations may account for these results. One is that the removal of Fgfr1 slows the cell cycle. Against this hypothesis is that no changes in proliferation rate were previously noted in cell cycle studies that we performed in Fgf2 knock-out mice in the VZ and SVZ (Vaccarino et al., 1999a; Zheng et al., 2004). A second possibility is that Fgfr1 increases the probability of precursor cells to re-enter S phase, and its loss causes premature depletion of the precursor pool. In support of this interpretation, the number of cells in the hippocampal VZ more than doubled between E16.5 and P0 in control mice but decreased in Fgr1 knock-outs during the same time period (Table 2). Although it is tempting to conclude that the disruption of Fgfr1 within VZ cells may be sufficient to cause this phenotype, FGFR1 is also expressed in their progeny. Hence, we cannot exclude that FGFR1 may also exert other effects at a later time point in the lineage. Conclusive proof that FGFR1 acts directly within radial glia-like cells of the VZ to regulate their proliferative potential will require a rescue experiment reintroducing wild-type Fgfr1 in these cells.
FGFR1 is required for the proliferation-survival of FGF2-sensitive hippocampal neural stem cells
FGF2 increases the self-renewal of stem and progenitor cells in the hippocampal and cortical primordium in vitro (Cattaneo and McKay, 1990; Kilpatrick and Bartlett, 1993; Ray et al., 1993; Vescovi et al., 1993; Ghosh and Greenberg, 1995; Vaccarino et al., 1995; Vicario-Abejon et al., 1995). Whether FGF ligands are required for neural stem cell growth in vivo is not clear. Although Fgf2 knock-out mice have decreased generation of hippocampal granule neurons (Cheng et al., 2002) and of cortical pyramidal neurons (Raballo et al., 2000), whether these phenotypes are caused by a lack of neural stem cells is uncertain. Here, we demonstrate that the Fgfr1Δflox hippocampal primordium yielded no FGF-sensitive primary neurospheres, whereas hundreds of neurospheres could be obtained under the same experimental conditions from the hippocampus of littermate control embryos. These data indicate that FGFR1 is absolutely required for the proliferative-survival effect of FGF2 on neural stem cells in vitro. However, we do not know whether FGFR1 exerts a cell-autonomous effect within neurosphere-forming cells or whether this is an indirect effect.
Despite the fact that FGF2-sensitive neural stem cells fail to grow in vitro, hippocampal development was not completely prevented in Fgfr1Δflox mice in vivo, possibly because the pool of neural stem cells sensitive to EGF was unaffected in Fgfr1Δflox mice. However, the pool of EGF-sensitive hippocampal neurospheres was exceedingly small, emphasizing that other compensatory factors are likely to be involved. Nonetheless, a failure in self-maintenance of a portion of hippocampal stem cells may explain the decrease in the hippocampal progenitor pool and the hippocampal atrophy that we observed in the Fgfr1 conditional knock-out mice.
The action of FGFR1 may be substantially modified by the local cellular context
Two Fgfr1 conditional mutant phenotypes were recently generated. Pirvola et al. (2002) reported that FGFR1 increases the proliferation of precursor cells for the sensory epithelium of the organ of Corti. In contrast, Herbert et al. (2003) suggested that FGFR1 decreases cell proliferation in the olfactory bulb primordium. We hypothesize that heterodimeric combinations of different FGF receptors activated by different ligands may bring about these regional differences in FGFR signaling. The olfactory bulb arises from a region where high levels of FGF8 were reported to induce Spry genes (Crossley et al., 2001; Storm et al., 2003). Spry decrease Foxg1 mRNA and act as intracellular antagonists of the Ras/MAPK (mitogen-activated protein kinase) pathway, which is normally activated by FGFR1 (Kouhara et al., 1997; Chambers et al., 2000; Furthauer et al., 2001; Yusoff et al., 2002). The olfactory bulb may have the unique competence to negatively regulate FGFR1 signaling via a pathway activated by high dosages of FGF8. In contrast, hippocampal VZ progenitors do not express Fgf8 or Spry mRNAs, and our Fgfr1Δflox mice do not exhibit changes in Foxg1 expression (Fig. 1).
Similarly to Herbert et al. (2003), we observed that cerebral cortical growth was not significantly affected by decreased Fgfr1 function. However, we recently reported that transgenic mice expressing a dominant negative FGFR1 exhibited defective cortical growth and a decrease in cortical pyramidal cell number (Shin et al., 2004). These results show that several FGFRs may function in a mutually compensatory manner to regulate cortical development. The different roles of Fgfr1 in these two regions could reflect a higher proportion of FGFR1 versus other FGFRs in the developing hippocampal formation and depend on the distribution of other signaling molecules.
Footnotes
This work was supported by National Institutes of Health Grants NS37709 and MH067715, National Science Foundation Grant NSF0083104, and the National Alliance for Research on Schizophrenia and Depression Foundation. We are extremely grateful to Dr. Albee Messing for generously making the hGFAP-Cre transgenic mice available to us and to Dr. Michael Brenner for valuable insights regarding the hGFAP promoter. We thank Drs. Claudio Basilico and Joseph Schessinger for the gifts of cell lines. We thank Drs. Lewis Williams, Nathaniel Heintz, Masahiko Watanabe, and Joseph Schlessinger for the gifts of antibodies and Drs. Alessandro Bulfone, Elizabeth Grove, Ryoichiro Kageyama, Antonello Mallamaci, John Rubenstein, and Antonio Simeone for the gifts of probes. We are grateful to Drs. Karen Smith, Nenad Sestan, Janice Naegele, Paul Lombroso, and David Ornitz for helpful comments on a previous version of this manuscript. We thank Yuchun Zhang for expert technical assistance in unbiased morphometric methods and Western blotting.
Correspondence should be addressed to Flora Vaccarino, Child Study Center, Yale University, 230 South Frontage Road, New Haven, CT 06520. E-mail: Flora.vaccarino@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246057-13$15.00/0
References
- Altman J, Bayer SA (1990) Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J Comp Neurol 301: 365-381. [DOI] [PubMed] [Google Scholar]
- Anderson S, Mione M, Yun K, Rubenstein JLR (1999) Differential origins of neocortical projectionn and local circuit neurons: role of Dlx genes in neocortical interneurogenesis. Cereb Cortex 9: 647-654. [DOI] [PubMed] [Google Scholar]
- Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P (1998) Targeted disruption of fibrobast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA 95: 5082-5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai T, Wanaka A, Kato H, Masana Y, Seo M, Tohyama M (1993) Differential expression of two members of FGF receptor gene family, FGFR-1 and FGFR-2 mRNA, in the adult rat central nervous system. Brain Res Mol Brain Res 17: 174-178. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, McKay RDG (1990) Proliferation and differentiation of neuronal stem cells regulated by nerve growth factor. Nature 347: 762-765. [DOI] [PubMed] [Google Scholar]
- Chambers D, Medhurst AD, Walsh FS, Price J, Mason I (2000) Differential display of genes expressed at the midbrain-hindbrain junction identifies sprouty2: an FGF8-inducible member of a family of intracellular FGF antagonists. Mol Cell Neurosci 15: 22-35. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Black IB, DiCicco-Bloom E (2002) Hippocampal granule neuron production and population size are regulated by levels of bFGF. Eur J Neurosci 15: 3-12. [DOI] [PubMed] [Google Scholar]
- Crossley P, Martinez S, Ohkubo Y, Rubenstein JLR (2001) Coordinate expression of Fgf8, Otx2, Bmp4, and Shh in the rostral prosencephalon during development of the telencephalic and optic vesicles. Neuroscience 108: 183-206. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martin GR (1995) The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121: 439-451. [DOI] [PubMed] [Google Scholar]
- Crossley PH, Martinez S, Martin GR (1996) Midbrain development induced by FGF8 in the chick embryo. Nature 380: 66-68. [DOI] [PubMed] [Google Scholar]
- Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P (1996) Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 84: 911-921. [DOI] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P (1994) Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8: 3045-3057. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lonnerberg P, Ayer-LeLievre C, Persson H (1990) Developmental and regional expression of basic fibroblast growth factor mRNA in the rat central nervous system. J Neurosci Res 27: 10-15. [DOI] [PubMed] [Google Scholar]
- Fukuchi-Shimogori T, Grove EA (2001) Neocortex patterning by the secreted signaling molecule FGF8. Science 294: 1071-1074. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Reifers F, Brand M, Thisse B, Thisse C (2001) sprouty4 acts in vivo as a feedback-induced antagonist of FGF signaling in zebrafish. Development 128: 2175-2186. [DOI] [PubMed] [Google Scholar]
- Gadisseux JF, Evrard P, Mission JP, Caviness VSJ (1992) Dynamic changes in the density of radial glial fibers of the developing murine cerebral wall: a quantitative immunohistological analysis. J Comp Neurol 322: 246-254. [DOI] [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer T, Peterson D, Ray J (1998) Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol 36: 249-266. [DOI] [PubMed] [Google Scholar]
- Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R (2000) Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127: 469-482. [DOI] [PubMed] [Google Scholar]
- Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM (2002) Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience 112: 977-991. [DOI] [PubMed] [Google Scholar]
- Garel S, Huffman KJ, Rubenstein JLR (2003) Molecular regionalization of the neocortex is disrupted in Fgf8 hypomorphic mutants. Development 130: 1903-1914. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Related articles, links distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89-103. [DOI] [PubMed] [Google Scholar]
- Grove EA, Tole S, Limon J, Yip LW, Ragsdale CW (1998) The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 125: 2315-2325. [DOI] [PubMed] [Google Scholar]
- Hartfuss E, Galli R, Heins N, Gotz M (2001) Characterization of CNS precursors subtypes and radial glia. Dev Biol 229: 15-30. [DOI] [PubMed] [Google Scholar]
- Hebert JM, Lin M, Partanen J, Rossant J, McConnell SK (2003) FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development 15: 1101-1111. [DOI] [PubMed] [Google Scholar]
- Heuer JG, von Bartheld CS, Kinoshita Y, Evers PC, Bothwell M (1990) Alternating phases of FGF receptor and NGF receptor expression in the developing chicken nervous system. Neuron 5: 283-296. [DOI] [PubMed] [Google Scholar]
- Kilpatrick TJ, Bartlett PF (1993) Cloning and growth of multipotential neural precursors: requirements for proliferation and differentiation. Neuron 10: 255-265. [DOI] [PubMed] [Google Scholar]
- Korada S, Zheng W, Basilico C, Schwartz ML, Vaccarino FM (2002) Fibroblast growth factor 2 is necessary for the growth of glutamate projection neurons in the anterior neocortex. J Neurosci 22: 863-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J (1997) A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89: 693-702. [DOI] [PubMed] [Google Scholar]
- Kriegstein AR, Gotz M (2003) Radial glia diversity: a matter of cell fate. Glia 43: 37-43. [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG (1999) The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 19: 7881-7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove EA, McMahon AP (2000) A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127: 457-467. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M (2000) Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 127: 5253-5263. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchnoff F, Gotz M (2003) Neuronal or glial progeny: regional differences in radial glia fate. Neuron 37: 751-764. [DOI] [PubMed] [Google Scholar]
- Moos M, Tacke R, Scherer H, Teplow D, Fruh K, Schachner M (1988) Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature 334: 701-703. [DOI] [PubMed] [Google Scholar]
- Nery S, Fishell G, Corbin JG (2002) The caudal ganglionic eminence is a source of distinct cortical and subcortical cell populations. Nat Neurosci 5: 1279-1287. [DOI] [PubMed] [Google Scholar]
- Niswander L, Tickle C, Vogel A, Booth I, Martin GR (1993) FGF-4 replaces the apical ectodermal ridge and directs outgrowth and patterning of the limb. Cell 5: 579-587. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Chiang C, Rubenstein JLR (2002) Coordinate regulation and synergistic action of BMP4, SHH and FGF8 in the rostral prosencephalon regulate morphogenesis of the telencephalic and optic vesicles. Neuroscience 111: 1-17. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Sakamoto M, Guillemot F, Kageyama R (2001) Roles of the basic helix-loop-helix genes Hes1 and Hes5 in expansion of neural stem cells of the developing brain. J Biol Chem 276: 30467-30474. [DOI] [PubMed] [Google Scholar]
- Ornitz DM (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. BioEssays 22: 108-112. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P (1991) Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development 113: 1419-1434. [DOI] [PubMed] [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L (1993) Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol 155: 423-430. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J (2002) FGFR1 is required for the development of the auditory sensory epithelium. Neuron 35: 671-680. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Anderson S, Hevner R, Bagri A, Marin O, Lowenstein DH, Rubenstein JL (2000) Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron 28: 727-740. [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM (2000) Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 20: 5012-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale CW, Grove EA (2001) Patterning the mammalian cerebral cortex. Curr Opin Neurobiol 11: 50-58. [DOI] [PubMed] [Google Scholar]
- Ray J, Peterson DA, Schinstine M, Gage FH (1993) Proliferation, differentiation, and long-term culture of primary hippocampal neurons. Proc Natl Acad Sci USA 90: 3602-3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds BA, Weiss S (1992) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255: 1707-1710. [DOI] [PubMed] [Google Scholar]
- Rickmann M, Amaral DG, Cowan WM (1987) Organization of radial glial cells during the development of the rat dentate gyrus. J Comp Neurol 264: 449-479. [DOI] [PubMed] [Google Scholar]
- Shibata T, Yamada K, Watanabe M, Ikenaka K, Wada K, Tanaka K, Inoue Y (1997) Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. J Neurosci 17: 9212-9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamura K, Rubenstein JLR (1997) Inductive interactions direct early regionalization of the mouse forebrain. Development 124: 2709-2718. [DOI] [PubMed] [Google Scholar]
- Shin DM, Korada S, Raballo R, Shashikant CS, Simeone A, Taylor JR, Vaccarino F (2004) Loss of glutamatergic pyramidal neurons in frontal and temporal cortex resulting from attenuation of FGFR1 signaling is associated with spontaneous hyperactivity in mice. J Neurosci 24: 2247-2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers J, Hartmann D, Pehlemann FW, Berry M (1992) Development of astroglial cells in the proliferative matrices, the granule cell layer, and the hippocampal fissure of the hamster dentate gyrus. J Comp Neurol 320: 1-32. [DOI] [PubMed] [Google Scholar]
- Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70-71. [DOI] [PubMed] [Google Scholar]
- Storm EE, Rubenstein JL, Martin GR (2003) Dosage of Fgf8 determines whether cell survival is positively or negatively regulated in the developing forebrain. Proc Natl Acad Sci USA 18: 1757-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Super H, Martinez A, Del Rio JA, Soriano E (1998) Involvement of distinct pioneer neurons in the formation of layer-specific connections in the hippocampus. J Neurosci 18: 4616-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Jauch R, Zhuo L, Speidel D, Wallraff A, Doring B, Frisch C, Sohl G, Teubner B, Euwens C, Huston J, Steinhauser C, Messing A, Heinemann U, Willecke K (2003) Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J Neurosci 23: 766-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tole S, Christian C, Grove EA (1997) Early specification and autonomous development of cortical fields in the mouse hippocampus. Development 124: 4959-4970. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Sibilia M, Ciruna BG, Rossant J, Wagner EF, van der Kooy D (1999) Distinct neural stem cells proliferate in response to EGF and FGF in the developing mouse telencephalon. Dev Biol 208: 166-188. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Hartigan D, Leckman JF (1995) Basic fibroblast growth factor increases the number of excitatory neurons containing glutamate in the cerebral cortex. Cereb Cortex 5: 64-78. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT (1999a) Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci 2: 848. [DOI] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R (1999b) Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr Top Dev Biol 46: 179-200. [DOI] [PubMed] [Google Scholar]
- Vescovi AL, Reynolds BA, Fraser DD, Weiss S (1993) bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 11: 951-966. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Johe KK, Hazel TG, Collazo D, McKay RD (1995) Functions of basic fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron 15: 249-253. [DOI] [PubMed] [Google Scholar]
- Wanaka A, Milbrandt J, Johnson Jr EM (1991) Expression of FGF receptor gene in rat development. Development 111: 455-468. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ (1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. J Mol Neurosci 231: 482-497. [DOI] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C (1998) Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125: 753-765. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J (1994) Fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 15: 3032-3044. [DOI] [PubMed] [Google Scholar]
- Yusoff P, Lao DH, Ong SH, Wong ES, Lim J, Lo TL, Leong HF, Fong CW, Guy GR (2002) Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of Raf. J Biol Chem 277: 3195-3201. [DOI] [PubMed] [Google Scholar]
- Zhao S, Hung FC, Colvin JS, White A, Dai W, Lovicu FJ, Ornitz DM, Over-beek PA (2001) Patterning the optic neuroepithelium by FGF signaling and Ras activation. Development 128: 5051-5060. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Sheng HZ, Amini R, Grinberg A, Lee E, Huang S, Taira M, Westphal H (1999) Control of hippocampal morphogenesis and neuronal differentiation by the LIM homeobox gene Lhx5. Science 284: 1155-1158. [DOI] [PubMed] [Google Scholar]
- Zheng W, Nowakowski R, Vaccarino FM (2004) Fgf2 is required for maintaining the neural stem cell pool in the mouse subventricular zone. Dev Neurosci, in press. [DOI] [PubMed]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A (2001) hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo Genesis 31: 85-94. [DOI] [PubMed] [Google Scholar]