

Abstract
Molecular mechanisms underlying C-fiber stimulation-induced ERK (extracellular signal-regulated kinase) activation in dorsal horn neurons and its contribution to central sensitization have been investigated. In adult rat spinal slice preparations, activation of C-fiber primary afferents by a brief exposure of capsaicin produces an eightfold to 10-fold increase in ERK phosphorylation (pERK) in superficial dorsal horn neurons. The pERK induction is reduced by blockade of NMDA, AMPA/kainate, group I metabotropic glutamate receptor, neurokinin-1, and tyrosine receptor kinase receptors. The ERK activation produced by capsaicin is totally suppressed by inhibition of either protein kinase A (PKA) or PKC. PKA or PKC activators either alone or more effectively together induce pERK in superficial dorsal horn neurons. Inhibition of calcium calmodulin-dependent kinase (CaMK) has no effect, but pERK is reduced by inhibition of the tyrosine kinase Src. The induction of cAMP response element binding protein phosphorylation (pCREB) in spinal cord slices in response to C-fiber stimulation is suppressed by preventing ERK activation with the MAP kinase kinase inhibitor 2-(2-diamino-3-methoxyphenyl-4H-1-benzopyran-4-one (PD98059) and by PKA, PKC, and CaMK inhibitors. Similar signaling contributes to pERK induction after electrical stimulation of dorsal root C-fibers. Intraplantar injection of capsaicin in an intact animal increases expression of pCREB, c-Fos, and prodynorphin in the superficial dorsal horn, changes that are prevented by intrathecal injection of PD98059. Intrathecal PD98059 also attenuates capsaicin-induced secondary mechanical allodynia, a pain behavior reflecting hypersensitivity of dorsal horn neurons (central sensitization). We postulate that activation of ionotropic and metabotropic receptors by C-fiber nociceptor afferents activates ERK via both PKA and PKC, and that this contributes to central sensitization through post-translational and CREB-mediated transcriptional regulation in dorsal horn neurons.
Keywords: ERK, C-fiber, CREB, dorsal horn, capsaicin, spinal cord slices, pain
Introduction
Increasing evidence indicates the involvement of the MAP kinase ERK (extracellular signal-regulated protein kinase) in neural plasticity (Impey et al., 1999; Sweatt 2001). Activation of ERK in dorsal horn neurons contributes to pain hypersensitivity (Ji et al., 1999; Karim et al., 2001), because of both a role in promoting acute central sensitization, an activity-dependent increase in the excitability of dorsal horn neurons (Woolf, 1983; Woolf and Salter, 2000; Ji et al., 2003), and alteration in gene transcription in the spinal cord (Ji et al., 2002a).
Noxious stimulation releases neurotransmitter glutamate and neuromodulators substance P and brain-derived neurotrophic factor (BDNF) from primary afferents in the spinal cord. Activation of the corresponding NMDA and metabotropic glutamate receptors (mGluRs) neurokinin-1 (NK-1) and tyrosine receptor kinase B (TrkB) receptors in postsynaptic dorsal horn neurons induce central sensitization (Mannion et al., 1999; Woolf and Salter, 2000; Hunt and Mantyh, 2001; Karim et al., 2001). Multiple neurotransmitter receptors and protein kinases are associated with ERK activation in the brain (Otani et al., 1999; Roberson et al., 1999), and activation of NMDA and metabotropic glutamate receptors are implicated in noxious stimulation-evoked ERK activation in dorsal horn neurons (Ji et al., 1999; Karim et al., 2001; Pezet et al., 2002; Lever et al., 2003). However, the molecular pathways responsible for ERK activation in the spinal cord in response to peripheral noxious stimuli and the downstream actions of ERK phosphorylation (pERK) are not well defined.
Noxious stimulation and peripheral inflammation induces gene expression in dorsal horn neurons (Wisden et al., 1990; Dubner and Ruda, 1992; Ji et al., 1994, 2003; Abbadie et al., 1996; Samad et al., 2001). ERK activation is required for the inflammation-induced upregulation of prodynorphin and NK-1 in the superficial dorsal horn and for maintaining persistent inflammatory pain (Ji et al., 2002a). ERK activation is associated with the transcription factor cAMP response element binding protein (CREB) in cultured hippocampal neurons and brain slices (Impey et al., 1998; Obrietan et al., 1999). Phosphorylation of CREB (pCREB) at serine 133 activates cAMP response element (CRE)-mediated gene expression (Impey et al., 1999; Obrietan et al., 1999). Many inflammation-induced genes, such as c-fos, prodynorphin, Cox-2, NK-1, and TrkB, contain CRE sites in their promoter regions (Lonze and Ginty, 2002), and c-fos is regulated by the ERK/CREB pathway in pheochromocytoma 12 cells and cortical neurons (Ginty et al., 1994; Xia et al., 1996). Noxious stimulation and inflammation induce CREB phosphorylation in dorsal horn neurons (Ji and Rupp, 1997; Messersmith et al., 1998; Wu et al., 2002), but it is not known whether this is ERK dependent.
We used two different protocols to activate C-fiber primary afferents in an adult rat spinal cord slice preparation: electrical stimulation of an attached dorsal root (Ji et al., 1999) and bath application of capsaicin. We found that glutamate, substance P, and BDNF are involved via ionotropic, metabotropic, and tyrosine kinase receptors in the C-fiber-induced activation of ERK, and that this involves protein kinase A (PKA), PKC and Src. ERK activation is required, moreover, for C-fiber-evoked induction of pCREB and the establishment of central sensitization.
Materials and Methods
Animals and reagents. Male adult Sprague Dawley rats (200–300 gm) were used according to Harvard Medical School Animal Care guidelines. The rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). Capsaicin, prepared in saline containing 10% Tween 80, was injected into the plantar surface of the left hindpaw. For intrathecal injection, a polyethylene 10 catheter was implanted at L3–L5 spinal level, and saline, NMDA, substance P, and BDNF as well as specific MAP kinase kinase (MEK) inhibitor 2-(2-diamino-3-methoxyphenyl-4H-1-benzopyran-4-one (PD98059) (1μg) were intrathecally injected (10μl) and flushed with 10 μl of saline. PD98059 was initially dissolved in 20% DMSO at the concentration of 1μg/μl (3.7 mm) as stock solution and further diluted to 0.1μg/μl in 10% DMSO for intrathecal injection. The 10 μl of 10% DMSO was rapidly diluted by CSF in the intrathecal space after injection. The DMSO concentration used in the spinal cord slice incubation buffer was 0.2%. Capsaicin, NMDA, substance P, APV, (+)-5-methyl-10, 11-dihydro-5H-dibenzo [a,d] cyclohepten-5, 10-imine maleate (MK-801), CNQX, PD98059, and L733061 were purchased from Sigma (St. Louis, MO), and 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)amino-N-(4-chlorocinnamyl)-N-methylbenzylamine (KN93), N-[2-(p-bromocinnamyl-amino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89), bisindolylmaleimide (Ro-31-8425), K252a, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), genistein, genistin, forskolin, and phorbol 12-myristate 13-acetate (PMA) were purchased from Calbiochem (La Jolla, CA). NK-1 antagonist GR205171A and BDNF were kindly provided by GlaxoSmithKline Pharmaceuticals (Harlow, UK) and Regeneron Pharmaceuticals (Tarrytown, NY), respectively. 7-(Hidroxyimino)cyclopropachromen-1a-carboxylate ethyl ester (CPCCOEt) was purchased from Tocris Cookson (Ballwin, MO).
Spinal cord slice preparation and C-fiber stimulation. Adult rat transverse spinal cord slices were prepared as described previously (Baba et al., 1999, 2003; Ji et al., 1999; Moore et al., 2002). Briefly, the lumbar spinal cord was removed and immersed in cold Krebs' solution, and a 700-μm-thick transverse slice with attached L4 dorsal root (15–20 mm long) was cut on a vibrating microslicer. The slice was perfused with Krebs' solution saturated with 95% O2 and 5% CO2 at 36–37°C. For electrical stimulation, the L4 dorsal root was stimulated using a suction electrode, and the current for C-fiber stimulation was 1000 μA (0.5 msec) (Ji et al., 1999). Three trains, separated by a 10 sec interval, were applied. Each train consisted of 50 pulses at 50 Hz for 1 sec. The threshold stimulation intensity and duration of C-fiber for this type of suction electrode (200 μA; 0.5 msec) have been established previously by compound action potential recordings (Baba et al., 1999, 2003; Moore et al., 2002; Kohno et al., 2003). Although this intensity will activate Aβ and Aδ fibers, pERK is not activated by Aβ-fiber stimulation and only very slightly by Aδ-fiber stimulation (Ji et al., 1999).
The spinal cord slices were perfused with Krebs' solution for at least 3 hr before electrical stimulation. Two minutes after the stimulation, the slices were fixed in 4% paraformaldehyde for 60 min, replaced with sucrose overnight, and cut in cryostat at 15 μm. Because the surfaces of the slices were likely to be damaged during the preparation, we trimmed the slices (100 μm each side) in a cryostat and collected 20–25 serial sections (15 μm thick) only from the middle of the slices. Six to eight nonadjacent sections of these were randomly picked for analysis. These sections were then processed for immunohistochemistry using ABC method or immunofluorescence (described below). Some slices were incubated with drugs at least 5–15 min before the dorsal root stimulation.
For capsaicin study, 8–10 adjacent slices (700 μm) without dorsal roots attached were prepared from the same spinal fragment, and at least one slice was used for control and one for capsaicin stimulation alone (3 μm; 5 min). The remaining slices were treated with drugs for 5–15 min followed by capsaicin stimulation. This arrangement would allow negative and positive control for each animal.
Immunohistochemistry. After separate survival times (10 min after intrathecal injection of NMDA, substance P, or BDNF) and 5, 60, and 180 min after capsaicin injection, the rats were perfused through the ascending aorta with saline followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 m phosphate buffer, pH 7.2–7.4 (4°C). After the perfusion, the L4–L5 spinal cord segments were removed and postfixed in the same fixative for 4 hr and then replaced with 15% sucrose overnight. All of the spinal cords for each experiment were arranged on the same blocks with optimal cutting temperature embedding medium and mounted on the same slides after sectioning. Transverse spinal sections (15 μm) were cut in a cryostat and processed for pERK, pCREB, and c-Fos immunostaining. Free-floating sections (30 μm) were also cut in cryostat for pERK/pCREB or pERK/c-Fos double staining.
Spinal sections were processed for immunohistochemistry using the ABC method or immunofluorescence (Ji et al., 1994, 1999). Briefly, spinal sections were blocked with 2% goat serum in 0.3% Triton X-100 for 1 hr at room temperature (RT) and incubated overnight at 4°C with anti-pERK (anti-rabbit, 1:300; Cell Signaling Technology, Beverly, MA), anti-pCREB (anti-rabbit, 1:1000; Cell Signaling Technology), anti-c-Fos (anti-rabbit, 1:1500; Santa Cruz Biotechnology, Santa Cruz, CA), or antiprodynorphin (anti-guinea pig, 1:3000; Chemicon, Temecula, CA) antibodies. For ABC staining, the sections were incubated for 90 min with biotinylated secondary antibody (1:200) and 30 min with ABC complex (1:75; Vector Laboratories, Burlingame, CA) at RT. Finally, the reaction product was visualized with 0.05% DAB/0.002–0.006% hydrogen peroxide in 0.1 m acetate buffer, pH 6.0, containing 2% ammonium nickel sulfate for 2–5 min. For immunofluorescence, the sections were incubated with FITC- or cyanin 3 (Cy3)-conjugated secondary antibody (anti-mouse or anti-rabbit, 1:300; Jackson ImmunoResearch, West-Grove, PA) for 90 min at RT after primary antibody incubation. For pERK/pCREB or pERK/c-Fos double immunofluorescence, free-floating spinal sections were incubated with a mixture of monoclonal pERK (1: 300; Cell Signaling Technology) and polyclonal pCREB (1:500) or c-Fos (1:1500) antibodies overnight at 4°C, followed by a mixture of FITC- and Cy3-congugated secondary antibodies for 90 min at RT. The sections (15 μm thick) from spinal cord slices were processed for immunostaining in the same way as described above. The Cell Signaling Technology pERK antibody recognizes two bands, ERK1 (p44) and ERK2 (p42) in Western blots, and they change to the same extent in response to peripheral noxious stimuli (Ji et al., 1999). The specificity of antibodies and immunostaining was tested by the omission of the primary antibody or preabsorption with the antigen protein–peptide (Ji et al., 1999, 2002a).
RNase protection assay. As described previously (Ji et al., 2002a,b), prodynorphin cDNA was generated by reverse transcription-PCR from rat DRG total RNA using primers 5′-TGGAAAAGCCCAGCTCCTAGACCCT-3′ and 5′-TTCCTCGTGGGCTTGAAGTGTGAAA-3′ and cloned into pCRII (Invitrogen, San Diego, CA). The plasmid was linearized with EcoRV, and an antisense probe was synthesized using Sp6 RNA polymerase and labeled with 32P-UTP (800 Ci/mmol; NEN, Boston, MA). RNase protection assays were performed using the RPA III (Ambion, Austin, TX) protocol. Ten micrograms of RNA samples was hybridized with labeled probe overnight at 42°C and then digested with RNase A/RNase T1 mix in RNase digestion buffer for 30 min at 37°C. Finally, samples were separated on denaturing acrylamide gel and exposed to x-ray films. Aβ-actin probe was used for each sample as a loading control.
Behavioral testing. Animals were habituated to the testing environment daily for at least 2 d before baseline testing. The room temperature, humidity, and time of testing remained unchanged for all experiments. For testing mechanical sensitivity, animals were placed in plastic boxes (11 × 13 × 24 cm) on an elevated metal mesh floor and allowed 30 min for habituation before examination. The plantar surface of each hindpaw was stimulated with a series of von Frey hairs (from 0.4 to 20 gm). The threshold was taken as the lowest force that evokes a brisk withdrawal response (Ji et al., 2002a,b; Jin et al., 2003). All testing was performed blind. Capsaicin (20 μg/20 μl) was intradermally injected to the heel of a hindpaw with a 30 ga needle, and secondary mechanical allodynia was tested in the middle plantar area (5–10 mm from the injection site).
Quantification and statistics. Six to eight nonadjacent sections from the lumbar spinal cord (L4–L5) segments were randomly selected. The region of the superficial dorsal horn sampled in all experiments using intraplantar capsaicin and electrical C-fiber stimulation was the medial two-thirds of the dorsal horn (laminas I and II) measured as a proportion of the distance from the medial border of lamina II with the dorsal column to its border with the dorsolateral column. This region receives input from the hindpaw, whereas the lateral third receives input from proximal territories (Swett and Woolf, 1985). This region in the superficial dorsal horn was captured inside the optic field (a square box, 450 × 338 μm) with a digital camera under 20× magnification. This size of the box, as shown in Figure 3a, was kept the same in all conditions. The numbers of immunoreactive (IR) neurons for pERK and prodynorphin and neuronal nuclei for pCREB and c-Fos in this region were counted. The spinal neurons were identified by their distinct morphology from glia under high magnification (20× and 40×) and confirmed by colocalization with neuronal-specific nuclear protein. However, intrathecal injection of NMDA, substance P, and BDNF and bath application of capsaicin, PKA, and PKC activators induced many pERK-IR neurons in the lateral one-third dorsal horn. In this case, all of the pERK-IR neurons (including both medial and lateral superficial dorsal horn) were counted in laminas I-II. The numbers of labeled neurons for every section in each slice or animal were averaged for that rat, and three to six slices or rats, each slice from a different rat, were included in each group. The data were represented as mean ± SEM. The difference between the groups was compared using ANOVA followed by Fisher's PLSD or using Student's t test, and the criterion for statistical significance was p < 0.05.
Figure 3.

a–g, Involvement of ERK, PKA, PKC, and CaMK in C-fiber stimulation-induced pCREB in the superficial dorsal horn of spinal cord slices. a–c, C-fiber electrical stimulation (Elec stim) induces pCREB in the medial superficial dorsal horn. c is a high magnification of the area in b indicated by the square. d, Inhibition of ERK activation with the MEK inhibitor PD98059 (50 μm) blocks the C-fiber-induced pCREB increase. PD98059 was incubated for 30 min before the electrical stimulation. The dotted lines indicate the top border of the gray matter of the superficial dorsal horn. Scale bars, 100 μm. e, Numbers of pCREB-IR nuclei in laminas I-II in the medial two thirds of the dorsal horn per 15-μm-thick section. **p < 0.01, compared with control; ++p < 0.01, compared with electrical stimulation without PD98059 (n = 4). PD98059 had no effect on basal pCREB levels. The spinal slices were fixed 5 min after electrical stimulation for pCREB immunostaining. f, Time course of capsaicin (3 μm; 5 min) induced pERK and pCREB increases in the superficial dorsal horn. At all of the time points, pERK (F(4, 15) = 16.982; p < 0.0001) and pCREB (F(4, 13) = 3.837; p = 0.0028) levels are significantly higher than control levels (p < 0.05; n = 3–5). g, Numbers of pCREB-IR neurons in the superficial dorsal horn (laminas I-II) after capsaicin stimulation (3 μm; 5 min) in the presence of MEK inhibitor PD98059 (PD), PKA inhibitor H89, PKC inhibitor Ro-31–84-25 (Ro), and CaMK inhibitor KN93. ANOVA indicates an overall effect of these inhibitors on pCREB expression (F(6,25) = 10.592; p < 0.0001). ++ p < 0.01, compared with control; *p < 0.05, **p < 0.01, compared with capsaicin (n = 4–6). Six to eight sections (15 μm) were analyzed for each slice. The slices were incubated with PD98059 for 30 min and the rest of the drugs for 10 min and fixed 10 min after the capsaicin stimulation.
Results
ERK activation by intrathecal NMDA, substance P, and BDNF
Intrathecal injection of NMDA, substance P, and BDNF was used to determine the effect of stimulation of NMDA, NK-1, and TrkB receptors on pERK levels in spinal cord neurons in vivo. Intrathecal injection of saline did not induce pERK (Fig. 1a), but intrathecal NMDA (0.5 μg, 2 nmol; 10 min) and BDNF (5 μg, 0.4 nmol; 10 min) induced pERK-IR in many neurons of the superficial dorsal horn (Fig. 1b,d,e). Intrathecal injection of substance P (10 μg, 7 nmol; 10 min) also resulted in pERK induction, although in this case restricted primarily to lamina I neurons (Fig. 1c) where the NK-1 receptor is localized (Mantyh et al., 1995). Intrathecal injection of the group I metabotropic glutamate receptor (mGluR1) agonist (RS)-3,5-dihydroxyphenylglycine markedly induces pERK in dorsal horn neurons (Karim et al., 2001). These data indicate that each primary afferent neurotransmitter or neuromodulator is sufficient to activate ERK in dorsal horn neurons.
Figure 1.

a–d, pERK immunostaining in the dorsal horn of intact rats after intrathecal injection of saline (a), NMDA (0.5 μg; b), substance P (SP) (10 μg; c), and BDNF (5 μg; d). Animals were perfused 10 min after the injection. Scale bars, 100 μm. e, Numbers of pERK-IR neurons per 15-μm-thick section in the superficial (laminas I-II) dorsal horn. ANOVA indicates an overall effect of these agents on pERK expression (F(3,12)=57.595; p<0.0001). *p<0.01, compared with control (n = 4).
ERK activation in spinal cord slices
To investigate the molecular pathways involved in ERK activation in the spinal cord by afferent inputs, we used an adult rat transverse spinal cord slice preparation (700 μm thick), which offers the opportunity for activating defined primary afferent inputs and controlled drug application. The slices were perfused for >3 hr before stimulation, and there were few pERK-IR neurons in control nonstimulated spinal cord slices (Fig. 2a,d). Bath application of NMDA (100 μm), substance P (100 μm), and BDNF (100 ng/ml) all increased pERK-labeled neurons in the superficial dorsal horn neurons to levels similar to that produced by intrathecal injections in vivo (Figs. 1e,2f). Direct depolarization of spinal cord neurons with KCl (90 mm; 5 min) also induced a robust ERK activation. In this case, the ERK was predominantly activated in superficial dorsal horn neurons, even though all spinal cord neurons were depolarized by the KCl (Fig. 2b).
Figure 2.

a–c, pERK immunofluorescence in control unstimulated (a), KCl depolarized (b), and capsaicin-stimulated (c) spinal cord slices. The slices were fixed 10 min after the capsaicin exposure (3μm; 5 min) and KCl (90 mm; 5 min) depolarization. The dotted lines indicate the top border of the gray matter of the spinal cord. d, e, pERK immunostaining by DAB in control unstimulated and electrical C-fiber stimulation (1 mA, 50 Hz; 50 msec; 150 pulses) of an attached dorsal root in spinal cord slices. The slices were fixed 2 min after electrical stimulation. Scale bars, 100μm. f, Numbers of pERK-IR neurons after exposure of spinal cord slices to NMDA (100 μm), substance P (SP; 100 μm), and BDNF (200 ng/ml). ANOVA indicates an overall effect of these agents on pERK expression (F(3, 11) = 18.489; p < 0.0001). *p < 0.01, compared with control (n = 4–5). All of the slices were fixed 10 min after exposure.
To examine the intracellular signal elements involved in nociceptor afferent-induced ERK activation, bath application of capsaicin (3 μm) was used to induce the release of neurotransmitters and neuromodulators from transient receptor potential vanilloid receptor subtype 1 (TRPV1)-expressing primary afferents (a subset of C-fibers) in the superficial dorsal horn. Brief exposure of capsaicin (5 min) to the spinal cord slice induced a large increase in pERK in the superficial dorsal horn (laminas I-II) (Fig. 2c) 2–10 min later (Fig. 3f).
To examine whether pERK is induced by monosynaptic or polysynaptic activation after capsaicin application, we applied TTX (1 μm) to block action potentials and thereby polysynaptic transmission. TTX did not decrease the capsaicin-induced pERK induction; the numbers of pERK-IR neurons were 28.2 ± 3.0 and 32.6 ± 8.1 (n = 6; p > 0.05) in capsaicin-treated and capsaicin-plus TTX-treated spinal sections, respectively. This result indicates that pERK is directly induced in postsynaptic neurons by those neurotransmitters released from primary afferents in response to activation of TRPV1 on their central terminals rather than by polysynaptic circuits. Electrical stimulation of the attached dorsal root at C-fiber intensity (1000 μA, 50 Hz; three trains with 10 sec interval, 150 pulses total) also induced an increase in pERK-positive neurons in the superficial layers (laminas I-II) of the dorsal horn, with the most prominent expression in lamina I (Fig. 2e).
Involvement of ionotropic and metabotropic glutamate receptors in C-fiber-induced ERK activation
Because glutamate is the fast neurotransmitter in primary afferents (for review, see Woolf and Salter, 2000), we tested whether ionotropic glutamate receptors are involved in ERK activation. Activation of NMDA receptors is implicated in ERK activation in cortical neurons (Xia et al., 1996; English and Sweatt, 1997). However, it is controversial whether this receptor is required for noxious stimulation-evoked ERK activation in dorsal horn neurons (Ji et al., 1999; Lever et al., 2003). In agreement with our previous in vivo study (Ji et al., 1999), pERK induction by capsaicin was only partially inhibited by the NMDA receptor antagonist MK-801 (100 μm; 44% inhibition; p < 0.001). The AMPA/kainate receptor antagonist CNQX (20 μm) also attenuated pERK induction (54% inhibition; p < 0.001) as did the group I metabotropic receptor mGluR1 antagonist CPCCOEt by 60% (p < 0.001) at 1 μm and 75% (p < 0.001) at 10 μm. CPCCOEt (10 μm) was more effective than MK-801 (100 μm; p < 0.01) and CNQX (20 μm; p < 0.01) in suppressing capsaicin-induced ERK activation (Fig. 4a).
Figure 4.

a, b, Involvement of ionotropic, metabotropic, and tyrosine kinase receptors in capsaicin-induced pERK induction in spinal cord slices. a, Number of pERK-IR neurons in the superficial dorsal horn (laminas I-II) after capsaicin stimulation (3 μm; 5 min) in the presence of the NMDA receptor antagonist MK-801 (50 and 100 μm), the AMPA receptor antagonist CNQX (10 and 20μm), and mGluR1 antagonist CPCCOEt (CP; 1 and 10μm). ANOVA indicates an overall effect of these agents on pERK expression (F(7, 33) = 29.782; p < 0.0001). Mean ± SEM; *p < 0.05; **p < 0.01, compared with capsaicin (n = 4–6). b, Number of capsaicin-induced pERK-IR neurons in the presence of the NK-1 receptor antagonist GR205171A (GR; 50 and 100 μm) and the Trk inhibitor K252a (50 and 100 nm). ANOVA indicates an overall effect of these agents on pERK expression (F(5, 25) = 16.442; p < 0.0001). *p < 0.05; **p < 0.01, compared with capsaicin (n = 4–6). Six to eight sections (15μm) were included for each slice. The slices were incubated with each drug for 5–10 min and fixed 10 min after the capsaicin stimulation.
C-fiber electrical stimulation of the dorsal root induced a fourfold increase in pERK levels, which, like the capsaicin experiments, was partially suppressed by blockade of NMDA (APV, 100 μm, 51% inhibition; p < 0.01) and AMPA/kainate receptors (CNQX, 20 μm, 37% inhibition; p < 0.01). A combination of NMDA and AMPA/kainate receptor antagonists (APV, 100 μm; CNQX, 20 μm) almost completely blocked the C-fiber-induced pERK (90% inhibition; p < 0.01) (Table 1).
Table 1.
Number of pERK-IR neurons in the medial two-thirds of superficial dorsal horn (laminas I-II) in spinal cord sections after C-fiber electrical stimulation of a dorsal root of an isolated spinal cord slice in the presence of the NMDA receptor antagonist APV (100 μm), AMPA receptor antagonist CNQX (20 μm), APV plus CNQX, NK-1 receptor antagonist GR205171A (100 μm) and its inactive form L733061 (100 μm), Trk inhibitor K252a (100 nm), PKC inhibitor H89 (1 μm), PKC inhibitor Ro-31-8425 (1 μm), H89 plus Ro-31-8425, CaMK inhibitor KN93 (20 μm), and general tyrosine kinase inhibitor genistein (100 μm) and its inactive form genistein (100 μm)
|
Treatment and drugs |
Receptor/kinase |
Number of pERK-IR neurons |
Percentage of inhibition over C-fiber |
p values versus C-fiber |
|---|---|---|---|---|
| Glutamate receptors | ||||
| Control | 3.2 ± 0.2 | p < 0.01 | ||
| C-fiber | 12.3 ± 0.7 | |||
| C plus APV | NMDA | 7.7 ± 0.5 | 51 | p < 0.01 |
| C plus CNQX | Non-NMDA | 8.9 ± 0.1 | 37 | p < 0.01 |
| C plus APV plus CNQX | NMDA plus non-NMDA | 4.1 ± 0.6 | 90 | p < 0.01 |
| NK-1 and Trk receptors | ||||
| Control | 3.0 ± 0.4 | p < 0.01 | ||
| C-fiber | 12.1 ± 0.6 | |||
| C plus GR205171A | NK-1 | 7.9 ± 0.5 | 46 | p < 0.01 |
| C plus L733061 | NK-1 (inactive form) | 12.0 ± 0.5 | 1 | p > 0.05 |
| C plus K252a | Trk | 11.2 ± 1.5 | 10 | p > 0.05 |
| Protein kinases | ||||
| Control | 3.0 ± 0.3 | p < 0.01 | ||
| C-fiber | 12.3 ± 0.4 | |||
| C plus H89 | PKA | 6.7 ± 0.2 | 60 | p < 0.01 |
| C plus Ro-31-8425 | PKC | 9.1 ± 0.6 | 34 | p < 0.01 |
| C plus H89 plus Ro | PKA plus PKC | 4.0 ± 0.5 | 89 | p < 0.01 |
| C plus KN93 | CaMK | 10.9 ± 0.6 | 15 | p > 0.05 |
| C plus genistein | TK | 6.0 ± 0.4 | 68 | p < 0.01 |
| C plus genistein |
TK (inactive form) |
12.7 ± 0.7 |
-4 |
p > 0.05 |
Data are represented as mean ± SEM. Six to eight sections (15 μm) were analyzed per slice with three to five slices. The slices were fixed 2 min after electrical stimulation. These drugs do not affect basal pERK levels, and thus percentage inhibition is calculated after subtraction of basal pERK levels. C, C-fiber; Ro, Ro-31-8425; TK, tyrosine kinase.
Involvement of NK-1 and Trk receptors in C-fiber-induced ERK activation
We first tested whether the G-protein-coupled NK-1 receptor plays a role in C-fiber-mediated ERK activation. The selective NK-1 antagonist GR205171A (100 μm) inhibited capsaicin-induced pERK but only by 27% (p < 0.05) (Fig. 4b). pERK induction by C-fiber electrical stimulation was suppressed by GR205171A to a limited extent (100 μm, 35% inhibition; p < 0.01). An inactive NK-1 antagonist, L733061 (100 μm), was ineffective (Table 1).
BDNF contributes to pain hypersensitivity via the TrkB receptor (Kerr et al., 1999; Mannion et al., 1999). Bath application of a general Trk inhibitor K252a at 50 and 100 nm, concentrations considered specific for Trk inhibition (Riccio et al., 1997), reduced capsaicin-induced pERK by 35 and 40%, respectively (Fig. 4b). However, K252a, even at 100 nm, had no effect on pERK induced by C-fiber electrical stimulation (10% inhibition; p > 0.05) (Table 1). This discrepancy may reflect the finding that BDNF is released only by very high-frequency (100 Hz) bursts of electrical stimulation of C-fibers (Lever et al., 2001, 2003). This result suggests that the involvement of BDNF and its TrkB receptor in C-fiber-induced ERK activation may depend on the frequency and intensity of C-fiber stimulation.
ERK activation by PKA and PKC
PKA and PKC are both activated by multiple transmitters and synaptic neuromodulators, and both kinases are coupled to ERK activation in hippocampal neurons (Roberson et al., 1999) and implicated in ERK-mediated neuronal excitability in dorsal horn neurons (Hu and Gereau, 2003; Hu et al., 2003) and central sensitization (Petersen-Zeitz and Basbaum, 1999; Ji and Woolf, 2001). To examine whether activation of either PKA or PKC is sufficient alone to induce pERK, spinal cord slices were exposed to the PKA activator forskolin (10 and 20 μm) or to the PKC activator PMA (1 and 5 μm) for 20 min. Both forskolin and PMA activated ERK, producing eightfold increases in pERK levels (Fig. 5b,c). Coapplication of forskolin and PMA produced a 16- to 20-fold increase in pERK levels, which is significantly higher than either alone (p < 0.05) (Fig. 5e). Most pERK-IR cells were localized in the superficial dorsal horn. The additive effect of PKA and PKC activators indicates that PKA and PKC are likely to activate ERK as independent parallel pathways.
Figure 5.

a–d, pERK immunofluorescence in control unstimulated (a) and after exposure to the PKA activator forskolin (20 μm)(b), the PKC activator PMA (5 μm)(c), and forskolin plus PMA (d). Scale bar, 100μm. The dotted lines indicate the top border of the gray matter of the spinal cord. e, Numbers of pERK-IR neurons in the superficial dorsal horn after each treatment. ANOVA indicates an overall effect of these agents on pERK expression (F(6, 22)=16.442;p<0.0001).*p<0.05;**p< 0.01 (n = 3–5). All of the slices were fixed 20 min after drug exposure.
We next investigated whether PKA and PKC are required for ERK activation in the dorsal horn. Capsaicin-induced pERK was not attenuated by a low concentration of either the PKA inhibitor H89 (0.1 μm, 25% inhibition; p > 0.05) or the PKC inhibitor Ro-31–8425 (0.1 μm, 15% inhibition; p > 0.05); however, it was suppressed by a combination of both inhibitors at this low concentration (0.1 μm, 63% inhibition; p < 0.001) (Fig. 6a). At a higher concentration, H89 (1 μm, 97% inhibition; p < 0.001) and Ro-31–8425 (1 μm, 96% inhibition; p < 0.001) independently blocked the pERK increase (Fig. 6a).
Figure 6.

a, b, Involvement of PKA, PKC, Src, and CaMK in capsaicin-induced pERK induction in spinal cord slices. a, Numbers of pERK-IR neurons in the superficial dorsal horn (laminas I-II) after capsaicin stimulation (3 μm; 5 min) in the presence of the PKA inhibitor H89 (0.1 and 1 μm), the PKC inhibitor Ro-31–84-25 (Ro; 0.1 and 1 μm), and H89 plus Ro-31–84-25 (0.1 μm). ANOVA indicates an overall effect of these inhibitors on pERK expression (F(6, 27) = 14.438; p < 0.0001). **p < 0.01, compared with capsaicin. Mean ± SEM (n = 3–6). b, Numbers of capsaicin-induced pERK-IR neurons in the presence of the general tyrosine kinase(TK) inhibitor genistein (50 and 100μm), the Srcinhibitor PP2 (5 and 20μm), and the CaMK inhibitor KN93 (20 μm).ANOVA indicates an overall effect of these agents on pERK expression (F(7, 23) = 4.327; p = 0.003). *p < 0.05; **p < 0.01, compared with capsaicin (n=3–6).Six to eight sections (15 μm) were analyzed for each slice. Mean ± SEM. The slices were incubated with each drug for 10 min and fixed 10 min after the capsaicin stimulation.
pERK induction by electrical C-fiber stimulation was also decreased by H89 (1 μm, 60% inhibition; p < 0.01) and by Ro-31–84-25 (1 μm, 34% inhibition; p < 0.01). pERK levels after cotreatment of H89 and Ro-31–84-25 were significantly lower (89%; p < 0.01) than H89 or Ro-31–84-25 treatment alone and not different from the levels in control slices (p > 0.05) (Table 1).
CaM kinase, Src, and C-fiber-induced ERK activation
Calcium calmodulin-dependent kinase (CaMK) is also involved in central sensitization (Fang et al., 2002; Garry et al., 2003). Although the CaMK is implicated in ERK activation in cortical neurons (Vanhoutte et al., 1999; Zhu et al., 2002), the CaMK inhibitor KN93, even at a high concentration (20 μm), had no effect on pERK induced either by capsaicin (0% inhibition) (Fig. 6b) or electrical C-fiber stimulation (15% inhibition; p > 0.05) (Table 1).
Tyrosine kinases, especially Src family members, couple to the MAPK (mitogen-activated protein kinase) cascade in neurons after intracellular calcium increase (Finkbeiner and Greenberg, 1996) and participate in central sensitization (Guo et al., 2002). PKC activation of ERK is mediated by Src (Lu et al., 1999). We found that a general tyrosine kinase inhibitor, genistein, inhibited the capsaicin-induced pERK induction at a concentration of 100 μm (57%; p < 0.01) but not 50 μm (25%; p > 0.05) (Fig. 6b). Genistein (100 μm) also inhibited pERK (68%; p < 0.01) induced by electrical stimulation, whereas its inactive form genistin (100 μm) was without effect (Table 1). A specific Src inhibitor, PP2, significantly inhibited capsaicin-induced pERK induction at a concentration of 20 μm (43%; p < 0.05) but not 5 μm (14%; p > 0.05) (Fig. 6b).
To test whether these antagonists and inhibitors alter basal pERK levels, spinal slices were incubated with each drug (at higher concentrations) in the absence of C-fiber stimulation. None of the drugs tested had an effect on basal pERK levels(F(13, 35) = 0.4781; p = 0.9925; n = 3–5 per group). The average numbers of pERK-IR neurons in laminas I-II of each spinal section were 2.9 ± 0.2 (control), 3.1 ± 0.4 (APV, 100 μm), 2.7 ± 0.2 (CNQX, 20 μm), 2.6 ± 0.3 (APV, 100 μm, plus CNQX, 20 μm), 3.0 ± 0.3 (MK-801, 100 μm), 3.2 ± 0.3 (CPCCOEt, 10 μm), 3.0 ± 0.2 (GR205171A, 100 μm), 2.8 ± 0.7 (K252a, 100 nm), 3.0 ± 0.2 (H89, 1 μm), 2.6 ± 0.3 (Ro-31–8425, 1 μm), 2.9 ± 0.5 (H89, 1 μm plus Ro-31–8425, 1 μm), 3.4 ± 0.7 (KN93, 20 μm), 3.4 ± 0.3 (genistein, 100 μm), and 3.0 ± 0.2 (PP2, 20 μm).
ERK activation and CREB phosphorylation after C-fiber stimulation
To explore the mechanisms underlying ERK-mediated increases in gene transcription (Ji et al., 2002a), we examined whether ERK is involved in CREB phosphorylation in dorsal horn neurons. In nonstimulated spinal cord slices, there is basal pCREB expression (Fig. 3a,e). Dorsal root C-fiber stimulation induced a significant increase in pCREB-IR in nuclei of superficial dorsal horn neurons beyond the basal levels (Fig. 3b,c,e). Bath application of a specific ERK upstream kinase (MEK) inhibitor, PD98059 (Alessi et al., 1995), at 50 μm completely blocked the C-fiber stimulationevoked increase in pCREB without altering the basal pCREB levels (Fig. 3d,e). Capsaicin exposure also induced a significant increase in pCREB levels, with the same time course as pERK, reaching a peak at 2 min and maintained at elevated levels for 1 hr (Fig. 3f). Preincubation with PD98059 also suppressed capsaicin-induced pCREB increase in a dose-dependent manner (Fig. 3g). Therefore, ERK is required for C-fiber stimulation-induced CREB phosphorylation.
Accordingly, capsaicin-induced pCREB was totally blocked by PKA inhibitor H89 (1 μm) and PKC inhibitor Ro-31–84-25 (1 μm) at the concentration (1 μm) that completely blocks pERK induction (Figs. 3g,6a). Although CaMK inhibitor KN93 (20μm) had no effect on capsaicin-induced pERK levels (Fig. 6b), it partially suppressed capsaicin-induced pCREB levels (p < 0.05) (Fig. 3g).
ERK activation, CREB phosphorylation, and gene expression after C-fiber stimulation in vivo
We next tested whether ERK activation causes pCREB induction in vivo. Injection of capsaicin (75 μg in 25 μl) into a hindpaw induced both pERK and pCREB in the superficial dorsal horn (Fig. 7a–d). Double immunofluorescence staining indicated that the pERK was colocalized primarily with pCREB in the nuclei of the medial superficial dorsal horn, with 89% pERK-IR neurons (170 of 192 neurons in 12 spinal sections obtained from two rats) expressing pCREB (Fig. 7e). Intrathecal PD98059 pretreatment (1 μg; 30 min before capsaicin injection) blocked both capsaicin-induced pERK and pCREB (p < 0.01) without affecting basal pCREB levels (Table 2). Control animals received intraplantar injection of capsaicin vehicle (saline with 10% Tween 80) and intrathecal injection of PD98059 vehicle (10% DMSO). Intrathecal vehicle had no effect (p > 0.05) on capsaicin-evoked pERK; the number of pERK-IR neurons in the superficial dorsal horn was 28.3 ± 5.6 (without DMSO) and 24.9 ± 2.5 (with DMSO).
Figure 7.
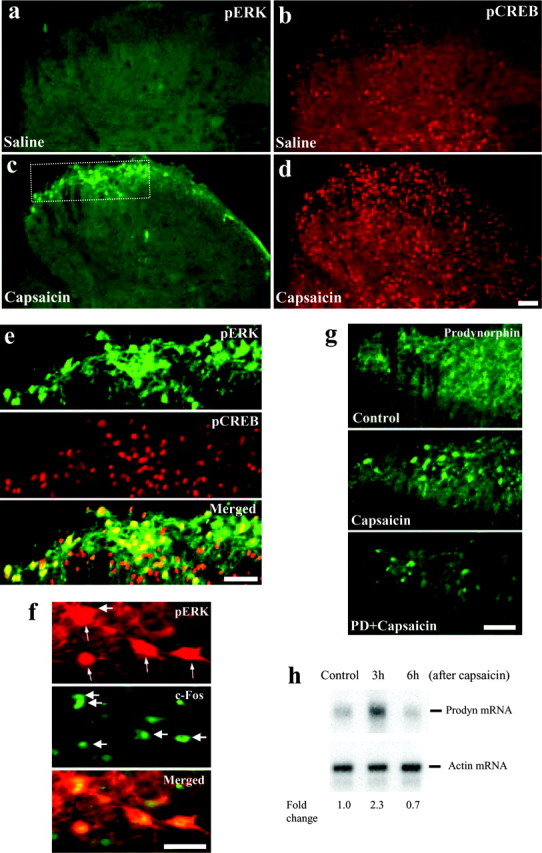
a–h, ERK activation contributes to the capsaicin-induced phosphorylation of CREB and the expression of c-fos and prodynorphin in vivo. a–d, Intraplantar injection of capsaicin induces pERK (a, c) and pCREB (b, d) in the ipsilateral dorsal horn. a–d were obtained from the same spinal sections. e, pERK is largely colocalized with pCREB in the nuclei of individual neurons in the medial laminas I and II of the dorsal horn. e is a high magnification of the areas in c and d indicated by the square. Animals were perfused 5 min after capsaicin injection. Scale bars, 100μm. f, Intraplantar capsaicin induces pERK and c-Fos in nuclei of neurons in medial laminas I and II. The arrows indicate double-stained neurons. Scale bar, 30 μm. g, Intraplantar capsaicin induces an increase in prodynorphin-IR neurons in the medial dorsal horn 3 hr after capsaicin injection, which is suppressed by intrathecal PD98059 (1μg) administered 30 min before capsaicin injection. Control animals received intrathecal vehicle (10% DMSO) and an intraplantar injection of saline with 10% Tween 80. Scale bar, 100μm. h, RNase protection assay showing a transient increase of prodynorphin mRNA levels in the ipsilateral dorsal horn after intraplantar capsaicin. Actin is used as the loading control. The fold change for the density of prodynorphin mRNA bands is calculated after normalization with actin.
Table 2.
Number of pERK-IR, pCREB-IR, c-Fos-IR, and prodynorphin-IR neurons in the medial two-thirds of superficial dorsal horn (laminas I-II) per spinal cord section at 5, 60, or 180 min after intraplantar capsaicin injection
|
|
Time point |
Vehicle |
Capsaicin |
PD98059 plus vehicle |
PD98059 plus capsaicin |
|---|---|---|---|---|---|
| pERK | 5 min | 2.7 ± 0.3 | 24.9 ± 2.5 | 2.2 ± 0.5 | 5.1 ± 0.6 |
| pCREB | 5 min | 24.8 ± 3.4 | 41.5 ± 1.9 | 21.4 ± 2.0 | 24.2 ± 1.7 |
| c-Fos | 60 min | 3.7 ± 0.2 | 13.0 ± 1.0 | 3.4 ± 0.8 | 5.3 ± 0.5 |
| Prodynorphin |
180 min |
6.9 ± 0.6 |
12.7 ± 0.8 |
6.1 ± 0.5 |
7.8 ± 0.4 |
Intrathecal injection of the MEK inhibitor PD98059 (1 μg) 30 min before capsaicin injection suppressed the capsaicin-induced increase in these signaling molecules (p<0.01) but had no effect on their basal levels in vehicle-treated animals (p>0.05). Vehicle-treated animals received intraplantar injection of saline with 10% Tween 80 and intrathecal injection of 10% DMSO. Mean ± SEM. Eight spinal sections (15 μm) were analyzed per spinal cord (n = 4).
Both the immediate early gene c-fos and the late response gene prodynorphin contain the CREB binding site CRE within their promoter regions (Lonze and Ginty, 2002). Because pERK contributes to CREB phosphorylation, we tested whether ERK activation leads to c-fos and prodynorphin expression after capsaicin treatment in vivo. Intraplantar capsaicin induced an increased expression in the medial superficial spinal cord neurons of c-Fos at 1 hr and prodynorphin at 3 hr after the stimulation, and the expression of both genes was suppressed by intrathecal PD98059 (1 μg) pretreatment (Fig. 7g, Table 2). PD98059 had no effect on basal prodynorphin levels (Table 2). Although the pERK level declined from its peak 1 hr after the intraplantar capsaicin injection (Ji et al., 1999), most pERK-positive neurons also labeled for c-Fos (Fig. 7f). An RNase protection assay shows that intraplantar capsaicin increased prodynorphin mRNA levels in the dorsal horn at 3 hr but not at 6 hr after the injection (Fig. 7h).
ERK activation and central sensitization generated pain hypersensitivity
Capsaicin injection into the skin induces an immediate shortlasting flicking response and a later secondary mechanical allodynia outside the area of injection that lasts for several hours. Because primary afferents innervating the area of secondary allodynia and hyperalgesia are not sensitized, the pain hypersensitivity in this area is an expression of central sensitization (LaMotte et al., 1992; Torebjork et al., 1992; Treede et al., 1992; Willis, 2002).
Injection of capsaicin (20 μg in 20 μl) into the heel of the hindpaw induced secondary mechanical allodynia in the center plantar surface of the paw. This mechanical hypersensitivity was present 15 min after the injection, maintained at 3 hr, and recovered toward baseline at 6 hr (Fig. 8). Intrathecal pretreatment with PD98059 (1 μg; 30 min before capsaicin injection) had no effect on basal mechanical pain sensitivity but significantly reduced secondary mechanical allodynia at both early (15–60 min) and later times (180 min) (Fig. 8).
Figure 8.

ERK activation is involved in mediating capsaicin-induced secondary mechanical allodynia. Capsaicin injection (20 μg in 20 μl) into the heel of a rat hindpaw induces a secondary mechanical allodynia in the center of the plantar surface of the hindpaw, which is inhibited by intrathecal injection of the MEK inhibitor PD 98059 (1 μg) 30 min before capsaicin. **p < 0.01, compared with vehicle control (10% DMSO); t test; n = 7. Data are expressed as percentage of precapsaicin control. The MEK inhibitor had no effect on basal pain sensitivity. Mechanical allodynia was measured with von Frey filaments.
Discussion
ERK/MAPK is activated in dorsal horn neurons after peripheral noxious stimulation and inflammation, and this plays a role in regulating the expression of prodynorphin and NK-1 as well as pain hypersensitivity (Ji et al., 1999, 2002a). In this study, we explored what is responsible for activating ERK and demonstrate that multiple nociceptor primary afferent transmitters and receptors contribute. We have further demonstrated a link between ERK and CREB/gene transcription in superficial dorsal horn neurons and a role for ERK in central sensitization.
Upstream regulators of ERK activation in the spinal cord differ from those in the cortex and hippocampus. CaMK is not involved in noxious stimulation-evoked pERK induction by capsaicin or electrical stimulation, although it contributes to ERK activation in brain slices (Vanhoutte et al., 1999; Zhu et al., 2002) and cortical neurons (Perkinton et al., 2002). CaMK is regarded as a major CREB kinase in cortical neurons (Lonze and Ginty, 2002), but it is only partially involved in C-fiber-induced CREB phosphorylation in spinal slices. Our data suggest that ERK and CaMK are two parallel pathways leading to CREB phosphorylation, and the ERK pathway appears to be more important for C-fiber phosphorylation of CREB in dorsal horn neurons (Fig. 3). Although blockade of the NMDA receptor completely blocks LTP-induced ERK induction in the hippocampus (English and Sweatt, 1997), it only partially inhibits C-fiber-evoked ERK activation after capsaicin or electrical stimulation (Fig. 4, Table 1) (Ji et al., 1999) (but see Lever et al., 2003).
Neurotransmitter–neuromodulator receptors involved in ERK activation
Multiple transmitter receptors are coupled to ERK activation in the superficial laminas of the spinal cord: ionotropic NMDA andnon-NMDA glutamate receptors, mGluR1, and G-protein-coupled NK-1 receptor. Extracellular Ca2+ appears essential for noxious stimulation-induced ERK activation (Lever et al., 2003). The NMDA receptor functions as a Ca2+ channel and is widely implicated in ERK activation in cortical neurons (Finkbeiner and Greenberg, 1996; Xia et al., 1996; English and Sweatt, 1997), and calcium permeable AMPA receptors also play a role (Perkinton et al., 1999) (but see Lever et al., 2003). We found that NMDA or AMPA receptor antagonists alone only partially blocked C-fiber stimulation-evoked pERK, but a combination of these receptor antagonists almost totally suppressed pERK (Table 1). Ca2+ influx after coactivation of these two ionotropic glutamate receptors may play a major role in ERK activation.
In agreement with previous in vivo (Karim et al., 2001) and in vitro studies (Lever et al., 2003), we found that the group I metabotropic glutamate receptor, mGluR1, plays a major role in nociceptor afferent-induced ERK activation in dorsal horn neurons. mGluR1 activates phospholipase C to produce DAG and increases in intracellular Ca2+, both of which activate PKC. BDNF induces pERK and pCREB in cortical and hippocampal neurons (Ji et al., 1998; Patterson et al., 2001; Ying et al., 2002), and in agreement with a recent study (Pezet et al., 2002), we found that BDNF induces pERK in dorsal horn neurons in vitro and in vivo. A general Trk inhibitor, K252a, at doses where its action is specific for the Trk receptors attenuated pERK induction by capsaicin stimulation.
Involvement of protein kinases in ERK activation
Increases in intracellular Ca2+ either by extracellular Ca2+ influx through NMDA, AMPA, or calcium channels as well as release from intracellular Ca2+ stores after activation of G-protein-coupled mGluR1 or NK-1 receptors will result in the direct activation of PKC. Ca2+ influx also increases intracellular cAMP through activation of Ca2+/CaM-sensitive adenylyl cyclases, which activate PKA. cAMP and PKA are coupled to the Raf/Ras/ERK pathway via a small G-protein Rap1 (Impey et al., 1999). Both PKA and PKC contribute to the activation of ERK in the dorsal horn by C-fiber inputs.
Furthermore, activation of either PKA or PKC alone is sufficient to activate ERK. A combined application of forskolin and PMA induced the strongest ERK activation that we found in the spinal cord, fivefold higher than that induced by NMDA, substance P, and BDNF but also twofold higher than that produced by depolarization of spinal neurons with KCl and stimulation of C-fibers with capsaicin. However, caution must be taken, because forskolin may activate ERK through cAMP-dependent but PKA-independent mechanisms. Nevertheless, in combination with the data from both activators and inhibitors, our results strongly suggest an essential role of PKA and PKC in regulating ERK activation in dorsal horn neurons. Recent studies on electrophysiology show that ERK integrates PKA and PKC signaling in superficial dorsal horn neurons to modulate A-type potassium currents (Hu and Gereau, 2003; Hu et al., 2003).
The tyrosine kinase Src is associated with ERK activation after both activation of G-protein-coupled receptors and intracellular Ca2+ increases (Lev et al., 1995; Widmann et al., 1999). A PKC/Pyk2/Src pathway contributes to synaptic plasticity, regulating NMDA receptor function (Lu et al., 1999; Huang et al., 2001), and Src is implicated in the phosphorylation of NMDA receptors in the spinal cord (Guo et al., 2002; Salter and Kalia, 2004). Our data show that both a general tyrosine kinase inhibitor genistein suppresses and a specific Src inhibitor PP2 attenuates pERK in the dorsal horn, suggesting a role for intracellular tyrosine kinases in activating ERK. Src and related tyrosine kinases may couple PKC signaling to ERK activation in dorsal horn neurons.
ERK activation, CREB phosphorylation, and gene transcription
In agreement with studies in cultured cortical and hippocampal neurons and in brain slices (Xia et al., 1996; Impey et al., 1998; Sgambato et al., 1998; Obrietan et al., 1999; Vanhoutte et al., 1999), we found that ERK activation in the dorsal horn is coupled to CREB phosphorylation and c-fos expression. pERK colocalizes with pCREB and c-Fos in most superficial dorsal horn neurons, providing a basis for ERK-mediated CREB phosphorylation and c-fos expression in the same neuron rather than through synaptic transmission. ERK is required for c-Fos expression in the spinal cord in a rat model of radicular neuritis (Kominato et al., 2003). Noxious stimulation and peripheral inflammation evoke ipsilateral pERK induction and c-fos expression but a bilateral induction of pCREB in the dorsal horn (Ji and Rupp, 1997; Messersmith et al., 1998). pERK is predominantly induced in superficial dorsal horn neurons, whereas pCREB and c-fos are also induced in the deep dorsal horn. In addition, there are high basal levels of pCREB but very low levels of pERK and c-fos. Therefore, pERK is not necessary for pCREB induction in the spinal cord, and other protein kinases must play a role in CREB phosphorylation (Lonze and Ginty, 2002). A direct coupling between pERK, pCREB, and c-fos appears to be restricted to superficial dorsal horn neurons.
The pERK/pCREB cascade regulates the expression of several late-response genes. ERK activation is required for the upregulation of prodynorphin in dorsal horn neurons after complete Freund's adjuvant-induced chronic inflammation (Ji et al., 2002) and capsaicin-induced acute inflammation (Fig. 7). In addition to c-fos and prodynorphin, CRE sites are found on the promoter regions of many other genes induced by inflammation in the dorsal horn, such as immediate-early gene zif268, Cox-2, and late-response gene NK-1 and TrkB (Wisden et al., 1990; Mannion et al., 1999; Samad et al., 2001; Ji et al., 2002a; Lonze and Ginty, 2002). pCREB is implicated in regulating NK-1 expression (Abrahams et al., 1999; Anderson and Seybold, 2000; Seybold et al., 2003). ERK can activate other transcription factors such as ELK-1 (Ets-like transcription factor), which can bind to the SRE (serum response element) to regulate gene transcription in a CREB-independent manner (Widmann et al., 1999).
ERK and central sensitization
ERK activation contributes to the second phase of sensitivity in the formalin model (Ji et al., 1999; Karim et al., 2001). This phase is a manifestation of an NMDA receptor-dependent central sensitization, as revealed by conditional deletion of the NR1 receptor subunit in the dorsal horn (South et al., 2003). Capsaicin-induced secondary mechanical allodynia is another model of central sensitization (LaMotte et al., 1992; Torebjork et al., 1992; Treede et al., 1992; Willis, 2002) and, as we now show, is also ERK dependent. In both models, ERK contributes to the early (<60 min) activity-dependent phase of central sensitization. This may be mediated through the phosphorylation of ion channels or receptors such as the A-type potassium channel Kv4.2 (Hu et al., 2003; Morozov et al., 2003), leading to increased neuronal excitability or to the trafficking of AMPA receptors from the cytoplasm to the subsynaptic membrane (Zhu et al., 2002; Ji et al., 2003).
In conclusion, C-fiber activation of multiple ionotropic, metabotropic, and tyrosine kinase receptors result in ERK activation in superficial dorsal horn neurons. Activation of these receptors is coupled to ERK through activation of PKA and PKC pathways. Activated ERK contributes to the acute phase of central sensitization and, by regulating CREB-mediated transcription, may produce long-lasting changes in sensory processing. We conclude that ERK is an important intracellular controller of activity-dependent plasticity in the spinal cord, mediating the coupling of diverse synaptic inputs to altered synaptic efficacy.
Footnotes
This study was supported by National Institutes of Health Grants NS40698 (R.-R.J.) and NS39518 (C.J.W.).
Correspondence should be addressed to Ru-Rong Ji, Department of Anesthesiology, Brigham and Women's Hospital, Medical Research Building, Room 604, 75 Francis Street, Boston, MA 02115. E-mail: rrji@zeus.bwh.harvard.edu.
DOI:10.1523/JNEUROSCI.2396-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/248310-12$15.00/0
References
- Abbadie C, Brown JL, Mantyh PW, Basbaum AI (1996) Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience 70: 201–209. [DOI] [PubMed] [Google Scholar]
- Abrahams LG, Reutter MA, McCarson KE, Seybold VS (1999) Cyclic AMP regulates the expression of neurokinin1 receptors by neonatal rat spinal neurons in culture. J Neurochem 73: 50–58. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR (1995) PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo J Biol Chem 270: 27489–27494. [DOI] [PubMed] [Google Scholar]
- Anderson LE, Seybold VS (2000) Phosphorylated cAMP response element binding protein increases in neurokinin-1 receptor-immunoreactive neurons in rat spinal cord in response to formalin-induced nociception. Neurosci Lett 283: 29–32. [DOI] [PubMed] [Google Scholar]
- Baba H, Doubell TP, Woolf CJ (1999) Peripheral inflammation facilitates Abeta fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J Neurosci 19: 859–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ (2003) Removal of GABAergic inhibition facilitates polysynaptic A fibermediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci 24: 818–830. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ruda MA (1992) Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci 15: 96–103. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272: 19103–19106. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD (2002) Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci 22: 4196–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME (1996) Ca2+-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron 16: 233–236. [DOI] [PubMed] [Google Scholar]
- Garry EM, Moss A, Delaney A, O'Neill F, Blakemore J, Bowen J, Husi H, Mitchell R, Grant SG, Fleetwood-Walker SM (2003) Neuropathic sensitization of behavioral reflexes and spinal NMDA receptor/CaM kinase II interactions are disrupted in PSD-95 mutant mice. Curr Biol 13: 321–328. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Bonni A, Greenberg ME (1994) Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation of CREB. Cell 77: 713–725. [DOI] [PubMed] [Google Scholar]
- Guo W, Zou S, Guan Y, Ikeda T, Tal M, Dubner R, Ren K (2002) Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor in the spinal cord during the development and maintenance of inflammatory hyperalgesia. J Neurosci 22: 6208–6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HJ, Gereau RW (2003) ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. II. Modulation of neuronal excitability. J Neurophysiol 90: 1680–1688. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Glauner KS, Gereau RW (2003) ERK integrates PKA and PKC signaling in superficial dorsal horn neurons. I: Modulation of A-type K currents. J Neurophysiol 90: 1671–1679. [DOI] [PubMed] [Google Scholar]
- Huang Y, Lu W, Ali DW, Pelkey KA, Pitcher GM, Lu YM, Aoto H, Roder JC, Sasaki T, Salter MW, MacDonald JF (2001) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29: 485–496. [DOI] [PubMed] [Google Scholar]
- Hunt SP, Mantyh PW (2001) The molecular dynamics of pain control. Nat Rev Neurosci 2: 83–91. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869–883. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR (1999) Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23: 11–14. [DOI] [PubMed] [Google Scholar]
- Ji RR, Rupp F (1997) Phosphorylation of transcription factor CREB in rat spinal cord after formalin-induced hyperalgesia: relationship to c-fos induction. J Neurosci 17: 1776–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ (2001) Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis 8: 1–10. [DOI] [PubMed] [Google Scholar]
- Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hokfelt T (1994) Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci 14: 6423–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Bose CM, Lesuisse C, Qiu D, Huang JC, Zhang Q, Rupp F (1998) Specific agrin isoforms induce cAMP response element binding protein phosphorylation in hippocampal neurons. J Neurosci 18: 9695–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ (1999) Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci 2: 1114–1119. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ (2002a) ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci 22: 478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002b) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36: 57–68. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ (2003) Central sensitization and longterm potentiation–do pain and memory share similar mechanisms? Trends Neurosci 26: 696–705. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 23: 4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW (2001) Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci 21: 3771–3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr BJ, Bradbury EJ, Bennett DL, Trivedi PM, Dassan P, French J, Shelton DB, McMahon SB, Thompson SW (1999) Brain-derived neurotrophic factor modulates nociceptive sensory inputs and NMDA-evoked responses in the rat spinal cord. J Neurosci 19: 5138–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Moore KA, Baba H, Woolf CJ (2003) Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol (Lond) 548: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominato Y, Tachibana T, Dai Y, Tsujino H, Maruo S, Noguchi K (2003) Changes in phosphorylation of ERK and Fos expression in dorsal horn neurons following noxious stimulation in a rat model of neuritis of the nerve root. Brain Res 967: 89–97. [DOI] [PubMed] [Google Scholar]
- LaMotte RH, Lundberg LE, Torebjork HE (1992) Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol (Lond) 448: 749–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J (1995) Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature 376: 737–745. [DOI] [PubMed] [Google Scholar]
- Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M (2001) Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci 21: 4469–4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M (2003) The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci 24: 259–270. [DOI] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605–623. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF (1999) G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci 2: 331–338. [DOI] [PubMed] [Google Scholar]
- Mannion RJ, Costigan M, Decosterd I, Amaya F, Ma QP, Holstege JC, Ji RR, Acheson A, Lindsay RM, Wilkinson GA, Woolf CJ (1999) Neurotrophins: peripherally and centrally acting modulators of tactile stimulus-induced inflammatory pain hypersensitivity. Proc Natl Acad Sci USA 96: 9385–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE, Simone DA (1995) Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science 268: 1629–1632. [DOI] [PubMed] [Google Scholar]
- Messersmith DJ, Kim DJ, Iadarola MJ (1998) Transcription factor regulation of prodynorphin gene expression following rat hindpaw inflammation. Mol Brain Res 53: 260–269. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ (2002) Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22: 6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER (2003) Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron 39: 309–325. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Smith D, Athos J, Storm DR (1999) Circadian regulation of cAMP response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem 274: 17748–17756. [DOI] [PubMed] [Google Scholar]
- Otani S, Auclair N, Desce JM, Roisin MP, Crepel F (1999) Dopamine receptors and groups I and II mGluRs cooperate for long-term depression induction in rat prefrontal cortex through converging postsynaptic activation of MAP kinases. J Neurosci 19: 9788–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER (2001) Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32: 123–140. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Sihra TS, Williams RJ (1999) Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci 19: 5861–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ (2002) Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem 80: 239–254. [DOI] [PubMed] [Google Scholar]
- Petersen-Zeitz KR, Basbaum AI (1999) Second messengers, the substantia gelatinosa and injury-induced persistent pain. Pain Suppl 6: S5–S12. [DOI] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB (2002) Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci 21: 684–695. [DOI] [PubMed] [Google Scholar]
- Riccio A, Pierchala BA, Ciarallo CL, Ginty DD (1997) An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 277: 1097–1100. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5: 317–328. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410: 471–475. [DOI] [PubMed] [Google Scholar]
- Seybold VS, McCarson KE, Mermelstein PG, Groth RD, Abrahams LG (2003) Calcitonin gene-related peptide regulates expression of neurokinin1 receptors by rat spinal neurons. J Neurosci 23: 1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J (1998) Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci 18: 8814–8825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE (2003) A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J Neurosci 23: 5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweatt JD (2001) The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 76: 1–10. [DOI] [PubMed] [Google Scholar]
- Swett JE, Woolf CJ (1985) The somatotopic organization of primary afferent terminals in the superficial laminae of the dorsal horn of the rat spinal cord. J Comp Neurol 231: 66–77. [DOI] [PubMed] [Google Scholar]
- Torebjork HE, Lundberg LE, LaMotte RH (1992) Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol (Lond) 448: 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treede RD, Meyer RA, Raja SN, Campbell JN (1992) Peripheral and central mechanisms of cutaneous hyperalgesia. Prog Neurobiol 38: 397–421. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J (1999) Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol 19: 136–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev 79: 143–180. [DOI] [PubMed] [Google Scholar]
- Willis WD (2002) Long-term potentiation in spinothalamic neurons. Brain Res Brain Res Rev 40: 202–214. [DOI] [PubMed] [Google Scholar]
- Wisden W, Errington ML, Williams S, Dunnett SB, Waters C, Hitchcock D, Evan G, Bliss TV, Hunt SP (1990) Differential expression of immediate early genes in the hippocampus and spinal cord. Neuron 4: 603–614. [DOI] [PubMed] [Google Scholar]
- Woolf CJ (1983) Evidence for a central component of post-injury pain hypersensitivity. Nature 306: 686–688. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288: 1765–1769. [DOI] [PubMed] [Google Scholar]
- Wu J, Fang L, Lin Q, Willis WD (2002) The role of nitric oxide in the phosphorylation of cyclic adenosine monophosphate-responsive elementbinding protein in the spinal cord after intradermal injection of capsaicin. J Pain 3: 190–198. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME (1996) Calcium influx via the NMDA receptor induces immediate early gene transcription by a MAP kinase/ERK-dependent mechanism. J Neurosci 16: 5425–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying SW, Futter M, Rosenblum K, Webber MJ, Hunt SP, Bliss TV, Bramham CR (2002) Brain-derived neurotrophic factor induces long-term potentiation in intact adult hippocampus: requirement for ERK activation coupled to CREB and upregulation of Arc synthesis. J Neurosci 22: 1532–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110: 443–455. [DOI] [PubMed] [Google Scholar]