

Abstract
Although the PI3K (phosphatidylinositol 3-kinase) pathway typically regulates cell growth and survival, increasing evidence indicates the involvement of this pathway in neural plasticity. It is unknown whether the PI3K pathway can mediate pain hypersensitivity. Intradermal injection of capsaicin and NGF produce heat hyperalgesia by activating their respective TRPV1 (transient receptor potential vanilloid receptor-1) and TrkA receptors on nociceptor sensory nerve terminals. We examined the activation of PI3K in primary sensory DRG neurons by these inflammatory agents and the contribution of PI3K activation to inflammatory pain. We further investigated the correlation between the PI3K and the ERK (extracellular signal-regulated protein kinase) pathway. Capsaicin and NGF induce phosphorylation of the PI3K downstream target AKT (protein kinase B), which is blocked by the PI3K inhibitors LY294002 and wortmannin, indicative of the activation of PI3K by both agents. ERK activation by capsaicin and NGF was also blocked by PI3K inhibitors. Similarly, intradermal capsaicin in rats activated PI3K and ERK in C-fiber DRG neurons and epidermal nerve fibers. Injection of PI3K or MEK (ERK kinase) inhibitors into the hindpaw attenuated capsaicin- and NGF-evoked heat hyperalgesia but did not change basal heat sensitivity. Furthermore, PI3K, but not ERK, inhibition blocked early induction of hyperalgesia. In acutely dissociated DRG neurons, the capsaicin-induced TRPV1 current was strikingly potentiated by NGF, and this potentiation was completely blocked by PI3K inhibitors and primarily suppressed by MEK inhibitors. Therefore, PI3K induces heat hyperalgesia, possibly by regulating TRPV1 activity, in an ERK-dependent manner. The PI3K pathway also appears to play a role that is distinct from ERK by regulating the early onset of inflammatory pain.
Keywords: PI3K, ERK, capsaicin, NGF, dorsal root ganglion, inflammatory pain
Introduction
Tissue injury is normally associated with inflammation and inflammatory pain. Inflammatory pain is induced by inflammatory mediators released in injured tissue, such as prostaglandin E2, nerve growth factor (NGF), and bradykinin, acting on nociceptors in the peripheral nerve terminals (Woolf and Salter, 2000; Julius and Basbaum, 2001). This pain is characterized by hyperalgesia (increased response to noxious stimulation) and allodynia (noxious response to previous innocuous stimulation). The capsaicin receptor TRPV1 [transient receptor potential vanilloid receptor-1 (VR1)] is expressed in unmyelinated C-fiber sensory neurons and plays an important role in mediating heat sensitivity (Caterina et al., 1997) and inflammatory heat hyperalgesia (Caterina et al., 2000; Davis et al., 2000; Chuang et al., 2001). The TTX-resistant sodium channel (NaV1.8/1.9) is also important for inflammatory hyperalgesia (Porreca et al., 1999; Kerr et al., 2001).
Inflammatory mediators activate several signaling pathways, such as protein kinase A (PKA) and protein kinase C (PKC-ϵ) in the peripheral nervous system, leading to pain hypersensitivity (Aley and Levine, 1999; Cesare et al., 1999; Khasar et al., 1999). PKA and PKC appear to regulate hyperalgesia through phosphorylation and activation of TRPV1 (Premkumar and Ahern, 2000; Bhave et al., 2002) and NaV1.8/1.9 (Gold et al., 1998; McCleskey and Gold, 1999; Julius and Basbaum, 2001; Bhave and Gereau, 2003). ERK (extracellular signal-regulated protein kinase), a member of the MAPK (mitogen-activated protein kinase) family, is activated in primary sensory neurons and epidermal nerve fibers after peripheral inflammation and contributes to inflammatory pain (Aley et al., 2001; Dai et al., 2002; Dina et al., 2003; Ji, 2003).
PI3K (phosphatidylinositol 3-kinase) is a lipid kinase that phophorylates the D-3 position of phosphatidylinositol lipids to produce PI(3,4,5)P3, acting as a membrane-embedded second messenger (Toker and Cantley, 1997). The downstream protein kinase AKT (protein kinase B) is postulated to mediate most of the effect of PI3K (Franke et al., 1997; Chan et al., 1999). PI3K is typically activated by neurotrophins mediating neuronal survival and axonal growth (Atwal et al., 2000; Patapoutian and Reichardt, 2001; Markus et al., 2002). A growing body of evidence indicates that PI3K is also involved in regulating neural plasticity, including long-term potentiation (LTP) (Kelly and Lynch, 2000; Lin et al., 2001; Yang et al., 2001; Izzo et al., 2002). In particular, PI3K inhibitors suppress NMDA receptor-mediated ERK activation in neurons (Chandler et al., 2001; Perkinton et al., 2002). PI3K and ERK have overlapping but distinct roles in regulating LTP (Opazo et al., 2003) and have opposing effects on muscle cell hypertrophy (Rommel et al., 1999).
Although PI3K has been implicated in NGF-induced TRPV1 expression (Bron et al., 2003) and the potentiation of the capsaicin-induced [Ca2+]i rise after NGF stimulation (Bonnington and McNaughton, 2003), it is not clear whether this pathway actually regulates inflammatory pain. We show that capsaicin and NGF can activate PI3K and ERK in primary sensory dorsal root ganglion (DRG) neurons and that the activation of ERK requires PI3K. Thus, both PI3K and ERK pathways are necessary for capsaicin- and NGF-induced heat hyperalgesia, possibly through their regulation of TRPV1 activity.
Materials and Methods
Animals and drugs. Male adult Sprague Dawley rats (180-230 gm) were used under Harvard Medical School Animal Care Institutional Guidelines. The animal room was artificially illuminated from 7:00 A.M. to 7:00 P.M. MEK (ERK kinase) inhibitors PD98059 and U0126, PI3K inhibitors LY294002 and wortmannin, and capsaicin were all purchased from Sigma (St. Louis, MO). NGF was purchased from Roche Applied Science (Indianapolis, IN).
Primary DRG culture. Despite the apparent early anatomical and neurochemical maturity of C fibers, physiological function is not fully established until the second week of life (Fitzgerald and Gibson, 1984). NGF has been shown to exert a sensitizing effect on sensory neurons on the second week (Zhu et al., 2004). In this study, we used 2- to 3-week-old rats to obtain high quality DRG neuronal cultures without NGF while maintaining the properties of adult DRG neurons. We found no difference in the activation of PI3K and ERK pathways between young and adult cultures, in agreement with a previous study (York et al., 2000). DRGs were removed aseptically, first incubated with collagenase (1.25 mg/ml; Roche Products)/dispaseII (2.4 U/ml; Roche Products) at 37°C for 90 min, and digested with 0.25% trypsin (Cellgro, Herndon, VA) for 8 min at 37°C, followed by 0.25% trypsin inhibitor (Sigma). Cells were then mechanically dissociated with a flame-polished Pasteur pipette in the presence of 0.05% DNase I (Sigma). The cell suspension was layered on a cushion of 15% fatty acid-free BSA and Percoll gradient to remove connective tissue and debris. Cells were plated onto poly-d-lysine (Sigma) and laminin (σ) -coated slide chambers (eight-chamber slide for immunostaining, 3000 neurons per chamber) or plates (six-well plate for Western blot, 30,000 neurons per well) and grown in a neurobasal-defined medium (with 2% B27 supplement; Invitrogen, Gaithersburg, MD) in the presence of 5 μm AraC to decrease non-neuronal cell numbers, at 36.5°C, with 5% carbon dioxide. These neurons were grown in the culture for 24 hr before experiments.
Western blot analysis. DRG cells were homogenized in a lysis buffer containing a mixture of proteinase inhibitors and phosphatase inhibitors (Sigma). The protein concentrations of the lysate were determined using a BCA Protein Assay kit (Pierce, Rockford, IL), and 30 μg of protein was loaded for each lane. Protein samples were separated on SDS-PAGE gel (4-15% gradient gel; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride filters (Millipore, Bedford, MA). The filters were blocked with 5% dry milk and incubated overnight at 4°C with primary antibody, phosphorylated (p) AKT-T (1:500; Cell Signaling Technology, Beverly, MA) or pERK (1:1000; Cell Signaling Technology) for 1 hr at room temperature (RT) with HRP-conjugated secondary antibody (1: 5000; Amersham Biosciences, Arlington Heights, IL). The blots were visualized in ECL solution (NEN, Boston, MA) for 1 min and exposed onto hyperfilms (Amersham Biosciences) for 1-30 min. Nonphosphorylated AKT and ERK2 antibodies were used as loading controls.
Immunocytochemistry and immunohistochemistry. The DRG cultures were fixed with 4% paraformaldehyde for 30 min and blocked with 2% goat serum-0.3% Triton X-100 PBS for 1 hr. The cultures were incubated with pAKT-T (Thr308, anti-rabbit, 1:200) pAKT-S (Ser473, anti-rabbit, 1:100), or pERK (p44/42 MAPK, anti-rabbit, 1:500) antibodies. The cultures were then incubated for 1 hr at RT with Cy3-conjugated secondary antibody (1:300; Jackson ImmunoResearch, West Grove, PA). For double immunofluorescence, DRG cultures were incubated with a mixture of polyclonal pAKT-T and monoclonal pERK (1:200; Cell Signaling Technology) antibodies overnight at 4°C, followed by a mixture of FITC- and Cy3-congugated secondary antibodies for 1 hr at RT.
Five minutes after intredermal capsaicin injection, animals were terminally anesthetized with isoflurane and perfused through the ascending aorta with saline, followed by 4% paraformaldehyde with 1.5% picric acid in 0.16 m phosphate buffer, pH 7.2-7.4 (4°C). After the perfusion, the L5 DRGs were removed and postfixed in the same fixative for 3 hr and then replaced with 15% sucrose overnight. All of the DRGs were embedded in OCT, and DRG sections (14 μm) were cut in a cryostat and processed for immunofluorescence. All of the sections were blocked with 2% goat serum in 0.3% Triton X-100 for 1 hr at RT and incubated overnight at 4°C with anti-pAKT-T antibody (1:200; Cell Signaling Technology). The sections were then incubated for 1 hr at RT with Cy3-conjugated secondary antibody (1:300; Jackson ImmunoResearch). For double immunofluorescence, DRG sections were incubated with a mixture of polyclonal pAKT-T-monoclonal NF-200 (1:5000; Chemicon, Temecula, CA) or pAKT-T-polyclonal VR1 (1:5000, anti-guinea pig; Chemicon) antibodies overnight at 4°C, followed by a mixture of FITC- and Cy3-congugated secondary antibodies for 1 hr at RT. For immunostaining of epidermal nerves, a piece of skin (5 × 5 mm) was dissected from the plantar surface of a hindpaw and fixed in 4% paraformaldehyde for 16 hr. The skin sections (30 μm) were cut in cryostat and processed for immunofluorescence with pAKT-T (1:100) and pERK (1:200) antibodies as described above.
The specificity for pAKT and pERK antibodies and immunostaining was confirmed by (1) loss of staining in the absence of primary antibodies, (2) suppression of induced pAKT signal by blocking PI3K pathway, and (3) single bands for pAKT and pERK in Western blotting.
Behavioral testing and drug injection. Animals were habituated to the testing environment daily for at least 2 d before baseline testing. The room temperature and humidity remained stable for all experiments. For testing heat sensitivity, animals were put in a plastic box placed on a glass plate, and the plantar surface was exposed to a beam of radiant heat through a transparent Perspex surface (Hargreaves et al., 1988; Ji et al., 2002a,b). The baseline latencies were adjusted to 12-18 sec with a maximum of 25 sec as cutoff to prevent potential injury. The latencies were averaged over three trials, separated by a 3 min interval. Ten microliters of the inhibitors, including PD98059 (1 and 10 μg), U0126 (1 μg), LY294002 (1 and 10 μg), or wortmannin (0.1 and 0.2 μg), all dissolved in 20% DMSO as vehicle, were injected into the center of the plantar surface of a hindpaw with a 30 gauge needle. This vehicle did not produce any cell toxicity in the skin. Neither did the vehicle change pain behavior, because both LY294002 and PD98059 dissolved in this vehicle had no effect on normal pain perception (see Fig. 3d). Five minutes after the injection of LY294002 and wortmannin, or 5 and 20 min after U0126 and PD98059, 5 μl of capsaicin (15 μg) or NGF (0.5 μg) was injected into the same site, and heat sensitivity was measured 15, 30, 60, 180, and 360 min after the second injection. The latencies were averaged over two trials, separated by a 3 min interval.
Figure 3.

a-d, PI3K and ERK are required for the induction and expression of capsaicin-induced heat hyperalgesia without affecting basal sensitivity. a, b, Pretreatment, Intradermal injection of PI3K inhibitors (LY294002, 1 and 10 μg; wortmannin, 0.2 μg) or MEK inhibitors (PD98059, 1 and 10 μg; U0126, 1 μg) before capsaicin injection prevents heat hyperalgesia in a dose-dependent manner. *p < 0.01; +p < 0.05, compared with vehicle control (n = 6). c, Posttreatment, Intradermal injection of LY294002 (10 μg) or PD98059 (10 μg), 15 min after capsaicin injection, reverses heat hyperalgesia. *p < 0.01, compared with control (n = 6). d, LY294002 (10 μg) or PD98059 (10 μg) has no effect on basal heat sensitivity (n = 6). p > 0.05. Heat sensitivity was measured by PWL and plotted as a proportion of preinjection baseline. The basal PWL (in seconds) in a, b, c, and d is 15.4 ± 0.6 (n = 24), 16.4 ± 0.5 (n = 24), 14.4 ± 0.5 (n = 18), 17.0 ± 0.8 (n = 12), respectively (mean ± SEM). Cont, Control; Ly, LY294002; PD, PD98059; Wort, wortmannin.
Electrophysiology. Acutely dissociated neurons from 5-week-old rats were used as reported previously (Shu and Mendell, 2001). Cell cultures were made as above, using neurobasal-defined medium without NGF. Cells were plated onto coverslips, and whole-cell recordings were performed within 2-12 hr after plating. All of the recordings were made from small-diameter (15-35 pF) DRG neurons. Bath solution contained the following (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 HEPES, and 10 glucose, pH 7.4. BSA at 0.1%, which had no effect on capsaicin-induced current (ICap), was added to aid the delivery of NGF during perfusion. The pipette solution contained the following (in mm): 120 cesium methane-sulfonate, 8 NaCl, 10 cesium BAPTA, 2 Mg-ATP, and 20 HEPES, pH 7.4. Several initial experiments were conducted using an EGTA-based pipette solution containing the following (in mm): 147 cesium, 120 methane-sulfonate, 8 NaCl, 10 EGTA, 2 Mg-ATP, and 20 HEPES, pH 7.4. The effect of NGF was more robust when we used the BAPTA-based pipette solution, which prevented desensitization more effectively than the EGTA-based pipette solution. Whole-cell currents were recorded at -60 mV holding potential. Other voltage-gated currents were inactivated. Data were collected using an Axopatch 2A patch-clamp amplifier, Digidata 1320, and pClamp 8.0 software (Axon Instruments, Foster City, CA), filtered at 1 kHz, and sampled at 5 kHz.
Quantification and statistics. For the quantification of immunofluorescence signal in DRG cultures, three optical fields (20×, 450 × 338 μm square) were randomly selected in each chamber (8 × 8 mm), and the images of immunostained neurons were captured with a CCD camera. The intensity of all of the positive neurons in each field was measured with NIH Image software, and mean intensity was averaged for that chamber. Four chambers from different experiments were included. The backgrounds of the areas (cell free) nearby the positive neurons were subtracted. An internal control (non-treated chambers) was included for each experiment with DRG cultures, and the intensities of the treated chambers were normalized to the internal control (as 100%). The immunohistochemistry signal (number of positive neuronal profiles) in the DRG was quantified as described previously and expressed as the percentage of total neuronal profiles (Ji et al., 1996, 2002b). Four rats were included in each group for quantification of immunohistochemistry, and six rats were included in each group for behavioral studies. All of the data were expressed as mean ± SEM. Differences between groups were compared using ANOVA, followed by Fisher's PLSD or t test. The criterion for statistical significance was p < 0.05.
Results
Activation of PI3K and ERK by capsaicin in cultured DRG neurons
Capsaicin activates C-fibers via the TRPV1 channel, producing inflammation and hyperalgesia (Julius and Basbaum, 2001). We examined whether capsaicin activates PI3K and ERK pathways in cultured DRG neurons. Phosphorylation of the downstream kinase AKT at threonine 308 (pAKT-T) or at serine 473 (pAKT-S) is used as a marker of PI3K activation (Kuruvilla et al., 2000; Lin et al., 2001; Yang et al., 2001; Sanna et al., 2002; Opazo et al., 2003). Phosphorylation at these two sites is necessary for the catalytic activity of AKT (Datta et al., 1999). Bath application of capsaicin (3 μm) induced phosphorylation of both AKT-T and ERK within 2 min in dissociated DRG neurons (Fig. 1a-c). The induction of pAKT-T and pERK had similar time courses, reaching peak levels at 10 min and maintaining elevated levels for >90 min (Fig. 1d). Double immunofluorescence indicated that pAKT-T and pERK were colocalized in the same DRG neurons, providing a cellular basis for the interaction between the PI3K and ERK pathways (Fig. 1b,b′). A specific PI3K inhibitor, LY294002, blocked the capsaicin-induced increase in pAKT-T levels in a dose-dependent manner (Fig. 1e), with complete inhibition at 100 μm (Fig. 1e). This concentration (Kuruvilla et al., 2000; Sanna et al., 2002; Shi et al., 2003) does not cause cellular toxicity (Vlahos et al., 1994). Not surprisingly, our results confirm that AKT is downstream of PI3K in DRG neurons. Bath application of LY294002 (100 μm) completely blocked the capsaicin-evoked pERK induction (Fig. 1f,g), indicating that PI3K is required for the capsaicin-induced ERK activation in DRG neurons. Another PI3K inhibitor, wortmannin, at concentrations (100 nm) used in other studies (Lin et al., 2001; Perkinton et al., 2002), also completely abolished the capsaicin-induced increase in pAKT-T and pERK levels (Fig. 1e,f).
Figure 1.
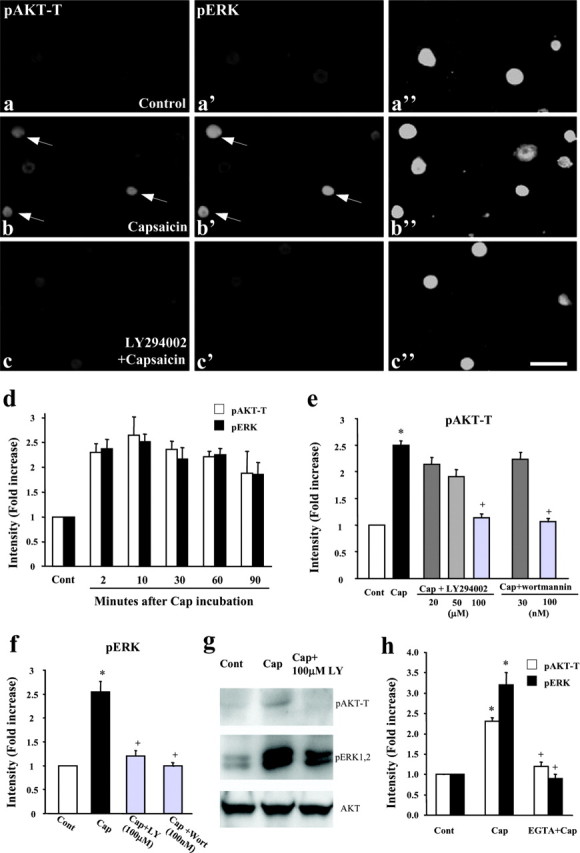
a-h, Capsaicin induces activation of AKT and ERK in cultured DRG neurons. a-c″, Bath application of capsaicin induces a rapid phosphorylation, within 2 min, of AKT at Thr308 (pAKT-T) and ERK. pAKT-T and pERK are colocalized in the same DRG neurons (b, b′', as indicated by arrows), and the capsaicin-induced phosphorylation of AKT-T (a, b) and ERK (a′, b′) is abolished by the PI3K inhibitor LY294002 (100 μm) (c, c′). a′, b′, and c′ are the same fields as a, b, and c, respectively, showing double staining. a″, b″, and c″ are images with high contrast to show all of the cells in each field. Scale bar, 50 μm. The DRG cultures were grown for 24 hr and stimulated with 3 μm capsaicin for 2 min. d, Time course of capsaicin (3 μm)-induced pAKT-T and pERK levels, as measured by the intensity of immunofluorescence. At all time points, pAKT-T and pERK levels are significantly higher (p < 0.01) than control (Cont) levels (n = 4). e, Concentration-dependent inhibition of capsaicin (Cap)-evoked pAKT-T induction by LY249002 and wortmannin. *p < 0.01, compared with control; +p < 0.01, compared with capsaicin group (n = 4). f, Inhibition of capsaicin-evoked pERK induction by LY294002 (LY) and wortmannin (Wort). *p < 0.01, compared with control; +p < 0.01, compared with capsaicin group (n = 4). g, Western blot reveals that the capsaicin-induced increase in pAKT-T and pERK levels is blocked by LY294002 (100 μm). Total AKT levels were used as loading controls. h, Capsaicin-induced pAKT-T and pERK is suppressed by EGTA (4 mm). *p < 0.01, compared with control; +p < 0.01, compared with capsaicin group (n = 4). The medium (containing 1.8 mm Ca2+) was incubated with 4 mm EGTA for 30 min in the culture incubator. The control medium, with or without capsaicin (3 μm), and EGTA-containing medium with capsaicin were added to DRG cultures for 2 min.
Activation of TRPV1 by capsaicin induces Ca2+ influx (Caterina et al., 1997). To test whether extracellular Ca2+ is required for the activation of PI3K and ERK, we added the Ca2+ chealater EGTA (4 mm) into the culture medium. EGTA primarily attenuated capsaicin-induced activation of PI3K and ERK (Fig. 1h), indicating that Ca2+ influx is essential for capsaicin activation of PI3K and ERK.
Activation of PI3K by capsaicin in the DRG and skin in vivo
In normal unstimulated DRG, in only 10% of neurons was pAKT-T detected. Five minutes after intradermal injection of capsaicin into the plantar surface of a hindpaw, 23% of neurons (p < 0.01) became pAKT-T immunoreactive (IR) (Fig. 2a-c). Double staining with NF-200, a marker for myelinated A-fiber DRG neurons, showed that pAKT-T was mainly found in NF-200-negative, small-sized C-fiber neurons after capsaicin stimulation (Fig. 2d). pAKT-T was also colocalized with TRPV1 (Fig. 2e). These results indicate that capsaicin can induce AKT activation in TRPV1-expressing C-fiber neurons.
Figure 2.

a-i, Intradermal capsaicin induces pAKT-T and pERK in the DRG and hindpaw skin. a, b, Capsaicin increases pAKT-T levels in the L5 DRG. c, Quantification of the percentage of all pAKT-T-positive neuronal profiles in each section. *p < 0.01, compared with vehicle control (n = 4). d, e, Double immunofluorescence indicates that capsaicin-induced pAKT-T is rarely colocalized with NF-200, a marker for myelinated A-fibers (d), but primarily colocalized with the TRPV1 capsaicin receptor. f-i, More pAKT-T- and pERK-positive nerve fibers are found in the capsaicin-stimulated epidermis of the hindpaw skin (g, i) than in controls (f, h). The same settings of contrast and brightness were used in control and capsaicin-stimulated images. Arrows indicate labeled single nerve fibers in the epidermis. Arrowheads show transected large nerve bundles in the border of the epidermis and dermis. The animals were fixed by perfusion 5 min after the capsaicin injection. Scale bars, 50 μm.
PI3K is activated in axons and growth cones (Kuruvilla et al., 2000; Markus et al., 2002; Shi et al., 2003), and hyperalgesia is mediated by nociceptors on the nerve fibers and terminals of DRG neurons after injection of inflammatory mediators into the hindpaw. The epidermis is mainly innervated by capsaicin-sensitive C-fibers (Simone et al., 1998). We examined whether AKT is activated in the hindpaw skin. pAKT-T-IR nerve fibers were barely detected in nonstimulated hindpaw skin (Fig. 2f), but many labeled fibers were found in the epidermis of the capsaicin-stimulated hindpaw (Fig. 2g). Intradermal capsaicin also induced pERK in epidermal nerve fibers (Fig. 2h,i) and induces pERK in small-sized neurons in the DRG (Dai et al., 2002).
Both PI3K and ERK pathways mediate capsaicin-induced heat hyperalgesia
Intradermal injection of capsaicin induced immediate spontaneous pain behavior, such as lifting and licking the affected paw. After ∼5 min, this was followed by heat hyperalgesia, as demonstrated by a decrease in paw-withdrawal latency (PWL) to radiant heat stimulation. Heat hyperalgesia by capsaicin was significant by 15 min and was maintained for <3 hr (Fig. 3a). To test whether PI3K contributes to heat hyperalgesia, we injected the PI3K inhibitor LY294002 (1 and 10 μg) intradermally 5 min before capsaicin injection. This pretreatment suppressed hyperalgesia at 15, 30, and 60 min, in a dose-dependent manner (Fig. 3a). Intradermal injection of another PI3K potent inhibitor, wortmannin (200 ng), 5 min before capsaicin injection also attenuated heat hyperalgesia at 15, 30, and 60 min (Fig. 3a). A lower dose of wortmannin (100 ng) had no detected effect (data not shown). Intradermal injection of the MEK inhibitor PD98059 (1 and 10 μg) 5 min before capsaicin reduced heat hyperalgesia at 30 and 60 min in a dose-dependent manner (Fig. 3b). Another potent MEK inhibitor, U0126 (1 μg), administrated 5 min before capsaicin also reduced hyperalgesia at 30 and 60 min (Fig. 3b). In contrast to PI3K inhibitors, MEK inhibitors had no effect on capsaicin-induced heat hyperalgesia at the first time point (15 min) (Fig. 3a,b). Because the delayed effect of ERK inhibition may be caused by the delayed action of MEK inhibitors, we also injected PD98059 20 min before capsaicin and still found no effect on heat hyperalgesia at 15 min after capsaicin (data not shown). Together, these pretreatment studies indicate that both PI3K and ERK are required for the generation of capsaicin-induced heat hyperalgesia.
To investigate whether PI3K and ERK pathway can reverse established heat hyperalgesia, LY294002 (10 μg) or PD98059 (10 μg) was intradermally injected 15 min after the capsaicin injection (posttreatment). Both LY294002 and PD98059 reversed the capsaicin induced-heat hyperalgesia (Fig. 3c). This result implies a therapeutic potential for developing PI3K and ERK inhibitors for treating clinical inflammatory pain. Furthermore, this result suggests that these two pathways play a role in both the induction and expression of heat hyperalgesia.
To determine whether PI3K or MEK inhibitor could attenuate hyperalgesia rather than inhibiting basal pain sensitivity, we injected these two inhibitors into the hindpaw of normal animals. Neither LY294002 (10 μg) nor PD98059 (10 μg) affected the paw-withdrawal latency to heat stimulation (Fig. 3d), suggesting that PI3K and ERK pathways do not regulate basal heat sensitivity.
Both PI3K and ERK pathways mediate NGF-induced heat hyperalgesia
PI3K and ERK pathways are two of the major signal cascades mediating NGF-induced cell survival and growth (Atwal et al., 2000, Patapoutian and Reichardt, 2001). Unlike capsaicin, NGF did not phosphorylate AKT at Thr308 (Fig. 4g) but instead phosphorylated AKT at Ser473 (pAKT-S) (Fig. 4a-f, h), as reported previously (Kuruvilla et al., 2000). LY294002 (100 μm) completely blocked NGF-induced increase in both pAKT-S and pERK levels (Fig. 4h-j). Wortmannin (100 nm) also abolished the NGF-induced increase in pAKT-S and pERK levels (data not shown).
Figure 4.
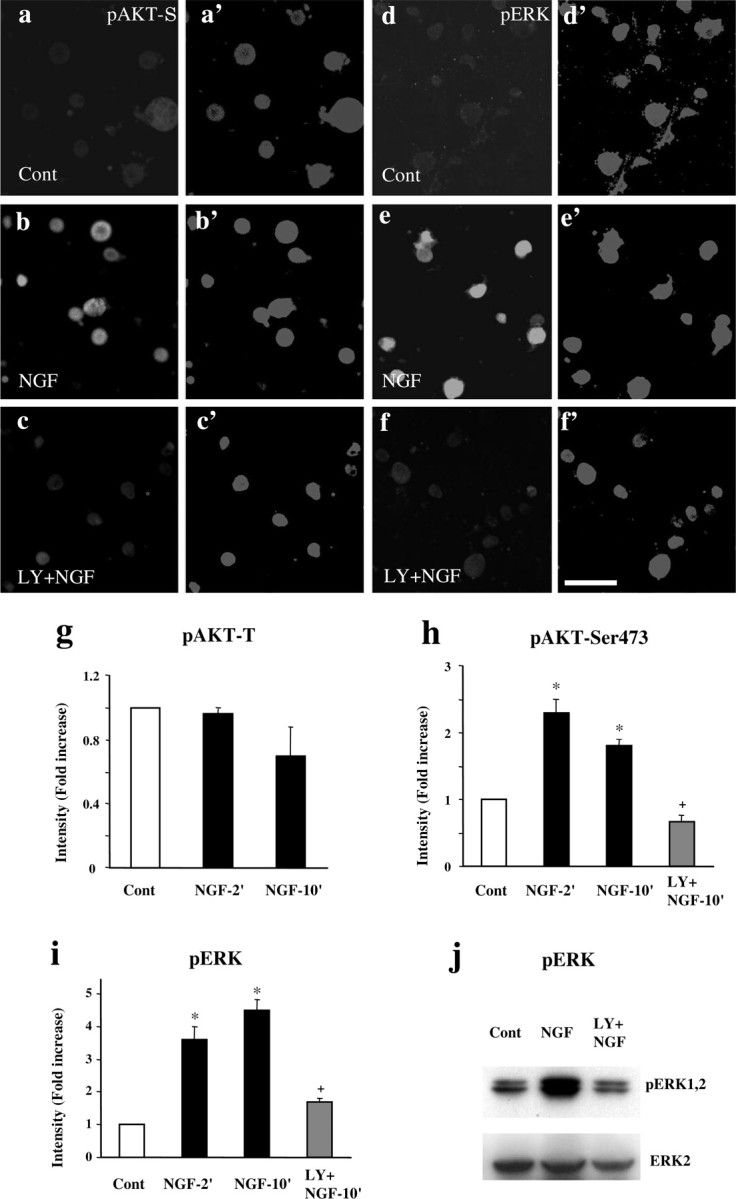
a-j, NGF induces activation of AKT and ERK in cultured DRG neurons. a-f′, Bath application of NGF (100 ng/ml) to DRG cultures results in phosphorylation of AKT at Ser473 (pAKT-S; a, b) and ERK (d, e). The induction of pAKT-S (c) and pERK (f) is abolished by LY294002 (100 μm; LY). a′-f′ are images of corresponding to a-f with high contrast to show all of the cells in each field. Scale bar, 50 μm. The DRG cultures were grown for 24 hr and stimulated with NGF for 10 min. g, NGF does not change AKT phosphorylation at Thr308 (pAKT-T). h, i, Intensity of pAKT-S and pERK immunofluorescence. NGF-evoked pAKT-S (h) and pERK (i) induction is effectively blocked by LY294002 (100 μm). *p < 0.01, compared with control; +p < 0.01, compared with NGF group (n = 4). j, Western blot confirms that NGF-induced increase in pERK levels is blocked by LY294002 (100 μm). Total AKT levels were used as loading controls. Cont, Control.
Intradermal injection of NGF (0.5 μg) induced slow-onset heat hyperalgesia, starting at 15 min, peaking at 1 hr, and returning to baseline after 6 hr (Fig. 5). Intradermal injection of LY294002 (10 μg) 5 min before NGF administration blocked the hyperalgesia at 15 and 30 min (Fig. 5). However, intradermal PD98059 (10 μg) injected 20 min before NGF suppressed the hyperalgesia at 30, 60, and 180 min (Fig. 5). These results suggest that, although PI3K induces hyperalgesia, ERK is involved in the maintenance of the hyperalgesia.
Figure 5.

PI3K and ERK are required for NGF-induced heat hyperalgesia. Intradermal injection of LY294002 (10 μg; LY) or PD98059 (10 μg; PD), 5 or 20 min before NGF injection, respectively, inhibits NGF-induced heat hyperalgesia at the time points indicated. *p < 0.01, compared with control (n = 6). Heat sensitivity was measured by PWL and plotted as a proportion of preinjection baseline. The basal PWL is 16.6 ± 1.0 sec (n = 18; mean ± SEM). Cont, Control.
Both PI3K and ERK pathways mediate NGF-induced potentiation of TRPV1 current
Previous studies in TRPV1 null mice have shown that TRPV1 current is an essential (if not the only) component of capsaicin-induced current in sensory neurons, and, more importantly, TRPV1 is required for inflammatory heat hyperalgesia (Caterina et al., 2000; Davis et al., 2000; Chuang et al., 2001; Bandell et al., 2004). To investigate the molecular mechanisms of PI3K- and ERK-mediated heat hyperalgesia, we measured ICap in acutely dissociated adult DRG cultures using whole-cell patch clamp. We found a capsaicin response in 78% of DRG neurons. Among these capsaicin-positive cells, capsaicin (40 nm) produced an inward current with mean current density of 7.2 ± 1.2 pA/pF (n = 40) [average capacitance per cell, 24.0 ± 0.4 pF (n = 63)]. ICap was outwardly rectifying, often with a negative slope at potentials more negative than -60 mV, and was blocked by 10 μm capsazapine. As reported previously (Shu and Mendell, 1999a, 2001; Bhave et al., 2002), ICap was characterized by both acute desensitization (desensitization during capsaicin application) and tachyphylaxis (a decreased response to subsequent capsaicin application compared with the initial application). We used the ratio of currents induced by the second capsaicin application (identical duration but 10 min apart) to those induced by the initial application, as an index for the degree of tachyphylaxis. In standard bath solution, the ratio was 80 ± 7% (n = 9) when the pipette solution contained 10 mm BAPTA (Fig. 6a,e). Tachyphylaxis was much stronger when the slower buffer, EGTA (10 mm) (Naraghi and Neher, 1997), was in the pipette solution (data not shown), suggesting a dependence on [Ca2+]i. In 75% of neurons (15 of 20), NGF (100 ng/ml for 10 min) not only reversed tachyphylaxis but also induced a striking potentiation (Fig. 6b) (2040 ± 670% of basal response; n = 15) of ICap. Interestingly, the potentiation was still maintained in many cells, even 10 min after NGF was washed out (Fig. 6b). ICap did not return to the basal level even after >30 min washout. The cells (5 of 20) that did not respond to NGF may reflect the fact that TrkA is not expressed in every DRG neuron (Snider and McMahon, 1998).
Figure 6.

a-e, PI3K and ERK are required for NGF-induced potentiation of capsaicin-induced current in DRG neurons. a, Desensitization (tachyphylaxis) of ICap during multiple bath applications of capsaicin (Cap). Standard bath solution and BAPTA-based pipette solutions were used. The cell was held at -60 mV. Interval between capsaicin (40 nm) applications was 10 min. b, Long-lasting NGF-evoked potentiation of ICap. In the absence of NGF, the second ICap (170 pA) was slightly smaller than the first ICap (200 pA). However, after exposure to 100 ng/ml NGF for 10 min, ICap increased by 14-fold to ∼2400 pA. Inset, Enhanced view of NGF-evoked augmentation of ICap. Note the continued potentiation (4th ICap) 10 min after removal of NGF from the bath. c, NGF failed to induce any potentiation of ICap in a neuron pretreated (>10 min) with the PI3K inhibitor LY294002 (20 μm). After washout of LY294002, NGF markedly potentiated ICap. d, Attenuation of NGF-induced potentiation by pretreatment with the MEK inhibitor PD98059 (20 μm). Washout of PD98059 was followed by a large NGF-induced potentiation. e, Summary of NGF effect on ICap under various conditions. NGF evoked strong potentiation in 15 cells; the other five cells exhibited desensitization. Because of the clear distinction between these two categories of responses, data were averaged separately. All cells that were included tested positive for NGF potentiation (see c, d) after washout of inhibitors. *p < 0.05. LY, LY294002; PD, PD98059.
Pretreatment with the PI3K inhibitor LY294002 (20 μm, for >10 min) completely prevented the NGF-induced potentiation of the TRPV1 current (-54 ± 11%, smaller than the basal response; n = 6; p < 0.05) (Fig. 6c,e). After washout of LY294002, NGF was applied again to determine whether these cells were NGF responsive. We found a strong potentiation of the TRPV1 current by NGF (Fig. 6c) in most cells (six of seven), indicating that these six cells are TrkA positive. NGF-induced potentiation was also blocked by pretreatment with another PI3K inhibitor, wortmannin (10 nm;30 ± 30%; n = 6; p < 0.05 compared with NGF alone) (Fig. 6e). Pretreatment with the MEK inhibitor PD98059 (20 μm, >10 min) markedly attenuated the NGF-induced potentiation of TRPV1 current (165 ± 112%; n = 5; p < 0.05 compared with NGF alone) (Fig. 5d,e). A similar result was obtained with another MEK inhibitor, U0126 (5 μm; 87 ± 57%; n = 3) (Fig. 6e).
Discussion
The PI3K pathway has been implicated in multiple biological responses, including membrane trafficking, proliferation, differentiation, and growth in non-neuronal cells (Rameh and Cantley, 1999). In neuronal cells, most of the previous studies on PI3K studied neuronal survival and axon outgrowth (Atwal et al., 2000, Patapoutian and Reichardt, 2001; Markus et al., 2002). Recent studies explored the roles of PI3K in regulating neural plasticity. The PI3K pathway has been shown to mediate LTP, fear memory, and cocaine-induced behavioral sensitization in the CNS (Kelly and Lynch, 2000; Lin et al., 2001, Izzo et al., 2002, Sanna et al., 2002). We now show that PI3K also regulates inflammatory hyperalgesia in the peripheral nervous system, an action that is likely to be mediated at least partially through PI3K-induced ERK activation. We further identified TRPV1 as a possible molecular target for this action.
Interaction between PI3K and ERK pathways
Both PI3K and ERK are major downstream targets of NGF. The prevailing view is that they represent distinct linear pathways and trigger distinct physiological roles (York et al., 2000; Kaplan and Cooper, 2001). Increasing evidence suggests that these two cascades are interconnected (Graness et al., 1998; York et al., 2000; Chandler et al., 2001; Perkinton et al., 2002). Our results support this link in DRG neurons. PI3K inhibitors blocked ERK activation induced by capsaicin and NGF. However, in several cases, PI3K was reported to inhibit, rather than increase, ERK activation (Rommel et al., 1999; Zimmermann and Moelling, 1999). Thus, it has been proposed that the ability of PI3K inhibitors to block ERK activation depends on the cell type, type of stimuli, and the strength of signal (Duckworth and Cantley, 1997; Wennstrom and Downward, 1999).
[Ca2+]i is important for the activation of the PI3K-ERK cascade. Glutamate induces a rapid Ca2+- and PI3K-dependent phosphorylation of AKT (Perkinton et al., 2002). Ca2+-permeable AMPA receptors activate ERK through a PI3K-dependent mechanism (Perkinton et al., 1999). NMDA receptor activation of ERK is also PI3K dependent (Chandler et al., 2001; Perkinton et al., 2002). We have shown that removal of extracelluar Ca2+ suppresses capsaicin activation of PI3K and ERK (Fig. 1h). Calmodulin activates PI3K by direct association with SH2 (Src homology 2) domains of the regulatory subunit (p85) of the PI3K (Joyal et al., 1997). In addition, PI3K may activate atypical PKC (PKCζ) through PDK1 (PI3K-dependent kinase-1) (Parekh et al., 1999), leading to ERK activation via Src family tyrosine kinases (Finkbeiner and Greenberg, 1996; Williamson et al., 2002).
Regulation of TRPV1 activity by PI3K and ERK
Although the requirement of TRPV1 for basal heat response in DRG neurons is still unclear (Caterina et al., 2000; Davis et al., 2000; Woodbury et al., 2004), accumulating evidence indicates that TRPV1 is essential for inflammatory heat hyperalgesia induced by NGF, bradykinin, cinnamaldehyde, ATP, and the protease-activated receptor agonists carrageenan and complete Freund's adjuvant (Caterina et al., 2000, Davis et al., 2000; Chuang et al., 2001; Moriyama et al., 2003; Amadesi et al., 2004; Bandell et al., 2004; Dai et al., 2004). NGF rapidly sensitizes DRG neuron response to capsaicin in acutely dissociated cultures (Shu and Mendell, 1999a, 2001). We used a similar preparation and found a more robust (20-fold) enhancement of TRPV1 current by NGF. However, the mechanisms underlying NGF sensitization are controversial. In neonatal DRG neurons, NGF enhances a capsaicin-induced [Ca2+]i rise in 37% of capsaicin-responsive neurons. This NGF-induced sensitization is abolished by the PI3K inhibitor wortmannin (20 nm) and significantly suppressed by the MEK inhibitor U0126 (10 μm) (Bonnington and McNaughton, 2003). However, a recent study did not see the sensitization effect of NGF in neonatal DRG neurons (Zhu et al., 2004). Using perforated whole-cell recording, Shu and Mendell (2001) failed to block the NGF-enhanced TRPV1 current by PD98059 (5-10 μm). In the present study, we found that the NGF-evoked sensitization was blocked by the PI3K inhibitors (LY294002 at 20 μm and wortmannin at 10 nm) and primarily suppressed by the MEK inhibitors (PD98059 at 20 μm and U0126 at 5 μm). Although differences in cell preparation, pipette solution, concentrations of the drugs, and the sensitizing protocol may all introduce variability, we found that a sufficient preincubation time (>10 min) is critical for an optimal effect of the MEK inhibitor PD98059.
A phospholipase C (PLC)-dependent mechanism has been widely implicated in regulating the activation of many transient receptor potential channels (Runnels et al., 2002; Clapham, 2003; Bandell et al., 2004). In heterologous Xenopus oocyte expressing TRPV1, PLC plays an essential role in regulating TRPV1 activity through phosphatidylinositol-4,5-biphosphate. A C-terminal residue critical for the sensitization of the channel has been identified (Chuang et al., 2001; Prescott and Julius, 2003). Although PLC is implicated for the NGF-mediated sensitizing effect of heat responses in DRG neurons (Galoyan et al., 2003), we still observed a robust sensitization in cells pretreated with the competitive antagonist of PLC, U73122 (10 μm). Another group has also demonstrated that NGF-induced sensitization in DRG neurons persists in the presence of the PI3K inhibitor neomycin (Bonnington and McNaughton, 2003). The discrepancy may be attributable to different cell types (oocytes vs DRG neurons) used or different temporal roles (transient vs sustained, constitutive vs induced) of these kinases.
Overlapping and distinct roles of PI3K and ERK in regulating hyperalgesia
Because PI3K inhibition in DRG neurons blocked capsaicin and NGF-induced ERK activation, it is reasonable to postulate that PI3K acts in an ERK-dependent manner. The following evidence further suggests an overlapping role of these two cascades. First, our results have shown that intradermal injection of PI3K and MEK inhibitors can prevent the heat hyperalgesia induced by intradermal capsaicin and NGF, indicating that both pathways are required for the generation of heat hyperalgesia. Second, PI3K and MEK inhibitors did not affect basal heat sensitivity, indicating that these drugs are antihyperalgesic but not antinociceptive. Third, inhibition of both PI3K and ERK could suppress NGF-induced sensitization of TRPV1 current. Fourth, posttreatment of PI3K and MEK inhibitors could reverse capsaicin-induced hyperalgesia, suggesting that both pathways are required for the expression of capsaicin-induced hyperalgesia. There is controversy regarding the role of PI3K in the induction versus expression of LTP. Although several studies support a role of PI3K in the induction of LTP (Kelly and Lynch, 2000; Lin et al., 2001; Opazo et al., 2003), Sanna et al. (2002) show that PI3K is only required for the expression of LTP in hippocampal CA1 region. Finally, it has been shown that both ERK and PI3K pathways are important for μ-opioid receptor agonist DAMGO (d-Ala2-N-Me-Phe4-Glycol5-enkephalin)-induced desensitization of high-voltage-activated Ca2+ currents(primarily N-type currents) in DRG neurons; combined application of PI3K and MAPK inhibitors was not additive, suggesting that these two kinases act in a common pathway to facilitate chronic desensitization (Tan et al., 2003).
PI3K and ERK also have distinct roles. Opazo et al. (2003) showed that PI3K can regulate the induction of LTP, thorough ERK-dependent and -independent mechanisms. Although both MEK and PI3K inhibitors suppress theta-frequency-induced LTP, only PI3K inhibitors blocked the LTP induced by high-frequency stimulation or low-frequency stimulation paired with postsynaptic depolarization (Opazo et al., 2003). In particular, PKC is likely to mediate some of the effects of PI3K. PI3K can activate the atypical isoform (PKCζ) and novel isoform (PKCϵ) (Parekh et al., 1999). PKC, in particular PKCϵ, is involved in sensitization of TRPV1 and in hyperalgesia (Cesare et al., 1999; Khasar et al., 1999; Premkumar and Ahern, 2000). Our results show that PI3K but not ERK inhibition can reduce the early event (onset, at 15 min) of the hyperalgesia by both capsaicin and NGF. Compared with capsaicin, NGF produced a slow but more sustained heat hyperalgesia (Fig. 6) (Rueff et al., 1996; Shu and Mendell, 1999b; Farquhar-Smith and Rice, 2003). In this model, whereas PI3K was involved in the early event (onset) of the hyperalgesia, ERK was involved in the late event (maintenance) of the hyperalgesia. ERK activation is strongly implicated in late phase of LTP and long-term memory through protein synthesis (Impey et al., 1998; Huang et al., 2000), as well as the induction of LTP (English and Sweatt, 1997; Winder et al., 1999; Watabe et al., 2000).
Concluding remarks
We have shown that the PI3K pathway affects inflammatory hyperalgesia in the peripheral nervous system and have further investigated its association with the ERK pathway. In DRG neurons, the inflammatory agents capsaicin and NGF activate the PI3K and ERK pathways, and both pathways mediate heat hyperalgesia produced by these agents. Following capsaicin and NGF, PI3K activates ERK. Therefore, PI3K could play a similar role as ERK in mediating heat hyperalgesia.
PI3K appears to be a common signaling cascade mediating the early induction of hyperalgesia. We explored the molecular mechanisms underlying PI3K and ERK-mediated heat hyperalgesia. Both kinases can sensitize TRPV1, possibly through post-translational regulation. They may also indirectly regulate the activity of this channel by inducing translation or trafficking-membrane insertion of the channel. In addition to TRPV1, other key channels implicated in sensory neuron sensitization (e.g., NaV1.8) are likely to be regulated by these pathways. Thus, the PI3K pathway is a potential new pharmaceutical target for the management of inflammatory pain in the peripheral nervous system.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS40698 (R.-R.J.).
Correspondence should be addressed to Ru-Rong Ji, Department of Anesthesiology, Brigham and Women's Hospital, 75 Francis Street, Medical Research Building, Room 604, Boston, MA 02115. E-mail: rrji@zeus.bwh.harvard.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248300-10$15.00/0
References
- Aley KO, Levine JD (1999) Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci 19: 2181-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO (2001) Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci 21: 6933-6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadesi S, Nie J, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis JB, Mayer EA, Bunnett NW (2004) Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci 24: 4300-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal JK, Massie B, Miller FD, Kaplan DR (2000) The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 27: 265-277. [DOI] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ, Patapoutian A (2004) Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41: 849-857. [DOI] [PubMed] [Google Scholar]
- Bhave G, Gereau RW (2003) Growing pains: the cytoskeleton as a critical regulator of pain plasticity. Neuron 39: 577-579. [DOI] [PubMed] [Google Scholar]
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW (2002) cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron 35: 721-731. [DOI] [PubMed] [Google Scholar]
- Bonnington JK, McNaughton PA (2003) Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol (Lond) 551: 433-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron R, Klesse LJ, Shah K, Parada LF, Winter J (2003) Activation of Ras is necessary and sufficient for upregulation of vanilloid receptor type 1 in sensory neurons by neurotrophic factors. Mol Cell Neurosci 22: 118-132. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816-824. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306-313. [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA (1999) Sepcific involvement of PKC-ϵ in sensitization of the neuronal response to painful heat. Neuron 23: 617-624. [DOI] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68: 965-1014. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D (2001) N-methyl-d-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem 276: 2627-2636. [DOI] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957-962. [DOI] [PubMed] [Google Scholar]
- Clapham DE (2003) TRP channels as cellular sensors. Nature 426: 517-524. [DOI] [PubMed] [Google Scholar]
- Dai Y, Iwata K, Fukuoka T, Kondo E, Tokunaga A, Yamanaka H, Tachibana T, Liu Y, Noguchi K (2002) Phosphorylation of extracellular signal-regulated kinase in primary afferent neurons by noxious stimuli and its involvement in peripheral sensitization. J Neurosci 22: 7737-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K (2004) Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci 24: 4293-4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in there akts. Genes Dev 13: 2905-2927. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405: 183-187. [DOI] [PubMed] [Google Scholar]
- Dina OA, McCarter GC, de Coupade C, Levine JD (2003) Role of the sensory neuron cytoskeleton in second messenger signaling for inflammatory pain. Neuron 39: 613-624. [DOI] [PubMed] [Google Scholar]
- Duckworth BC, Cantley LC (1997) Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin. Dependence on signal strength. J Biol Chem 272: 27665-27670. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem 272: 19103-19106. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Rice AS (2003) A novel neuroimmune mechanism in cannabinoid-mediated attenuation of nerve growth factor-induced hyperalgesia. Anesthesiology 99: 1391-1401. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME (1996) Ca2+-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron 16: 233-236. [DOI] [PubMed] [Google Scholar]
- Fitzgerald M, Gibson S (1984) The postnatal physiological and neurochemical development of peripheral sensory C fibres. Neuroscience 13: 933-944. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88: 435-437. [DOI] [PubMed] [Google Scholar]
- Galoyan SM, Petruska JC, Mendell LM (2003) Mechanisms of sensitization of the response of single dorsal root ganglion cells from adult rat to noxious heat. Eur J Neurosci 18: 535-541. [DOI] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM (1998) Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro J Neurosci 18: 10345-10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graness A, Adomeit A, Heinze R, Wetzker R, Liebmann C (1998) A novel mitogenic signaling pathway of bradykinin in the human colon carcinoma cell line SW-480 involves sequential activation of a Gq/11 protein, phosphatidylinositol 3-kinase beta, and protein kinase Cepsilon. J Biol Chem 273: 32016-32022. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77-88. [DOI] [PubMed] [Google Scholar]
- Huang YY, Martin KC, Kandel ER (2000) Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 20: 6317-6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR (1998) Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron 21: 869-883. [DOI] [PubMed] [Google Scholar]
- Izzo E, Martin-Fardon R, Koob GF, Weiss F, Sanna PP (2002) Neural plasticity and addiction: PI3-kinase and cocaine behavioral sensitization. Nat Neurosci 5: 1263-1264. [DOI] [PubMed] [Google Scholar]
- Ji RR (2003) Activation of MAP kinase in primary sensory neurons. Proceedings of the 10th World Congress on Pain 24: 81-87. [Google Scholar]
- Ji RR, Zhang Q, Pettersson RF, Hokfelt T (1996) aFGF, bFGF and NGF differentially regulate neuropeptide expression in dorsal root ganglia after axotomy and induce autotomy. Regul Pept 66: 179-189. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ (2002a) ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 up-regulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci 22: 478-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002b) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36: 57-68. [DOI] [PubMed] [Google Scholar]
- Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White MF, Sacks DB (1997) Calmodulin activates phosphatidylinositol 3-kinase. J Biol Chem 272: 28183-28186. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413: 203-210. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Cooper E (2001) PI-3 kinase and IP3: partners in NT-3-induced synaptic transmission. Nat Neurosci 4: 5-7. [DOI] [PubMed] [Google Scholar]
- Kelly A, Lynch MA (2000) Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology 39: 643-651. [DOI] [PubMed] [Google Scholar]
- Kerr BJ, Souslova V, McMahon SB, Wood JN (2001) A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. NeuroReport 12: 3077-3080. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO (1999) A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron 24: 253-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuruvilla R, Ye H, Ginty DD (2000) Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron 27: 499-512. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW (2001) A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron 31: 841-851. [DOI] [PubMed] [Google Scholar]
- Markus A, Zhong J, Snider WD (2002) Raf and akt mediate distinct aspects of sensory axon growth. Neuron 35: 65-76. [DOI] [PubMed] [Google Scholar]
- McCleskey EW, Gold MS (1999) Ion channels of nociception. Annu Rev Physiol 61: 835-856. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, Leon C, Suzuki N, Inoue K, Gachet C, Noguchi K, Tominaga M (2003) Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci 23: 6058-6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci 17: 6961-6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ (2003) Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci 23: 3679-3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ (1999) Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem 274: 34758-34764. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280. [DOI] [PubMed] [Google Scholar]
- Perkinton MS, Sihra TS, Williams RJ (1999) Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase-dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J Neurosci 19: 5861-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkinton MS, Ip JK, Wood GL, Crossthwaite AJ, Williams RJ (2002) Phosphatidylinositol 3-kinase is a central mediator of NMDA receptor signalling to MAP kinase (Erk1/2), Akt/PKB and CREB in striatal neurones. J Neurochem 80: 239-254. [DOI] [PubMed] [Google Scholar]
- Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC (1999) A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA 96: 7640-7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408: 985-990. [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300: 1284-1288. [DOI] [PubMed] [Google Scholar]
- Rameh LE, Cantley LC (1999) The role of phosphoinositide 3-kinase lipid products in cell function. J Biol Chem 274: 8347-8350. [DOI] [PubMed] [Google Scholar]
- Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286: 1738-1741. [DOI] [PubMed] [Google Scholar]
- Rueff A, Dawson AJ, Mendell LM (1996) Characteristics of nerve growth factor induced hyperalgesia in adult rats: dependence on enhanced bradykinin-1 receptor activity but not neurokinin-1 receptor activation. Pain 66: 359-372. [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE (2002) The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol 5: 329-336. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W (2002) Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci 22: 3359-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN (2003) Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell 112: 63-75. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM (1999a) Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett 274: 159-162. [DOI] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM (1999b) Neurotrophins and hyperalgesia. Proc Natl Acad Sci USA 96: 7693-7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu XQ, Mendell LM (2001) Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol 86: 2931-2938. [DOI] [PubMed] [Google Scholar]
- Simone DA, Nolano M, Johnson T, Wendelschafer-Crabb G, Kennedy WR (1998) Intradermal injection of capsaicin in humans produces degeneration and subsequent reinnervation of epidermal nerve fibers: correlation with sensory function. J Neurosci 18: 8947-8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, McMahon SB (1998) Tackling pain at the source: new ideas about nociceptors. Neuron 20: 629-632. [DOI] [PubMed] [Google Scholar]
- Tan M, Groszer M, Tan AM, Pandya A, Liu X, Xie CW (2003) Phosphoinositide 3-kinase cascade facilitates μ-opioid desensitization in sensory neurons by altering G-protein-effector interactions. J Neurosci 23: 10292-12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toker A, Cantley LC (1997) Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature 387: 673-676. [DOI] [PubMed] [Google Scholar]
- Vlahos CJ, Matter WF, Hui KY, Brown RF, Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269: 5241-5248. [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O'Dell TJ (2000) Coactivation of beta-adrenergic and cholinergic receptors enhances the induction of long-term potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci 20: 5924-5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wennstrom S, Downward J (1999) Role of phosphoinositide 3-kinse in activation of Ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol 19: 4279-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH (2002) Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: involvement of Src family protein kinases. J Neurosci 22: 10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER (1999) ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24: 715-726. [DOI] [PubMed] [Google Scholar]
- Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM (2004) Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci 24: 6410-6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW (2000) Neuronal plasticity: increasing the gain in pain. Science 288: 1765-1769. [DOI] [PubMed] [Google Scholar]
- Yang F, He X, Feng L, Mizuno K, Liu XW, Russell J, Xiong WC, Lu B (2001) PI-3 kinase and IP3 are both necessary and sufficient to mediate NT3-induced synaptic potentiation. Nat Neurosci 4: 19-28. [DOI] [PubMed] [Google Scholar]
- York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJ (2000) Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol 20: 8069-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Galoyan SM, Petruska JC, Oxford GS, Mendell LM (2004) A developmental switch in acute sensitization of small dorsal root ganglion (DRG) neurons to capsaicin or noxious heating by NGF. J Neurophysiol Jun 16 [Epub ahead of print]. [DOI] [PubMed]
- Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286: 1741-1744. [DOI] [PubMed] [Google Scholar]