

Abstract
Neurotrophins, such as NGF and BDNF, activate Trk receptor tyrosine kinases through receptor dimerization at the cell surface followed by autophosphorylation and intracellular signaling. It has been shown that activation of Trk receptor tyrosine kinases can also occur via a G-protein-coupled receptor (GPCR) mechanism, without involvement of neurotrophins. Two GPCR ligands, adenosine and pituitary adenylate cyclase-activating polypeptide (PACAP), can activate Trk receptor activity to increase the survival of neural cells through stimulation of Akt activity. To investigate the mechanism of Trk receptor transactivation, we have examined the localization of Trk receptors in PC12 cells and primary neurons after treatment with adenosine agonists and PACAP. In contrast to neurotrophin treatment, Trk receptors were sensitive to transcriptional and translational inhibitors, and they were found predominantly in intracellular locations particularly associated with Golgi membranes. Biotinylation and immunostaining experiments confirm that most of the transactivated Trk receptors are found in intracellular membranes. These results indicate that there are alternative modes of activating Trk receptor tyrosine kinases in the absence of neurotrophin binding at the cell surface and that receptor signaling may occur and persist inside of neuronal cells.
Keywords: NGF, tyrosine phosphorylation, basal forebrain, adenosine, PACAP, Golgi apparatus
Introduction
Neurotrophins are essential regulators of neuronal development, growth, and differentiation in the vertebrate nervous system (Huang and Reichardt, 2001). The actions of neurotrophins are dictated by two classes of cell surface receptors, the Trk receptor tyrosine kinase and the p75 neurotrophin receptor, a member of the tumor necrosis factor (TNF) receptor superfamily (Hempstead, 2002; Chao, 2003a; Huang and Reichardt, 2003). Although NGF binds exclusively to TrkA, BDNF and neurotrophin-4 (NT-4) bind to TrkB, and NT-3 binds to TrkC, all neurotrophins bind to p75. After binding, neurotrophins and each of their receptors undergo internalization and transport from axon terminals to neuronal cell bodies (Hendry et al., 1974; DiStefano et al., 1992; Bronfman et al., 2003), in which a number of transcriptional and enzymatic activities are activated. Engagement of Trk receptors results in increases in cAMP response element-binding protein (CREB) and extracellular signal-related kinase 5 (Erk5) activities (Riccio et al., 1997; Watson et al., 1999b, 2001), as well as phosphoinositide lipid phosphorylation and activation of GTPases, such as Ras and Rap1 (York et al., 1998).
Another mode of receptor activation is through transactivation of receptor tyrosine kinases in response to G-protein-coupled receptor signaling (Daub et al., 1996; Luttrell et al., 1999; Fischer et al., 2003). For example, receptors for epidermal growth factor (EGF), insulin-like growth factor-1, and platelet-derived growth factor can be transactivated through G-protein-coupled receptors to give proliferative and mitogen-activated protein kinase (MAPK) responses. Also, signaling through dopamine receptors leads to PDGF receptor activation that results in changes in synaptic transmission (Kotecha et al., 2002). Hence, transactivation can lead to effects on cell proliferation, differentiation, and synaptic plasticity; however, the cellular mechanisms that account for these transactivation effects are not well understood.
We reported previously that TrkA and TrkB receptors can be activated by ligands of the G-protein-coupled receptor (GPCR) family of transmembrane receptors in the absence of NGF or BDNF (Lee and Chao, 2001; Lee et al., 2002b). The ligands include the nucleoside adenosine or adenosine agonists, such as CGS 21680, and the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP). Adenosine and CGS 21680 recognize the A2A receptor (Jarvis et al., 1989), whereas PACAP binds to the PAC1 receptor (Ishihara et al., 1992). Not only do adenosine and PACAP activate Trk receptors, but effectors of the Trk tyrosine kinase are also phosphorylated in response to these ligands. Adenosine treatment of PC12 cells promotes the phosphorylation of Shc adaptor proteins, as well as phospholipase Cγ, analogous to the induction observed with NGF. Activation of phosphoinositide (PI)-3 kinase and Akt accounted for neuroprotective effects afforded by transactivation by adenosine and PACAP. The effects of adenosine and PACAP can be specifically blocked by K252a, an inhibitor of Trk tyrosine kinases.
In the present study, we report that Trk activation by adenosine and its agonist CGS 21680 or PACAP occurs exclusively in an intracellular location and partly involves the Golgi apparatus. Biotinylation assays indicate that CGS 21680 activates an intracellular pool of Trk receptors. Indirect immunofluorescence experiments confirm that only intracellular Trk receptors are activated by CGS 21680. Furthermore, colocalization of activated Trk receptors and markers of the Golgi apparatus, such as TGN-38 and GM130, suggest that Trk receptors can reside and be activated in Golgi membranes. Taken together with previous observations, these results suggest that intracellular membranes may potentially serve as a platform for Trk receptors to mediate cell survival through the PI3K-Akt pathway.
Materials and Methods
Reagents. Adenosine and CGS 21680 were obtained from Sigma (St. Louis, MO). PACAP 38 was from Peninsula Laboratories (San Carlos, CA). K252a and brefeldin A were purchased from Calbiochem (La Jolla, CA). Cycloheximide and actinomycin D were purchased from Sigma. Sulfo-NHS-LC-biotin, sulfo-NHS-acetate, and streptavidin-agarose were purchased from Pierce Biotechnology (Rockford, IL). NGF was obtained from Harlan Bioproducts (Indianapolis, IN), and BDNF was from PeproTech (Rocky Hill, NJ). Anti-pan-Trk rabbit antiserum raised against the C-terminal region of the Trk receptor was from Barbara Hempstead (#45, Weill Medical College, New York, NY), and C-14 pan-Trk was from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-Trk antibodies against the C terminus of Trk were obtained from Santa Cruz Biotechnology (B-3), and the TrkE7 antibody against the extracellular domain of TrkA was from Zymed (San Francisco, CA). Phosphotyrosine antibodies (pY99), monoclonal Akt antibodies (Akt B-1), and Erk 1/2 antibodies were from Santa Cruz Biotechnology. Antibodies to Y490 and Y674/Y675 of Trk as well as antibodies to phosphorylated Akt (pAkt S473) and phosphorylated ERK1/2 were purchased from Cell Signaling Technology (Beverly, MA). Mouse monoclonal antibodies to Golgi matrix 130 (GM130) and early endosome antigen-1 (EEA-1) were from BD Biosciences (San Diego, CA). Mouse monoclonal antibodies to trans Golgi network-38 (TGN-38) were from Affinity Bioreagents (Golden, CO).
Immunoprecipitation and immunoblotting. PC12-615 cells (Hempstead et al., 1992) were maintained in DMEM containing 5% fetal bovine serum, 10% horse serum supplemented with 2 mm glutamine plus 250 μg/ml G418. For all experiments, cells were serum-starved by washing twice with PBS and placing in serum-free media for 24 hr. In some cases, cells were pretreated with 5 μg/ml brefeldin A, 20 μg/ml cycloheximide, 200 ng/μl actinomycin D, or vehicle. Cell lysates from PC12 cells were prepared by incubating in lysis buffer (1% Nonidet P-40, 150 mm NaCl, 1mm EDTA, 10 mm Tris, pH 8.0, 10% glycerol, 10 mm NaF, 1 mm sodium orthovanadate, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 25 μg/ml phenylmethylsulfonyl fluoride) for 30 min on ice. Clarified lysates were immunoprecipitated by incubating overnight at 4°C with anti-pan-Trk polyclonal antibody (Trk C-14) followed by incubation with protein A-Sepharose beads. Equivalent amounts of protein were analyzed for each condition. The beads were washed three times with lysis buffer, and the immune complexes were boiled in SDS-sample buffer and loaded on SDS-PAGE gels for immunoblot analysis. The immunoreactive protein bands were detected by enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL).
RT-PCR analysis. Total RNA from PC12-615 cells or from submandibular gland dissected from adult Sprague Dawley rats was isolated using TRIZOL (Invitrogen, Carlsbad, CA). Five hundred nanograms of total RNA were subjected to reverse transcription using avian myeloblastosis virus reverse transcriptase (Roche Applied Science, Penzberg, Germany) for 30 min at 50°C. Total RNA not incubated with reverse transcriptase was used as a negative control. cDNA products were incubated with TaqDNA polymerase (Roche Applied Science) and primers for rat NGF or for glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers used for NGF amplification were designed as described previously (Donovan et al., 1995): NGF, 5′-CGCTCATCCACCCACCCAGTCTTC, 3′-CTTGACAAAGGTGTGAGTCGTGGT. These primers yielded a product of 267 base pairs. After an initial 95°C hot start, 35 cycles of 94°C for 30 sec, 60°C for 1 min, and 68°C for 1 min were performed, followed by a final extension of 68°C for 5 min. Products were resolved by electrophoresis using a 2% agarose gel and visualized using ethidium bromide.
Cell surface biotinylation. Serum-starved cells were rinsed twice with PBS containing 1 mm CaCl2 and 0.5 mm MgCl2 (PBS-Ca-Mg) and then incubated with 0.5 mg/ml sulfo-NHS-LC-biotin dissolved in biotinylation buffer (0.01 m TEA, pH 7.4, 2 mm CaCl2, 150 mm NaCl) for 30 min at 4°C. Cells were then quenched with PBS-Ca-Mg containing 100 mm glycine for 20 min at 4°C. After warming to 37°C, cells were treated with 10 nm CGS 21680, 100 ng/ml NGF, or vehicle media for the indicated times. Cells were then lysed in RIPA buffer (1% NP-40, 0.1% SDS, 0.1% deoxycholate, 150 mm NaCl, 1 mm EDTA, 10 mm Tris, pH 8.0, 10 mm NaF, 1 mm sodium orthovanadate, 2 μg/ml aprotinin, 1 μg/ml leupeptin, and 25 μg/ml phenylmethylsulfonyl fluoride) for 30 min on ice. Clarified lysates were then split into two equal portions, one that was immunoprecipitated with streptavidin-agarose and the other with Trk C-14.
Receptor insertion assay. Serum-starved PC12 cells were rinsed twice with PBS-Ca-Mg and incubated with 1.5 mg/ml sulfo-NHS-acetate in blocking buffer (0.1 m sodium phosphate buffer, pH 7.4, 150 mm NaCl) for 30 min at 4°C. The blocking reaction was quenched for 20 min at 4°C using 100 mm glycine dissolved in PBS-Ca-Mg. Cells were subsequently allowed to warm to 37°C and then treated with CGS 21680 or NGF as indicated. After treatment, surface biotinylation and lysis were performed as outlined above. Cell lysates were subjected to immunoprecipitation with streptavidin-agarose. Material obtained from this immunoprecipitation was labeled “cell surface pool.” Supernatants from the initial immunoprecipitation were subjected to a second immunoprecipitation with streptavidin-agarose to clear the lysate of biotinylated material. The second supernatant was confirmed to be free of biotinylated proteins by blotting with streptavidin-HRP. TrkA was then immunoprecipitated from the biotin-cleared lysate using Trk C-14 antibodies and labeled “internal pool.” Immunoprecipitates were analyzed by blotting with phosphotyrosine antibodies, phospho-Trk antibodies, or pan-Trk antibodies.
Generation and affinity purification of antibodies against phosphorylated TrkA. A peptide containing phosphorylated tyrosine 794 of rat TrkA, previously shown to be a site for phospholipase C-γ (PLC-γ) recruitment in response to NGF (Loeb et al., 1994), was used to immunize rabbits (LQALAQAPPSpYLDVC). Crude serum was first passed through an affinity column made by coupling the nonphosphorylated version of the peptide to cyanogen bromide-activated Sepharose beads. Flow through from this column was then passed though a column made by coupling the phospho-peptide to cyanogen bromide-activated Sepharose. The column was washed extensively with Tris-buffered saline containing 0.1% Tween 20, and antibodies were then eluted with 0.1 m glycine, pH 2.5. Fractions were collected from the eluate, neutralized with Tris buffer, pH 9.5, and then tested for specificity by blotting NGF-treated PC12 cell lysates. Specificity was further confirmed by a peptide competition assay in which a fivefold weight excess of either phosphorylated or nonphosphorylated TrkA peptide was preincubated with antibody for 2 hr at 4°C and then used for Western blotting.
Basal forebrain cell cultures. Dissociated primary cultures of basal forebrain neurons from embryonic day 18 (E18) rats were prepared from timed-pregnant Sprague Dawley rats as described previously (Aibel et al., 1998). Fetuses were removed under sterile conditions and kept in PBS on ice for microscopic dissection of the basal forebrain. The meninges were removed, and the tissue was placed in HBSS supplemented with 0.37% glucose. The tissue was minced briefly with fine forceps and then tritutated with a fire-polished Pasteur pipette. Cells were counted and plated on culture wells coated with 0.01 mg/ml poly-l-lysine overnight in Neurobasal media containing B27 supplement and l-glutamine (0.5 mm). Experiments were conducted 10 d after plating.
Immunofluorescence and confocal microscopy. Cells were cultured on Nunc Permanox eight-well chamber slides coated with poly-l-lysine. Twenty-four hours after plating, cells were serum-starved by washing twice with PBS and placing in serum-free media for 24 hr. Cells were fixed directly after treatment in 4% paraformaldehyde for 10 min at room temperature. In some cases, cells were pretreated with 5 μg/ml brefeldin A. After two rinses in PBS, cells were permeabilized with 0.1% Triton X-100 for 10 min on ice. Cells were blocked in 5% BSA/PBS for 30 min at room temperature before incubation for 3 hr at room temperature with primary antibody diluted in blocking buffer. Cells were washed three times with PBS and then incubated with secondary antibody (FITC-conjugated anti-rabbit; Cy3-conjugated anti-mouse; Jackson ImmunoResearch, West Grove, PA; 1:100) diluted in blocking buffer for 1 hr at room temperature. Cells were washed three times in PBS and then mounted in Vectashield mounting medium containing 4′,6′-diamidino-2-phenylindole (DAPI). Standard fluorescence microscopy was performed using a Nikon Eclipse E800 microscope equipped with a Nikon Plan Apo 60×, 1.4 numerical aperture (NA) oil immersion objective. Images were acquired with a Zeiss AxioCam HRc digital camera. Confocal images were acquired using a Bio-Rad Radiance 2000 confocal unit coupled to an Olympus BX5OWI microscope equipped with an Olympus PlanApo 60×, 1.4 NA oil immersion objective.
Results
GPCR ligands activate TrkA isoforms
Treatment of hippocampal neurons or PC12 cells with adenosine resulted in an activation of Trk activity and increased cell survival in the absence of BDNF or NGF (Lee and Chao, 2001). Transactivation of Trk neurotrophin receptors leads to selective induction of the PI3-kinase-Akt pathway that accounts for the survival response. The increase in Trk activity was inhibited by protein kinase inhibitors, such as PP1, specific for the Src family members. The Trk-dependent activation of Akt could be blocked with LY294002, a PI3-kinase inhibitor. PACAP also transactivated Trk receptors in a manner very similar to adenosine (Lee et al., 2002b), although many other GPCR ligands failed to transactivate Trk receptors.
To determine which residues of the Trk receptor are phosphorylated after GPCR stimulation, we performed phosphorylation analysis of Trk in PC12-615 cells (Hempstead et al., 1992). These cells express elevated levels of TrkA and have been widely used to study signal transduction. TrkA was immunoprecipitated from cells treated with 10 μm adenosine and analyzed with antibodies to specific Trk phosphorylation sites, including the activation loop of the kinase domain phosphotyrosines (amino acids 683 and 684 of rat TrkA) and the Shc-binding phosphotyrosine site (amino acid 490). Indeed, all of these tyrosine residues were phosphorylated in response to adenosine treatment (Fig. 1A). The increase in specific phosphorylation sites is consistent with the finding that Trk enzymatic activity is enhanced after adenosine or PACAP treatment (Lee et al., 2002b). Furthermore, the phosphorylation events occurred in a similar time interval for all three residues, which involved at least 60 min to observe the activation of TrkA.
Figure 1.

Adenosine activates TrkA receptors at multiple phosphotyrosine residues. A, PC12 cells were treated with 10 μm adenosine for the indicated times. Trk was immunoprecipitated from cell lysates using a pan-Trk antibody. Phosphorylation of Trk was assessed with antibodies against phosphotyrosine 499 of rat TrkA [pTrk 490 (Cell Signaling)] or antibodies against phosphotyrosines 683 and 684 of rat TrkA [pTrk 674/675 (Cell Signaling)]. Immunoblot with pan-Trk antibody (B3) was used to ensure equal loading. B, PC12 cells were pretreated with either 20 μg/ml cycloheximide or 200 ng/μl actinomycin D for 30 min before incubation with 10 nm CGS 21680 for 1 or 3 hr. Trk was immunoprecipitated from cell lysates and analyzed with phosphotyrosine antibodies. Trk protein levels were determined by blotting with pan-Trk antisera. C, Quantitation of phospho-Trk and total Trk levels from lysates of CGS 21680-treated PC12-615 cells. Three independent experiments were quantified using ImageJ analysis software. Levels of band intensity relative to untreated control are represented on the y-axis. Error bars reflect SEM. D, RT-PCR for NGF mRNA was performed on total RNA extracted from untreated PC12 cells or cells treated with 10 nm CGS 21680 for 3 hr. RNA extracted from adult rat submandibular gland was used as a positive control; GAPDH was used as an internal loading control.
Phosphorylation of the activation-loop and Shc-binding residues was observed in both the 140 and 110 kDa isoforms of Trk. The phosphotyrosine analysis clearly indicated that the immature 110 kDa form of Trk was activated first, followed by the mature 140 kDa form (Fig. 1A). The Trk receptor exists in a number of different glycosylation states within the cell, but only the fully glycosylated 140 kDa receptor is thought to be targeted to the plasma membrane (Watson et al., 1999a; Jullien et al., 2002). At the cell surface, NGF engages only the fully mature receptor found to initiate signaling. The appearance of immature, underglycosylated forms of TrkA early in the time course of transactivation suggests that intracellular processing events may play an important role in the mechanism and that a distinct pool of Trk receptors is involved in this process.
To assess whether the activation and processing of Trk receptors depended on transcriptional or posttranscriptional events, we performed analyses using inhibitors of transcription and translation. PC12 cells were pretreated with either actinomycin D (0.2 μg/μl) or cycloheximide (20 μg/ml) for 30 min and then treated with CGS 21680 (10 nm). CGS 21680 was used instead of adenosine because it is a specific and stable agonist that interacts with the A2A receptor (Jarvis et al., 1989). Previous experiments indicated that CGS 21680 elicited identical responses as adenosine (Lee et al., 2001).
Phosphorylation of Trk was assessed by immunoprecipitation from lysates using a pan-Trk antibody followed by blotting with a phosphotyrosine antibody, pY99. After 1 and 3 hr of CGS 21620 treatment, we consistently observed phosphorylation of the 110 kDa TrkA species. Pretreatment with actinomycin D, a potent inhibitor of cellular transcription, or cycloheximide, a translation inhibitor, prevented the transactivation of Trk by the adenosine agonist CGS 21680 (Fig. 1B). Hence, GPCR-mediated transactivation of Trk receptors requires transcriptional and protein synthesis events that may influence the processing and activation of the receptor. An increase in Trk receptor levels was observed after 3 hr of treatment with CGS 21680; however, it should be noted that activation of TrkA occurred before this increase. Quantitation of multiple experiments clearly indicates that TrkA activity was induced after 1 hr of CGS 21680 treatment (Fig. 1C), a time at which TrkA receptors were not significantly upregulated.
To determine whether the gene up-regulation requirement for transactivation directly involved production of NGF, we performed RT-PCR analysis on PC12 cells treated with CGS 21680. As shown in Figure 1D, NGF mRNA was not detected in either untreated or CGS-treated PC12 cells. In contrast, NGF mRNA was readily detected in adult rat submandibular gland. Previous experiments also failed to detect any NGF biological activity in cells treated with GPCR ligands (Lee et al., 2002). Therefore, it is unlikely that transactivation of TrkA receptors involves NGF.
GPCR ligands activate an internal pool of Trk receptors
The finding that immature forms of Trk were activated preferentially early during the time course of transactivation (Fig. 1A) suggested than an intracellular pool Trk receptors is targeted. To localize the site of Trk receptor transactivation, we performed a cell surface biotinylation procedure, as outlined in Figure 2A. PC12 cells were subjected to surface labeling with sulfo-NHS-biotin and then treated with 10 nm CGS 21680 or 100 ng/ml NGF.
Figure 2.

Transactivated Trk receptors are not detected at the cell surface. A, A schematic diagram of the biotinylation procedure is shown. PC12 cells were biotinylated as described in Materials and Methods and then treated with 10 nm CGS 21680 for the indicated times or with 100 ng/ml NGF for 10 min. Cell lysates were prepared and split into two equal portions. Half of the lysate was subjected to immunoprecipitation with streptavidin-agarose, and the other half was immunoprecipitated with pan-Trk antibodies. B, Immunoprecipitates were analyzed by blotting with phosphotyrosine antibodies and with pan-Trk antibodies.
After 10 min of NGF treatment, a considerable amount of activated TrkA receptors could be found in the biotinylated cell surface pool of proteins (Fig. 2B); however, at no point after CGS 21680 treatment was phosphorylated TrkA detected in the cell surface pool, although transactivated Trk appeared in the total Trk pool after 2 hr of CGS 21680 treatment (Fig. 2B). This delayed time course was consistent with previous observations (Lee and Chao, 2001). The lack of receptors in the biotinylated fraction suggests that most of the transactivated receptors did not arise from the cell surface pool.
Although these biotinylation results revealed that preexisting surface Trk receptors were not involved in the transactivation mechanism, the possibility still remained that newly synthesized Trk receptors induced by GPCR activity could be inserted into the plasma membrane and become activated. To assess whether the newly synthesized Trk receptors are correctly processed and inserted into the cell membrane after CGS 21680 treatment, we performed a membrane receptor insertion assay using a modified cell surface biotinylation procedure (Peng et al., 2001). Cells were first blocked from biotinylation using sulfo-NHS-acetate and then treated with 10 nm CGS 21680 for the indicated times (Fig. 3A). After treatment, cells were biotinylated with sulfo-NHS-LC-biotin and then subjected to immunoprecipitation. Nonbiotinylated Trk was isolated by immunoprecipitation with pan-Trk antibodies from the biotin-depleted lysate.
Figure 3.

TrkA is transactivated on internal membranes. A, A receptor insertion assay was performed as described in Materials and Methods and is depicted in the schematic diagram. PC12 cells were treated with the biotinylation-blocking reagent S-NHS-acetate and then treated with either 10 nm CGS 21680 or 100 ng/ml NGF for 3 hr. Cell lysates were immunoprecipitated with avidin-agarose. Supernatants from the avidin-agarose immunoprecipitation that were depleted of biotinylated material were subsequently subjected to immunoprecipitation with pan-Trk antibodies. B, Immunoprecipitates were analyzed by blotting with phosphotyrosine antibodies, antibodies to phosphorylated Trk, or pan-Trk antibodies. Untreated cells that were biotinylated immediately after block (lane 1) or 3 hr after block (lane 2) were used as controls for basal levels.
This procedure allowed us to measure the newly inserted membrane proteins during a treatment period after the blocking step. The level of receptor insertion was then determined by subsequent SDS-PAGE and immunoblotting with pan-Trk antibodies. The basal level of receptor insertion was determined by comparing cells that were blocked and then immediately biotinylated (Fig. 3B, lane 1) with cells that were blocked and left in serum-free media for 3 hr before biotinylation (lane 2). All conditions of treatment were compared with cells that were left untreated for 3 hr (lane 2).
Treatment with NGF gave rise to a detectable increase in activated Trk receptors at the cell surface (Fig. 3B, lane 7). An increase in the amount of biotinylated Trk was observed with increasing periods of incubation with CGS 21680 (top panel). Insertion of receptors can be seen as early as after 1 hr of treatment (lane 4), and maximal insertion is detected at 3 hr of treatment (lane 6). Notably, NGF did not induce significant Trk receptor insertion after 3 hr of treatment (Fig. 3B, compare lane 7 and lane 2).
To determine whether newly inserted Trk receptors induced by CGS 21680 treatment are activated, we reblotted the top panel of Figure 3B with a phosphotyrosine antibody (pY). Surprisingly, none of the Trk receptors at the cell surface were tyrosine phosphorylated after treatment with CGS 21680. After NGF treatment, however, surface receptors were robustly activated. These results were confirmed by blotting with an antibody specific to phosphotyrosine 490 site of Trk (pTrk).
These results indicate that the activation of Trk receptors by CGS 21680 is restricted to an internal pool of receptors that do not reach the cell membrane. To confirm that Trk receptors transactivated by CGS 21680 are found inside the cell, we analyzed the nonbiotinylated lysate by phosphotyrosine blotting. Indeed, activation of TrkA by CGS 21680 was detected in the internal pool of receptors after 2 hr (Fig. 3B, bottom panel), which is consistent with previous measurements with adenosine and PACAP (Lee et al., 2001, 2002b).
Immunostaining with phospho-specific TrkA antibody
Previous experiments in PC12 cells and sympathetic neurons in compartmentalized cultures have provided evidence that Trk receptor signaling occurs in intracellular locations (Grimes et al., 1997; Tsui-Pierchala and Ginty, 1999; Jullien et al., 2002; Ye et al., 2003). To address the intracellular localization of transactivated Trk receptors, we subjected PC12 cells to indirect immunofluorescence to determine the intracellular location of Trk activation by GPCR ligands.
To follow activated Trk receptors, we generated a new phospho-specific TrkA antibody for this purpose. The antibody was made against a phosphopeptide spanning the PLC-γ site at Y785. Western blot analysis of the affinity-purified antibody showed that it specifically recognized the activated TrkA receptor and not the unstimulated receptor in PC12 cells (Fig. 4). Specificity for the phosphorylated form of TrkA was confirmed using peptide competition assays. As shown in Figure 4, the phosphopeptide, and not the unphosphorylated peptide, effectively blocked binding of the pTrkA antibody to immunoblots from NGF-treated PC12 cell lysates. This antibody did not recognize TrkB receptors in transfected and hippocampal cells or other tyrosine phosphorylated proteins (data not shown).
Figure 4.

Specificity of phospho-TrkA antibodies. Affinity purification of polyclonal antibodies raised against a short peptide in the C-terminal region of rat TrkA containing tyrosine 794, which is phosphorylated in response to NGF (Loeb et al., 1994), was performed as described in Materials and Methods. To assess specificity of purified antibodies, lysates (40 μg each well) obtained from PC12 cells treated with 50 ng/ml NGF for 10 min were probed with affinity-purified serum as well as purified sera that was preincubated with either a phosphorylated or nonphosphorylated TrkA peptide competitor.
To image TrkA receptors by immunofluorescent microscopy, we serum-starved PC12 cells for 24 hr before treatment with either CGS 21680 or 100 ng/ml NGF for 5 min. Cells were fixed, permeabilized, and stained with the phospho-TrkA-specific antibody generated against the C-terminal phosphotyrosine. Cells were simultaneously costained with pan-Trk antibodies. As seen in Figure 5, untreated PC12 cells display Trk localization that spans the entire cell. Trk (red) was detected uniformly at the plasma membrane as well as in numerous small-diameter intracellular vesicles and in a perinuclear compartment; however, untreated cells display only a negligible amount of phospho-Trk immunoreactivity (pTrkA) that is mostly limited to the small intracellular vesicles. On the other hand, cells treated with NGF for 5 min displayed punctate staining for phospho-Trk along the plasma membrane but not within the cell, where there is an extensive amount of nonactivated Trk as shown by the pan-Trk staining in red.
Figure 5.
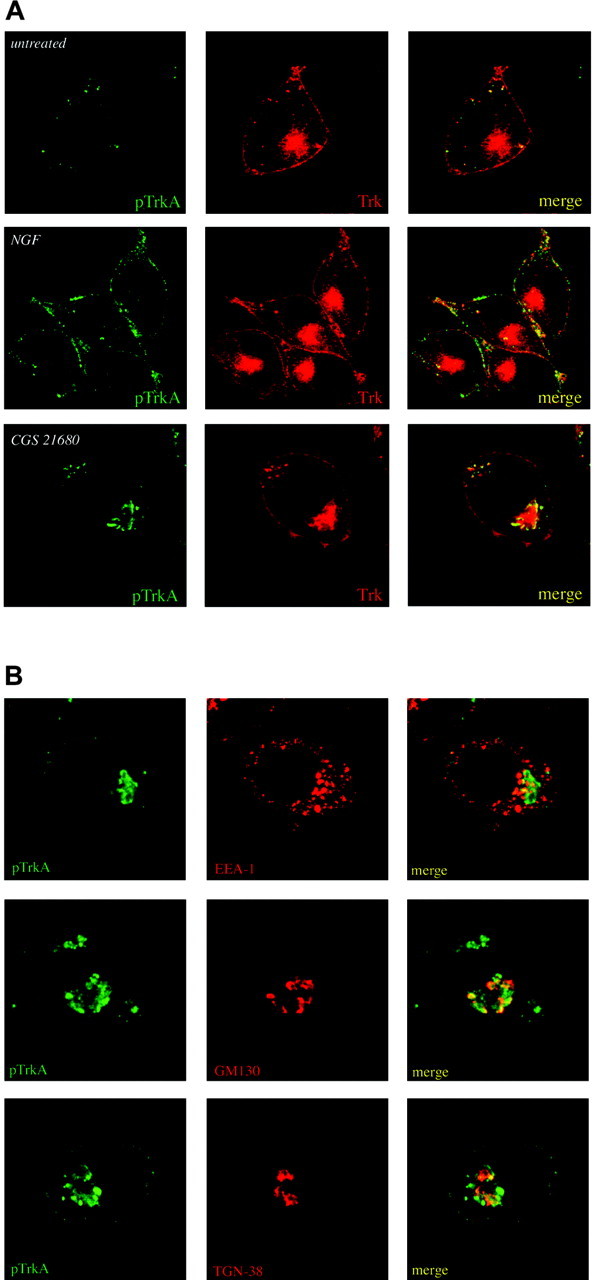
Localization of transactivated TrkA receptors. Subcellular localization of transactivated TrkA in PC12 cells was examined by confocal microscopy. A, Cells were treated with 10 nm CGS 21680 for 3 hr or with 100 ng/ml NGF for 5 min and then double labeled for phospho-TrkA (rabbit polyclonal pTrkA; FITC-labeled goat anti-rabbit) and pan-Trk (monoclonal Trk B3; Cy3-goat anti-mouse). B, To further characterize the specific location of transactivation events, double labeling of CGS 21680-treated cells was performed with antibodies directed against EEA-1, GM130, or TGN-38 (red) and phospho-TrkA (green).
The staining pattern of phospho-Trk that is observed after CGS 21680 treatment is quite distinct from that observed in NGF-treated cells. As seen in Figure 5A (bottom panels), phospho-Trk staining in cells treated with CGS 21680 for 3 hr was concentrated to closely apposed large vesicles in the perinuclear region of the cell. Staining was absent along the plasma membrane, consistent with our findings that GPCR-activated Trk is limited to an intracellular pool of receptors.
The staining pattern that we observed was reminiscent of Golgi staining from PC12 cells (Kim and von Zastrow, 2003). To confirm the Golgi localization, we analyzed PC12 cells treated with CGS 21680 by confocal microscopy using anti-phospho-Trk (green) and antibodies against GM130 or TGN38 (red), both of which are well established markers of the Golgi apparatus (Luzio et al., 1990; Nakamura et al., 1995). As shown, phospho-Trk partially colocalizes with both of these Golgi markers (Fig. 5B). On the other hand, cells stained with a marker for early endosomes, EEA-1 (Fig. 5B), or for late endosomes, LAMP1, displayed much less colocalization with phospho-Trk after GPCR stimulation (data not shown). These results suggest that GPCR-activated Trk is restricted to an intracellular region that is not associated with the endocytic trafficking pathway and suggests that part of the activated pool is detected close to or contiguous with the Golgi apparatus.
Brefeldin A treatment
To verify that the Golgi is involved in transactivation by GPCRs, we disrupted cells with brefeldin A, a fungal metabolite that is a potent inhibitor of Golgi vesicle fusion (Pelham, 1991). PC12 cells were treated with either 5 μg/ml brefeldin A or vehicle for 3.5 hr. During this period of time, cells were simultaneously treated with 10 nm CGS 21680 or 100 ng/ml NGF, such that the treatment period would coincide with the end of brefeldin A treatment. Untreated cells were used for control. Cell lysates were prepared and used for Trk immunoprecipitation and subsequent analysis by SDS-PAGE and blotting with phosphotyrosine antibodies. As shown in Figure 6 (top panel), brefeldin A pretreatment completely abolished activation of Trk by CGS 21680 at both time points of treatment, whereas NGF-mediated Trk activation was left unimpaired.
Figure 6.

The Golgi apparatus is involved in Trk transactivation. A, PC12 cells were pretreated for 30 min with 5 μg/ml brefeldin A or vehicle and then treated with CGS 21680 or NGF as indicated. In all cases of brefeldin A treatment, cells were exposed to the drug for a total duration of 3.5 hr. Trk immunoprecipitates were then analyzed by blotting with phosphotyrosine or pan-Trk antibodies. Phosphorylation of Akt was examined by blotting cell lysates with either phospho-Akt or anti-Akt antibodies for activated and total Akt levels. B, PC12 cells pretreated with brefeldin A or vehicle were treated with 10 nm CGS 21680 for 3 hr or 100 ng/ml NGF for 5 min and then stained for pTrkA (green) or total Trk (red) and imaged by standard immunofluorescent microscopy. Cell nuclei were visualized with DAPI.
In addition, Akt activation was also blocked when cells were pretreated with brefeldin, indicating that activation and downstream signaling of Trk mediated by GPCRs requires an intact Golgi apparatus. Interestingly, TrkA glycosylation was impaired by brefeldin as indicated by the pan-Trk blot shown in Figure 6A. The 110 kDa isoform of Trk accumulated when cells were treated with brefeldin. These results imply that an intact Golgi apparatus is a potential location for Trk transactivation and that the positive phospho-Trk staining observed in cells (Fig. 3A) may have arisen from this organelle.
To determine how brefeldin A affected TrkA subcellular localization, we subjected PC12 cells to indirect immunofluorescence for TrkA and pTrkA after treatment with brefeldin A. As depicted in Figure 6B, brefeldin A abolished perinuclear TrkA immunoreactivity in cells treated with CGS 21680 but did not affect the ability of NGF to activate TrkA at the plasma membrane. This indicated that Trk localization at the plasma membrane was not greatly disrupted by brefeldin A.
Effect of transactivation on primary neurons
PACAP displays pronounced effects on the survival of septal cholinergic neurons (Takei et al., 2000) as well as many other neuronal cells, such as mesencephalic dopaminergic neurons (Takei et al., 1998) and sensory neurons (Lioudyno et al., 1998). PACAP also is capable of triggering transactivation of Trk receptors in primary neurons in a time course identical to adenosine or CGS 21680 treatment (Lee et al., 2002b). To determine the distribution of transactivated TrkA receptors in primary neurons, we established E18 basal forebrain cultures. It was established previously that TrkA receptors are expressed by a subset of these neurons (Bernd et al., 1988). After treatment with PACAP, the localization of Trk receptors and phospho-Trk receptors was followed using pan-Trk antibodies and the phospho-Trk-specific antibody. Similar to what was observed in cultured PC12 cells, the phosphorylated form of Trk resided predominantly in an intracellular location in basal forebrain neurons, whereas Trk receptors could be found throughout the cell body and processes (Fig. 7). The phospho-Trk staining was heavily concentrated in a perinuclear region of the cell body, as illustrated by the DAPI and phospho-Trk distribution. This pattern of staining was inhibited by both K-252a and brefeldin A (Fig. 7), confirming that the observed immunoreactivity represents activated TrkA in a Golgi compartment. The finding of activated Trk receptors in response to PACAP treatment in basal forebrain neurons confirms that transactivation occurs in an intracellular localization in primary neurons.
Figure 7.

Localization of transactivated TrkA in basal forebrain neurons. Indirect immunofluorescence analysis was used to examine localization of transactivated TrkA in primary basal forebrain neuronal cultures. Cultures were treated with PACAP38 as indicated and labeled with phospho-TrkA (green) and pan-Trk (red) antibodies. Where indicated, K-252a (100 nm) or brefeldin A (5 μg/ml) was used to pretreat cells before addition of PACAP38. Cell nuclei were stained with DAPI (blue).
Discussion
Cross talk between GPCR ligands and receptor tyrosine kinases has been described as a means of conveying proliferative signals in cells (Downward, 2003; Fischer et al., 2003). In contrast, transactivation of neurotrophic receptors can result in survival responses and neuronal differentiation. In this study, we have shown that Trk receptors undergo activation in intracellular membranes and not at the cell surface. Biotinylation and immunofluorescence measurements indicate that transactivation events displayed a delayed time course and absence of involvement of cell surface receptors. These features are in direct contrast to activation of mitogenic growth factor receptors (Daub et al., 1997; Luttrell et al., 1999; Prenzel et al., 1999).
The location of signaling events representing cross talk between GPCR ligands and Trk receptor tyrosine kinases plays a crucial role in the kinetics and mechanism of transactivation. Although EGF receptor transactivation by GPCR ligands is rapid and requires endocytosis and close proximity of EGF receptor and GPCRs (Maudsley et al., 2000; Pierce et al., 2001), the time course of Trk receptor transactivation is longer and involves intracellular processing events. For example, processing of immature forms of Trk is involved in transactivation.
Several other aspects of GPCR-mediated transactivation of Trk receptors distinguish it from other transactivation and neurotrophin signaling events. A noticeable difference between adenosine and NGF Trk is that the phosphorylation of Trk substrates, such as the Shc adaptor proteins PLC-γ and PI3 kinase, requires an extended period of time, >1 hr of adenosine treatment (Lee and Chao, 2001), in contrast to NGF treatment, which requires <1 min. Second, GPCR-mediated activation of Trk results in downstream signaling to phosphoinositide 3-kinase and Akt but not MAPK, unlike neurotrophin signaling, which can robustly activate both pathways. The specific activation of Akt by transactivated Trk receptors provides a survival response in neurons (Lee et al., 2001, 2002a). MAPK activation is the hallmark of many growth factor receptors, such as EGF and PDGF receptors, that provide proliferative signals after transactivation (Luttrell et al., 1999; Wetzker and Bohmer, 2003).
Transactivation by adenosine or PACAP does not involve the production or release of newly synthesized or processed neurotrophins, as has been observed for the EGF receptor (Prenzel et al., 1999). One mechanism for transactivation of the EGF receptor involves the action of matrix metalloproteases. Cleavage of membrane-bound proHB-EGF by metalloproteases results in the release of EGF that binds and activates the EGF receptor (Prenzel et al., 1999). Experiments performed in PC12 cells indicate that GPCR ligands did not result in an increase in the expression of NGF that might contribute to TrkA receptor activation (Fig. 1C) (Lee et al., 2002a). Also, binding experiments show that adenosine and PACAP do not displace NGF from binding to TrkA receptors, indicating that the effects of these GPCR ligands occur through an intracellular pathway.
What is the relevance of these transactivation events? During retrograde transport, complexes of NGF with TrkA in clathrin-coated vesicles and endosomes have been envisioned, giving rise to the model of NGF and TrkA as components of a retrograde signal (Riccio et al., 1997; Senger and Campenot, 1997; Ginty and Segal, 2002; Grimes and Miettinen, 2003). A number of signaling molecules have been found to be associated with the TrkA receptor during retrograde transport (Delcroix et al., 2003). This suggests that signaling by trophic factors persists after internalization of their receptors in intracellular locations. Implicit in this model is that internalization of NGF is necessary for eliciting survival signals at the cell body. The evidence comes from the activation of the CREB transcription factor (Riccio et al., 1997) and Erk5 (Watson et al., 2001) by retrograde signals at distal terminals.
Retrograde signaling events, however, may also be transmitted in the absence of NGF internalization and transport. Survival of sympathetic neurons can be mediated by transported Trk receptors in the cell body after stimulation at nerve terminals by NGF linked to beads (MacInnis and Campenot, 2002). This raises the possibility that mechanisms other than endocytosed NGF may propagate signals from distal axons to TrkA receptors at the cell body (Campenot and MacInnis, 2004). A number of arguments have been raised both for and against the hypothesis that retrograde signaling can occur in the absence of NGF uptake (Chao 2003b; Ye et al., 2003), and several diverse models have been proposed to account for the discrepancies in retrograde signaling mechanisms (Ginty and Segal, 2002; Heerssen and Segal, 2002; Miller and Kaplan, 2003).
One common feature of retrograde transport of neurotrophin signaling is the ability of internalized Trk receptors to signal in a productive manner. Activation of tyrosine kinase receptors from GPCR transactivation illustrates an alternative way in which signals may be transmitted by intracellular receptors in the absence of ligand. This is exemplified by the findings in this study in which adenosine is capable of activating Trk receptors in intracellular membranes and not at the cell surface. Activation of intracellular Trk receptors may depend on Src kinase activity, recruitment of adaptor proteins, or an increase in the local concentration of Trk, which could lead to constitutive activation. The results demonstrate that internal Trk receptors may be found in discrete organelles (Grimes et al., 1997; Jullien et al., 2002; Delcroix et al., 2003), where they are fully active and promote Akt survival signals (Lee et al. 2001, 2002a,b). Also, it has been established that TrkA receptors found in sympathetic neurons may be activated by engagement of the Ret tyrosine kinase by GDNF in the absence of NGF (Tsui-Pierchala et al., 2002). Hence, these observations solidify the growing realization that Trk receptors function not only at the cell membrane but provide potent signals from discrete intracellular locations without neurotrophin binding. Trafficking of neurotrophin receptors in different membrane compartments must play a decisive role in regulating various fundamental signaling pathways in neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS21072 and HD23315 (M.V.C.) and the Whitehall Foundation and the National Alliance for Research on Schizophrenia and Depression (F.S.L.). We thank Juan Carlos Arevalo for generating Trk antibodies, Lani Mustacchi for primary cultures, Daniela Pereira for advice, and Wen-biao Gan for advice on confocal microscopy.
Correspondence should be addressed to Moses V. Chao, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, 540 First Avenue, New York, NY 10016. E-mail: chao@saturn.med.nyu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246650-09$15.00/0
References
- Aibel L, Martin-Zanca D, Perez P, Chao M (1998) Functional expression of TrkA receptors in hippocampal neurons. J Neurosci Res 54: 424-431. [DOI] [PubMed] [Google Scholar]
- Bernd P, Martinez H, Dreyfus C, Black I (1988) Localization of high-affinity and low-affinity nerve growth factor receptors in cultured rat basal forebrain. Neuroscience 26: 121-129. [DOI] [PubMed] [Google Scholar]
- Bronfman F, Tcherpakov M, Jovin T, Fainzilber M (2003) Ligand-induced internalization of the p75 neurotrophin receptor: a slow route to the signaling endosome. J Neurosci 23: 3209-3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campenot RB, MacInnis BL (2004) Retrograde transport of neurotrophins: fact and function. J Neurobiol 58: 217-229. [DOI] [PubMed] [Google Scholar]
- Chao MV (2003a) Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci 4: 299-309. [DOI] [PubMed] [Google Scholar]
- Chao MV (2003b) Retrograde transport redux. Neuron 39: 1-2. [DOI] [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A (1996) Role of transactivation of the EGF receptor in signaling by G-protein coupled receptors. Nature 379: 557-560. [DOI] [PubMed] [Google Scholar]
- Daub H, Wallash C, Lankenau A, Herrlich A, Ullrich A (1997) Signal characteristics of G protein-transactivated EGF receptor. EMBO J 16: 7032-7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix J-D, Valetta J, Wu C, Hunt S, Kowal A, Mobley W (2003) NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron 39: 69-84. [DOI] [PubMed] [Google Scholar]
- DiStefano PS, Friedman B, Radziejewski C, Alexander C, Boland P, Schick CM, Lindsay RM, Wiegand SJ (1992) The neurotrophins BDNF, NT-3, and NGF display distinct patterns of retrograde axonal-transport in peripheral and central neurons. Neuron 8: 983-993. [DOI] [PubMed] [Google Scholar]
- Donovan MJ, Miranda RC, Kraemer R, McCaffrey TA, Tessarollo L, Mahadeo D, Sharif S, Kaplan DR, Tsoulfas P, Parada L, Toran-Allerand CD, Hajjar DP, Hempstead BL (1995) Neurotrophin and neurotrophin receptors in vascular smooth muscle cells. Regulation of expression in response to injury. Am J Pathol 147: 309-324. [PMC free article] [PubMed] [Google Scholar]
- Downward J (2003) Role of receptor tyrosine kinases in G-protein-coupled receptor regulation of Ras: transactivation or parallel pathways. Biochem J 376: e9-e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer OM, Hart S, Gschwind A, Ullrich A (2003) EGFR signal transactivation in cancer cells. Biochem Soc Trans 31: 1203-1208. [DOI] [PubMed] [Google Scholar]
- Ginty D, Segal R (2002) Retrograde neurotrophin signaling: Trk-ing along the axon. Curr Opin Neurobiol 12: 268-274. [DOI] [PubMed] [Google Scholar]
- Grimes M, Miettinen H (2003) Receptor tyrosine kinase and G-protein coupled receptor signaling and sorting within endosomes. J Neurochem 84: 905-918. [DOI] [PubMed] [Google Scholar]
- Grimes M, Beattie E, Mobley W (1997) A signaling organelle containing the nerve growth factor-activated receptor tyrosine kinase, TrkA. Proc Natl Acad Sci USA 94: 9909-9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerssen HM, Segal RA (2002) Location, location, location: a spatial view of neurotrophin signal transduction. Trends Neurosci 25: 160-165. [DOI] [PubMed] [Google Scholar]
- Hempstead B (2002) The many faces of p75NTR. Curr Opin Neurobiol 12: 260-267. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR (1992) Overexpression of the Trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 9: 883-896. [DOI] [PubMed] [Google Scholar]
- Hendry I, Stoeckel K, Thoenen H, Iversen L (1974) The retrograde axonal transport of nerve growth factor. Brain Res 68: 103-121. [DOI] [PubMed] [Google Scholar]
- Huang E, Reichardt L (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, Reichardt L (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609-642. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Shigemoto R, Mori K, Takahashi K, Nagata S (1992) Functional expression and tissue distribution of a novel receptor for vasoactive intestinal polypeptide. Neuron 8: 811-819. [DOI] [PubMed] [Google Scholar]
- Jarvis M, Schulz R, Hutchinson A, Do U, Sills M, Williams M (1989) [3H]CGS21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J Pharmacol Exp Ther 251: 888-893. [PubMed] [Google Scholar]
- Jullien J, Guilli V, Reichardt L, Rudkin B (2002) Molecular kinetics of nerve growth factor receptor trafficking and activation. J Biol Chem 277: 38700-38708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, von Zastrow M (2003) Neurotrophin-regulated sorting of opioid receptors in the biosynthetic pathway of neurosecretory cells. J Neurosci 23: 2075-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha S, Oak J, Jackson M, Perez Y, Orser B, Van Tol H, MacDonald J (2002) A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron 35: 1111-1122. [DOI] [PubMed] [Google Scholar]
- Lee F, Rajagopal R, Chao M (2002a) Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev 13: 11-17. [DOI] [PubMed] [Google Scholar]
- Lee F, Rajagopal R, Kim A, Chang P, Chao M (2002b) Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J Biol Chem 277: 9096-9102. [DOI] [PubMed] [Google Scholar]
- Lee FS, Chao MV (2001) Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci USA 92: 3555-3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioudyno M, Skoglosa Y, Takei N, Lindholm D (1998) Pituitary adenylate cyclase-activating polypeptide (PACAP) protects dorsal root ganglion neurons from death and induces calcitonin gene-related peptide (CGRP) immunoreactivity in vitro. J Neurosci Res 51: 243-256. [DOI] [PubMed] [Google Scholar]
- Loeb DM, Stephens RM, Copeland T, Kaplan DR, Greene LA (1994) A Trk nerve growth-factor (NGF) receptor point mutation affecting interaction with phospholipase c-gamma-1 abolishes NGF-promoted peripherin induction but not neurite outgrowth. J Biol Chem 269: 8901-8910. [PubMed] [Google Scholar]
- Luttrell L, Daaka Y, Lefkowitz R (1999) Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol 11: 177-183. [DOI] [PubMed] [Google Scholar]
- Luzio J, Brake B, Banting G, Howell K, Braghetta P, Stanley K (1990) Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38). Biochem J 270: 97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacInnis B, Campenot R (2002) Retrograde support of neuronal survival without retrograde transport of nerve growth factor. Science 295: 1536-1539. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Pierce K, Musa Zamah A, Miller W, Ahn S, Daaka Y, Lefkowitz R, Luttrell L (2000) The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem 275: 9572-9580. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2003) Neurobiology. Trk makes the retrograde. Science 295: 1471-1473. [DOI] [PubMed] [Google Scholar]
- Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis T, Warren G (1995) Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 131: 1715-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham H (1991) Multiple targets for brefeldin A. Cell 67: 449-451. [DOI] [PubMed] [Google Scholar]
- Peng Y, Amemiya M, Yang X, Fan L, Moe O, Yin H, Preisig P, Yanagisawa M, Alpern R (2001) ETB receptor activation causes exocytic insertion of NHE3 in OKP cells. Am J Physiol Renal Physiol 280: F34-F42. [DOI] [PubMed] [Google Scholar]
- Pierce K, Tohgo A, Ahn S, Field M, Luttrell L, Lefkowitz R (2001) Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors. J Biol Chem 276: 23155-23160. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A (1999) EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402: 884-888. [DOI] [PubMed] [Google Scholar]
- Riccio A, Pierchala B, Ciarallo C, Ginty D (1997) An NGF-TrkA-mediated retrograde signal to transcription factor CREB in sympathetic neurons. Science 277: 1097-1100. [DOI] [PubMed] [Google Scholar]
- Senger D, Campenot R (1997) Rapid retrograde tyrosine phosphorylation of TrkA and other proteins in rat sympathetic neurons in compartmented cultures. J Cell Biol 138: 411-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, Skoglosa Y, Lindholm D (1998) Neurotrophic and neuroprotective effects of pituitary adenylate cyclase-activating polypeptide (PACAP) on mesencephalic dopaminergic neurons. J Neurosci Res 54: 698-706. [DOI] [PubMed] [Google Scholar]
- Takei N, Torres E, Yuhara A, Jongsma H, Otto C, Korhonen L, Abiru Y, Skoglosa Y, Schultz G, Hatanaka H, Sofroniew M, Lindholm D (2000) Pituitary adenylate cyclase-activating polypeptide promotes the survival of basal forebrain cholinergic neurons in vitro and in vivo: comparison with effects of nerve growth factor. Eur J Neurosci 12: 2273-2280. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Milbrandt J, Johnson E (2002) NGF utilizes c-Ret via a novel GFL-independent, inter-RTK signaling mechanism to maintain the trophic status of mature sympathetic neurons. Neuron 33: 261-273. [DOI] [PubMed] [Google Scholar]
- Tsui-Pierchala BA, Ginty DD (1999) Characterization of an NGF-P-TrkA retrograde-signaling complex and age-dependent regulation of TukA phosphorylation in sympathetic neurons. J Neurosci 19: 8207-8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson F, Porcionatto M, Bhattacharyya A, Stiles C, Segal R (1999a) Trk glycosylation regulates receptor localization and activity. J Neurobiol 39: 323-336. [DOI] [PubMed] [Google Scholar]
- Watson F, Heerssen H, Moheban D, Lin M, Sauvageot C, Bhattacharyya A, Pomeroy S, Segal R (1999b) Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci 19: 7889-7900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson F, Heerssen H, Bhattacharyya A, Klesse L, Lin M, Segal R (2001) Neurotrophins use the Erk5 pathway to mediate a retrograde survival response. Nat Neurosci 4: 981-988. [DOI] [PubMed] [Google Scholar]
- Wetzker R, Bohmer FD (2003) Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol 4: 651-657. [DOI] [PubMed] [Google Scholar]
- Ye H, Kuruvilla R, Zeifel L, Ginty D (2003) Evidence in support of signaling endosome-based retrograde survival of sympathetic neurons. Neuron 39: 57-68. [DOI] [PubMed] [Google Scholar]
- York R, Yao H, Dillon T, Ellig C, Eckert S, McCleskey E, Stork P (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392: 622-626. [DOI] [PubMed] [Google Scholar]