

Abstract
Cyclic nucleotide levels within extending growth cones influence how navigating axons respond to guidance cues. Pharmacological alteration of cAMP or cGMP signaling in vitro dramatically modulates how growth cones respond to attractants and repellents, although how these second messengers function in the context of guidance cue signaling cascades in vivo is poorly understood. We report here that the Drosophila receptor-type guanylyl cyclase Gyc76C is required for semaphorin-1a (Sema-1a)-plexin A repulsive axon guidance of motor axons in vivo. Our genetic analyses define a neuronal requirement for Gyc76C in axonal repulsion. Additionally, we find that the integrity of the Gyc76C catalytic cyclase domain is critical for Gyc76C function in Sema-1a axon repulsion. Our results support a model in which cGMP production by Gyc76C facilitates Sema-1a-plexin A-mediated defasciculation of motor axons, allowing for the generation of neuromuscular connectivity in the developing Drosophila embryo.
Keywords: semaphorin-1a, plexin A, Gyc76C, axon guidance, receptor guanylyl cyclase, cGMP
Introduction
During neural development, axons extend along complex, but precisely defined, routes to contact their appropriate targets and establish the connectivity of the adult nervous system. Guidance cues belonging to several families have been identified that direct axons along these pathways through attractive and repulsive mechanisms (Tessier-Lavigne and Goodman, 1996). For many of these extracellular cues, including ephrins, netrins, slits, and semaphorins, cell surface receptors have been identified that are required for the establishment of these neuronal trajectories (Huber et al., 2003). The signal transduction pathways by which these guidance cue receptors direct the cytoskeletal alterations critical for attractive or repulsive steering events, however, are only now beginning to be understood.
Two well characterized intracellular effectors that can dictate how an axon responds to extracellular signals are the second messengers cAMP and cGMP. Experiments with cultured Xenopus spinal neurons, in an in vitro growth cone turning assay, showed that changing the intracellular levels of cAMP or cGMP alters how an axon responds to extracellular guidance cues (Song and Poo, 1999). For example, the attractive response of an axon to the guidance cue netrin-1 can be converted to repulsion by decreasing the effective levels of cAMP within the responding neuron (Ming et al., 1997). Conversely, the axonal response to the potent chemorepellent semaphorin 3A (Sema3A) can be converted from repulsion to attraction by increasing cGMP levels within the neuron (Song et al., 1998). More recently, cAMP and cGMP have been shown to function together to influence how an axon responds to a particular attractant or repellent. The ratio of cAMP to cGMP determines how extending axons respond to netrin-1 in the growth cone turning assay: high cAMP to cGMP ratios produce an attractive response, whereas low ratios lead to repulsion (Nishiyama et al., 2003). Related observations have been made for Sema3A. When cAMP levels are raised in cultured neurons, the potent growth cone collapsing effect of Sema3A is neutralized (Dontchev and Letourneau, 2002; Chalasani et al., 2003). However, in these same cultures, raising the levels of cGMP potentiates the growth cone collapsing effect of Sema3A, suggesting that both cyclic nucleotides can modulate the response to a single axon guidance cue (Dontchev and Letourneau, 2002).
The molecular mechanisms underlying axon guidance effects caused by pharmacologically altering cyclic nucleotide levels are still unclear. Insight into how cAMP dictates axonal steering responses has been gained from the identification of Nervy, a protein that couples plexin A (PlexA), the receptor for the invertebrate transmembrane semaphorin-1a (Sema-1a), with the cAMP-dependent protein kinase A (PKA) (Terman and Kolodkin, 2004). Sema-1a is present on motor axons in the developing Drosophila nervous system and through its receptor PlexA acts as a repellent and directs individual axons away from the tightly fasciculated bundles in which they travel (Winberg et al., 1998; Yu et al., 1998). Nervy tethers PKA to the PlexA, positioning PKA to antagonize Sema-1a-mediated repulsion in response to local increases in cAMP.
Several studies provide hints as to which proteins involved in cGMP signaling may be involved in modulating or supporting axon guidance events (Gibbs and Truman, 1998; Polleux et al., 2000; Seidel and Bicker, 2000; Schmidt et al., 2002; Nishiyama et al., 2003). How specific proteins function in particular axon guidance signaling pathways to alter cGMP levels is, however, unknown. Using a novel Sema-1a-dependent forward genetic screening approach, we found that the Drosophila receptor guanylyl cyclase (rGC) Gyc76C, a member of the phylogenetically conserved family of single transmembrane domain guanylyl cyclases (Wedel and Garbers, 2001), is necessary for Sema-1a-mediated repulsive signaling in the developing Drosophila embryonic nervous system. Furthermore, our data strongly suggest that cGMP production by Gyc76C is essential for its function in vivo. Together, these findings provide a functional link between local production of cGMP within the growth cone and Sema-1a repulsive axon guidance signaling.
Materials and Methods
Molecular characterization of Gyc76C genomic structure
We used an 800 bp KpnI/EcoRI fragment from expressed sequence tag (EST) LD28142 that corresponds to an exon of Gyc76C close to the ORF start site to probe a Lambda Zap II embryonic Drosophila cDNA library. We identified two contiguous clones that contain all of the defined Gyc76C 5′ sequences as well as that of the CG32215 gene located 20 kb upstream of the Gyc76C ORF start site. Alignments were done using Sequencher software (Gene Codes Corporation, Ann Arbor, MI). The extents of deletions caused by P-element mobilization were identified by PCR using a battery of primers corresponding to the regions of the Gyc76C gene that flanked the insertion site of the P element.
In situ hybridization
RNA in situ hybridization analysis for Gyc76C was performed as described (Terman et al., 2002) on whole-mount Drosophila embryos using a 2.4 kb EcoRI fragment of the Gyc76C cDNA to make Gyc76C-specific antisense and sense probes.
Drosophila genetics
Genetic reagents. Culturing of Drosophila was performed as described (Terman et al., 2002). All crosses and embryo collections were done in a humidified incubator maintained at 25°C. The Gyc76C transgenes were created by compiling sequences from Gyc76C ESTs SD05894 and LD28142 into a contiguous ORF downstream of the IgK-leader sequence in the pSecTag vector (Invitrogen, Carlsbad, CA). The N-terminal fragment of Gyc76C was engineered by PCR to remove the Gyc76C signal sequence and to include two myc epitopes (EQLISEEDL) in frame with the Gyc76C sequence. The D945A mutation was engineered by PCR-mediated site-directed mutagenesis, and a fragment containing the mutated sequence was swapped into the vector containing the wild-type cDNA. The Gyc76C cDNAs were then subcloned into the pUASt vector for transformation into embryos (Terman et al., 2002). Transgene expression in embryos was confirmed by anti-myc immunocytochemistry on embryos expressing the upstream activation sequence transgenes under control of the neuron-specific elav-Gal4 transactivator. Transgenic lines used in this study showed comparable expression levels. Deficiencies and mutations used in the genetic screen were obtained from the Bloomington Stock Center. The KG03723 line was a generous gift from the Berkeley Gene Disruption Project (Bellen et al., 2004). Lines containing excisions of KG03723 were generated by crossing to a fly line containing a Δ2-3 transposase source (Robertson et al., 1988). All other stocks were described previously: sema1aP1, UAS:Sema-1a (Yu et al., 1998), elav-Gal4 (Yao and White, 1994), Df(4)C3 (Winberg et al., 1998), Df(3R)swp2MICAL (Terman et al., 2002), UAS:PlexA (Winberg et al., 1998).
Genetic screen. Male Drosophila containing deficiencies or individual mutations on the third chromosome were crossed with “PUP” (P-52 Gal4, UAS:Sema-1a, sema1aP1)/CyO, wg-lacZ females. The resulting F1 males were crossed with sema1aP1 females to generate PUP/sema1aP1; Df or mut/+ embryos. Embryos were fixed, stained with the BP102 antibody (Seeger et al., 1993), and assayed for β-galactosidase activity (Yu et al., 1998). After sorting based on the presence or absence of a blue precipitate, the population scored contained a mixture of PUP/sema1aP1 embryos with and without the deficiency or mutation on the third chromosome. We staged and scored these pools for the number of commissures (two, one or zero) present in each segment. Compared to PUP/sema1aP1 embryos, which have a distribution centered around one commissure per segment, enhancers were identified as those pools that showed a significant shift to embryos with zero commissures per segment, whereas embryos containing suppressor elements were identified as those that more closely resembled wild-type with most segments containing two commissures.
Microscopy and imaging
All images were captured using Openlab software (Improvision, Boston, MA) with an ORCA-ER digital CCD camera (Hamamatsu Photonics, Shizouka, Japan) on an Axioplan upright microscope (Zeiss, Oberkochen, Germany) with 63× oil-immersion and 10× air objectives.
Results
A dominant enhancer and suppressor screen for Sema-1a downstream signaling components
The transmembrane semaphorin Sema-1a is a potent repellent for motor axons in the Drosophila embryonic nervous system. Sema-1a-mediated axonal repulsion is required for the establishment of neuromuscular connectivity and for the formation of a subset of longitudinal axonal pathways within the CNS (Winberg et al., 1998; Yu et al., 1998). Sema-1a is also highly expressed on most, if not all, commissural axons. Therefore, to address whether Sema-1a is required for the guidance of commissural axons, we examined midline-crossing axons in sema1a loss-of-function (LOF) mutant embryos. In Drosophila embryos, axons within the ventral nerve cord cross the midline in an anterior and a posterior commissure within each segment, giving rise to a stereotypical ladder-like architecture easily visualized using the monoclonal antibody BP102 (Fig. 1A) (Seeger et al., 1993). In sema1a mutants, commissural axon pathways are not grossly disturbed (Table 1). Likewise, ectopic expression of high levels of Sema-1a in midline glial cells using the Gal4-UAS system (UAS: Sema-1a, P52-Gal4) does not exert adverse effects on commissural axons (Table 1) (Brand and Perrimon, 1993; Zhou et al., 1997).
Figure 1.
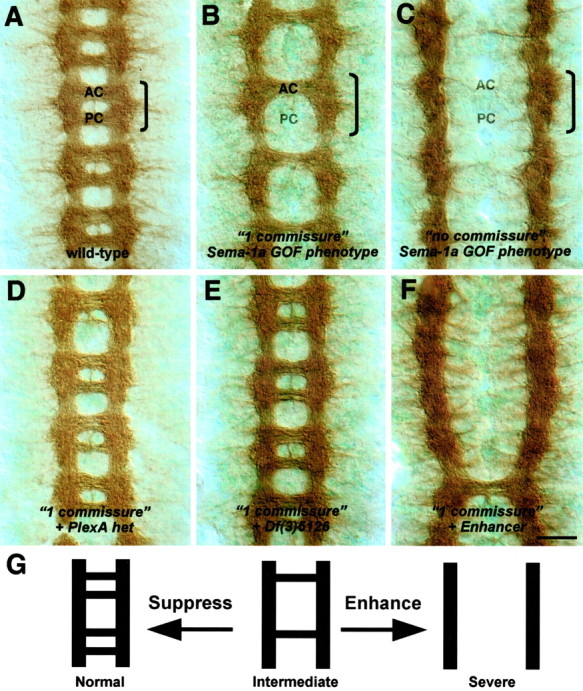
Dominant enhancer and suppressor screen of a Sema-1a-dependent phenotype. Filleted preparations of stage 14-16 embryos stained with the BP102 antibody to reveal commissural axons are shown. The anterior (AC) and posterior (PC) commissures are labeled in one segment that is outlined by a bracket. Anterior is up. A, In a wild-type embryo, axons cross the midline in the anterior and posterior commissures. B, Ectopic expression of low levels of Sema-1a in midline glia using the Gal4-UAS system in a sema1aP1 mutant background leads to the failure of axons within the posterior commissure to cross the midline (“1 commissure” Sema-1a GOF phenotype = P52-Gal4, UAS:Sema-1a, sema1aP1/sema1aP1). C, Ectopic expression of high levels of Sema-1a in midline glia in a sema1a mutant background enhances the 1 commissure Sema-1a GOF phenotype, resulting in the failure of all commissural axons to cross the midline (“no commissure” Sema-1a GOF phenotype = P52-Gal4, UAS:Sema-1a, sema1aP1/P52-Gal4, UAS:Sema-1a, sema1aP1). D, A deficiency, Df(4)C3, removing one copy of PlexA, the Sema-1a receptor, suppresses the 1 commissure phenotype in B. E, A deficiency, Df(3)5126, removing one copy of Gyc76C, suppresses the 1 commissure phenotype in B. F, A deficiency removing one copy of the genomic region at cytolocation 95F enhances the 1 commissure phenotype in B. G, Summary of Sema-1a GOF commissural axon phenotypes from the dominant enhancer and suppressor screen. Suppression of the intermediate 1 commissure phenotype yields a more wild-type-like architecture, whereas enhancement results in a more severe phenotype in which most axons do not cross the midline. Scale bar: A-F, 10 μm.
Table 1.
Sema-1a CNS phenotypes
|
Genotype |
Percentage of abnormal CNS segments (n)a |
Percentage of segments with [2, 1, 0] commissuresb |
|---|---|---|
| Wild type | 0 (500) | [100, 0, 0] |
| sema1aP1/sema1aP1 | 0 (350) | [100, 0, 0] |
| UAS:Sema-1a, P52-GAL4/UAS:Sema-1a, P52-GAL4 | 0 (300) | [100, 0, 0] |
| sema1aP1, UAS:Sema-1a, P52-GAL4/sema1aP1, UAS:Sema-1a, P52-GAL4 | 100 (830) | [0, 2, 98] |
| sema1aP1, UAS:Sema-1a, P52-GAL4/sema1aP1 | 92 (530) | [8, 72, 20] |
| sema1aP1, UAS:Sema-1a, P52-GAL4/sema1aP1; Df(4)C3/+ | 31 (90) | [69, 28, 3] |
|
sema1aP1, UAS:Sema-1a, P52-GAL4/sema1aP1; Df(3)5126/+ |
42 (102) |
[60, 39, 3] |
Abnormal CNS segments represent the percentage of the number (n) of segments scored that do not contain two commissural axon tracts.
Values represent the percentage of segments that contain two, one, or zero commissural axon tracts.
Strikingly, ectopically expressing high levels of Sema-1a in midline glia in a sema1a mutant embryo (sema1aP1, UAS:Sema1a, P52-Gal4) prevents all commissural axons from crossing the midline (Fig. 1C, Table 1). This “no-commissure” phenotype is similar to roundabout gain-of-function (GOF) and commissureless LOF phenotypes that result from the increased sensitivity of commissural axons to the midline repellent Slit (Tear et al., 1996; Kidd et al., 1999). Expressing lower levels of ectopic Sema-1a in midline glia in a sema1a mutant embryo results in the repulsion of fewer commissural axons away from the midline. This is reflected in the loss of only the posterior commissure and not the anterior commissure in most segments, producing a “one-commissure” phenotype (Fig. 1B, Table 1). This dosage sensitivity of commissural axons to ectopically expressed Sema-1a suggests that a Sema-1a receptor is present on all commissural axons. Indeed, decreasing the levels of the Sema-1a receptor plexin A (PlexA) results in a suppression of the one-commissure phenotype to a more wild-type-like two-commissure state (Fig. 1D, Table 1). These results demonstrate that commissural axons are responsive to the repulsive effects of Sema-1a and that this response is dependent on the presence of the Sema-1a receptor plexin A.
In addition to revealing that Sema-1a is a potent repellent for commissural axons, our findings also provide a fully penetrant and easily observable axon guidance phenotype that is dependent on the repulsive effects of Sema-1a. This phenotype served as the basis for an enhancer and suppressor mutagenesis screen to identify components of the Sema-1a-PlexA signaling pathway. Because the Sema-1a receptor PlexA is required for this axonal response, the molecules that signal the repulsive effects of PlexA should also be required for commissural axon repulsion by Sema-1a. Therefore, the intermediate one-commissure phenotype (Fig. 1B) is a sensitized genetic background that allowed us to identify genes that dominantly modify this phenotype. To screen for genes involved in the Sema-1a-PlexA signaling pathway, we looked for mutants that either suppressed or enhanced the Sema-1a-dependent one-commissure phenotype (Fig. 1G). Mutants that suppress this phenotype to a more wild-type state should disrupt genes that normally facilitate Sema-1a repulsion (Fig. 1E,G), whereas mutants that enhance this phenotype, causing an increase in the number of axons repelled from the midline, should disrupt genes that normally antagonize Sema-1a repulsion (Fig. 1F,G).
Our initial screen covered 85% of the third chromosome using deficiencies to rapidly screen for chromosomal regions that, when heterozygous, suppress or enhance the intermediate Sema-1a GOF phenotype. Of the 79 deficiencies screened, we identified five that modified this dosage-sensitive Sema-1a-dependent phenotype; three act as suppressors, and two are enhancers. The identification of these large genomic regions, and subsequent genetic analyses to narrow down these regions to a single gene, led to the discovery of several candidate Sema-1a repulsive axon guidance signaling components. One of these effectors is the gene Gyc76C.
Gyc76C mutants suppress a Sema-1a GOF phenotype
We identified a deficiency line that removes a portion of the Drosophila third chromosome at the cytological region from 76B4 to 77B, Df(3)5126, which suppresses the Sema-1a-dependent one-commissure midline phenotype (Fig. 1E, Table 1). Examining candidate mutations within this genomic region revealed that a gene disrupted by P-element insertion l(3)L0090-a, at cytolocation 76C, could suppress the one-commissure phenotype (data not shown). Furthermore, embryos homozygous for the l(3)L0090-a P-element insertion displayed motor axon phenotypes resembling sema1a homozygous mutant embryos, and additional genetic tests examining embryos doubly heterozygous for l(3)L0090-a and sema1a (sema1a/+; l(3)L0090-a/+) revealed that this P-element was likely disrupting a gene important for Sema-1a repulsive signaling (Table 2 and see below).
Table 2.
ISNb and SNa phenotypes of Gyc76C LOF and GOF mutants
|
Genotype |
Percentage of abnormal ISNb pathways (n)a |
Percentage of abnormal SNa pathways (n)b |
|---|---|---|
| Wild type | 12.2 (254) | 4.7 (254) |
| Loss of function | ||
| I(3)L0090-a/I(3)L0090-a | 22.0 (303) | 36.5 (307) |
| gyc76CKG03723/gyc76CKG03723 | 43.0 (144) | 64.0 (147) |
| gyc76CKG03723ex33/gyc76CKG03723ex33 | 45.9 (183) | 32.2 (180) |
| gyc76CKG03723ex173/gyc76CKG03723ex173 | 52.4 (208) | 37.7 (204) |
| gyc76CKG03723ex33/gyc76CKG03723ex173 | 50.0 (100) | 41.8 (98) |
| gyc76CKG03723ex173/Df(3)5126 | 52.1 (169) | 31.3 (166) |
| gyc76CKG03723ex144/gyc76CKG03723ex144 | 8.1 (135) | 14.6 (130) |
| UAS:Gyc76C, elav-Gal4/+;gyc76CKG03723ex173/gyc76CKG03723ex173 | 23.4 (184)c | 21.5 (186)c |
| UAS:Gyc76CD945A, elav-Gal4/+;gyc76CKG03723ex173/gyc76CKG03723ex173 | 60.0 (130) | 50.4 (121) |
| sema1aP1/sema1aP1 | 84.1 (138) | 90.0 (138) |
| Df(4)C3plexA/Df(4)C3plexA | 86.0 (50) | 88.0 (50) |
| Genetic interactions | ||
| sema1aP1/+;I(3)L0090-a/+ | 14.0 (140) | 45.0 (174) |
| sema1aP1/+;gyc76CKG03723ex173/+ | 54.1 (111) | 39.8 (103) |
| gyc76CKG03723ex173/+;Df(4)C3plexA/+ | 48.6 (105) | 32.3 (102) |
| gyc76CKG03723ex173/Df(3R)swp2MICAL | 56.3 (87) | 30.2 (86) |
| gyc76CKG03723ex173/+ | 27.6 (362) | 10.7 (355) |
| enaGC10/+;gyc76CKG03723ex173/+ | 24.6 (207) | 13.6 (206) |
| Gain of function | ||
| UAS:Gyc76C, elav-Gal4/UAS:Gyc76C, elav-Gal4d | 57.4 (148) | 68.0 (150) [26.0]e |
|
UAS:Gyc76CD945A, elav-Gal4/UAS:Gyc76CD945A, elav-Gal4f
|
59.1 (110) [11.8]g
|
56.1 (107) |
Abnormal ISNb phenotype defined as failure of ISNb axons from the RP5, V, or RP3 neurons to properly innervate ventral lateral muscles 12/13 or 6/7. Phenotypes include weak or absent innervations, target bypasses, and axon bundle stalling.
Abnormal SNa phenotype defined as failure of SNa axons to make two characteristic turns at choice points along the lateral transverse muscles 22, 23, and 24 and the failure of axons to reach muscle 24.
Statistically different from values for gyc76CKG03723ex173 homozygous mutants. Fisher's exact test using a two-by-two contingency table; p <0.0001.
Longitudinal bundles of CNS axons disrupted in 18 of 20 embryos.
Percentage of total SNa pathways that incorrectly navigate between muscles 21 and 22 instead of muscles 22 and 23.
Longitudinal bundles of CNS axons disrupted in 6 of 11 embryos.
Percentage of total hemisegments in which the RP3 axon extends an exuberant process in the cleft between muscles 6 and 7.
Molecular analysis indicated that the l(3)L0090-a P-element is inserted in the 5′ untranslated region of the Gyc76C gene (Fig. 2A). Gyc76C encodes a Drosophila transmembrane rGC. Gyc76C was first identified because of its high degree of sequence conservation to vertebrate cyclases and was found to be expressed in the Drosophila adult nervous system (Liu et al., 1995; McNeil et al., 1995). Gyc76C contains the hallmark domains of the phylogenetically conserved single transmembrane receptor guanylyl cyclases, including a putative ligand-binding extracellular domain (Fig. 2B) (Lucas et al., 2000). At present, no ligands have been identified for Gyc76C or any of the other six rGCs in Drosophila, but certain vertebrate rGCs are receptors for natriuretic peptides, a heat-stable enterotoxin, guanylin, and uroguanylin (Lucas et al., 2000). Intracelluarly, Gyc76C contains a catalytically inactive kinase homology domain (KHD), a dimerization domain, a well conserved catalytic cyclase domain, and a large region at the C terminus unique to Gyc76C (Fig. 2B).
Figure 2.

Gyc76C structure and localization. A, Scale representation of the genomic organization of Gyc76C. l(3)L0090-a and KG03723 indicate the locations of P-elements within the Gyc76C gene. Vertical bars and filled boxes represent exons. The extents of the lesions in gyc76CKG03723ex173 and gyc76CKG03723ex33 generated by imprecise excision of the KG03723 transposable element are indicated. B, Protein domain organization of Gyc76C. A putative ligand-binding domain is located at the N terminus (Ecto). A transmembrane (TM) domain anchors the protein in the plasma membrane. The KHD, dimerization (DD), guanylyl cyclase (G-Cyc), and C-terminal (C-term) domains are intracellular. C, A filleted preparation of a wild-type stage 14 embryo hybridized with a cRNA probe specific for a region of Gyc76C. The Gyc76C transcript is broadly distributed but is enriched in cells within the ventral nerve cord (VNC). Scale bar, 35 μm.
In line with genetic interaction data suggesting a role for Gyc76C in Sema-1a-PlexA signaling, we found that Gyc76C is expressed in the Drosophila embryonic CNS. In situ hybridization with a Gyc76C-specific cRNA probe reveals a broad distribution of Gyc76C transcript in later stage embryos but with clear enrichment in the ventral nerve cord (Fig. 2C). Hybridization of this same in situ probe to gyc76C mutant embryos reveals little to no Gyc76C transcript (data not shown). This is similar to the background seen when the corresponding Gyc76C sense probe is hybridized to wild-type embryos (data not shown). The Gyc76C nervous system expression is similar to that seen for the Sema-1a receptor PlexA (Winberg et al., 1998) and two of its previously identified downstream signaling effectors, MICAL (Terman et al., 2002) and Off-track (Otk) (Pulido et al., 1992; Winberg et al., 2001). As has been reported previously (Liu et al., 1995; McNeil et al., 1995), we also observe a large Gyc76C maternal contribution (data not shown) and expression in somatic musculature (Fig. 2C).
Our identification of the Gyc76C rGC from a Sema-1a-dependent genetic screen, our initial phenotypic analyses, and Gyc76C nervous system expression make Gyc76C a good candidate for a downstream effector of the Sema-1a-PlexA signaling pathway.
Gyc76C is an essential gene required for embryonic motor axon guidance
To determine whether Gyc76C plays a role in Sema-1a-PlexA signaling, we next examined Gyc76C function in motor axon guidance. The original P-element line identified in our screen, l(3)L0090-a, is homozygous viable and appears to be a hypomorphic allele (data not shown). To generate a Gyc76C null allele, we excised a different P-element present in the Gyc76C gene KG03723 (Bellen et al., 2004) and generated fly lines with both precise and imprecise removal of the P-element as defined by molecular analysis of the Gyc76C genomic region (data not shown and see Materials and Methods). Two lethal imprecise excision lines that contain deletions of ∼8 kb of genomic sequence 3′ to the P-element including a neighboring Gyc76C exon (Fig. 2A, gyc76CKG03723ex33 and gyc76CKG03723ex173) fail to complement a deficiency line lacking this region. These alleles provide null, or severely hypomorphic, Gyc76C alleles for subsequent analyses (Table 2).
The establishment of neuromuscular connectivity in Drosophila is an excellent paradigm for identifying and characterizing axon guidance cues and their downstream signaling components (Araujo and Tear, 2003). Approximately 40 identified motor neurons that reside in the ventral nerve cord innervate ∼30 muscles that line the body wall in each hemisegment of the Drosophila embryo (Landgraf et al., 1997). Staining late stage 16/17 embryos with the anti-fasciclin II antibody (mAb1D4), which labels all motor axons, allows for the visualization of motor axon pathways emanating from the ventral nerve cord as two large bundled fascicles, the intersegmental nerve (ISN) and the segmental nerve (SN) (VanVactor et al., 1993). Axons within the “b” branch of the ISN (ISNb) defasciculate from axons within the main ISN branch, enter the ventral longitudinal muscle field, and further defasciculate to innervate muscles 6, 7, 12, and 13 (Fig. 3A,H). Axons within the “a” branch of the SN (SNa) navigate past the ventral longitudinal muscle field and defasciculate, sending one branch posteriorly and another that continues dorsally between muscles 22 and 23. An axon within this branch further defasciculates and makes two characteristic turns to innervate its target, muscle 24 (Fig. 3A,H).
Figure 3.
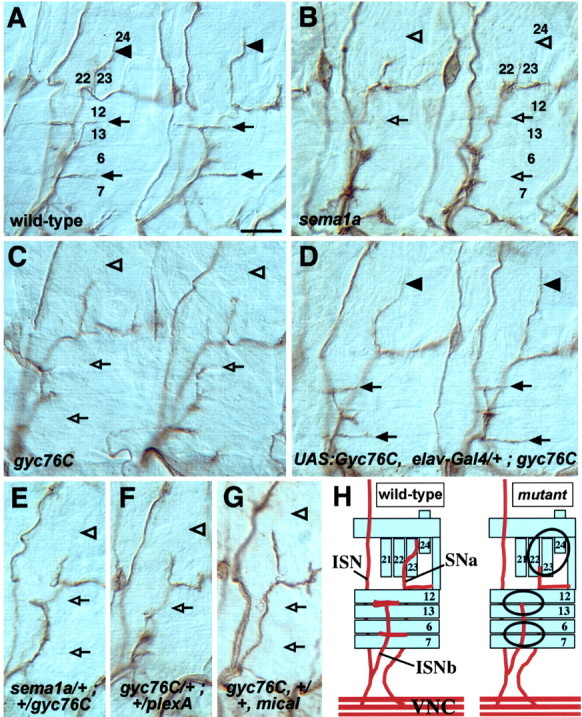
Gyc76C guides motor axons and genetically interacts with Sema-1a, PlexA, and MICAL. Filleted preparations of late stage 16 embryos stained with the anti-fasciclin II monoclonal antibody to reveal motor axons in abdominal segments. Two hemisegments are displayed in A-D; one hemisegment is shown in E-G. Anterior is left, and dorsal is up. A, In a wild-type embryo, axons within the ISNb innervate the ventral lateral muscles 12, 13, 6, and 7 (arrows), and an axon from the SNa innervates the lateral transverse muscle 24 (arrowheads). B, C, In sema1aP1 (B) and gyc76CKG03723ex173 (C) mutants, axons within the ISNb (open arrows) and SNa (open arrowheads) often fail to reach their proper targets. D, Neuronal expression of Gyc76C in a gyc76CKG03723ex173 mutant background restores the proper innervation of the musculature by ISNb (arrows) and SNa (arrowheads) axons. E-G, Gyc76C genetically interacts with Sema-1a, PlexA, and MICAL. Axons within the ISNb (open arrows) and SNa (open arrowheads) often fail to reach their targets in embryos heterozygous for sema-1aP1/+; gyc76CKG03723ex173/+ (E); gyc76CKG03723ex173/+; Df(4)C3PlexA/+ (F); gyc76CKG03723ex173, +/+, Df(3R)swp2MICAL (G). H, Summary of normal ISNb and SNa axon guidance seen in wild-type embryos (left) and the ISNb and SNa axon guidance defects seen in sema-1a and gyc76C mutants (right). Scale bar: A-G, 10 μm.
In sema1a mutants, motor axons often fail to defasciculate from one another and subsequently fail to innervate their proper muscle targets because of a decrease in Sema-1a-dependent axon-axon repulsive signaling (Fig. 3B, Table 2) (Yu et al., 1998, 2000). Homozygous gyc76C mutants (gyc76CKG03723ex33 or gyc76CKG03723ex173) have similar defects in motor axon guidance as those seen in sema1a mutants. In these gyc76C mutants, ISNb and SNa motor axons often fail to defasciculate and do not innervate their proper muscle targets (Fig. 3C, Table 2).
Although gyc76CKG03723 also displays a motor axon guidance phenotype (Table 2), a fly line from which this transposable element was excised precisely, gyc76CKG03723ex144, does not show motor axon guidance defects (Table 2). Moreover, expression of a Gyc76C transgene in all neurons under the control of the elav-Gal4 transactivator significantly rescues the motor axon guidance defects observed in gyc76CKG03723ex173 mutant embryos (Fig. 3D, Table 2). Neuronal expression also partially rescues the lethality of gyc76C mutants in 20% of all progeny, which translates into a 60% rescue of expected homozygous adult progeny (Table 3). These data show that Gyc76C expression in the nervous system is essential for adult viability and for the proper defasciculation of motor axon bundles.
Table 3.
Lethality rescue of gyc76C mutants
|
Males × females cross |
Homozygous viable progeny (n)a |
|---|---|
| gyc76CKG03723ex173/TM3, Sb | 0 (318) |
| UAS:Gyc76C, elav-Gal4/CyO;gyc76CKG03723ex173/TM3, Sb | 20% (321) |
| UAS:Gyc76CD945A, elav-Gal4/CyO;gyc76CKG03723ex173/TM3, Sb | 2% (304) |
| gyc76CKG03723ex33/TM3, Sb | 0 (354) |
| UAS:Gyc76C, elav-Gal4/CyO; gyc76CKG03723ex33/TM3, Sb | 24% (307) |
| UAS:Gyc76CD945A, elav-Gal4/CyO; gyc76CKG03723ex33/TM3, Sb | 4% (273) |
|
gyc76CKG03723ex173/TM3, Sb × gyc76CKG03723ex33/TM3, Sb
|
<1% (417) |
Homozygous viable progeny scored as progeny without the Stubble (Sb) phenotype, a dominant marker on the TM3 balancer chromosome. For a balanced viable mutation, the expected non-Sb progeny from a sibling cross would be 33%.
Overexpression of Gyc76C perturbs motor and CNS axon guidance
In light of our finding that disruption of the Gyc76C gene gives rise to axon guidance phenotypes resembling sema1a mutants, we asked whether overexpressing Gyc76C in all neurons mimics the phenotypes observed when PlexA is overexpressed in all neurons (Winberg et al., 1998). Expressing PlexA in all neurons in a wild-type background produces phenotypes consistent with increased repulsion of axons. For example, fasciclin II-positive axon bundles aligning each side of the midline within the CNS (Fig. 4A) misproject as a consequence of PlexA overexpression and send axonal projections away from the midline (Winberg et al., 1998). Expressing high levels of a Gyc76C transgene in all neurons in a wild-type background yields a similar phenotype in which the same subset of axons within the CNS are repelled away from the other axons (Fig. 4B).
Figure 4.
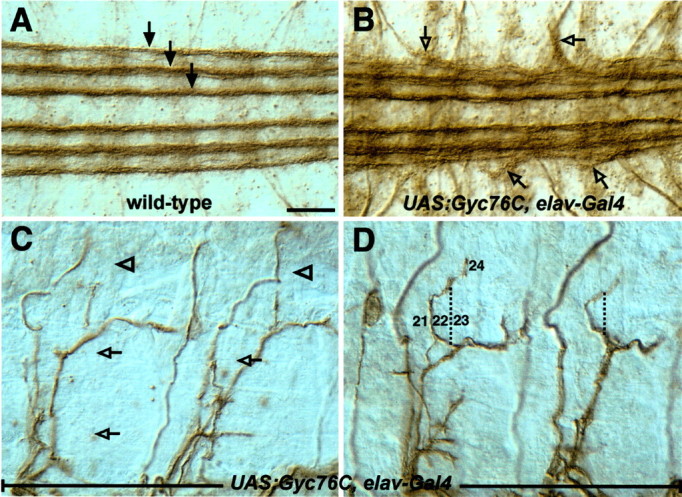
Neuronal overexpression of Gyc76C perturbs CNS and motor axon guidance. Shown are filleted preparations of late stage 16 embryos stained with the anti-fasciclin II monoclonal antibody 1D4 to reveal a subset of CNS axons (A, B) and motor axons (C, D). Anterior is left, and dorsal is up. A, In a wild-type embryo, three well separated bundles of axons are present on each side of the CNS midline (arrows). B, Overexpression of Gyc76C in all neurons with the Gal4-UAS system in a wild-type background results in the third/outermost longitudinal axon fascicle defasciculating abnormally and projecting axons away from the CNS (open arrows). C, Overexpressing Gyc76C in all neurons produces motor axon guidance defects in which axons often fail to defasciculate from the ISNb and SNa axon bundles. D, Overexpressing Gyc76C in all neurons also results in a novel phenotype affecting the dorsal branch of the SNa, which now projects incorrectly between muscles 21 and 22 and then extends toward its proper target, muscle 24. The path that this bundle of SNa axons normally follows is indicated by the dashed line. Scale bar: A-D, 10 μm.
Interestingly, overexpressing PlexA in all neurons also gives rise to axon guidance phenotypes reminiscent of decreased axon-axon repulsion (Winberg et al., 1998). Similarly, overexpressing Gyc76C in all neurons results in these same hyperfasciculation phenotypes. ISNb and SNa axons in half of the hemisegments scored do not defasciculate and fail to innervate their proper muscle targets (Fig. 4C, Table 2). We also observed a novel phenotype, not observed in any gyc76C LOF embryos, that affects SNa axons. In one-quarter of the hemisegments examined, the dorsal branch of the SNa misprojects between muscles 21 and 22 instead of between muscles 22 and 23 (Fig. 4D). Following this improper anterior wandering, the axon bundle often makes an aberrant double turn toward its proper muscle target, muscle 24.
Combined with LOF data from our analysis of the gyc76C mutants, our Gyc76C GOF analyses show that proper levels of Gyc76C protein are necessary for normal guidance of motor and CNS axons responsive to the effects of Sema-1a.
Gyc76C genetically interacts with Sema-1a, PlexA, and MICAL
The identification of Gyc76C as a suppressor of a Sema-1a GOF midline phenotype, and the fact that Gyc76C LOF and GOF mutants phenocopy Sema-1a and PlexA LOF and GOF mutants, suggests that Gyc76C is necessary for proper axon guidance decisions mediated by Sema-1a-PlexA signaling. To further test the role played by Gyc76C in Sema-1a-mediated motor axon guidance events, we examined embryos doubly heterozygous for Gyc76C and Sema-1a (sema1a/+; gyc76C/+) in a trans-heterozygous genetic analysis. When two gene products function together in the same pathway, it is often possible to see a dosage-dependent genetic interaction between them (Artavanis-Tsakonas et al., 1995; Winberg et al., 1998). Although the gyc76C/+ heterozygous embryos show a slight increase of axonal pathfinding errors over that seen for wild-type embryos (Table 2), the sema1a/+; gyc76C/+ embryos reveal a dominant, synergistic genetic interaction between Sema-1a and Gyc76C; ISNb and SNa axons often fail to innervate their muscle targets at frequencies similar to those seen for gyc76C homozygous embryos (Fig. 3E, Table 2). As a control, we examined the trans-heterozygous interaction between Gyc76C and ena, a gene encoding a protein that, at present, is not thought to be in the Sema-1a pathway but is important for proper motor axon guidance (Wills et al., 1999). We do not observe a genetic interaction between these two genes (Table 2). Gyc76C also genetically interacts with PlexA and MICAL, the receptor and a signal transducer, respectively, of the Sema-1a repulsive signaling cascade (Fig. 3 F, G; Table 2). Qualitatively and quantitatively, these trans-heterozygous mutant embryos display phenotypes similar to what is seen in gyc76C homozygous mutant embryos, supporting a role for Gyc76C in Sema-1a-mediated axon-axon repulsion.
To provide additional evidence that Gyc76C functions with PlexA to mediate axon guidance events, we examined how altering Gyc76C expression levels affects a novel PlexA-dependent GOF phenotype. Overexpressing high levels of PlexA in all neurons causes thick axon bundles of ipsilaterally projecting fasciclin II-positive axons to cross the midline at an average of two crossings per embryo (Fig. 5B,F). Removal of Gyc76C from embryos expressing high levels of PlexA significantly suppresses this phenotype and restores the proper pathfinding of these CNS axons (Fig. 5C,F). Furthermore, simultaneously increasing the levels of Gyc76C and PlexA in all neurons causes a severe augmentation of the PlexA-dependent phenotype (Fig. 5D,F). On average, four thick axon bundles cross the midline per embryo, with some embryos having five or more bundles aberrantly decussating. The requirement of Gyc76C to observe high penetrance of this PlexA-dependent phenotype and the ability of ectopic Gyc76C to increase the severity of this phenotype, in conjunction with the trans-heterozygous analyses, provide additional support for Gyc76C functioning as a component of the Sema-1a-PlexA signaling cascade.
Figure 5.
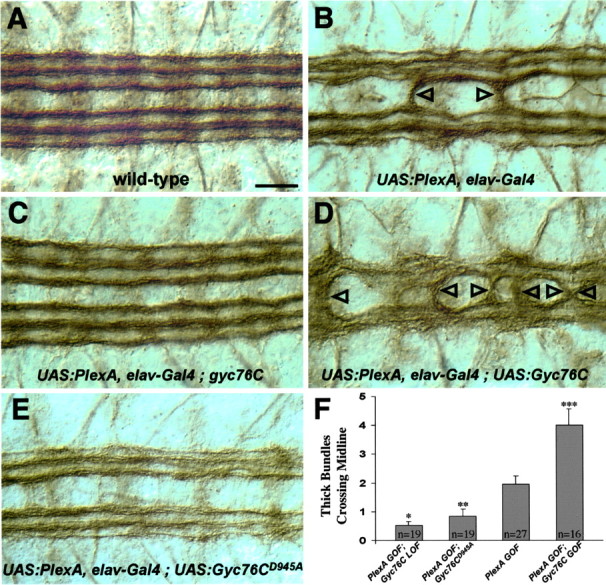
Altering levels of Gyc76C modifies a Plexin A GOF phenotype. Shown are filleted preparations of late stage 16 embryos stained with the anti-fasciclin II monoclonal antibody to reveal a subset of CNS axons, as in Figure 4. A, In a wild-type embryo, three bundles of ipsilaterally projecting axons are present on each side of the CNS midline that they do not cross. B, Overexpressing high levels of PlexA in all neurons in an otherwise wild-type embryo causes thick bundles of axons to abnormally cross the midline (arrowheads). C, An embryo overexpressing high levels of PlexA in all neurons, but also homozygous for the gyc76CLOF mutant, exhibits a more wild-type phenotype in which few axon bundles cross the midline. D, An embryo overexpressing both PlexA and Gyc76C results in a severe axon guidance phenotype in which many axon bundles cross the midline. E, An embryo overexpressing both PlexA and the Gyc76CD945A mutant transgene that lacks the catalytic aspartate residue displays a more wild-type phenotype in which few axon bundles cross the midline. F, Graph displaying the average number of thick axon bundles crossing the midline per embryo with the following genetic backgrounds: (PlexA GOF; Gyc76CLOF = UAS:PlexA, elav-Gal4; gyc76CKG03723ex173); (PlexA GOF; Gyc76CD945A = UAS:PlexA, elav-Gal4; UAS:Gyc76CD945A); (PlexA GOF = UAS:PlexA, elav-Gal4); (PlexA GOF; Gyc76C GOF = UAS:PlexA, elav-Gal4; UAS:Gyc76C). Error bars indicate SEM. The asterisks indicate values of two-tailed t tests with equal variance comparing the values of each phenotype to those for PlexA GOF. *p < 0.0005; **p < 0.02; ***p < 0.0002. Scale bar, A-E, 10 μm.
The integrity of the Gyc76C catalytic cyclase domain is required for its function in Sema-1a-mediated axonal repulsion
rGCs, including Gyc76C, contain a catalytic cyclase domain that is highly conserved among all family members. A conserved aspartate residue in the cyclase domain is essential for the conversion of GTP to cGMP. Mutating this catalytic aspartate to an alanine inactivates the cyclase without disrupting its ability to form homodimers (Thompson and Garbers, 1995). To begin to analyze the importance of this cyclase activity for Gyc76C function in Sema-1a signaling, we made a Gyc76C transgene with the corresponding amino acid substitution, mutating the Asp945 residue to an alanine residue (Fig. 6A). The expression level of this mutant transgene in embryos is similar to the expression level of the wild-type transgene (data not shown and see Materials and Methods). We then asked whether this mutant Gyc76C transgene (Gyc76CD945A) with a single amino acid change designed to disrupt the Gyc76C cyclase activity could substitute for the wild-type protein. In contrast to the effects of expresssing the wild-type Gyc76C transgene in all neurons in homozygous mutant gyc76C embryos, ectopically expressing the Gyc76CD945A transgene in all neurons fails to rescue both the gyc76C mutant axon guidance phenotypes (Fig. 6B, Table 2) and the lethality of the gyc76C mutant line (Table 3). Increasing the expression levels of this mutant transgene does not rescue the gyc76C mutant phenotypes (data not shown). Furthermore, neuronal expression of Gyc76CD945A in embryos overexpressing high levels of neuronal PlexA suppresses the PlexA GOF CNS phenotype, showing that the Gyc76CD945A transgene is indeed expressed and that it can function in a dominant-negative manner (Fig. 5E,F). In fact, overexpression of Gyc76CD945A in a wild-type background gives rise to CNS and motor axon guidance phenotypes similar to those observed in sema1a and plexA mutants (Fig. 6C,D). Overexpression of the Gyc76CD945A transgene in all neurons also yields novel phenotypes qualitatively distinct from the novel phenotype produced by overexpression of the wild-type Gyc76C transgene (compare Figs. 4D,6E-G). In ∼12% of hemisegments, an exuberant process innervating muscles 6 and 7 spans most of the segment (Fig. 6E, Table 2). Overexpressing a modified form of MICAL also produces a similar phenotype (Terman et al., 2002, their Fig. 6G) Some motor axon bundles that have not properly defasciculated take circuitous routes as they navigate toward their targets (Fig. 6F,G). Taken together, these results demonstrate that the function of Gyc76C is dependent on an intact cyclase domain for proper Sema-1a-dependent motor axon guidance, and they further suggest that cGMP production is critical for these guidance events.
Figure 6.
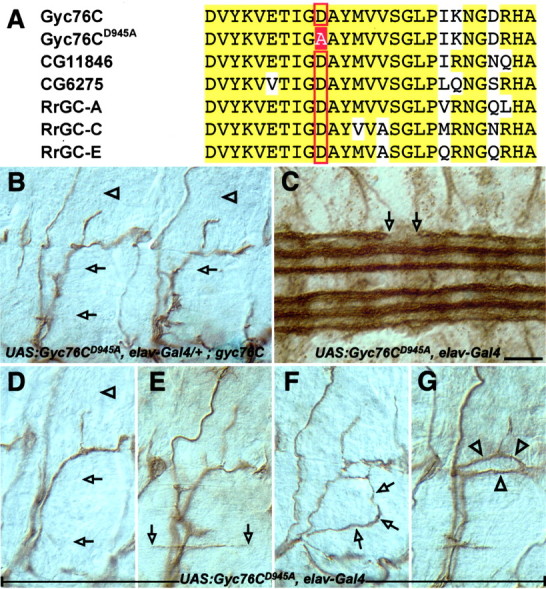
A catalytically inactive form of Gyc76C fails to rescue the gyc76C mutant phenotype and perturbs motor and CNS axon guidance. A, Alignment of a portion of the catalytic regions of three Drosophila and three rat rGCs. The red box indicates the catalytic aspartate (D945) that was converted to alanine to make the Gyc76CD945A transgene. B-G, Filleted preparations of late stage 16 embryos stained with the anti-fasciclin II monoclonal antibody to label motor and CNS axons, as in Figure 4. B, An embryo expressing the mutant Gyc76CD945A in all neurons in a mutant gyc76C background exhibits the same defects observed in gyc76C homozygous mutants. C-G, Overexpressing the mutant Gyc76CD945A in all neurons in a wild-type background disrupts normal CNS and motor axon pathfinding. C, An embryo overexpressing the mutant Gyc76CD945A in a wild-type background shows an interruption of a CNS axon tract with a large gap in the outermost bundle of longitudinal axons within the CNS (open arrows). D, A wild-type embryo overexpressing Gyc76CD945A in all neurons displays LOF-like phenotypes in which axons of the ISNb (open arrowheads) and SNa (open arrows) fail to reach their proper targets. E-G, Overexpressing Gyc76CD945A in all neurons in a wild-type background also produces novel phenotypes. E, The RP3 axon (open arrowheads) extends exuberantly in the cleft between muscles 6 and 7. The ISNb (F, open arrows) and SNa (G, open arrowheads) axon bundles wander inappropriately as they extend toward their targets. Scale bar: B-G, 10 μm.
Discussion
The activities and levels of intracellular signaling components determine how navigating axons interpret extracellular environmental signals. Cyclic nucleotide levels within neurons have been shown to be important determinants for regulating neurite responses to axon guidance cues in vitro. We present experiments here that provide an important molecular link between semaphorin-mediated repulsion and cGMP signaling in vivo. We show that Gyc76C is critical for Sema-1a-Plexin A-mediated selective defasciculation of axon bundles in the developing Drosophila neuromuscular system. We also show that a conserved amino acid residue within the Gyc76C cyclase domain, a residue required for rGC catalytic activity, is also required in Gyc76C for correct motor axon pathfinding. The identification of Gyc76C as an essential component of the Sema-1a-PlexA repulsive axon guidance signaling pathway provides insight into how cyclic nucleotide production is linked to the cascade of events downstream of semaphorin-mediated repulsion. These observations also provide a potential target for modulating repulsive semaphorin signaling by alterations of cGMP levels directly through rGCs.
Gyc76C plays a role in Sema-1a-PlexA-mediated axonal repulsion
Our analyses demonstrate a role for the rGC Gyc76C in Sema-1a-mediated axon-axon repulsion. We generated LOF mutations in the Gyc76C gene and observed highly penetrant phenotypes similar to the motor axon guidance defects observed in sema1a, plexA, and mical mutants (Winberg et al., 1998; Yu et al., 1998; Terman et al., 2002). Neuronal expression of a Gyc76C cDNA restores the wild-type innervation pattern in gyc76C mutant embryos and also restores viability to the lethal gyc76C mutant line, demonstrating a requirement for Gyc76C in neurons for correct axonal pathfinding. Neuronal overexpression of wild-type Gyc76C also results in phenotypes resembling PlexA GOF phenotypes (Winberg et al., 1998). Our genetic interaction analyses confirm a role for Gyc76C in Sema-1a-PlexA repulsive signaling. Embryos heterozygous for both Gyc76C and other members of this signaling cascade, including Sema-1a, PlexA, and MICAL, display motor axon pathway disruptions. These phenotypes are qualitatively similar to LOF mutant phenotypes observed in sema1a, plexA, and mical LOF mutants and are seen at comparable frequencies (Winberg et al., 1998; Yu et al., 1998; Terman et al., 2002). In addition to suppressing the Sema-1a- dependent midline phenotype, loss of Gyc76C function also suppresses a PlexA- dependent phenotype. However, increasing the levels of Gyc76C enhances this PlexA GOF phenotype. Finally, a Gyc76C transgene lacking a key conserved aspartate residue required for cyclase catalytic activity does not rescue either the gyc76C embryonic motor axon guidance defects or the lethality associated with gyc76C mutants and appears to function in a dominant-negative manner. Taken together, our results link Gyc76C to the proper generation of neuromuscular connectivity in Drosophila through its role in mediating semaphorin-plexin signaling events associated with axonal repulsion. In addition, our results strongly suggest that cGMP production is critical for Gyc76C participation in Sema1a neuronal signaling events.
Gyc76C and cyclic nucleotide modulation of semaphorin guidance
Initial in vitro observations demonstrating the importance of cGMP levels in semaphorin-mediated repulsion showed that increasing cGMP signaling reverses the repulsive signal from the secreted vertebrate semaphorin Sema3A, resulting in Sema3A acting as an attractant in the single growth cone steering assay (Song et al., 1998). Recent studies show that Sema3A growth cone collapse requires increased cGMP signaling and also that cAMP signaling acts in opposition to cGMP signaling in the modulation of Sema3A-mediated growth cone collapse (Dontchev and Letourneau, 2002; Chalasani et al., 2003). Support for cAMP signaling cascades modulating semaphorin-mediated repulsion in vivo is provided by a demonstration that the A-kinase anchoring protein Nervy serves to antagonize Sema-1a-mediated axonal repulsion in Drosophila motor axons. Presumably, Nervy acts by localizing cAMP activation of PKA to the Plexin receptor and decreases Sema-1a repulsive signaling (Terman and Kolodkin, 2004). Our identification of Gyc76C as a positive effector in vivo of Sema-1a-PlexA-mediated repulsion is consistent with these Sema3A growth cone collapse studies. A model recently proposed for cyclic nucleotide modulation of netrin-1-mediated attraction and repulsion provides insight into how cGMP might effect semaphorin-mediated steering, collapse, and in vivo axonal repulsion. Using the in vitro growth cone steering assay, Hong and colleagues (Nishiyama et al., 2003) show that the [cAMP]/[cGMP] ratio determines whether netrin-1 acts in an attractive or a repulsive manner: high ratios promote attraction, whereas lower ratios promote repulsion. Importantly, a basal level of cGMP signaling is required for both netrin-mediated attractive and repulsive responses in this system. Although it remains to be determined, it is tempting to speculate that, like the observations for netrin-1-mediated guidance, the [cAMP]/[cGMP] ratio also serves to modulate semaphorin signaling events. In Drosophila motor axons, Gyc76C and Nervy could function antagonistically to regulate Sema-1a signaling in this manner. Gyc76C production of cGMP would lower a [cAMP]/[cGMP] ratio and thus promote repulsion, whereas increases in cAMP levels would decrease repulsion through PKA tethered to PlexA by Nervy. A loss of Gyc76C altogether would result in abolition of Sema-1a repulsion because of a cGMP requirement for any guidance response, and this is what we observe in our gyc76C mutants. Future experiments will determine how raising or lowering Gyc76C activity affects the guidance response to Sema-1a in vivo.
Specificity of rGCs in semaphorin signaling
We describe here a role for a receptor-type guanylyl cyclase in axon guidance as an effector of transmembrane Sema1a axonal repulsion. Soluble guanylyl cyclases in both vertebrates and invertebrates have been implicated in axonal (Seidel and Bicker, 2000; Gibbs et al., 2001) and dendritic (Polleux et al., 2000) guidance. However, in our GOF genetic screen for Sema-1a signaling components, we assayed genomic regions containing genes encoding all of the identified Drosophila soluble guanylyl cyclase subunits, including one known to be expressed in the nervous system (Tomancak et al., 2002), yet found that heterozygosity at these loci did not suppress or enhance the Sema-1a GOF phenotype (data not shown). This may reflect a requirement for cGMP production at or near the PlexA receptor to provide a local increase in cGMP levels essential for semaphorin-mediated axonal repulsion and suggests that basal cGMP signaling provided by soluble gyanylyl cyclases is not essential for semaphorin-mediated repulsion. Our initial genetic screen covered an additional two of the seven Drosophila rGCs, however, neither of the deficiencies that remove these rGCs genetically interacted with our Sema-1a GOF phenotype. Taken together, these results from our genetic screen suggest that Gyc76C is an integral component of the semaphorin signaling cascade and that cGMP production by other sources may not contribute to this repulsion. These results also motivate future experiments to investigate specific interactions between Gyc76C and PlexA.
Vertebrate receptor guanylyl cyclases that have a single transmembrane domain like Gyc76C are best known for their roles as receptors for natriuretic peptides that regulate blood pressure and volume and also for their role in the visual phototransduction cascade (Wedel and Garbers, 2001). The other vertebrate rGCs, however, have no known ligands or functions. In addition, very little is known about what roles, if any, these vertebrate rGCs play during neural development. It will be of great interest to investigate whether any vertebrate rGCs participate in semaphorin repulsive signaling.
Potential roles for the other domains of Gyc76C
Because Gyc76C is a multidomain protein, it is likely that regions other than the cyclase domain are important for its function. Interestingly, like the transmembrane protein Off-track, which is also required for Sema1a-mediated motor axon repulsion in Drosophila (Winberg et al., 2001), Gyc76C contains a catalytically inactive KHD. In the vertebrate receptor guanylyl cyclase GC-A, this region has been shown to play a regulatory role by inhibiting the catalytic cyclase domain (Chinkers and Garbers, 1989). The KHD of Gyc76C, or possibly Off-track, may function as an important modulator of cyclase activity.
The portion of Gyc76C that is C terminal to the conserved cyclase domain is unique among rGC family members; it is much longer than the same region in other rGCs and shares no amino acid similarity with these regions or with sequences of any known proteins. However, the last four amino acids of Gyc76C fit the consensus for a PDZ (PSD-95, Discs-large, zona occludens-1) domain binding motif (Suh et al., 2001). A similar motif is also found in MICAL, another component of the Sema-1a signaling cascade (Terman et al., 2002), raising the possibility that, as has been observed for other assemblages of signaling components (Li and Montell, 2000), PDZ domain-containing scaffolding proteins may serve an important role in semaphorin signaling.
Gyc76C may provide a direct physical link between the leading edge of the growth cone and the motile machinery of the actin cytoskeleton. Vertebrate rGCs in photoreceptors are able to bind actin filaments (Hallett et al., 1996), and the C-terminal domains of intestinal rGCs have also been implicated in interactions with the actin cytoskeleton (Kuno et al., 1986; Waldman et al., 1986). Perhaps the large C-terminal extension of Gyc76C functions in a similar manner to bridge the regions of signal reception and output. Whether or not Gyc76C cyclase activity is ligand gated remains unknown, and like all other Drosophila rGCs and the majority of vertebrate rGCs, Gyc76C is an orphan receptor. Future experiments will address whether Sema-1a triggers Gyc76C catalytic activity and also whether Gyc76C is indeed part of the receptor complex for Sema-1a.
In conclusion, using a novel genetic screening paradigm for identifying semaphorin signaling cascade components, we found an in vivo link between Sema-1a-mediated repulsive guidance and cGMP signaling pathways. Characterization of other candidates from this screen will likely provide additional insight into the mechanisms of repulsive axon guidance signaling.
Footnotes
This work was supported by National Institutes of Health (NIH)-National Institute of Mental Health Grant MH069787 (J.R.T.), Paralyzed Veterans of America Spinal Cord Research Foundation Grant 2050 (J.R.T.), the Kirsch Foundation (A.L.K.), and NIH-National Institute of Neurological Disorders and Stroke Grant NS35165 (A.L.K.). We thank Katie Peer for expert technical assistance; David Ginty, Natalia Glebova, David Kantor, Rejji Kuruvilla, Tianyi Mao, and Jeroen Pasterkamp for helpful comments on this manuscript; David Forsthoefel and Mark Seeger for the enaGC10 fly line; the Bloomington Stock Center, the Berkeley Drosophila Gene Disruption Project, and the Berkeley Drosophila Genome Project for fly lines and cDNAs; and members of the Kolodkin and Ginty laboratories for helpful discussions.
Correspondence should be addressed to Dr. Alex L. Kolodkin, Department of Neuroscience, The Johns Hopkins University School of Medicine, 1001 Preclinical Teaching Building, 725 North Wolfe Street, Baltimore, MD 21205. E-mail: kolodkin@jhmi.edu.
H.-H. Yu's present address: Division of Basic Sciences, Fred Hutchinson Cancer Research Center, 1100 Fairview Avenue North, Seattle, WA 98109.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246639-11$15.00/0
References
- Araujo SJ, Tear G (2003) Axon guidance mechanisms and molecules: lessons from invertebrates. Nat Rev Neurosci 4: 910-922. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Matsuno K, Fortini M (1995) Notch signaling. Science 268: 225-232. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Lewis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, Hoskins RA, Spradling AC (2004) The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics, in press. [DOI] [PMC free article] [PubMed]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401-415. [DOI] [PubMed] [Google Scholar]
- Chalasani SH, Sabelko KA, Sunshine MJ, Littman DR, Raper JA (2003) A chemokine, SDF-1, reduces the effectiveness of multiple axonal repellents and is required for normal axon pathfinding. J Neurosci 23: 1360-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinkers M, Garbers DL (1989) The protein kinase domain of the ANP receptor is required for signaling. Science 245: 1392-1394. [DOI] [PubMed] [Google Scholar]
- Dontchev VD, Letourneau PC (2002) Nerve growth factor and semaphorin 3A signaling pathways interact in regulating sensory neuronal growth cone motility. J Neurosci 22: 6659-6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs SM, Truman JW (1998) Nitric oxide and cyclic GMP regulate retinal patterning in the optic lobe of Drosophila Neuron 20: 83-93. [DOI] [PubMed] [Google Scholar]
- Gibbs SM, Becker A, Hardy RW, Truman JW (2001) Soluble guanylate cyclase is required during development for visual system function in Drosophila J Neurosci 21: 7705-7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett MA, Delaat JL, Arikawa K, Schlamp CL, Kong F, Williams DS (1996) Distribution of guanylate cyclase within photoreceptor outer segments. J Cell Sci 109: 1803-1812. [DOI] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF (2003) Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci 26: 509-563. [DOI] [PubMed] [Google Scholar]
- Kidd T, Bland KS, Goodman CS (1999) Slit is the midline repellent for the Robo receptor in Drosophila Cell 96: 785-794. [DOI] [PubMed] [Google Scholar]
- Kuno T, Kamisaki Y, Waldman SA, Gariepy J, Schoolnik G, Murad F (1986) Characterization of the receptor for heat-stable enterotoxin from Escherichia coli in rat intestine. J Biol Chem 261: 1470-1476. [PubMed] [Google Scholar]
- Landgraf M, Bossing T, Technau GM, Bate M (1997) The origin, location, and projections of the embryonic abdominal motorneurons of Drosophila J Neurosci 17: 9642-9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Montell C (2000) TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. J Cell Biol 150: 1411-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Yoon J, Burg M, Chen L, Pak WL (1995) Molecular characterization of two Drosophila guanylate cyclases expressed in the nervous system. J Biol Chem 270: 12418-12427. [DOI] [PubMed] [Google Scholar]
- Lucas KA, Pitari GM, Kazerounian S, Ruiz-Stewart I, Park J, Schulz S, Chepenik KP, Waldman SA (2000) Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 52: 375-414. [PubMed] [Google Scholar]
- McNeil L, Chinkers M, Forte M (1995) Identification, characterization, and developmental regulation of a receptor guanylyl cyclase expressed during early stages of Drosophila development. J Biol Chem 270: 7189-7196. [DOI] [PubMed] [Google Scholar]
- Ming G-L, Song H-J, Berninger S, Holt CE, Tessier-Lavigne M, Poo M-M (1997) cAMP-dependent growth cone guidance by netrin-1. Neuron 19: 1225-1235. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K (2003) Cyclic AMP/GMP-dependent modulation of Ca2+ channels sets the polarity of nerve growth-cone turning. Nature 424: 990-995. [DOI] [PubMed] [Google Scholar]
- Polleux F, Morrow T, Ghosh A (2000) Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature 404: 567-573. [DOI] [PubMed] [Google Scholar]
- Pulido D, Campuzano S, Koda T, Modolell J, Barbacid M (1992) Dtrk, a Drosophila gene related to the trk family of neurotrophin receptors, encodes a novel class of neural cell adhesion molecule. EMBO J 11: 391-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, Benz WK, Engels WR (1988) A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118: 461-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Werner M, Heppenstall PA, Henning M, More MI, Kuhbandner S, Lewin GR, Hofmann F, Feil R, Rathjen FG (2002) cGMP-mediated signaling via cGKIalpha is required for the guidance and connectivity of sensory axons. J Cell Biol 159: 489-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M, Tear G, Ferres-Marco D, Goodman CS (1993) Mutations affecting growth cone guidance in Drosophila: genes necessary for guidance toward or away from the midline. Neuron 10: 409-426. [DOI] [PubMed] [Google Scholar]
- Seidel C, Bicker G (2000) Nitric oxide and cGMP influence axonogenesis of antennal pioneer neurons. Development 127: 4541-4549. [DOI] [PubMed] [Google Scholar]
- Song HJ, Poo MM (1999) Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol 9: 355-363. [DOI] [PubMed] [Google Scholar]
- Song H-j, Ming G-l, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M-m (1998) Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science 281: 1515-1518. [DOI] [PubMed] [Google Scholar]
- Suh PG, Hwang JI, Ryu SH, Donowitz M, Kim JH (2001) The roles of PDZ-containing proteins in PLC-beta-mediated signaling. Biochem Biophys Res Commun 288: 1-7. [DOI] [PubMed] [Google Scholar]
- Tear G, Harris R, Sutaria S, Kilomanski K, Goodman CS, Seeger MA (1996) Commissureless controls growth cone guidance across the CNS midline in Drosophila and encodes a novel membrane protein. Neuron 16: 501-514. [DOI] [PubMed] [Google Scholar]
- Terman JR, Kolodkin AL (2004) Nervy links protein kinase a to plexin-mediated semaphorin repulsion. Science 303: 1204-1207. [DOI] [PubMed] [Google Scholar]
- Terman JR, Mao T, Pasterkamp RJ, Yu HH, Kolodkin AL (2002) MICALs, a family of conserved flavoprotein oxidoreductases, function in Plexin-mediated axonal repulsion. Cell 109: 887-900. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Goodman CS (1996) The molecular biology of axon guidance. Science 274: 1123-1133. [DOI] [PubMed] [Google Scholar]
- Thompson DK, Garbers DL (1995) Dominant negative mutations of the guanylyl cyclase-A receptor. Extracellular domain deletion and catalytic domain point mutations. J Biol Chem 270: 425-430. [DOI] [PubMed] [Google Scholar]
- Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, Rubin GM (2002) Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol 3: 0088.1-0088.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanVactor D, Sink H, Fambrough D, Tsoo R, Goodman CS (1993) Genes that control neuromuscular specificity in Drosophila Cell 73: 1137-1153. [DOI] [PubMed] [Google Scholar]
- Waldman SA, Kuno T, Kamisaki Y, Chang LY, Gariepy J, O'Hanley P, Schoolnik G, Murad F (1986) Intestinal receptor for heat-stable enterotoxin of Escherichia coli is tightly coupled to a novel form of particulate guanylate cyclase. Infect Immun 51: 320-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel B, Garbers D (2001) The guanylyl cyclase family at Y2K. Annu Rev Physiol 63: 215-233. [DOI] [PubMed] [Google Scholar]
- Wills Z, Bateman J, Korey CA, Comer A, Van Vactor D (1999) The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron 22: 301-312. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Noordermeer JN, Tamagnone L, Comoglio PM, Spriggs MK, Tessier-Lavigne M, Goodman CS (1998) Plexin A is a neuronal semaphorin receptor that controls axon guidance. Cell 95: 903-916. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Tamagnone L, Bai J, Comoglio PM, Montell D, Goodman CS (2001) The transmembrane protein Off-track associates with Plexins and functions downstream of semaphorin signaling during axon guidance. Neuron 32: 53-62. [DOI] [PubMed] [Google Scholar]
- Yao KM, White K (1994) Neural specificity of elav expression: defining a Drosophila promoter for directing expression to the nervous system. J Neurochem 63: 41-51. [DOI] [PubMed] [Google Scholar]
- Yu HH, Araj HH, Ralls SA, Kolodkin AL (1998) The transmembrane semaphorin Sema I is required in Drosophila for embryonic motor and CNS axon guidance. Neuron 20: 207-220. [DOI] [PubMed] [Google Scholar]
- Yu HH, Huang AS, Kolodkin AL (2000) Semaphorin-1a acts in concert with the cell adhesion molecules fasciclin II and connectin to regulate axon fasciculation in Drosophila Genetics 156: 723-731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Schnitzler A, Agapite J, Schwartz LM, Steller H, Nambu JR (1997) Cooperative functions of the reaper and head involution defective genes in the programmed cell death of Drosophila central nervous system midline cells. Proc Natl Acad Sci USA 94: 5131-5136. [DOI] [PMC free article] [PubMed] [Google Scholar]