

Abstract
Multiple lines of evidence implicate β-amyloid (Aβ) in the pathogenesis of Alzheimer's disease (AD), but the mechanisms whereby Aβ is involved remain unclear. Addition of Aβ to the extracellular space can be neurotoxic. Intraneuronal Aβ42 accumulation is also associated with neurodegeneration. We reported previously that in Tg2576 amyloid precursor protein mutant transgenic mice, brain Aβ42 localized by immunoelectron microscopy to, and accumulated with aging in, the outer membranes of multivesicular bodies, especially in neuronal processes and synaptic compartments. We now demonstrate that primary neurons from Tg2576 mice recapitulate the in vivo localization and accumulation of Aβ42 with time in culture. Furthermore, we demonstrate that Aβ42 aggregates into oligomers within endosomal vesicles and along microtubules of neuronal processes, both in Tg2576 neurons with time in culture and in Tg2576 and human AD brain. These Aβ42 oligomer accumulations are associated with pathological alterations within processes and synaptic compartments in Tg2576 mouse and human AD brains.
Keywords: Alzheimer, amyloid, synapse, oligomer, transgenic, pathology
Introduction
Alzheimer's disease (AD) neuropathology is characterized by loss of synapses and neurons and by accumulation of senile plaques and neurofibrillary tangles. The β-amyloid peptide (Aβ) is the principal component of plaques and was further linked to AD by familial AD (FAD) mutations in the amyloid precursor protein (APP) near Aβ cleavage sites (Selkoe, 2001; Golde, 2003). Transgenic mice harboring human APP with FAD mutations develop AD-like amyloidosis and synaptic, oxidative, physiological, and behavioral abnormalities (Holcomb et al., 1998; Chapman et al., 1999; Moechars et al., 1999; Pratico et al., 2001).
Extracellular addition of Aβ to cultured neurons or injection into brain can be neurotoxic (Yankner et al., 1989; Geula et al., 1998). Evidence also indicates that accumulation of Aβ42, the first Aβ species found in plaques, occurs intracellularly (Echeverria and Cuello, 2002; Tabira et al., 2002) in AD (Gouras et al., 2000; D'Andrea et al., 2001), Down's syndrome (Gyure et al., 2001; Busciglio et al., 2002; Mori et al., 2002), and transgenic mice that develop Aβ plaques (Wirths et al., 2001; Oddo et al., 2003; Sheng et al., 2003). Additionally, extracellular Aβ42 can increase levels of intraneuronal Aβ42 (Glabe, 2001). We demonstrated in Tg2576 transgenic mice that intraneuronal Aβ42 accumulates with aging in the outer limiting membranes of multivesicular bodies (MVBs), especially in distal processes and synaptic compartments (Takahashi et al., 2002). Moreover, before and with the development of plaques, marked accumulation of Aβ42 in MVBs was associated with subcellular morphological alterations within processes and synapses. Thus, understanding Aβ biology within endosomal vesicles, such as MVBs, may be important in unraveling the mechanism of AD pathogenesis. In the present study, we set out to establish an in vitro primary neuronal culture system in which to investigate endosomal Aβ accumulation. Analogous to increases of intracellular Aβ42 reported by ELISA in NT2 cells with time in culture (Skovronsky et al., 1998), we now report intracellular accumulation of Aβ42 within cultured primary neurons.
Increasing evidence indicates that the biophysical state of the most toxic form of Aβ may be its protofibrillar rather than β-pleated conformation (Selkoe, 2001; Klein, 2002). Levels of soluble, and potentially oligomeric Aβ42, rather than plaques, are the best Aβ correlate of cognitive dysfunction in AD (McLean et al., 1999; Naslund et al., 2000). In vitro evidence indicates that Aβ monomers are converted to oligomers before aggregation into β-pleated amyloid (Kirkitadze et al., 2002). It was reported that Aβ oligomers are preferentially generated intracellularly rather than extracellularly (Walsh et al., 2000). We now present evidence that Aβ42 oligomers arise especially within processes of cultured Tg2576 primary neurons with time in culture. Moreover, in Tg2576 and human AD brain, accumulation of Aβ42 oligomers within neuronal processes and synaptic profiles is associated with pathological alterations. Both in vitro and in vivo we observe a redistribution of Aβ42, as it aggregates from monomer to oligomer, from the outer membranes of endosomal vesicles– MVBs, where it normally resides, to the inner membranes of morphologically abnormal endosomal organelles and to microtubules. Our data further support the scenario that Aβ42 aggregation occurs within neuronal processes and results in synaptic degeneration.
Materials and Methods
Antibodies. MBC42, kindly provided by Dr. Haruyasu Yamaguchi (Gunma University, Gunma, Japan), is a monoclonal antibody (mAb) to the C terminus of Aβ42 and has been characterized previously (Takahashi et al., 2002). Results with MBC42 were additionally confirmed using a commercially available polyclonal anti-Aβ42 antibody (Chemicon, Temecula, CA) [characterized in Kamal et al. (2001)]. Aβ42 oligomers were detected using the rabbit polyclonal antibody, M16, kindly provided by Dr. Charles Glabe (University of California, Irvine, CA); M16 was raised against synthetic Aβ1–42 and preferentially recognizes Aβ42 oligomers on Western blot (Yang et al., 1995). For biochemical analyses, Aβ, APP C-terminal fragments, and full-length APP were recognized by antibodies 4G8 (directed at Aβ17–24; Signet Laboratories, Dedham, MA) and 6E10 (directed at Aβ5–10; Signet Laboratories). Polyclonal rabbit Ab against lysobisphosphatidic acid (LBPA) was kindly provided by Dr. J. Gruenberg (University of Geneva, Geneva, Switzerland) (Kobayashi et al., 1998). Anti-α-tubulin Ab was from Sigma (St. Louis, MO), and anti-neuron-specific nuclear protein (NeuN) Ab was from Chemicon.
Preparation of aggregated synthetic Aβ42 peptides. Fibrillar Aβ42 was made according to the method described by Fezoui et al. (2000). Briefly, lyophilized Aβ42 synthetic peptides (American Peptide Company, Sunnyvale, CA) were dissolved in 20 mm NaOH, pH 10.5, at a concentration of 1 mg/ml, sonicated, and lyophilized. NaOH-treated Aβ42 was dissolved in water at a concentration of 1 mg/ml and filtered through a 0.22 μm Ultrafree-MC filter (Millipore, Bedford, MA). Aβ peptide (0.5 mg/ml) was buffered at a final concentration of 50 mm phosphate, 100 mm sodium chloride and incubated for 54 hr. Two hundred nanograms of aggregated Aβ42 peptide were loaded in each lane for Western blot (see below).
Mice. Tg2576 mice harboring human APP with the Swedish 670/671 mutation (Hsiao et al., 1996) were used in this study, as described previously (Takahashi et al., 2002). All methods were approved by the Weill Medical College of Cornell University Institutional Animal Care and Use Committee and conformed to National Institutes of Health guidelines.
Human brain tissue. Human cortical brain tissue from neurologically normal controls (n = 2; ages 44 and 54) and subjects with AD (n = 2; ages 54 and 62) was obtained from the Department of Pathology, Weill Medical College of Cornell University, as a result of neurosurgical procedures unrelated to this study, as described previously (Takahashi et al., 2002).
Primary murine neuronal cultures. Primary neuronal cultures were derived from cerebral cortex and hippocampus of embryonic day (E) 16 Tg2576 or wild-type mice, as described previously (Gouras et al., 2000) with modifications. Cortices and hippocampi were incubated with 0.25% trypsin and then triturated in glass pipettes until neurons were dissociated. Neurons were plated in serum-free Neurobasal media with B27 supplement (Invitrogen, Gaithersburg, MD) and 2 mm l-glutamine on poly-d-lysine-treated (0.1 mg/ml; Sigma) 100 mm dishes. To maintain elevated levels of extracellular Aβ, media was not changed. No differences in cell viability were detected by trypan blue staining or lactate dehydrogenase release between wild-type and transgenic neurons at 12 or 19 d in culture. Primary neurons are viable for >3–4 weeks under our culturing conditions. For immunofluorescence, ∼4 × 105 neurons were plated per well in six-well plates containing coverslips, and for immunoprecipitation–Western blot, ∼3.3 × 106 neurons were plated per 100-mm-diameter dish. Genotyping was performed on cerebellum from the same embryo.
Immunofluorescence microscopy. Immunofluorescence was performed on E16 primary neuronal cultures grown for 5, 12, or 19 d in vitro on poly-d-lysine-coated round glass coverslips (EM Science). Cultures were fixed for 20 min with 4% paraformaldehyde in PBS containing 0.12 m sucrose, permeabilized in 0.3% Triton X-100 in PBS for 5 min, and rinsed twice in PBS. The cells were preincubated in 10% BSA in PBS for 1 hr at 37°C, exposed to primary antibodies (diluted in 1% BSA in PBS) overnight at 4°C, and then rinsed in PBS and incubated with secondary antibodies for 1 hr at 37°C. MBC42 (1:150), M16 (1:1,000), LBPA (1:50), and α-tubulin (1:300) were used as primary antibodies. Fluorescent secondary antibodies were Alexa 488 goat anti-mouse and Alexa 546 goat anti-rabbit (1:200; Molecular Probes, Eugene, OR). Immunoreactivity was captured with a Leica DM IRB microscope with a Hamamatsu digital camera or a Zeiss LSM 510 confocal microscope and analyzed using Meta-Morph software (Universal Imaging, West Chester, PA) for quantitative analysis. To keep neuronal cell numbers constant for comparisons of Tg2576 neurons at 12 versus 19 d in culture, several coverslips were cocultured in the same well. We randomly picked five different areas from a coverslip for analysis. Evaluation was done using a 20× objective on five different cell bodies and processes, and average intensities were measured in selected areas. The intensities of processes were divided by the intensities of cell bodies. The process-to-cell body intensity ratio in Tg2576 neurons was compared between 12 and 19 d in culture using Student's t test for statistical analysis. Data were expressed as the mean ± SEM, and the significance threshold was p < 0.05.
Immunogold electron microscopy. Mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused via the ascending aorta with 3.75% acrolein (Polyscience, Warrington, PA) and 2% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. Human AD brain biopsy tissue was rapidly immersion-fixed with 1.875% acrolein and 2% paraformaldehyde in 0.1 m PB as described previously (Takahashi et al., 2002). Brain tissue was cut (40 μm thick) on a vibrating microtome and treated with 1% sodium borohydride, as described previously (Takahashi et al., 2002). For immunogold labeling, free-floating sections were labeled with MBC42 (1:50), M16 (1:100), or Chemicon Aβ42 (1:50) antibodies by the immunogold–silver procedure of Chan et al. (1990). Sections were then incubated in secondary antibodies conjugated to 1 nm gold particles (Amersham Biosciences, Arlington Heights, IL) in 0.01% gelatin and 0.08% BSA in PBS. The conjugated gold particles were enhanced by treatment with silver solution (IntenSE, Amersham Biosciences). Sections were fixed in 2% osmium tetroxide in PB, embedded in EMBed 812, sectioned (65–76 nm thick), and counterstained with uranyl acetate and Reynolds' lead citrate (Milner et al., 1998). Final preparations were examined with a Philips CM10 electron microscope. Morphological terminology is consistent with Peters et al. (1991). Final illustrations were generated from a high-resolution digital imaging CCD camera system (Advanced Microscopy Techniques, Danvers, MA) and processed using Adobe Photoshop 7.0 (Adobe System, Mountain View, CA).
For cell immunoelectron microscopy (EM), primary neurons were fixed for 45 min in 0.1% glutaraldehyde, 2% paraformaldehyde, and 0.005% saponin in 0.1 m PB at room temperature. Neurons were then rinsed in 0.1 m PB with 0.005% saponin and treated with 1% sodium borohydride. For immunogold labeling, the neurons were labeled with either MBC42 or M16 Ab by the immunogold–silver procedure as described above.
Immunoprecipitation–Western blot for Aβ. Cultured primary neurons from Tg2576 mice were assayed for determination of Aβ levels as described previously for cell lysates, using 4G8 for immunoprecipitation and 6E10 for blotting (Takahashi et al., 2002). To maintain equivalent neuronal numbers and density, equal numbers of dissociated Tg2576 neurons from a given mouse brain were cultured in three separate dishes, and dishes of each Tg2576 pup were cultured for 12 or 19 d and later compared with neurons derived from an equivalent set of cultures derived from different Tg2576 mouse pups. Neurons were lysed in 6% SDS, vortexed, and heated at 95°C for several minutes, followed by sonication. Equivalent amounts of protein, as determined by the Bradford technique (Bio-Rad Laboratories, Hercules, CA), were spun at 15,000 × g, and supernatants were adjusted to 190 mmol/l NaCl, 50 mmol/l Tris-HCl, pH 8.3, 6 mmol/l EDTA, and 2.5% Triton X-100 and incubated overnight with Ab 4G8 (Signet Laboratories), followed by addition of secondary rabbit anti-mouse Ab (Cappel, West Chester, PA) for 1 hr and then addition of protein A-Sepharose beads (Amersham Biosciences) for 2 hr (all at 4°C). Samples were run on 10–20% Tris-Tricine SDS PAGE gels followed by electrophoretic transfer to polyvinylidene-difluoride membrane (Millipore). Polyvinylidene-difluoride membranes were boiled in PBS for 5 min, followed by Western blotting with 6E10 for human Aβ and visualization after enhanced chemiluminescence on Kodak X-OMAT AR5 film (Eastman-Kodak, Rochester, NY).
All experiments were repeated at least three times, and different primary cultures derived from at least three different embryos were averaged. For characterization of antibodies against Aβ42, Aβ42 synthetic peptides (America Peptide Company) were prepared as described above, and 200 ng was loaded into each lane. We used the following antibodies for blotting: MBC42 (1:300), 6E10 (1:1000), M16 (1:1000), anti-α-tubulin (1:1000), and anti-NeuN (1:1500) by a semidry blotting method. Intensity of bands was analyzed by Scion Image, followed by Student's t test for statistical analysis. Data were expressed as the mean ± SEM, and significance was assessed at p < 0.03.
Results
Aβ42 immunofluorescence in cultured neurons has a vesicular pattern of staining
We previously used immunogold EM to localize Aβ42 especially to the outer membranes of MVBs and smaller tubulovesicular organelles within the cytoplasm of neurons in normal, young mouse, rat, and human brains (Takahashi et al., 2002). Importantly, the specificity of our immuno-EM was confirmed by lack of labeling in APP knock-out mouse brain (Takahashi et al., 2002). In the present study, we used well characterized monoclonal antibodies against Aβ42, which we had used previously for immuno-EM, for immunofluorescence microscopy of cultured primary neurons. We observed a vesicular pattern of Aβ42 staining (Fig. 1A), consistent with Aβ42 immunofluorescence in cultured Down's syndrome astrocytes (Busciglio et al., 2002) and neurons infected with Semliki Forest virus harboring human APP (Runz et al., 2002). We obtained analogous immunofluorescence using monoclonal (MBC42) or commercially available polyclonal anti-Aβ42 antibodies (Chemicon) (Kamal et al., 2001). Colocalization with markers of subcellular organelles indicated significant overlap with markers of late endosomes, such as LBPA (Fig. 1A). LBPA has been reported previously to label late endosomal vesicles by immunofluorescence and immuno-EM (Kobayashi et al., 1998; Mobius et al., 2003).
Figure 1.

Aβ42 within cultured neurons localizes to late endosomal vesicles and increases with time in culture, especially within processes. A, Confocal immunofluorescence microscopy of primary neurons at 19 d in culture. Aβ42 fluorescence, with the Chemicon Aβ42 Ab, partially localized with LBPA, which is known to be associated especially with late endosomes. Scale bar, 10 μm. B, Immunofluorescence of Tg2576 mouse primary neurons stained with mAb MBC42 against the C terminus of Aβ42. Note the Aβ42 increases in Tg2576 (Tg) neurons between 12 d (left) and 19 d (right) in culture. There is less Aβ42 immunofluorescence in wild-type (WT) neurons cultured for 19 d (below). Scale bar, 50 μm. C, Tg2576 primary neurons: ratio of Aβ42 immunofluorescence intensity in neuronal processes divided by that in cell bodies (processes/cell bodies) between 12 and 19 d in culture, with the ratio standardized to 1.0 for 12 d in culture. The Aβ42 ratio of processes/cell bodies increased 1.65 ± 0.06-fold between 12 and 19 d in culture; p < 0.001. D, Representative images of α-tubulin immunofluorescence in Tg2576 mouse primary neurons at 12 and 19 d in culture. There was no significant increase inα-tubulin fluorescence, compared with that of Aβ42 fluorescence (B), within processes between 12 and 19 d in culture. Scale bar, 50 μm. E, Cell body Aβ42 detected by immunogold electron microscopy using Ab MBC42. Representative Aβ42 in the outer membrane of a MVB, containing characteristic internal vesicles, in the cell body of a Tg2576 primary neuron at 21 d in culture. Scale bar, 500 nm. F, Aβ42 immuno-EM of a neurite process. Representative image of Aβ42 accumulation in outer membranes and rarely inner vesicles of MVBs in a neurite process of a Tg2576 primary neuron at 21 d in culture, detected by immunogold EM with MBC42. Note also an Aβ42 gold particle at the plasma membrane (arrowhead). Scale bar, 500 nm.
Aβ42 accumulates and redistributes to neuronal processes within Tg2576 neurons with time in culture
To determine whether intraneuronal Aβ42 increases with time in culture and to assess for changes in Aβ42 distribution within neurons, we used immunofluorescence microscopy for Aβ42 in Tg2576 neurons at 12 and 19 d in culture. As expected, neurons overexpressing the human APP mutant transgene had increased Aβ42 immunofluorescence compared with wild-type neurons at 19 d in culture (Fig. 1B). Remarkably, we observed with time in culture a gradual shift of Aβ42 immunofluorescence from cell bodies to neurites in Tg2576 neurons (Fig. 1B). Primary neurons at 19 d in culture had a more extensive network of processes than at 12 d. In addition, the number of processes was greater in “older” neurons, as was the intensity of Aβ42 staining. The ratio of Aβ42 fluorescence intensity per unit area of processes to that of cell bodies increased 1.65 ± 0.06-fold (p < 0.001) in Tg2576 neurons between 12 and 19 d in culture (Fig. 1C). Immunofluorescence for α-tubulin confirmed an increase in the network of processes between 12 and 19 d in culture, but in contrast to Aβ42, there was no significant increase inα-tubulin immunofluorescence within processes (Fig. 1D). To determine the subcellular site(s) of these Aβ42 increases in primary neurons, we used immuno-gold EM. Aβ42 gold particles were most prominent in the outer membranes of endosomal vesicles, consistent with MVBs, in the cytoplasm of Tg2576 primary neurons cultured for 3 weeks, both in cell bodies (Fig. 1E) and within processes (Fig. 1F).
Aβ accumulates in Tg2576 primary neurons with time in culture by Western blot
To confirm biochemically whether intraneuronal levels of Aβ in Tg2576 cultured primary neurons increase with time in culture, we used immunoprecipitation using anti-Aβ Ab 4G8 and Western blot using anti-Aβ Ab 6E10. Immunoprecipitation–Western for human Aβ demonstrated increases in levels of intracellular Aβ with time in culture (Fig. 2A). Specifically, between 12 and 19 d in culture there was a 3.36 ± 0.51-fold increase (p < 0.03) in intracellular Aβ levels by densitometry (Fig. 2B). There was a less pronounced 1.46 ± 0.29-fold increase, not reaching statistical significance (p = 0.19), in levels of full-length APP between 12 and 19 d in culture (Fig. 2B). There were no significant differences in levels of NeuN or α-tubulin between 12 and 19 d in culture (Fig. 2A).
Figure 2.

Biochemical analysis of intraneuronal Aβ accumulation in primary neurons with time in culture. A, Increased levels of Aβ with time in culture in primary neurons from Tg2576 mouse by immunoprecipitation–Western blot. Equal amounts of 12- and 19-d-old cultured neuron cell lysates were immunoprecipitated with 4G8 and immunoblotted with 6E10 for determination of Aβ and full-length APP levels. After immunoprecipitation with 4G8, supernatants were immunoblotted with antibodies against NeuN or α-tubulin. B, Aβ levels increased 3.36 ± 0.51-fold from 12 to 19 d in culture (p < 0.03). A slight increase in APP levels (1.46 ± 0.29-fold) from 12 to 19 d in culture was not statistically significant (p = 0.19). Data are expressed as mean ± SEM of three independent experiments. C, Aggregation-state specificity of antibodies against Aβ42. Aggregated Aβ42 (200 ng) was separated by gel electrophoresis, transferred to polyvinylidene-difluoride membrane, and blotted with MBC42, 6E10, and M16. MBC42 detected especially Aβ42 monomers and some dimers but did not recognize oligomers. 6E10 recognized most aggregation states of Aβ42, being most sensitive for monomers. M16 mainly recognized larger Aβ42 oligomers, with relatively minor reactivity to Aβ monomers.
Aggregation state specificity of anti-Aβ antibodies
Increasing numbers of reports are emphasizing the potentially critical role of Aβ oligomers rather than Aβ monomers or Aβ fibrils in AD (Hardy and Selkoe, 2002; Klein, 2002). Because anti-Aβ antibodies have selective affinities for the conformational state of Aβ, a given Ab will not efficiently visualize the total pool of Aβ. To characterize which conformations of Aβ42 are recognized by the anti-Aβ antibodies that we used, we blotted preaggregated synthetic Aβ42 peptides with MBC42 and 6E10. MBC42 detected mainly Aβ42 monomers and also some Aβ42 dimers without detecting larger Aβ42 oligomers (Fig. 2C, top). 6E10 detected monomeric Aβ42 but also a wide variety of Aβ42 oligomers (Fig. 2C, middle). To investigate changes in Aβ42 oligomers we used Ab M16, which has been previously well characterized as binding Aβ42 oligomers (Yang et al., 1995). We also found that M16 mainly detected oligomers of Aβ42 (Fig. 2C, bottom).
Aβ42 oligomerization within processes of Tg2576 neurons with time in culture
Because the monoclonal MBC42 predominantly detects Aβ42 monomers (Fig. 1), we used the M16 Ab, which predominantly detects Aβ42 oligomers, to more fully investigate Aβ42 increases in Tg2576 neurons. We studied wild-type and Tg2576 neurons with time in culture by M16 immunofluorescence microscopy. M16 staining was not observed in wild-type mouse neurons at 5, 12, and 19 d in culture, or in primary neurons from Tg2576 mice cultured for 5 d (data not shown). In Tg2576 neurons at 12 d in culture, Aβ42 oligomers were observed in cell bodies and rarely in processes (Fig. 3A, left column). In contrast, at 19 d in culture, Aβ42 oligomers were rarely observed in cell bodies (Fig. 3A, arrows in top right panel) but were now numerous in neuronal processes, especially as punctate accumulations (Fig. 3A, arrow-heads in right column). The ratio of Aβ42 oligomer intensity per area of processes compared with cell bodies increased 1.46 ± 0.09-fold between 12 and 19 d in culture (p < 0.01) (Fig. 3B).
Figure 3.

Aβ42 oligomers accumulate within neuronal processes with time in culture. A, Increased oligomeric Aβ42 (M16) immunofluorescence in processes of Tg2576 primary neurons with time in culture. Top panel, Localization of Aβ42 in cell bodies (arrows) and processes (arrowheads) at 12 d (left) and 19 d (right) in culture. Bottom panel, Localization of Aβ42 oligomers in neuronal processes. Scale bar, 10 μm. B, Ratio of oligomeric Aβ42 immunofluorescence intensity in processes to that in cell bodies (processes/cell bodies) at 12 and 19 d in culture in Tg2576 primary neurons, with intensity ratio standardized as 1.0 at day 12 in culture. There was a 1.46 ± 0.09-fold increase in the process-to-cell body ratio of Aβ42 oligomer fluorescence between 12 and 19 d in culture; p < 0.01. C, Aβ42 oligomer accumulation in neuronal processes of Tg2576 neurons at 19 d in culture, especially within endosomal-like vesicles (thick arrows), and less prominently in their outer membranes (thin arrows). These processes are associated with morphological alterations, including fibrillar dark material (arrowheads) that appears associated with intravesicular Aβ42 oligomers. Scale bar, 250 nm. D, Aβ42 oligomers (arrows) also localized and accumulated along microtubules (arrowheads) within neuronal processes of Tg2576 neurons at 19 d in culture. Scale bar, 250 nm.
To assess the subcellular localization of Aβ42 oligomer increases in Tg2576 primary neurons with time in culture, we turned to immunogold EM using M16. In Tg2576 primary neurons at 19 d in culture, gold particles labeling Aβ42 oligomers were found associated with amorphous floccular, fibrillar-appearing dark material within large endosomal-like organelles (Fig. 3C). We did not observe Aβ42 oligomer labeling gold particles on normal-appearing endosomal organelles, as we had with antibodies directed mainly at Aβ42 monomers (Fig. 1E,F). The darkened-appearing organelles containing the Aβ42 oligomers may reflect altered or even “degenerating” endosomes, because they appear at times to contain MVB-characteristic smaller internal vesicles. Aβ42 oligomer gold particles were mainly inside darkened endosomal vesicles, whereas Aβ42 monomers were mainly on outer membranes (Fig. 1E,F). Interestingly, Aβ42 oligomer immunogold labeling also prominently localized along microtubules within neuronal processes (Fig. 3D).
Aβ42 oligomerization in Tg2576 mouse brain with aging
A previous report showed that microdeposits of Aβ42 appear as early as 7–8 months in Tg2576 brain (Kawarabayashi et al., 2001). Although these microdeposits increase in number between 7 and 10 months, consistent initiation of AD-like plaque pathology begins at 11–13 months (Irizarry et al., 1997). In an attempt to observe the earliest stages of Aβ42 aggregation and plaque formation, we labeled sections from 3-, 11-, and 20-month-old Tg2576 brains with the M16 Ab against Aβ42 oligomers. We did not observe M16 immunolabeling of 11-month-old wild-type (Fig. 4A, top left panel) or 3-month-old Tg2576 mouse cerebral cortex (Fig. 4A, top right panel). In cortices from 11-month-old Tg2576 brains, M16 Aβ42 oligomer staining was detected as microdeposits (Fig. 4A, bottom left panel; at higher magnification, Fig. 4B). At 20 months, Aβ42 oligomer deposits were increased in number and size (Fig. 4A, bottom right panel). The early stage of Aβ42 oligomer aggregation was then investigated in Tg2576 brain by immuno-EM using M16 immunoperoxidase labeling (Fig. 4C). In the absence of overt neuritic plaques, we observed intense Aβ42 oligomer immunoreactivity in elongated patterns that could be consistent with degenerated neuronal processes (Fig. 4C). In the presence of amyloid plaques, M16 immunoreactivity, indicative of Aβ42 oligomers, surrounded and was concentrated along the edges of the amyloid core at the center of plaques (Fig. 4D). The amyloid core was surrounded by swollen and distended M16 immunolabeled degenerating neuritic-appearing profiles (Fig. 4D, arrows).
Figure 4.
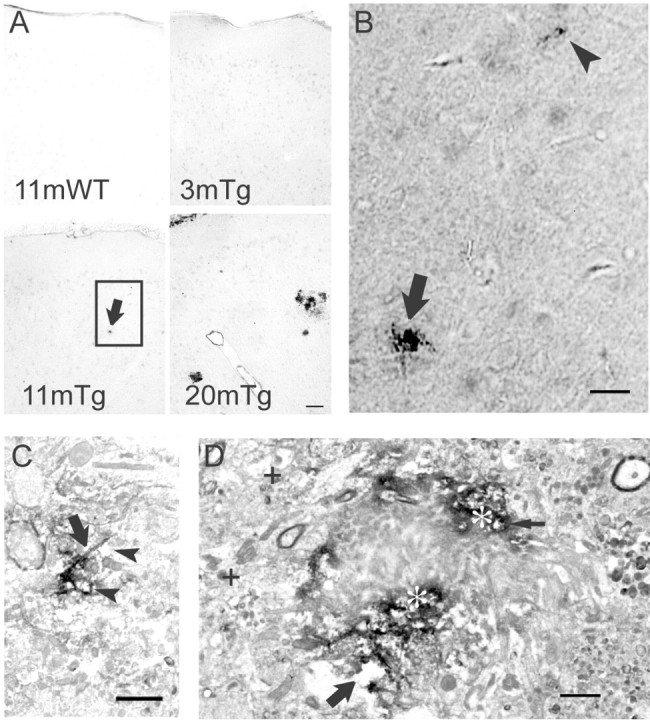
Aβ42 oligomers accumulate in Tg2576 mouse brain. A, Low-magnification light microscopy images of M16 Aβ42 oligomer immunoperoxidase labeling of wild-type (WT) and Tg2576 (Tg) mouse brains at different ages. Aβ42 oligomers were not detected in 11-month-old wild-type (top left) or 3-month-old Tg2576 mouse brains (top right) but were faintly detected as dense micro-deposits (bottom left, arrow) in an 11-month-old Tg2576 mouse. By 20 months, M16 Aβ42 oligomer staining showed increased number and size of microdeposits (bottom right) and also surrounded neuritic plaques. Scale bar, 50 μm. B, High-magnification light microscopic image of the rectangle in A (bottom left), showing a microdeposit (arrow) and even smaller Aβ42 oligomer aggregation before plaques in an 11-month-old Tg2576 mouse (arrowhead). Scale bar, 30 μm. C, Immunoperoxidase EM of Aβ42 oligomer aggregations distant from an obvious plaque. Aβ42 oligomer immunoreactivity appears concentrated along processes (arrow), and structures around the aggregation sites are degenerating (vacuolated spaces, arrowheads). Scale bar, 1μm. D, Ultrastructural localization of Aβ42 oligomers around a plaque. Marked accumulation of immunoperoxidase reaction product (appearing black; asterisks) representing Aβ42 oligomers surrounds an unlabeled plaque core. Areas around the plaque core are degenerated and include degenerating neurites (thin arrow) and abnormal empty spaces (large arrow). For reference, more normal neurites distant from the plaque are also seen (plus signs). Scale bar, 1 μm.
To better define the subcellular location(s) of Aβ42 aggregation and oligomer accumulation we used immunogold EM with M16. Adjacent to the outer rim of Aβ plaques staining with M16, we could detect dystrophic neurites containing M16 Aβ oligomer gold particles (Fig. 5A,C). At times, these oligomer-containing neurites could be seen to contain subcellular organelles, such as mitochondria, although the cytoarchitecture invariably was abnormal in the presence of Aβ oligomer labeling (Fig. 5A,C, for higher magnification of dystrophic Aβ oligomer-containing neurites). Because M16 Aβ oligomer labeling was associated with morphological–pathological alterations, adjacent nonpathological tissue is represented to underscore the fact that the surrounding normal tissue lacking M16 labeling demonstrates a normal subcellular cytoarchitecture (Fig. 5A,B; B is a higher magnification image of the normal cytoarchitecture seen in the rectangle in A). At high magnification even isolated areas of M16-labeled Aβ42 oligomer aggregations distant from plaques were observed. We found Aβ42 oligomer accumulations close to synaptic active zones (Fig. 5D). The normal microtubular network was absent in areas surrounding even isolated Aβ42 oligomer accumulations (Fig. 5D). Distended-appearing processes with multiple M16 gold particles were observed that were markedly abnormal, with atypical electron-dense fibril-like material, large empty spaces, and lack of any well defined normal organelles, such as mitochondria (Fig. 5E).
Figure 5.
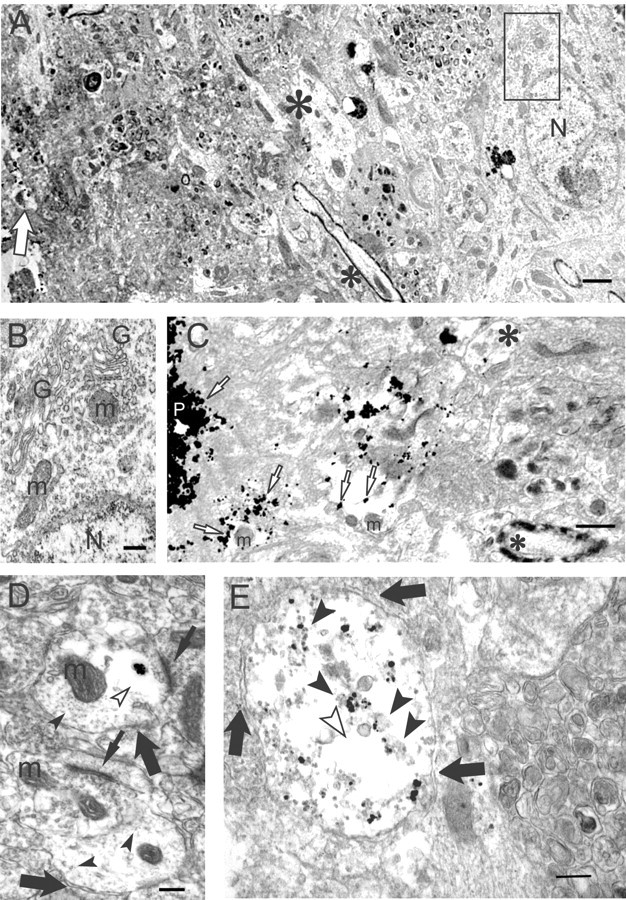
Aβ oligomers by immunogold EM accumulate in dystrophic processes and synapses of Tg2576 mouse brain. A, A lower-magnification image of a degenerated, morphologically abnormal area around a plaque core (left) surrounded by normal-appearing neuropil (right). A degenerating neurite (empty arrow) is observed close to the plaque at the left edge of the figure. Normal dendrite (large asterisk) and a myelinated axon (small asterisk) are at the border of the normal and abnormal areas. A normal neuron is seen distant from the plaque (right); the rectangle within this normal-appearing neuron is enlarged in B, below. N, Nucleus. Scale bar, 1 μm. B, Normal organelles and cytoarchitecture can be seen in this higher-magnification image of the small rectangle within the neuron in A. G, Golgi apparatus; m, mitochondrion; N, nucleus. Scale bar, 250 nm. C, A higher-magnification image of an Aβ oligomer containing degenerating neurites and abnormal empty spaces just around a plaque core (P). Aβ oligomers (empty arrow) are associated with the plaque core, and degenerating neurites are associated with empty spaces. A relatively normal dendrite with an isolated M16 gold particle (large asterisk) and a myelinated axon (small asterisk) are observed distant from the plaque core. m, Mitochondrion. Scale bar, 500 nm. D, A cluster of gold particles representing Aβ42 oligomers in a postsynaptic profile near an active zone of a synapse (top thin arrow). The plasma membranes of two postsynaptic profiles are indicated (large arrows). The cluster of Aβ42 oligomers is surrounded by a clear area (empty arrowhead), in which the normal microtubular network (filled arrowheads) is absent. The lower synaptic profile, lacking Aβ42 oligomers, shows a normal microtubular network throughout. Synaptic active zones are indicated by thin arrows. m, Mitochondrion. Scale bar, 250 nm. E, M16 immunogold EM of a Tg2576 neuritic process. Vertical section through a neuritic process in an 18-month-old Tg2576 mouse brain. Distant from a plaque, substantial Aβ42 oligomer gold particles are associated with granular and fibril-like electron-dense material (arrowheads) in an abnormal pathological-appearing process with poorly defined cytoarchitecture, including empty spaces (empty arrowhead) and lacking any normal organelles, such as mitochondria. Arrows outline the plasma membrane of the neuritic process. Scale bar, 250 nm.
Aβ42 oligomer aggregation in human AD brain
Aβ42 oligomers were not observed in normal, non-AD cortical biopsy tissue (data not shown). Aβ42 oligomer accumulations, however, were observed in human AD cortical brain tissue, within abnormal processes and synaptic compartments, but not in more normal-appearing surrounding neuropil (Fig. 6A). Within dystrophic-appearing processes and synaptic compartments, Aβ42 oligomers seemed at times to localize to tubulovesicular-like structures (Fig. 6B,C) or membranous profiles (Fig. 6B,D). Secondary to the altered cytoarchitecture associated with M16 labeling, these abnormal subcellular structures cannot be adequately classified as being related to endosomes, endoplasmic reticulum, or other organelles. Aβ42 oligomer-containing synaptic compartments were markedly abnormal. They contained open spaces and poorly defined membrane fragments (Fig. 6B–D), and there was associated electron-dense fibril-like material (Fig. 6B). Figure 6D shows a markedly abnormal postsynaptic profile containing multiple Aβ42 oligomer gold particles. The profile is devoid of any normal organelles and contains dark areas, consistent with degeneration. In addition, this abnormal postsynaptic profile synapses with an abnormally darkened presynaptic profile in the bottom left (Fig. 6D).
Figure 6.
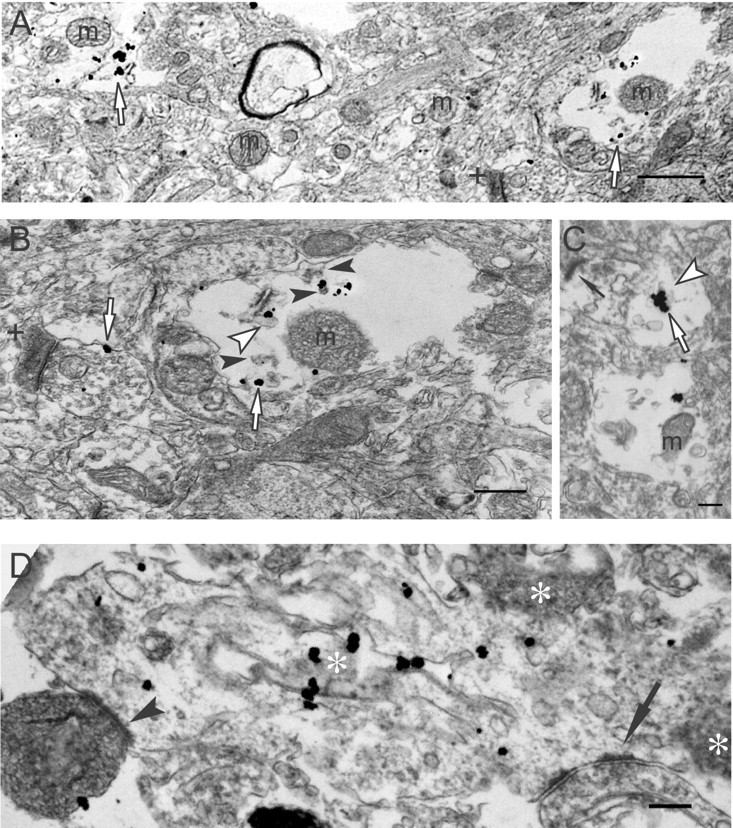
Aβ42 oligomerization within dystrophic processes of human AD brain. A, Aβ42 oligomers (empty arrows) are observed in degenerating neurites and are not associated with more normal-appearing neuropil (middle area of image). A darkened, abnormal-appearing postsynaptic profile is also observed (plus sign). m, Mitochondrion. Scale bar, 1μm. B, A higher-power image of a degenerating neurite in A (A, right empty arrow). Aβ42 oligomers (empty arrows) are associated with a tubulovesicular-like organelle (empty arrowhead) and granular fibril-like electron-dense material (filled arrowheads). Isolated Aβ42 oligomers localize to the membrane of presynaptic profile (left empty arrow) associated with a darkened, degenerating-appearing postsynaptic profile (plus sign). m, Mitochondrion. C, Aggregated Aβ42 oligomers (empty arrow) appear associated with a tubulovesicular-like organelle (empty arrowhead) in a postsynaptic compartment with an active zone (thin arrow). m, Mitochondrion. Scale bar, 300 nm. D, M16 Aβ42 oligomers aggregate on tubular-like membranous structures within a disrupted, swollen dendrite containing abnormal dark profiles (asterisks), consistent with degeneration; no normal organelles can be found in this postsynaptic profile. Presynaptic profiles are seen on the right (arrow) and left (arrowhead) corners of the image; the left one is darkened, indicative of degeneration. Scale bar, 250 nm.
Discussion
We reported previously that MVBs are the major subcellular site of Aβ42 accumulation within neurons and that accumulating Aβ42 in MVBs within distal neuronal processes and synaptic profiles was associated with morphological alterations (Takahashi et al., 2002). We now provide data demonstrating that Aβ42 accumulates also within processes of Tg2576 primary neurons in vitro with time in culture. These tissue culture results are consistent with our previous in vivo results in brains of Tg2576 mice with aging and human subjects with AD and extend these observations now to a cell culture system that can be manipulated experimentally. Moreover, we provide evidence that accumulating Aβ42 within processes aggregates as Aβ42 oligomers both in primary Tg2576 neurons with time in culture and in aging Tg2576 mouse brain and human AD brain.
Increasing evidence suggests that Aβ42 oligomers are the most toxic form of Aβ. We found that Aβ42 oligomer accumulation occurred in processes and synaptic compartments. These processes and synaptic compartments were markedly abnormal, often lacking any normal cytoskeletal architecture or subcellular organelles. Because normal synaptic functioning requires these subcellular structures, these Aβ42 oligomer accumulating processes and synaptic compartments are unlikely to allow for functional synapses. Synaptic dysfunction is the most established correlate of cognitive decline in AD (Masliah, 2001). Our results therefore provide direct evidence for the importance of Aβ42 oligomers in the pathogenesis of AD.
Interestingly, M16 oligomers localized not only to the outer membranes of endosomal–MVB-like organelles, reminiscent of monomeric Aβ42, but localized prominently inside darkened late endosomal-like organelles. The occurrence of early abnormalities in the endosomal–lysosomal system is well established in AD brain (Cataldo et al., 2000). Interestingly, late endosomal MVBs are increasingly being studied in the ubiquitin–proteasome system (UPS)-dependent recycling and degradation of various cellular receptors and proteins (Katzmann et al., 2002), including those involved in the regulation of synaptic plasticity (Ehlers, 2003). We hypothesized previously that intracellular Aβ42 accumulation in AD may parallel a growing list of neurodegenerative diseases in which aberrant intracellular protein accumulation and impairment of the UPS may be involved. Interestingly, the translocation of EGF receptor–EGF complexes from the outer limiting membrane of MVBs to the inner vesicles was shown by immuno-EM to be proteasome dependent (Longva et al., 2002). Our observations associating Aβ42 oligomer accumulation in the inner vesicles of darkened MVB-like organelles with neuritic pathology suggest that aberrant intraendosomal trafficking of Aβ42 may be involved in the pathogenesis of AD. A better understanding of the cell biology and pathology of late endosomal Aβ42 may be important in better understanding the early events in the pathogenesis of AD.
We also observed Aβ42 oligomers not associated with vesicular organelles and appearing to reside along microtubules. It is well known that endosomal vesicles such as MVBs, which normally contain Aβ42 in their outer membranes (Takahashi et al., 2002), are transported along microtubules. Vesicles containing APP, β-site APP-cleaving enzyme, and presenilin were reported to be transported along microtubules in axons of peripheral nerve that can generate Aβ (Kamal et al., 2001). The accumulation of Aβ42 on microtubules provides an intracellular association between Aβ and the microtubule-associated protein tau and thereby a more direct association for the two neuropathological hallmarks of AD, Aβ plaques, and tau tangles. Of note, the Aβ42 oligomers along microtubules were within the cytoplasm and outside the normal lipid bilayer environment for Aβ. It was reported previously that transgenic mice overexpressing Aβ intracellularly, but not transgenic mice overexpressing Aβ extracellularly, demonstrate neurodegeneration (LaFerla et al., 1995). Furthermore, it was reported that Aβ1–42, but not Aβ1–40 or Aβ42–1, is exquisitely neurotoxic when introduced into the cytosol of cultured primary neurons (Zhang et al., 2002).
The mechanisms by which extracellular Aβ can modulate intracellular Aβ42 remain unclear, as do the mechanisms by which Aβ42 monomers accumulate in the outer membranes of MVBs, are translocated within endosomal vesicles as oligomers, and then are converted to amyloid-like electron-dense fibrils. Aβ42 might gradually cause MVBs to degenerate as it oligomerizes. In support of this, Aβ42 oligomers at times appear to be associated with vesicular organelles that sometimes even contain internal vesicles, characteristic of MVBs (Fig. 3C). Another possibility is that accumulating Aβ42 monomers or oligomers on MVBs in processes impair vesicular transport of vital cargo by interfering with kinesin-based transport mechanisms. Release of Aβ42 monomers or oligomers into the cytoplasm and subsequent aggregation on microtubules may also be critical determinants of neurotoxicity. The detailed mechanism by which neurite processes and synaptic compartments degenerate and the role of aggregated Aβ42 in these events remain to be elucidated.
Footnotes
This work was supported by National Institutes of Health Grants HL18974 (T.A.M.), NS02037 (G.K.G.), and NS045677 (G.K.G.), Paul Beeson awards (M.T.L., G.K.G.), and the American Academy of Neurology Education and Research Foundation (G.K.G.). We appreciate the advice of Dr. Asao Hirano (Department of Pathology, Albert Einstein College of Medicine) and Dr. Frederick Maxfield (Department of Biochemistry, Weill Medical College of Cornell University).
Correspondence should be addressed to Dr. Gunnar K. Gouras, Laboratory of Alzheimer's Disease Neurobiology, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, 525 East Sixty-Eighth Street, New York, NY 10021. E-mail: gkgouras@med.cornell.edu.
DOI:10.1523/JNEUROSCI.5167-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243592-08$15.00/0
References
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan ML, Mori H, Yankner BA (2002) Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33: 677–688. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA (2000) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Aoki C, Pickel VM (1990) Optimization of differential immunogold-silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods 33: 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271–276. [DOI] [PubMed] [Google Scholar]
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38: 120–134. [DOI] [PubMed] [Google Scholar]
- Echeverria V, Cuello AC (2002) Intracellular A-beta amyloid: a sign for worse things to come? Mol Neurobiol 26: 299–316. [DOI] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231–242. [DOI] [PubMed] [Google Scholar]
- Fezoui Y, Hartley DM, Harper JD, Khurana R, Walsh DM, Condron MM, Selkoe DJ, Lansbury Jr PT, Fink AL, Teplow DB (2000) An improved method of preparing the amyloid beta-protein for fibrillogenesis and neurotoxicity experiments. Amyloid 7: 166–178. [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA (1998) Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med 4: 827–831. [DOI] [PubMed] [Google Scholar]
- Glabe C (2001) Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci 17: 137–145. [DOI] [PubMed] [Google Scholar]
- Golde TE (2003) Alzheimer disease therapy: can the amyloid cascade be halted? J Clin Invest 111: 11–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyure KA, Durham R, Stewart WF, Smialek JE, Troncoso JC (2001) Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125: 489–492. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97–100. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997) APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965–973. [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414: 643–648. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3: 893–905. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB (2002) Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res 69: 567–577. [DOI] [PubMed] [Google Scholar]
- Klein WL (2002) Abeta toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int 41: 345–352. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J (1998) A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392: 193–197. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G (1995) The Alzheimer's A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet 9: 21–30. [DOI] [PubMed] [Google Scholar]
- Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002) Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156: 843–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E (2001) Recent advances in the understanding of the role of synaptic proteins in Alzheimer's disease and other neurodegenerative disorders. J Alzheimers Dis 3: 121–129. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860–866. [DOI] [PubMed] [Google Scholar]
- Milner TA, Lee A, Aicher SA, Rosin DL (1998) Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol 395: 310–327. [PubMed] [Google Scholar]
- Mobius W, van Donselaar E, Ohno-Iwashita Y, Shimada Y, Heijnen HFG, Slot JW, Geuze HJ (2003) Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic 4: 222–231. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F (1999) Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem 274: 6483–6492. [DOI] [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguchi H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA (2002) Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9: 88–102. [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283: 1571–1577. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles. Intracellular abeta and synaptic dysfunction. Neuron 39: 409–421. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster Hd (1991) The fine structure of nervous system. Oxford: Oxford UP.
- Pratico D, Uryu K, Leight S, Trojanoswki JQ, Lee VM (2001) Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci 21: 4183–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T (2002) Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci 22: 1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE (2003) Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis 14: 133–145. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Doms RW, Lee VM (1998) Detection of a novel intraneuronal pool of insoluble amyloid beta protein that accumulates with time in culture. J Cell Biol 141: 1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabira T, Chui DH, Kuroda S (2002) Significance of intracellular Abeta42 accumulation in Alzheimer's disease. Front Biosci 7: a44–a49. [DOI] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161: 1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ (2000) The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry 39: 10831–10839. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306: 116–120. [DOI] [PubMed] [Google Scholar]
- Yang AJ, Knauer M, Burdick DA, Glabe C (1995) Intracellular Aβ1–42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J Biol Chem 270: 14786–14792. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL (1989) Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 245: 417–420. [DOI] [PubMed] [Google Scholar]
- Zhang Y, McLaughlin R, Goodyer C, LeBlanc A (2002) Selective cytotoxicity of intracellular amyloid β peptide 1–42 through p53 and Bax in cultured primary human neurons. J Cell Biol 156: 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]