

Abstract
Vanilloid receptor 1 (TRPV1), a nonspecific cation channel expressed primarily in small sensory neurons, mediates inflammatory thermal pain sensation. The function and expression of TRPV1 are enhanced during inflammation and certain neuropathies, leading to sustained hyperalgesia. Activation of TRPV1 in the spinal cord and periphery promotes release of adenosine, which produces analgesia by activating A1 and A2A adenosine receptor (AR) on central and peripheral neurons. This study provides evidence of a direct interaction of AR analogs with TRPV1. Adenosine analogs inhibit TRPV1-mediated Ca2+ entry in human embryonic kidney (HEK293) cells stably expressing TRPV1 (HEK/TRPV1) and DRG neurons. This inhibition was independent of A2AAR activation. Specific binding of [3H]resiniferatoxin (RTX) in plasma membrane preparations was inhibited by CGS21680, an A2AAR agonist. Similar degrees of inhibition were observed with both agonists and antagonists of ARs. Adenosine analogs inhibited [3H]RTX binding to affinity-purified TRPV1, indicative of a direct interaction of these ligands with the receptor. Furthermore, specific capsaicin-sensitive binding of [3H]CGS21680 was observed in Xenopus oocyte membranes expressing TRPV1. Capsaicin-induced inward currents in DRG neurons were inhibited by adenosine and agonist and antagonist of A2AAR at nanomolar concentrations. Increasing the concentrations of capsaicin reversed the inhibitory response to capsaicin, suggesting a competitive inhibition at TRPV1. Finally, exposure of HEK/TRPV1 cells to capsaicin induced an ∼2.4-fold increase in proapoptotic cells that was abolished by adenosine analogs. Together, these data suggest that adenosine could serve as an endogenous inhibitor of TRPV1 activity by directly interacting with the receptor protein.
Keywords: TRPV1, adenosine, adenosine receptor, capsaicin, dorsal root ganglion, apoptosis
Introduction
Vanilloid receptor 1 (TRPV1) is a nonspecific cation channel that mediates thermal inflammatory pain. These receptors are expressed primarily in small sensory neurons type C and Aδ fibers and, to a lesser extent, in certain brain nuclei and non-neuronal tissues (Caterina et al., 2000). Activation of TRPV1 by capsaicin, the active ingredient of chili peppers, underlies its ability to induce a burning sensation (Caterina et al., 1997; Tominaga et al., 1998). In contrast, mice deficient in TRPV1 demonstrate reduced thermal hyperalgesia and inflammatory pain sensitization (Caterina et al., 2000; Davis et al., 2000). These studies support an integral role of TRPV1 in mediating certain modalities of pain perception.
Both the function and expression of TRPV1 are enhanced during inflammation and neuropathic pain. Various mediators produced during the inflammatory response, such as ATP, bradykinin, and NGF, have been shown to increase temperature and proton sensitivity and also contribute to enhanced TRPV1 channel activity (Premkumar and Ahern, 2000; Chuang et al., 2001; Tominaga et al., 2001). Increased expression of TRPV1 in dorsal root ganglion (DRG) neurons has also been observed in conditions of inflammation and neuropathic pain, which may contribute to sustained hyperalgesia (Hudson et al., 2001; Amaya et al., 2003). As such, drugs that specifically target TRPV1 may prove valuable in the management of pain associated with chronic neuropathic conditions and inflammation.
Activation of TRPV1 in the spinal cord and periphery promotes increased release of the nucleoside adenosine (for review, see Liu et al., 2001; Sawynok and Liu, 2003), possibly through increased intracellular Ca2+ entry through the TRPV1 channels (Sweeney et al., 1989; Cahill et al., 1993). It is believed that this rise in intracellular Ca2+ leads to activation of protein kinase C and ultimately 5′nucleotidase, an enzyme responsible for the production of adenosine from adenosine monophosphate (Kitakaze et al., 1995; Obata et al., 2001, Obata, 2002). Adenosine administered both systemically and centrally produces analgesia in a variety of rodent pain models (Sawynok, 1998; Dickenson et al., 2000). The analgesic actions of adenosine are produced through activation of the A1 and A2A adenosine receptor (AR) subtypes, present both centrally and peripherally (Sawynok and Liu, 2003). Adenosine analogs mediate the antiallodynic effects against L5/L6 spinal nerve ligation in rats (Lee and Yaksh, 1996). In addition, intrathecal adenosine has been shown to attenuate pain perception in different models in human (Rane et al., 1998). The A1AR is present throughout the spinal cord, with higher levels present in lamina 2, the intermediolateral cell column, and the ventral horn (Deuchars et al., 2001). The A2AAR is present mainly on DRG neurons and on presynaptic terminals of sensory afferent neurons in the spinal cord (Kaelin-Lang et al., 1998). On the basis of the data presented above, we speculate that adenosine released via TRPV1 activation could mediate feedback inhibition of TRPV1 function.
The present study tested the hypothesis that adenosine regulates TRPV1 function. We show that adenosine and adenosine analogs inhibit capsaicin-mediated TRPV1 activation in cultures of DRG neurons and in human embryonic kidney (HEK293) cells stably expressing TRPV1 (HEK/TRPV1) through a direct interaction of these agents with TRPV1.
Materials and Methods
Materials. ZM241385 [4-(2-[7-amino-2-(2-furyl)(1,2,4)triazolo(2,3-a)(1,3,5)triazin-5-ylamino] ethyl)phenol] was obtained from Tocris Cookson (Bristol, UK). R-PIA (R-phenylisopropyladenosine) and DPCPX (8-cyclopentyl-1, 3-dipropylxanthine) were obtained from Boehringer Mannheim (Indianapolis, IN). [3H]CGS21680 (2-[4-(2-p-carboxy-ethyl) phenylamino]-5′-N-ethylcarboxamidoadenosine) was obtained from DuPont-NEN (Boston, MA), and [3H]resiniferatoxin (RTX) was from PerkinElmer Life Sciences (Boston, MA). All other chemicals, unless otherwise stated, were obtained from Sigma (St. Louis, MO).
Cell culture. HEK293 cells were obtained from American Tissue Culture Collection (Rockville, MD). Growth medium consisted of 90% DMEM, 10% fetal bovine serum, 50 U/ml penicillin, 25 μg/ml streptomycin (all from Invitrogen, Grand Island, NY), and 0.2 mg/ml G418 sulfate (Calbiochem, La Jolla, CA) for selection of resistance. Cells were cultured at 37°C, in the presence of 5% CO2 and 95% ambient air. HEK293 cells were transfected with rat TRPV1 cDNA (a gift from D. Julius, University of California, San Francisco, San Francisco, CA) using Lipofectamine 2000 (Invitrogen) according to the protocol of the manufacturer. Cells were cultured in the presence of G418 sulfate, neomycin-resistant cells were expanded, and frozen stocks were kept in liquid nitrogen. For all experiments, these HEK/TRPV1 cells were cultured in G418 sulfate to ensure propagation of the transgene.
DRGs were isolated from rat fetuses, triturated, and cultured in Neurobasal medium (Invitrogen) with addition of 100 ng/ml murine NGF 2.5s (Promega, Madison, WI), 2% serum-free B27 supplement (Invitrogen), and 0.5 mm l-glutamine. Cells were cultured for 5 d before performing the experiments.
Oocyte isolation and preparation. Stage V and VI oocytes were isolated from tricaine-anesthetized Xenopus laevis. Depth of anesthesia was monitored and oocytes were removed through an incision made in the abdomen. Ovaries were collagenase treated (1–2 mg/ml) in OR2 solution (in mm: 82.5 NaCl, 2 KCl, 1 MgCl2, and 5 HEPES, pH 7.4) at 18°C. Defolliculated oocytes were injected with 50–70 nl (1 μg/ml) of TRPV1 cRNA and cultured at 18°C in ND96 buffer solution (in mm: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, and 2.5 Na pyruvate, pH 7.6) until use. A batch of oocytes was tested for capsaicin-induced currents before radioligand binding.
Membrane preparation. HEK/TRPV1 cells were detached in ice-cold PBS containing 5 mm EDTA for TRPV1 binding. Samples were prepared by lysing whole cells in ice-cold buffer (in mm: 5 KCl, 5.8 NaCl, 0.75 CaCl2, 2 MgCl2, 320 sucrose, and 10 HEPES, pH 7.4) with the help of a tissue homogenizer. Homogenates were centrifuged for 10 min at 1000 × g (4°C), and the supernatant from the first centrifugation was further centrifuged at 35,000 × g for 30 min (4°C) to obtain a partially purified membrane fraction. This membrane preparation was treated with 1 U/ml adenosine deaminase (Boehringer Mannheim) for 15 min at 37°C and then used for radioligand binding. The addition of adenosine deaminase increased specific radioligand binding to both A2AAR and TRPV1. DRGs were isolated from mice in ice-cold 1× PBS and stored frozen at –80°C until use. Excised DRGs were thawed out, lysed in ice-cold buffer, and sonicated for 15 sec, after which, whole-cell lysate was used for TRPV1 radioligand binding.
For A2AAR binding, HEK/TRPV1 cells were similarly detached in ice-cold PBS. Samples were prepared by lysing cells in 50 mm Tris-HCl buffer, pH 7.4, containing 10 mm MgCl2, 5 mm EDTA, 10 μg/ml soybean trypsin inhibitor, 10 μg/ml benzamidine, and 2 μg/ml pepstatin with sonication for 15 sec. Membranes were obtained by centrifugation of the homogenates at 14,000 × g for 15 min (4°C). The final pellet was resuspended in the same buffer. Endogenously released adenosine was degraded using adenosine deaminase and incubating the mixture for 15 min at 37°C. This mixture was then used for radioligand binding. Xenopus oocyte membranes for [3H]CGS21680 binding were prepared similar to that described above for HEK/TRPV1 A2AAR binding.
Immunoprecipitation. TRPV1 antibody (Ab) affinity columns were prepared using a C-terminal Ab (Neuromics, Northfield, MN) that was cross-linked to immobilized protein G (50% slurry) according to the Seize X Mammalian Immunoprecipitation kit (Pierce, Rockford, IL). Columns were stored in binding buffer at 4°C until ready for use and were not used more than four times. HEK/TRPV1 cells were used as a source of TRPV1 for immunoprecipitation. Cells were first solubilized and incubated with the TRPV1 affinity column at 4°C overnight. Columns were washed several times, and the bound TRPV1 was eluted in 200 μl of elution buffer, pH 2. The pH of the eluate was adjusted to ∼7.0 using 5–7 μl of a 1 m Tris-HCl, pH 7.4, stock solution and used for [3H]RTX binding and Western blotting.
Radioligand binding. Quantification of TRPV1 was performed using [3H]RTX (43 Ci/mmol). Sample preparations (∼70 μg of protein) were incubated with 1.2 nm [3H]RTX in the absence or presence of cold RTX (100 nm) or capsaicin (100 μm) to estimate nonspecific binding, in a total volume of 450 μl of binding buffer. The tubes were incubated for 1 hr at 37°C, followed by immediate cooling on ice. For the competition binding assays, most compounds (at close to the effective concentrations used) were soluble in water. These compounds include R-PIA and CGS21680. ZM241385 and DPCPX were diluted from a stock solution of 10 mm in dimethylsulfoxide (DMSO). At the concentrations of these agents used for the assay, the concentrations of DMSO were ≤0.1%. We did not observe any effect of 0.1% DMSO on TRPV1 as determined by [3H]RTX binding assays. Quantification of A2AAR was performed using the antagonist 125I-ZM241385 (2200 Ci/mmol) or the agonist radioligand [3H]CGS21680 (30 Ci/mmol). Membrane preparations (∼50 μgofprotein) were incubated with the radioligands in the absence or presence of 1 mm theophylline to define nonspecific binding. The total volume in each assay tube of 250 μl was made up with 50 mm Tris-HCl, 10 mm MgCl2, and 1 mm EDTA, pH 7.4. Incubations were performed for 1 hr at 37°C. After incubation, samples were filtered over Whatman GF/B glass fiber filters (Brandel, Gaithersburg, MD) pretreated with polyethyleneimine (0.05%) and washed with 10 ml of ice-cold 20 mm Tris-HCl buffer, pH 7.4, containing 0.01% of CHAPS (3-[(3-cholamidopropyl) dimethylammonio]-1 propanesulfonate). The radioactive content of each filter was determined using a Beckman Instruments (Fullerton, CA) liquid scintillation counter (LS5801) or a Packard (5780) gamma counter. Radioligand binding data were plotted using the Prism software (GraphPad Software, San Diego, CA).
SDS-PAGE and Western blotting. TRPV1 immunoprecipitate was used for Western blotting to confirm the presence of the receptor protein. Samples were electrophoresed using SDS-PAGE (Laemmli, 1970). Proteins were transferred to nitrocellulose membranes and blocked in buffer containing 130 mm NaCl, 2.7 mm KCl, 1.8 mm Na2HPO4, 1.5 mm KH2PO4, 0.1% NaN3, 0.1% Triton X-100, and 5% low-fat skim milk for 2 hr, followed by incubation with primary TRPV1 Ab at 4°C overnight. After washes in blocking solution, blots were processed using an Enhanced Chemiluminescence kit (ECL Plus; Amersham Biosciences, Piscataway, NJ) and visualized by exposure to Eastman Kodak (Rochester, NY) XAR film.
Immunocytochemistry. HEK/TRPV1 cells were plated on glass coverslips and fixed with 4% paraformaldehyde. Cells were permeabilized with 0.5% Triton X-100, and nonspecific binding was reduced by 5% normal donkey serum. Cells were then treated with a primary polyclonal antibody (pAb) (Santa Cruz Biotechnology, Santa Cruz, CA) to detect TRPV1 at a dilution of 1:100 in 5% donkey serum along with 0.05% Triton X-100 in PBS for 1 hr. For detection of the A2AAR, cells were treated with a pAb for the A2AAR (Santa Cruz Biotechnology), at a 1:50 dilution. This was followed by incubation with rhodamine-labeled secondary antibodies (1:100). Coverslips were then mounted on glass microscope slides using Aquamount and imaged by confocal microscopy (Fluoview confocal laser-scanning microscope; Olympus Optical, Tokyo, Japan) with krypton laser at 568 nm using a 40× objective.
Ca2+ imaging. HEK/TRPV1 cells and embryonic rat DRGs were grown on glass coverslips to detect Ca2+ entry induced by TRPV1 activation. Adherent cells were washed with HEPES-buffered saline (in mm: 130 NaCl, 4 KCl, 10 HEPES, 5 glucose, 2 CaCl2, and 2 MgCl2, pH 7.3) and loaded with 5 μm Fluo-4 AM (Molecular Probes, Eugene, OR). After incubation at 37°C for 20 min, the cells were washed with physiologic buffer and treated with adenosine analogs immediately before imaging by confocal microscopy using an argon laser at 488 nm. Images were recorded at baseline (F0), and the image was collected every 5 sec after addition of 2 μm capsaicin to obtain the relative F values. Data was analyzed as the ratio of F/F0 for each cell and was obtained using the Fluoview software.
Electrophysiology. Cultured DRG neurons grown on poly-d-lysine-coated coverslips were used for recording TRPV1 currents. For perforated-patch recording, the bath solution contained 140 mm NaGlu, 2.5 mm KCl, 10 mm HEPES, 2 mm MgCl2, and 1 mm EGTA, pH 7.35, and the pipette solution contained 130 mm NaGlu, 10 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 1 mm MgCl2, 0.2 mm EGTA, and 240 μg/ml amphotericin B. Currents were recorded using a WPC 100 patch-clamp amplifier (E.S.F. Electronic, Goettingan, Germany). Data were digitized (VR-10B; InstruTech, Great Neck, NY) and stored on videotape. For analysis, data were filtered at 2.5 kHz (–3 dB frequency with an eight-pole low-pass Bessel filter; LPF-8; Warner Instruments, Hamden, CT) and digitized at 5 kHz. Current amplitudes were measured using Channel 2 (software kindly provided by Michael Smith, Australian National University, Canberra, Australia). The traces and graphs were made using Origin (Microcal Software, Northampton, MA).
Apoptosis assay. Cell death was determined by the annexin V-FITC apoptosis assay kit (Oncogene Research Products, San Diego, CA). The HEK/TRPV1 cells were harvested using 0.5% trypsin solution at 37°C for 1 min and centrifuged at 220 × g for 5 min. The cells were resuspended in 0.5 ml of 1× annexin-binding buffer, and 1.25 μl of annexin V-FITC was added to the cell suspension. The mixture was then incubated at room temperature for 15 min. After centrifugation, 0.5 ml of 1× annexin-binding buffer and 10 μl of propidium iodide were added. Samples were placed on ice away from light and analyzed immediately. Quantitation of annexin V-FITC and propidium iodide signals was performed by a flow cell-based bench top FACS Calibur Cytometer (Becton Dickinson, Mountain View, CA) equipped with an argon ion laser light source emitting an excitation light at 488 nm, leading to high FITC emission, and a 530/30 bandpass filter that transmits wavelengths of light between 515 and 545 nm. Fluorescence from annexin V-FITC positive and propidium iodide negative (early apoptotic cell populations) and annexin V-FITC and propidium iodide positive (necrotic or late apoptotic cells) was determined.
Protein determination. Protein concentrations in samples were determined by the established Bradford protein assay (Bradford, 1976) using bovine serum albumin to prepare standard curves.
Statistical analyses. Statistical analyses were performed using Student's t test or ANOVA, followed by Tukey's honestly significant difference post hoc test to compensate for multiple pairwise comparisons. Data is represented as mean ± SEM.
Results
Characterization of TRPV1 and A2AAR in HEK293 membranes
Expression of TRPV1 in HEK/TRPV1 cells was demonstrated by immunocytochemistry using a pAb derived from the C-terminal portion of TRPV1 (Fig. 1A). Immunolabeling was observed in the plasma membranes and also in cytosol. The latter localization is probably a result of cell permeabilization to ensure Ab access to the C-terminal portion of the receptor. In addition, specific labeling of TRPV1 was observed using [3H]resiniferatoxin (Szallasi et al., 1999). Using 1.2 nm [3H]RTX and 100 nm cold RTX to define nonspecific binding, we detected ∼600 fmol/mg protein TRPV1 binding sites. Because the concentration of the radioligand used is fivefold higher than the Kd, it is estimated that this level of receptor is close to the maximal receptor density in these cells. As expected, untransfected HEK293 cells showed no [3H]RTX binding, and specific binding was inhibited by known TRPV1 ligands, such as capsaicin and capsazepine (Fig. 1B), suggesting that [3H]RTX binding in our experiments labeled the TRPV1 receptor.
Figure 1.

Characterization of TRPV1 and A2AAR in HEK293 cells. A, TRPV1 expression. Immunocytochemistry was performed in HEK/TRPV1 cells using a goat pAb and a tetramethylrhodamine isothiocyanate (TRITC)-labeled secondary Ab. Immunolabeled preparations were visualized by confocal microscopy, which shows the presence of TRPV1 within cytoplasm and on plasma membrane. The corresponding phase microscopic image is shown (right). B, [3H]RTX binding to membranes obtained from HEK/TRPV1 cells was inhibited by known TRPV1 ligands, such as capsaicin (100 μm), RTX (1 μm), and capsazepine (10 μm). Asterisks indicate statistically significant difference from untreated membranes (control); p < 0.05. C, A2AAR expression. A2AAR immunoreactivity in HEK/TRPV1 cells was detected using a rabbit polyclonal primary Ab, followed by a TRITC-labeled donkey anti-rabbit IgG. Images were visualized by confocal microscopy using a 40× objective. D, 125I-ZM241385 binding assays were performed using crude plasma membranes (∼50 μg of protein per assay tube) and 1 mm theophylline to detect nonspecific binding. The saturation curve shown was fitted by GraphPad Prism software and is a representative of three independent experiments.
A fortuitous observation was that HEK293 cells also expressed the A2AAR. Immunolabeling studies using A2AAR Ab demonstrated intense red membrane and cytoplasmic staining when visualized by confocal microscopy (Fig. 1C). Because the Ab targets the cytosolic C-terminal tail of the receptor, the cytosolic expression likely reflects the labeling of pools of A2AAR receptors in intracellular compartments and/or nonspecific labeling of the primary Ab. Binding of 125I-ZM241385, a selective antagonist of this receptor subtype, was used to confirm the presence of A2AAR (Palmer et al., 1995). Saturation binding curve (Fig. 1D) shows the total number of the A2AAR (Bmax) as 82.8 ± 40.1 fmol/mg protein and an equilibrium dissociation constant (Kd) of 2.9 ± 2.6 nm. On the other hand, no significant levels of the adenosine A1AR or A3AR were detected. Additional confirmation for the presence of A2AAR was provided using the selective agonist ligand [3H]CGS21680. We obtained specific binding of this radioligand in HEK293 cells using 30 nm [3H]CGS21680, a concentration that is equivalent to the Kd of this ligand in pheochromocytoma (PC12) cultures (data not shown). Thus, our data indicate that HEK/TRPV1 cells express both TRPV1 and the A2AAR and would serve as a good model to study potential interaction between these two receptors.
Adenosine analogs inhibit capsaicin-induced increase in intracellular Ca2+
Capsaicin induces a rapid rise in intracellular Ca2+ in HEK/TRPV1 cells, as detected by Ca2+ imaging with Fluo-4 AM using confocal microscopy. The increase in Ca2+ peaked within 15 sec after the addition of capsaicin and was maintained for at least 2 min (Fig. 2A). Cells pretreated with capsazepine (10 μm), a competitive antagonist of TRPV1, or ruthenium red (20 μm), a specific pore blocker and a functional antagonist of TRPV1, failed to show any response to capsaicin. To determine whether adenosine analogs could regulate TRPV1 activation, HEK293 cells were pretreated for 2 min with CGS21680 (10 μm), a selective agonist of the A2AAR, before the application of capsaicin. The addition of CGS21680 alone had no effect on the basal cytoplasmic Ca2+ fluorescence but produced an almost complete inhibition of capsaicin-induced Ca2+ entry, observed up to 2 min after drug addition (Fig. 2B, top panel). The addition of capsaicin (2 μm) to the cultures resulted in a significant and time-dependent increase in intracellular Ca2+, with an F/F0 value of 3.0 within 15 sec of agonist exposure (Fig. 2B, bottom panel). The addition of CGS21680 (10 μm) thus completely abolished the entry of Ca2+ elicited by capsaicin. We observed similar results when capsaicin was used to activate TRPV1 in DRG neurons (Fig. 2C), which express both the TRPV1 and A2AAR.
Figure 2.
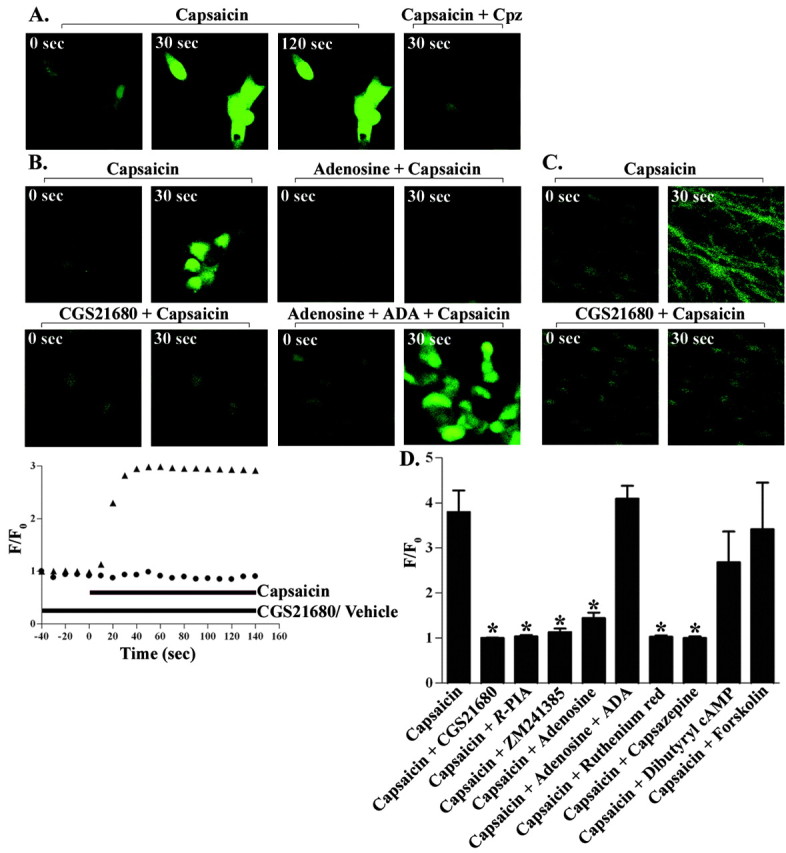
Adenosine analogs inhibit TRPV1-induced Ca2+ entry. HEK/TRPV1 cells (A, B) or DRG neurons (C) were loaded with 5 μm Fluo-4 AM for 20 min in 1 ml of physiologic buffer, pretreated with vehicle, TRPV1 antagonists, or adenosine analogs (10 μm), and then followed by capsaicin (2 μm) after 30 sec of baseline recording by confocal microscopy. A, Capsaicin-induced Ca2+ fluorescence was reduced by capsazepine (10 μm; CPZ). B, Pretreatment with CGS21680 (10 μm) or adenosine (50 μm) attenuated the rise in intracellular Ca2+ observed in 30 sec. Adenosine deaminase (0.2 U/ml; ADA) reversed the inhibition observed with adenosine. Bottom row, Graphical representation of capsaicin-induced TRPV1 activation over time using F/F0 values (▴, capsaicin; •, CGS21680 plus capsaicin). D, Comparison of the effectiveness of different agents in inhibiting TRPV1 activity. HEK/TRPV1 cells were pretreated with CGS21680 (10 μm), R-PIA (10 μm), ZM241385 (10 μm), adenosine (50 μm), adenosine deaminase (0.2 U/ml), ruthenium red (20 μm), capsazepine (10 μm), forskolin (10 μm), or dibutyryl cAMP (1 mm) before the administration of capsaicin (2 μm). Data are presented as the mean ± SEM of four to six cells each from three coverslips. *p < 0.05 indicates statistically significant difference from capsaicin-treated cells.
Another adenosine analog tested, R-PIA, also reduced the response to capsaicin by ∼70% (p < 0.05). Furthermore, addition of adenosine (50 μm) to the cultures inhibited capsaicin-mediated Ca2+ entry, and this inhibition was reversed in cultures pretreated with adenosine deaminase (0.2 U/ml) 5 min before the addition of capsaicin and 5 min after the addition of adenosine (Fig. 2B,D). In these experiments, adenosine deaminase was able to completely reverse the inhibition produced by adenosine. The addition of adenosine deaminase alone did not alter capsaicin-induced Ca2+ entry in HEK293 cultures (data not shown), suggesting that the basal release of adenosine in these cultures was not high enough to produce tonic suppression of TRPV1 activity. To determine whether inhibition of capsaicin-induced Ca2+ entry by CGS21680 was mediated via the A2AAR, we determined whether coadministration of the A2AAR antagonist ZM241385 would reduce the inhibitory effect of CGS21680. However, we observed that the addition of ZM241385 (10 μm) by itself mimicked the response of CGS21680 on Ca2+ entry (Fig. 2D). For these latter experiments, ZM241385 was solubilized in dimethylsulfoxide. Vehicle containing a similar concentration of dimethylsulfoxide did not alter the binding of ligands to TRPV1 nor the capsaicin-induced Ca2+ entry. Together, these data suggest that the inhibition of TRPV1 activity by adenosine analogs was likely not mediated via the A2AAR but results from a direct interaction of these agents with TRPV1.
Additional experiments were performed to better understand the mechanism underlying inhibition of TRPV1-mediated Ca2+ entry by adenosine analogs. Because the A2AAR is coupled to increases in adenylyl cyclase activity (Guroff et al., 1981; Noronha-Blob et al., 1986), we determined whether increasing intracellular cAMP by administering dibutyryl cAMP (1 mm) to the coverslips or by the addition of forskolin (10 μm), an activator of adenylyl cyclase, would modulate capsaicin-induced intracellular Ca2+ accumulation. Neither dibutyryl cAMP nor forskolin affected capsaicin-induced Ca2+ entry, as observed for adenosine analogs (Fig. 2D), suggesting that inhibition of the capsaicin response is independent of cAMP generation. Furthermore, the addition of ATP (50 μm) and 2-methylthioATP (10 μm) did not inhibit, but appeared to potentiate, capsaicin-mediated Ca2+ entry, presumably by activating P2Y1 receptors (Tominaga et al., 2001).
Adenosine analogs decrease the TRPV1 current
Additional evidence supporting a functional interaction between adenosine analogs and TRPV1 was obtained by whole-cell patch-clamp recordings using cultured embryonic rat DRG neurons. Capsaicin (100 nm) induced an inward current that was dose dependently reduced by CGS21680, with complete inhibition of current observed with 1 μm CGS21680 (Fig. 3A). Moreover, inhibition of current was also observed with adenosine (Fig. 3B), albeit with lower potency. The inhibition produced by CGS21680 was reversible, as indicated by a lack of effect of CGS21680 when currents were activated with a higher (10 μm) concentration of capsaicin (Fig. 3C). Full dose–response curves for inhibition of TRPV1 were performed using CGS21680, the A2AAR antagonist ZM241385, and adenosine. When fitted to the Hill equation, the mean IC50 for CGS21680 from at least three different experiments for each concentration was 3.6 nm, for ZM241385 was 34.9 nm, and for adenosine was 160.0 nm (Fig. 3D). These results provide additional support for the hypothesis that adenosine and structurally similar compounds can inhibit the TRPV1 channel activity.
Figure 3.

Adenosine analogs decrease the TRPV1 current. A, Representative whole-cell patch-clamp recording showing inhibition of capsaicin-induced TRPV1 current by increasing concentrations of CGS21680 with complete blockade at 1 μm CGS21680. B, Inhibition of TRPV1 current by adenosine (1 μm). C, Capsaicin (10 μm) activated TRPV1 current to the same extent in the presence and absence of CGS21680 (10 μm). D, Dose-dependent inhibition of TRPV1 current by CGS21680 (IC50 of 3.6 nm), the A2AAR antagonist ZM241385 (IC50 of 34.9 nm) and adenosine (IC50 of 160.0 nm) compared at various concentrations (▪, CGS21680; •, ZM241385; ▴, adenosine).
Adenosine analogs inhibit the binding of [3H]resiniferatoxin to TRPV1
To explore the possibility that adenosine analogs bind directly to TRPV1, we performed competition radioligand binding experiments for [3H]RTX in HEK/TRPV1 cells, using CGS21680 as competitor. In initial studies, we observed that pretreatment of HEK/TRPV1 preparations with adenosine deaminase (to degrade adenosine) before performing [3H]RTX binding assays resulted in a substantial increase (approximately twofold) in specific radioligand binding, suggesting that adenosine released during sample preparation might be competing for the [3H]RTX binding site. As such, subsequent [3H]RTX binding studies were performed in the presence of adenosine deaminase. Different adenosine analogs tested for inhibition of capsaicin-induced Ca2+ influx (above) were also tested for their ability to interact with the [3H]RTX binding site. Nonspecific binding was estimated using 100 μm capsaicin to demonstrate the comparative inhibitory action of these agents and RTX. Incubation of membranes with [3H]RTX along with CGS21680 (10 μm), R-PIA (10 μm), ZM241385 (10 μm), or DPCPX (10 μm) inhibited the binding of [3H]RTX by ∼60% (p < 0.05; n = 4) for all of the analogs tested (Fig. 4A). Using different concentrations of CGS21680, we saw a decrease in the specific [3H]RTX binding with significant inhibition at 0.1, 1, 10, and 100 μm CGS21680 (p < 0.05) (data not shown). These results clearly indicate that adenosine analogs can interact directly with the [3H]RTX binding site on TRPV1.
Figure 4.
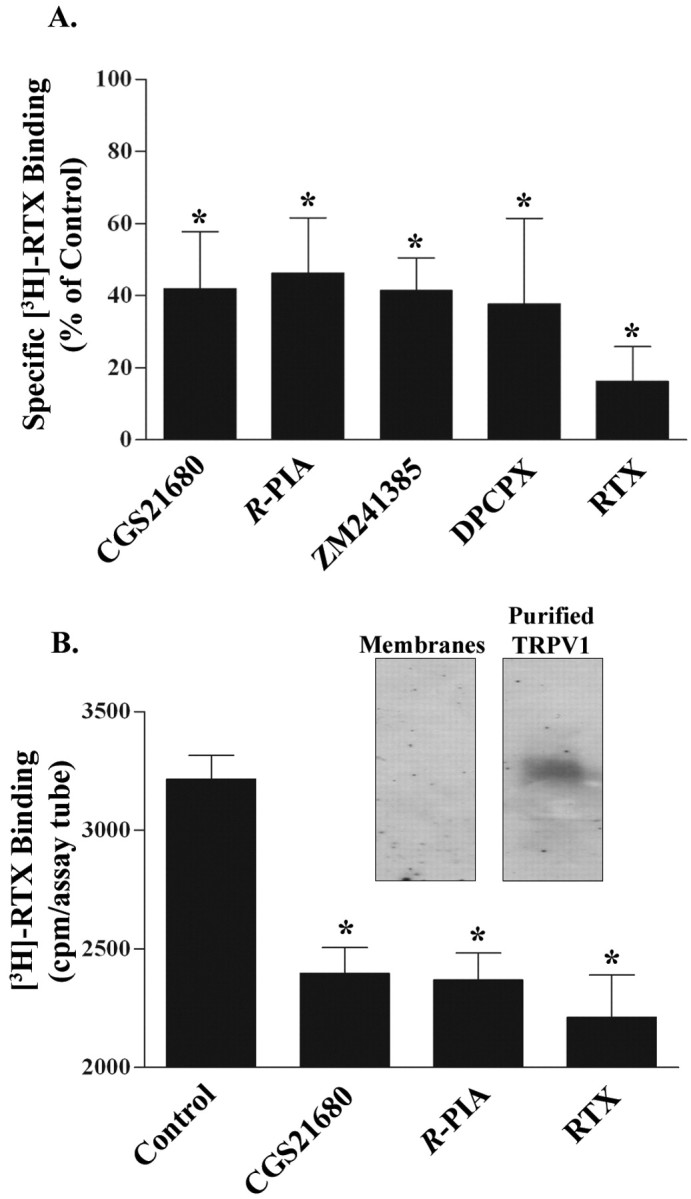
Adenosine analogs inhibit the binding of [3H] RTX to TRPV1. A, Plasma membranes (∼70 μg of protein) from HEK/TRPV1 cells were incubated with 1.2 nm [3H]RTX, and inhibition of [3H]RTX binding was assessed with CGS21680 (10 μm), R-PIA (10 μm), ZM241385 (10 μm), and DPCPX (10 μm) and compared with RTX (100 nm). *p < 0.05 indicate statistically significant difference from control. B, Inhibition of [3H]RTX binding in affinity-purified TRPV1 preparations. TRPV1 was purified from HEK/TRPV1 cells by Ab affinity columns (Pierce). Radioligand binding experiments were performed using different adenosine analogs: 1 μm CGS21680 and 1 μm R-PIA. Inset, TRPV1 protein detected by Western blotting in crude membranes versus purified preparations using a C-terminal Ab (Neuromics).
To further confirm that adenosine analogs interact directly with TRPV1, we obtained purified TRPV1 preparations using Ab affinity columns. Isolation of the receptor protein was confirmed by radioligand binding assays and by Western blotting (Fig. 4B, inset). No A2AAR was detected in the TRPV1 enriched preparation by Western blotting, indicating that the receptor did not copurify with TRPV1. Using the affinity-purified TRPV1, we showed inhibition of [3H]RTX binding by CGS21680 (1 μm) and R-PIA (1 μm) (Fig. 4B). These data support a direct interaction of adenosine analogs with TRPV1. Similar findings were observed when [3H]RTX binding was performed in lysates obtained from excised mouse DRGs. In these preparations, CGS21680 inhibited [3H]RTX binding at concentrations similar to that observed in HEK/TRPV1 cells (data not shown).
Additional experiments were performed to determine whether [3H]CGS21680 could label TRPV1 sites, because we observed high-affinity interaction of this compound with TRPV1 sites, as assessed by [3H]RTX binding. Xenopus oocytes were used for these studies because these cells do not express native specific [3H]CGS21680 binding sites. Radioligand binding performed with 40 nm [3H]CGS21680 showed specific capsaicin-sensitive binding in membrane preparations of oocytes injected with the TRPV1 cRNA, averaging 54 fmol/mg protein (mean of two experiments). We confirmed that these oocytes exhibit capsaicin-induced currents by electrophysiology. In contrast, uninjected oocytes, which did not show any capsaicin current, also did not show any specific capsaicin-sensitive [3H]CGS21680 binding.
Adenosine analogs protect against capsaicin-induced cell death
Agonists of ARs confer protection against oxidative stress (Ramkumar et al., 2001). Because capsaicin has been shown to produce cell killing by increasing intracellular Ca2+ influx (Caterina et al., 1997; Jambrina et al., 2003), we investigated whether pretreatment with adenosine analogs could protect against capsaicin-induced cell death in HEK/TRPV1 cells. Cell killing was assessed by an annexin V-FITC assay in HEK/TRPV1 cells. Cells exposed to 5 μm capsaicin for 1 hr showed an ∼2.4-fold increase in the number of proapoptotic cells (235.1 ± 30.7% of control). Capsazepine pretreatment protected these cells from capsaicin-induced apoptosis, indicative of the involvement of TRPV1 in this process. Similarly, pretreatment of cells with R-PIA (10 μm) led to a significant reduction in the number of early apoptotic cells (Fig. 5A,B). The percentage of early apoptotic cells in the R-PIA-treated group were 107.0 ± 24.6% of control. We observed some late apoptotic and necrotic cells after treatment with capsaicin, the levels of which were also decreased by R-PIA pretreatment. To test whether the protective action of R-PIA was mediated via the A2AAR, cells were pretreated with ZM241385 and R-PIA before the administration of capsaicin. We observed no reversal of the protective effect of R-PIA by ZM241385. In fact, the level of protection was similar to that obtained with R-PIA administered alone, suggesting that the direct protective effect of adenosine analogs in inhibiting TRPV1-mediated apoptosis was independent of A2AAR activation.
Figure 5.

Adenosine analogs protect against capsaicin-induced cell death. HEK/TRPV1 cells were pretreated with R-PIA (10 μm), R-PIA (10 μm) plus ZM241385 (10 μm), or capsazepine (10 μm), followed by capsaicin (5 μm) for 1 hr. Apoptosis was detected using an annexin V-FITC apoptosis kit (Oncogene Research Products) and quantitated by flow cytometry. A, Representative histograms derived from the low right quadrant of the flow chart, depicting early apoptotic cells (as shown by the bars). The number of early apoptotic cells in each histogram, defined as annexin positive and propidium iodide negative, is indicated as a percentage. B, The results from annexin V-FITC assay are presented as mean ± SEM of three independent experiments. *p < 0.05 a statistically significant difference in apoptosis produced by capsaicin treatment compared with control cells. **p < 0.05 indicates protection from capsaicin-mediated apoptosis obtained with capsazepine, R-PIA, and the combination of ZM241385 and R-PIA.
Discussion
A number of studies have shown that adenosine analogs are potential analgesic agents that can increase nociceptive thresholds in response to noxious stimulation. For example, adenosine reduces hypersensitivity to thermal stimuli in animals after peripheral inflammation or nerve injury (Lee and Yaksh, 1996; Poon and Sawynok, 1998). Intrathecal administration of adenosine in human reduces allodynia and hyperalgesia induced by capsaicin (Eisenach et al., 2002). Furthermore, increasing extracellular adenosine by inhibition of adenosine metabolism produces a peripheral antinociceptive response in a neuropathic pain model (Jarvis et al., 2002). The release of adenosine from both spinal and peripheral compartments inhibits pain transmission (Sawynok, 1998). This nucleoside also plays an important modulatory role in inflammatory and neuropathic pain (Dickenson et al., 2000). The mechanism(s) underlying this antinociceptive action is not clear.
This study provides novel insight into a possible mechanism underlying the peripheral antinociceptive action of adenosine. We show that adenosine analogs inhibit TRPV1 function through a direct interaction with the receptor protein. This conclusion is based on several pieces of data obtained in this study. First, adenosine analogs compete for [3H]RTX binding sites in membranes prepared from HEK293 cells, DRGs, and purified preparations of TRPV1. Reductions in [3H]RTX were also obtained at concentrations of these agents in the nanomolar range, which are similar to those required for interaction at the A2AAR. Second, different adenosine analogs tested inhibited [3H]RTX binding, as well as capsaicin-induced Ca2+ entry. Agonists and antagonists for the A2AAR were equally effective in mediating these responses, suggesting that inhibition was independent of A2AAR activation. Third, adenosine and adenosine analogs decreased capsaicin-induced inward current by interacting competitively with this receptor, because a diminution of the inhibition was observed with increasing capsaicin concentrations. Unlike adenosine, increasing cellular cAMP by addition of forskolin or by addition of dibutyryl cAMP to the cells did not alter capsaicin-induced Ca2+ entry. Because A2AAR activation is generally linked to increases in cAMP, these data support the contention that inhibition of TRPV1 was independent of the A2AAR activation. Furthermore, these data suggest that cAMP does not possess the structural requirements necessary for inhibition of TRPV1. Fourth, we detected capsaicin-sensitive [3H]CGS21680 binding in Xenopus oocytes injected with TRPV1 cRNA, whereas naive oocytes did not show any capsaicin-sensitive binding to this radioligand.
The use of [3H]RTX to label TRPV1 binding sites in HEK293 cells expressing the receptor has been documented previously (Szallasi et al., 1999). These investigators demonstrated saturable and high-affinity binding of the radioisotope in membranes prepared from HEK/TRPV1 cells, which was reversed by unlabeled TRPV1 ligands. Similarly, we observed significant inhibition of [3H]RTX binding using classical TRPV1 ligands, such as capsaicin, capsazepine, and RTX. Inhibition of [3H]RTX binding was observed at nanomolar concentrations of CGS21680, concentrations at which demonstrable interaction at the A2AAR is generally observed in radioligand binding studies (Nie et al., 1999). Again, we observed inhibition of [3H]RTX binding with both agonists and antagonists of the A2AAR and even with DPCPX, an antagonist of the A1AR, indicating that inhibition was independent of AR activation. Thus, it appears that a similar structural core common to all adenosine analogs might allow for the specific interaction with TRPV1. This conclusion was further supported by data showing inhibition of [3H]RTX binding to affinity-purified TRPV1 by adenosine analogs. The affinity-purified preparations were enriched in TRPV1, because we were able to detect the receptor by Western blotting in these preparations compared with crude cell membrane preparations. We could not detect the A2AAR in the affinity-purified preparations by Western blotting, suggesting that these two receptors did not comigrate during purification.
The interaction of adenosine analogs with the [3H]RTX binding site translates into functional inhibition of TRPV1 activity. All of these agents effectively inhibited capsaicin-mediated Ca2+ entry into HEK293 cells and DRG neurons. Similar inhibitory responses of these adenosine analogs at the TRPV1 channels were observed when channel activity was measured using patch-clamp recordings. However, higher concentrations of these analogs were required for inhibition of Ca2+ as determined by Fluo-4 AM fluorescence than for inhibition of capsaicin-induced inward current. A likely explanation for this apparent discrepancy is that a higher concentration of capsaicin (2 μm) was used to produce measurable Fluo-4 AM fluorescence in HEK/TRPV1 cells and DRG neurons than that required for eliciting an inward current (100 nm). Because of the apparent competitive nature of the interaction between adenosine and TRPV1, inhibition of the effects of higher concentrations of TRPV1 agonists would necessarily require higher concentrations of the adenosine analogs. In contrast, relatively low levels of capsaicin were used for eliciting an inward current, and relatively low concentrations of adenosine analogs were required for inhibiting this response. Another likely explanation for this discrepancy is that the application of capsaicin in culture leads to increased adenosine release, as observed in the spinal cord and periphery (Liu et al., 2001; Sawynok and Liu, 2003). This could lead to partial inhibition of Ca2+ entry, before the application of adenosine analogs. Such an effect would mask the inhibitory action of lower concentrations of adenosine analogs. Support for this is provided by our data that show reversal of adenosine-mediated inhibition of Ca2+ entry in HEK/TRPV1 cells by exogenously applied adenosine deaminase.
Stimulation of TRPV1 by capsaicin produces excitotoxicity, leading ultimately to destruction of primary afferent nerve fibers (Caterina et al., 1997). Cell death displays mixed features of both apoptosis and necrosis, as described previously in TRPV1 transfected HEK293 cells (Jambrina et al., 2003). The endocannabinoid anandamide induces a similar form of excitotoxic cell death by stimulating TRPV1 (Maccarrone et al., 2000; Jambrina et al., 2003). Because TRPV1-mediated cell death is linked to peripheral neuropathy, we determined whether adenosine analogs could inhibit capsaicin-mediated apoptosis. Our data clearly demonstrate a reduction in apoptotic cells in cultures pretreated with R-PIA before administration of capsaicin. In addition, the number of necrotic cells was also decreased, suggesting that adenosine analogs provide protection from cell killing induced by both the apoptotic and necrotic pathways. Although initial studies showed that TRPV1-mediated killing was mediated primarily by necrosis, recent studies using shorter treatment durations indicate the involvement of both signaling pathways in cell killing (Jambrina et al., 2003). A similar ligand inhibition profile was observed for apoptosis as was observed for inhibition of Ca2+ and [3H]RTX binding, indicating involvement of a common mechanism. We did not observe an expected additive effect of R-PIA and ZM241385, suggesting that the response of R-PIA was already maximal or that the A2AAR antagonist was blocking a cytoprotective action mediated via the activation of the endogenous A2AAR. This suggests that two cytoprotective mechanisms are likely activated by adenosine (agonist) analogs to block apoptosis. The first involves blockade of TRPV1, whereas the second involves activation of A2AAR cytoprotective mechanisms.
One obvious in vivo extension of these findings is the implication that adenosine serves as an endogenous inhibitor of TRPV1 activity. The levels of endogenous adenosine under normoxic conditions are in the submicromolar range (Pedata et al., 2001), levels at which we observed direct inhibition of TRPV1 channel activity. Furthermore, the levels should be higher in the vicinity of adenosine release sites, where it could interact with TRPV1. Because adenosine is released in response to TRPV1 activation in addition to a number of physiological processes (Sawynok and Liu, 2003), it is possible that this nucleoside mediates negative feedback inhibition of TRPV1 activity, independent of activation of ARs. We observed direct interaction of adenosine with TRPV1 in vitro using several different techniques. Such an interaction manifested in vivo would occur at sites along the pain pathway where TRPV1 are localized, namely small-diameter sensory afferents C and Aδ fibers and neurons in laminas 1 and 2 of the spinal cord (Caterina et al., 1997; Guo et al., 1999). Certainly, additional modulation of TRPV1 activity could be mediated via activation of A1 and/or A2AAR present in the spinal cord, DRG, and peripheral nerve terminals. The release of adenosine is augmented in inflammation and during ischemia (Pedata et al., 2001), conditions linked to increased TRPV1 activity. It is possible that adenosine mediates tonic suppression of TRPV1 activity in these conditions and thereby reduces the sensory inputs triggered by activation of this receptor.
Small variations in ATP levels within the cell can result in a large increase in AMP and adenosine, because of a high ratio of ATP to AMP concentration. Thus, adenosine serves as a sensitive indicator of increased cellular metabolic activity and oxidative stress (Latini and Pedata, 2001). Extracellular ATP produces a sensation of pain in vivo mediated through ionotropic and metabotropic purinoceptors (Chizh and Illes, 2001; Tominaga et al., 2001). Increased levels of extracellular ATP under pathophysiological conditions have been shown to enhance TRPV1 activity (Tominaga et al., 2001). Adenosine, produced as a byproduct of ATP metabolism (Burnstock, 1978), could suppress TRPV1 activity and thereby counter the generation and propagation of pain impulses.
In summary, we demonstrated that adenosine and its analogs can directly inhibit TRPV1 channel activity, supporting a role of this nucleoside as an endogenous modulator of TRPV1. During conditions of inflammation and neuropathic pain, the increase in endogenous adenosine observed may counter the excessive TRPV1 activation and pain sensation. Our findings also suggest that peripherally acting adenosine analogs may act as analgesics by blockade of TRPV1 channel activity.
Footnotes
This research was supported by grants from Southern Illinois University Central Research Committee and National Institute of Neurological Diseases and Stroke Grant NS042296 (L.S.P.). We thank Dr. Satyanarayan Bhat for his assistance in cell culturing.
Correspondence should be addressed to Dr. Vickram Ramkumar, Department of Pharmacology, Southern Illinois University School of Medicine, P.O. Box 19629, Springfield, IL 62794-9629. E-mail: vramkumar@siumed.edu.
DOI:10.1523/JNEUROSCI.4773-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243663-09$15.00/0
References
- Amaya F, Oh-hashi K, Naruse Y, Iijima N, Ueda M, Shimosato G, Tominaga M, Tanaka Y, Tanaka M (2003) Local inflammation increases vanilloid receptor 1 expression within distinct subgroups of DRG neurons. Brain Res 963: 190–196. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Burnstock G (1978) Cell membrane receptors for drugs and hormones: a multidisciplinary approach (Bolis L, Straub RW, eds), pp 107–118. New York: Raven.
- Cahill CM, White TD, Sawynok J (1993) Involvement of calcium channels in depolarization-evoked release of adenosine from spinal cord synaptosomes. J Neurochem 60: 886–893. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306–313. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Illes P (2001) P2X receptors and nociception. Pharmacol Rev 53: 553–568. [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957–962. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405: 183–187. [DOI] [PubMed] [Google Scholar]
- Deuchars SA, Brooke RE, Deuchars J (2001) Adenosine A1 receptors reduce release from excitatory but not inhibitory synaptic inputs onto lateral horn neurons. J Neurosci 21: 6308–6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson AH, Suzuki R, Reeve AJ (2000) Adenosine as a potential analgesic target in inflammatory and neuropathic pains. CNS Drugs 13: 77–85. [Google Scholar]
- Eisenach JC, Hood DD, Curry R (2002) Preliminary efficacy assessment of intrathecal injection of an Am formulation of adenosine in humans. Anesthesiology 96: 29–34. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R (1999) Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11: 946–958. [DOI] [PubMed] [Google Scholar]
- Guroff G, Dickens G, End D, Londos C (1981) The action of adenosine analogs on PC12 cells. J Neurochem 37: 1431–1439. [DOI] [PubMed] [Google Scholar]
- Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J (2001) VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci 13: 2105–2114. [DOI] [PubMed] [Google Scholar]
- Jambrina E, Alonso R, Alcalde M, del Carmen RM, Serrano A, Martinez A, Garcia-Sancho J, Izquierdo M (2003) Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J Biol Chem 278: 14134–14145. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Yu H, McGaraughty S, Wismer CT, Mikusa J, Zhu C, Chu K, Kohlhaas K, Cowart M, Lee CH, Stewart AO, Cox BF, Polakowski J, Kowaluk EA (2002) Analgesic and anti-inflammatory effects of A-286501, a novel orally active adenosine kinase inhibitor. Pain 96: 107–118. [DOI] [PubMed] [Google Scholar]
- Kaelin-Lang A, Lauterburg T, Burgunder JM (1998) Expression of adenosine A2a receptor gene in rat dorsal root and autonomic ganglia. Neurosci Lett 246: 21–24. [DOI] [PubMed] [Google Scholar]
- Kitakaze M, Hori M, Morioka T, Minamino T, Takashima S, Okazaki Y, Node K, Komamura K, Iwakura K, Itoh T (1995) Alpha 1-adrenoceptor activation increases ecto-5′-nucleotidase activity and adenosine release in rat cardiomyocytes by activating protein kinase C. Circulation 91: 2226–2234. [DOI] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- Latini S, Pedata F (2001) Adenosine in the central nervous system: release mechanisms and extracellular concentrations. J Neurochem 79: 463–484. [DOI] [PubMed] [Google Scholar]
- Lee YW, Yaksh TL (1996) Pharmacology of the spinal adenosine receptor which mediates the antiallodynic action of intrathecal adenosine agonists. J Pharmacol Exp Ther 277: 1642–1648. [PubMed] [Google Scholar]
- Liu XJ, White TD, Sawynok J (2001) Involvement of primary sensory afferents, postganglionic sympathetic nerves and mast cells in the formalin-evoked peripheral release of adenosine. Eur J Pharmacol 429: 147–155. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Lorenzon T, Bari M, Melino G, Finazzi-Agro A (2000) Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J Biol Chem 275: 31938–31945. [DOI] [PubMed] [Google Scholar]
- Nie Z, Mei Y, Malek RL, Marcuzzi A, Lee NH, Ramkumar V (1999) A role of p75 in NGF-mediated down-regulation of the A2A adenosine receptors in PC12 cells. Mol Pharmacol 56: 947–954. [DOI] [PubMed] [Google Scholar]
- Noronha-Blob L, Marshall RP, Kinnier WJ, U'Prichard DC (1986) Pharmacological profile of adenosine A2 receptor in PC12 cells. Life Sci 39: 1059–1067. [DOI] [PubMed] [Google Scholar]
- Obata T (2002) Adenosine production and its interaction with protection of ischemic and reperfusion injury of the myocardium. Life Sci 71: 2083–2103. [DOI] [PubMed] [Google Scholar]
- Obata T, Kubota S, Yamanaka Y (2001) Histamine increases interstitial adenosine concentration via activation of ecto-5′-nucleotidase in rat hearts in vivo. J Pharmacol Exp Ther 298: 71–76. [PubMed] [Google Scholar]
- Palmer TM, Poucher SM, Jacobson KA, Stiles GL (1995) 125I-4-(2-[7-amino-2-[2-furyl][1,2,4]triazolo[2,3-a][1,3,5] triazin-5-yl-amino]ethyl)phenol, a high affinity antagonist radioligand selective for the A2a adenosine receptor. Mol Pharmacol 48: 970–974. [PMC free article] [PubMed] [Google Scholar]
- Pedata F, Corsi C, Melani A, Bordoni F, Latini S (2001) Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann NY Acad Sci 939: 74–84. [DOI] [PubMed] [Google Scholar]
- Poon A, Sawynok J (1998) Antinociception by adenosine analogs and inhibitors of adenosine metabolism in an inflammatory thermal hyperalgesia model in the rat. Pain 74: 235–245. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408: 985–990. [DOI] [PubMed] [Google Scholar]
- Ramkumar V, Hallam DM, Nie Z (2001) Adenosine, oxidative stress and cytoprotection. Jpn J Pharmacol 86: 265–274. [DOI] [PubMed] [Google Scholar]
- Rane K, Segerdahl M, Goiny M, Sollevi A (1998) Intrathecal adenosine administration: a phase 1 clinical safety study in healthy volunteers, with additional evaluation of its influence on sensory thresholds and experimental pain. Anesthesiology 89: 1108–1115. [DOI] [PubMed] [Google Scholar]
- Sawynok J (1998) Adenosine receptor activation and nociception. Eur J Pharmacol 347: 1–11. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Liu XJ (2003) Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol 69: 313–340. [DOI] [PubMed] [Google Scholar]
- Sweeney MI, White TD, Sawynok J (1989) Morphine, capsaicin and K+ release purines from capsaicin-sensitive primary afferent nerve terminals in the spinal cord. J Pharmacol Exp Ther 248: 447–454. [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM, Annicelli LL, Krause JE, Cortright DN (1999) The cloned rat vanilloid receptor VR1 mediates both R-type binding and C-type calcium response in dorsal root ganglion neurons. Mol Pharmacol 56: 581–587. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D (1998) The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21: 531–543. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98: 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]