

Abstract
Although synapsins are abundant synaptic vesicle proteins that are widely used as markers of presynaptic terminals, the mechanisms that target synapsins to presynaptic terminals have not been elucidated. We have addressed this question by imaging the targeting of green fluorescent protein-tagged synapsins in cultured hippocampal neurons. Whereas all synapsin isoforms targeted robustly to presynaptic terminals in wild-type neurons, synapsin Ib scarcely targeted in neurons in which all synapsins were knocked-out. Coexpression of other synapsin isoforms significantly strengthened the targeting of synapsin Ib in knock-out neurons, indicating that heterodimerization is required for synapsin Ib to target. Truncation mutagenesis revealed that synapsin Ia targets via distributed binding sites that include domains B, C, and E. Although domain A was not necessary for targeting, its presence enhanced targeting. Domain D inhibited targeting, but this inhibition was overcome by domain E. Thus, multiple intermolecular and intramolecular interactions are required for synapsins to target to presynaptic terminals.
Keywords: localization, presynaptic, synapsin, synaptic vesicles, synaptic transmission, synaptic vesicle trafficking, presynaptic localization, vesicle pools
Introduction
Synapsins are a family of neuron-specific proteins that associate with synaptic vesicles (SVs) (Greengard et al., 1993; Hilfiker et al., 1999; Dresbach et al., 2001). Synapsins are thought to have multiple functions within presynaptic terminals, including recruitment of SVs to a reserve pool and regulation of the fusion of SVs (Hilfiker et al., 1999). Synapsins also have been implicated in neuronal development, synaptogenesis, and maintenance of mature synapses (Ferreira and Rapoport, 2002; Kao et al., 2002).
These physiological functions are thought to be mediated by the rich biochemical properties of synapsins. Synapsins bind to phospholipid and protein constituents of the SV as well as to components of the cytoskeleton, such as actin, tubulin, and spectrin (Hilfiker et al., 1999). Synapsins are the most abundant phosphoproteins in the brain and interact with several protein kinases (Matsubara et al., 1996; Hilfiker et al., 1999) as well as with other elements of cellular signaling pathways (Cousin et al., 2003; Tu et al., 2003). Structural similarity to ATP-using enzymes suggests that synapsins possess an as yet unidentified enzymatic function (Esser et al., 1998; Hosaka and Südhof, 1998a,b).
Three mammalian synapsin genes, synapsins I, II, and III, have been identified (Südhof et al., 1989; Kao et al., 1999). Synapsins are expressed throughout the central and peripheral nervous systems, although their expression varies across synapse types and brain regions (Fried et al., 1982; Südhof et al., 1989; Mandell et al., 1992; Matus-Leibovitch et al., 1997). In contrast to synapsins I and II, which are stably expressed in adult presynaptic terminals, synapsin III expression is developmentally controlled and is not confined to synaptic terminals (Ferreira et al., 2000; Pieribone et al., 2002). Each of these genes is subject to differential splicing, producing at least nine isoforms: synapsins Ia, Ib, IIa, IIb, IIIa, IIIb, IIIc, and the atypical IIId and IIIe (Porton et al., 1999). These isoforms arise from the combinatorial arrangement of numerous domains (Fig. 1A) (Südhof et al., 1989; Kao et al., 1999). The N-terminal portion of all synapsins is highly conserved and consists of domains A, B, and C, whereas the C-terminal portion is variable because of heterogeneous combinations of domains D–J.
Figure 1.
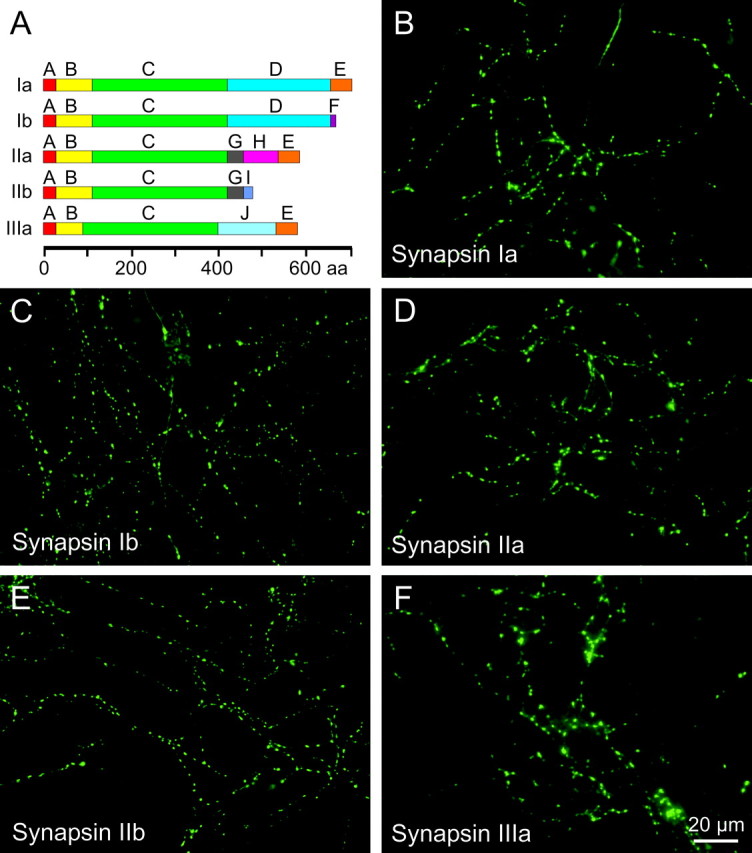
Targeting of GFP-tagged synapsins in wild-type neurons. A, Domain structure of synapsin isoforms considered in our study. B–F, In 2-week-old cultured neurons, GFP-tagged synapsins are located in punctuate structures.
Although synapsins clearly are important for presynaptic function, it is not known how these proteins target to presynaptic terminals. Here, we examined this question by fusing green fluorescent protein (GFP) to synapsins and examining the distribution of these fluorescent synapsins within living neurons. With this approach, we defined the molecular determinants within synapsin I that enable synapsin targeting and find that correct localization requires multiple sites, specifically domains B, C, and E. Furthermore, domain D provides a negative influence on the targeting of synapsins, whereas domain A enhances targeting. Remarkably, we found that an abundant splice variant of synapsin I, synapsin Ib, targets very weakly to presynaptic terminals in the absence of other synapsins. Taking advantage of this result, we established that heterodimerization plays an unexpected yet important role in localization of synapsins and is required for synapsin Ib to fully target to presynaptic terminals.
Materials and Methods
Generation of synapsin triple knock-out mice. Synapsin triple knock-out (TKO) mice were generated by mating synapsin I knock-outs (Chin et al., 1995) with synapsin II knock-outs (Ferreira et al., 1998) and then mating these synapsin I/II double knock-out mice with synapsin III knock-outs (Feng et al., 2002). Genotyping using tail DNA was performed by PCR using primers directed toward the targeting vector and genomic DNA. The genotype of synapsin TKO mice was verified by immunoblot analyses that indicated the absence of all three synapsins using synapsin-type-specific antibodies (Czernik et al., 1991; Ferreira et al., 2000; Menegon et al., 2000). Immunolabeling of cultured neurons with polyclonal antibody G304 (raised against the E domain common to all “a” isoforms) (Pieribone et al., 1995) did not detect endogenous synapsins, although synaptotagmin I immunofluorescence revealed the presence of synapses in the cultured neurons. A line of matching wild-type mice was derived in parallel to the TKO mice; these were termed “triple wild-type” mice.
Mammalian expression constructs. The vectors pEGFP-C1, -C2, and -C3 (representing three reading frames and collectively referred to as pEGFP-C; Clontech, Palo Alto, CA) use the cytomegalovirus promoter to drive expression of enhanced GFP (EGFP) fusion proteins in mammalian cells. Plasmids expressing rat synapsins Ia, Ib, IIa, IIb (Südhof et al., 1989), and IIIa (Kao et al., 1998) fused to the C terminus of EGFP were constructed by ligating the corresponding synapsin cDNA into pEGFP-C. To express EGFP fusions with synapsin domains or truncated synapsins, individual domains or portions of synapsins were amplified by PCR (Pfu DNA polymerase; Stratagene, Cedar Creek, TX), restricted, and ligated (Takara, Shigag, Japan) into the same vector. Following is a list of the synapsin I fragments used in this study. The numbers indicate the amino acid residues, within the synapsin Ia (or Ib) sequence, constituting the fragment: ABCD 1–659, ABCD1 1–617 (contains an inadvertent mutation P598H), ABCD4 1–538, ABC 1–416, ABC1 1–244, BC 31–416, C 115–414, C′ 243–416, CD 115–654, CDE 113–704, DE 415–704, E 656–704, CDF 113–668 (from synapsin Ib). Synapsins and synapsin fragments were additionally subcloned into either pEYFP-C1 or pECFP-C1 vectors that express EYFP and ECFP fusion proteins, respectively, either by PCR amplification or by direct restriction–ligation, when possible. Phosphorylation site point mutants of synapsin Ia were produced by using the QuickChange mutagenesis kit (Stratagene). Serines 9, 566, or 603 (phosphorylation sites 1, 2, and 3) were mutated to either alanine or glutamate, and double mutations were performed serially. All constructs were verified by automated sequencing at the Rockefeller University DNA Sequencing Resource Center or the Duke University DNA analysis facility.
Cell culture. Postnatal day (P) 0–2 pups were anesthetized with isofluorane (Abbott Laboratories, North Chicago, IL) according to approved animal care protocols. Pups were decapitated, and the hippocampi were removed, treated with papain (Worthington, Lakewood, NJ), and serially triturated. The dissociated neurons were plated onto poly-d-lysine (Sigma, St. Louis, MO)-coated glass coverslips (Fisher Scientific, Pittsburgh, PA) at 60,000–100,000 cells per well. Initially, cells were plated in plating medium consisting of Neurobasal-A medium (Invitrogen, Carlsbad, CA) supplemented with 5% defined FBS (Hyclone, Logan, UT), 2% B27, 2 mm Glutamax I, and 1 μg/ml gentamicin (Invitrogen). After 24 hr, the plating medium was replaced by serum-free culture medium that consisted of Neurobasal-A, 2 mm Glutamax I, and 2% B27. Cultures were typically maintained at 37°C in a 5% CO2 humidified incubator for 12–18 d before imaging. Depending on the glial cell density, 32 μm 5-fluoro-2-deoxyuridine (Floxuridine) and 80 μm uridine (Sigma) were added to the medium 7–14 d after plating to inhibit additional growth of glial cells. Neuronal ages are expressed as the sum of the age of the pups at the time of neuron harvesting and the number of days spent in culture.
Transfection. Neurons were transfected after 2–3 d in culture by lipofection, using Lipofectamine 2000 (Invitrogen) as described by the manufacturer. Cells were transfected using 200–400 ng of plasmid DNA per milliliter of culture medium (additive if a multiple transfection). The lipofection reagent and the DNA were diluted in OPTIMEM (Invitrogen). After 4 hr, the cells were returned to culture medium that was previously preconditioned in the same wells and saved.
Immunochemistry. Cultured cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) in PBS (20 mm NaHPO4 and 80 mm Na2HPO4, pH 7.4) for 10 min. The cells were permeabilized by incubating in ice-cold methanol for 5 min, followed by extensive washing with PBS (for PSD-95, permeabilization was achieved by incubating fixed cells for 5 min with a 0.1% Triton X-100 PBS solution). Blocking was done with 10% goat serum (Sigma) in PBS overnight at 4°C or for 4 hr at room temperature. The preparation was then incubated with the primary antibody solution (see below) for 2 hr at room temperature, and after extensive washing with PBS, the coverslips were overlaid with the secondary antibody solution for 1 hr at room temperature. After washing, the samples were imaged in PBS. Antibodies were diluted in blocking solution. The primary antibodies used were: monoclonal antisynaptotagmin-I (10 μg/ml; clone 1D12; gift from Dr. Masami Takahashi, Kitasato University, Sagamihara, Japan), monoclonal anti-synaptophysin-I (1:1000; clone SY38; Chemicon, Temecula, CA), monoclonal anti synaptobrevin-2 (1:2000; clone 69.1; gift from Dr. Reinhart Jahn, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), monoclonal anti PSD-95 (1:100; clone 6G6-1C9; ABR, Golden, CO), Alexa-Fluor 488-conjugated rabbit anti-GFP (1:500; Molecular Probes, Eugene, OR), and mouse anti-growth-associated protein 43 (1:500; clone 9-1E12; Chemicon). The secondary antibody was Rhodamine Red-X-conjugated goat anti-mouse IgG (1:500; Molecular Probes).
Microscopy. Cultured cells were imaged in a sterile chamber on an inverted microscope (IX70; Olympus, Tokyo, Japan), using a 40× UP-lanFl 0.75 numerical aperture objective. Exposure to the light source, a mercury arc lamp (HB075W), was controlled by a Unibilitz shutter (Vincent Associates, Rochester, NY). Fluorescence filter sets were obtained from Chroma (Brattleboro, VT) and were 470 ± 20/505/530 ± 20 nm for GFP and Alexa 488, 436 ± 10/455/480 ± 20 nm for cyan fluorescent protein (CFP), 500 ± 10/515/535 ± 15 nm for yellow fluorescent protein (YFP), and 540 ± 10/570/LP590 nm for DsRed2 and Rhodamine Red-X (expressed as excitation range/dichroic mirror cutoff/emission range). Cross-talk between CFP/YFP, GFP/DsRed2, or Alexa 488/Rhodamine Red-X channels was insignificant. Images were detected with a Coolsnap FX CCD camera (Roper Scientific, Trenton, NJ) and acquired and processed with ISEE software (Isee Imaging Systems, Raleigh, NC).
Quantification of targeting. To quantify the degree of targeting of synapsin constructs to the synapses, we defined a “targeting factor.” The tagged targeting factor is a measure of the specific association of the GFP-tagged protein with the synapse and is related to the ratio of the concentration of a GFP-tagged protein in a synapse to the concentration of the same protein in the axon. For this purpose, both a GFP-tagged protein of interest and soluble DsRed2 (pDsRed2-C1; Clontech) were expressed in cultured neurons. Fluorescence intensity was measured, at both wavelengths, along lines drawn on the axonal segments and across the varicosities. The lines were continued outside of the axon to measure the local background. The peak fluorescence of the bright spot and the fluorescence of the axon were determined by fitting Gaussian functions to the resultant intensity profiles and were corrected for the measured local background values. To calculate the mean targeting factor for each condition (reported as mean ± SEM), we averaged targeting factors obtained from 7–20 different neurons. For each neuron, the value was determined from the mean of 15–40 synapses. The targeting factor value was stable across different cultures. One caveat of this technique is that measurements rely on the assumption that GFP fluorescence can be used to identify synaptic varicosities. This is true, except for cases of proteins that do not target or do so weakly; in these cases, it is possible that occasional nonsynaptic structures were included in the analysis. However, this effect is negligible in the case of moderately or strongly targeting proteins for which synapses are easily identifiable (see below), allowing a clear delineation of the levels of targeting of relevant proteins. Statistical comparison of the average targeting factors of the various constructs was performed by Tukey's post hoc HSD (honestly significant difference) analysis of an ANOVA test.
Results
Targeting of fluorescent synapsins
Immunocytochemical studies have demonstrated that endogenous synapsin isoforms localize to presynaptic terminals (Südhof et al., 1989; but see Ferreira et al., 2000; Pieribone et al., 2002). Here, we visualize targeting by expressing exogenous, fluorescent synapsins in cultured neurons (Chi et al., 2001, 2003). We first asked whether these exogenous GFP–synapsin fusion proteins are capable of targeting to presynaptic terminals in cultured neurons. For this purpose, synapsins Ia, Ib, IIa, IIb, and IIIa were fused to GFP and transfected into cultured hippocampal neurons. Fluorescence imaging was then used to examine the location of GFP–synapsins at various times after transfection.
All five of the synapsin isoforms studied distributed in a punctate manner when expressed in neurons from wild-type mice (Fig. 1B–F). This pattern of localization was detectable as early as 5–6 d after neurons of P0–P2 pups were cultured, concurrent with the formation of functional synapses (Fletcher et al., 1991), and became more pronounced with time. The images shown in Figure 1 illustrate 12- to 18-d-old neurons, time points at which the punctuate localization pattern was very prominent.
To determine whether the sites of synapsin localization corresponded to presynaptic terminals, neurons transfected with GFP–synapsin Ia were immunolabeled with antibodies directed against synaptotagmin I, synaptobrevin 2, or synaptophysin I, all of which are integral proteins of the SV membrane (Wiedenmann and Franke, 1985; Trimble et al., 1988; Baumert et al., 1989; Perin et al., 1991). The vast majority of GFP–synapsin Ia puncta (green in center images of Fig. 2A–C) colocalized with the SV proteins (red in left images). This is evident in the merged images on the right in Figure 2, A–C, as a predominance of yellow puncta (representing overlap of red and green labels) over green puncta that reflect expression of GFP–synapsin Ia alone. The red puncta present in the merged images correspond to synapses that are formed by untransfected neurons and, thus, do not express GFP–synapsin Ia. This result suggests that exogenous GFP–synapsin Ia is associated with SVs accumulated in presynaptic terminals.
Figure 2.

Colocalization of GFP–synapsin Ia with synaptic marker proteins. Neurons expressing GFP–synapsin Ia were stained with antibodies that bind to synaptotagmin I (A), synaptobrevin 2 (B), synaptophysin I (C), or PSD-95 (D). The left column indicates the location of synaptic markers (red), the center column shows the location of GFP–synapsin Ia (green), and mergers of these two images are shown in the right column. Colocalization of synapsin and the synaptic markers is shown by the yellow areas in the merged images.
Because synapsin I has been identified as a component of cytoplasmic transport packets, which are mobile precursors of presynaptic sites (Ahmari et al., 2000), the punctuate distribution of GFP–synapsin Ia also might reflect these transport packets rather than fully formed synapses. We considered this possibility by immunolabeling synapsin-transfected neurons with antibodies for PSD-95, a well characterized constituent of the postsynaptic density of excitatory synapses (Hunt et al., 1996; Sheng, 2001). For most neurons, a great majority of GFP–synapsin Ia puncta were either apposed to or overlying PSD-95 puncta (Fig. 2D), indicating that GFP–synapsin Ia was indeed located at synapses. In a few neurons, we found no correlation between the location of any of the GFP–synapsin puncta and PSD-95. We presume that these were inhibitory neurons, the postsynaptic targets of which do not contain PSD-95 (Rao et al., 1998). Results similar to those described here were obtained for other synapsin isoforms (data not shown). Taken together, these observations indicate that GFP-tagged synapsins correctly target to presynaptic terminals.
Synapsin Ib targeting requires endogenous synapsins
Synapsins are known to interact with each other (Hosaka and Südhof, 1999). To determine whether such interactions with endogenous synapsins are responsible for the targeting of exogenous, GFP-tagged synapsins, we examined the targeting properties of GFP-tagged synapsins in neurons devoid of endogenous synapsins. For this purpose, mice lacking the three known synapsin genes (TKO mice) were generated by crossing previously established lines of mice: knock-outs of synapsin I (Chin et al., 1995), synapsin II (Ferreira et al., 1998), and synapsin III (Feng et al., 2002). The genotype of the TKO mice was verified by genomic PCR, by immunoblotting, and by immunocytochemistry of cultured TKO neurons; all procedures indicated the absence of expression of any synapsins.
Despite the absence of endogenous synapsins, synapsins Ia, IIa, IIb, and IIIa localized properly to the presynaptic terminals of neurons cultured from TKO mice (Fig. 3A–D). This indicates that these isoforms do not require endogenous synapsins to target to presynaptic terminals. In contrast, synapsin Ib remained widely distributed, with little concentration in synaptic varicosities (Fig. 3E). This was not attributable to the absence of synapses, because TKO neurons expressing synapsin Ib form both presynaptic terminals and functional synapses (data not shown). The fluorescence of GFP–synapsin Ib in the neurites was significantly higher than that observed in the cell body (Fig. 3F). In some cases, synapsin Ib was further enriched in a subset of neurites. To determine whether these processes were axons or dendrites, we immunostained neurons with an antibody against GAP-43, a protein that localizes exclusively to axons at the ages examined in this study (Goslin et al., 1990). In this experiment, we used neurons after 7–8 d in culture, because the lower density of processes at this age allowed us to examine neurites from transfected neurons without confounding overlap with nontransfected neurons. We found that neurites in which synapsin Ib was highly concentrated invariably were stained by GAP-43 (Fig. 3G,H), indicating that these processes are axons. Thus, synapsin Ib is transported along axons, yet does not visibly target to presynaptic terminals unless other synapsin isoforms are present.
Figure 3.
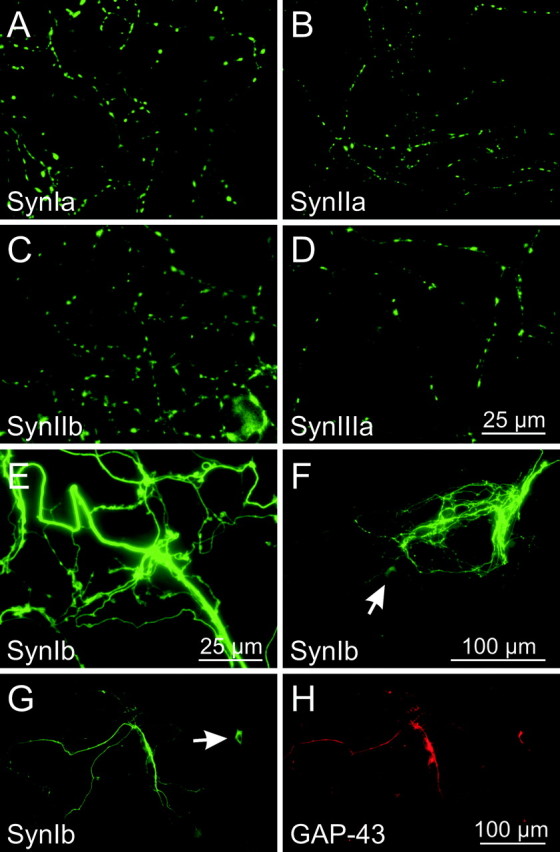
Targeting of GFP-tagged synapsins in synapsin TKO neurons. Although several isoforms of synapsin targeted properly in 2-week-old TKO neurons (A–D), no targeting of synapsin Ib was visible (E). F, Low magnification image of synapsin Ib distribution in a 3-week-old TKO neuron. The neurites of this neuron were substantially brighter than the cell body (arrow), indicating that synapsin Ib is not distributed homogeneously within the neuron. G, H, Axonal distribution of synapsin Ib. Neurons (7 d in vitro) expressing GFP–synapsin Ib were stained with anti-GAP-43 antibodies (red), a marker for axons of mature neurons. Neurites containing the highest levels of synapsin Ib also stained for GAP-43. The arrow in G indicates the cell body of the neuron.
Quantification of targeting
Targeting of GFP-tagged synapsins is evident as a punctuate accumulation of green fluorescence in presynaptic varicosities (Figs. 1, 2, 3). However, the intensity of GFP fluorescence is not a simple measure of targeting, because this value depends on other factors, particularly the volume of the varicosities and the level of protein expression. To quantify the synaptic targeting of GFP-tagged proteins, we devised a ratiometric imaging method (Fig. 4) (Tanasugarn et al., 1984; Grynkiewicz et al., 1985; Pennuto et al., 2003; Sankaranarayanan et al., 2003). For this purpose, neurons were cotransfected with the GFP-tagged synapsin of interest (Fig. 4A) and with a soluble fluorescent protein, DsRed2, which served as a measure of optical path-length and, therefore, the volume of the cellular compartment being imaged (Fig. 4B). The ratio of intensities of fluorescence produced by these two proteins then corrects for variations in cell volume. In addition, cell-to-cell variations in synapsin expression level were taken into account by comparing, within a given neuron, the relative amounts of synapsin (and DsRed2) found in varicosities in comparison to axons. Combining these two ratios yielded a synapsin targeting factor, defined as:
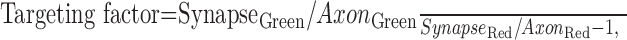 |
where Synapse and Axon indicate the intensity of GFP or DsRed2 fluorescence in the synaptic varicosities and axons, respectively. If a protein is not enriched in synaptic terminals, its targeting factor will be 0. For proteins that do target, the targeting factor should be >0 and proportional to the degree of presynaptic enrichment of the protein.
Figure 4.
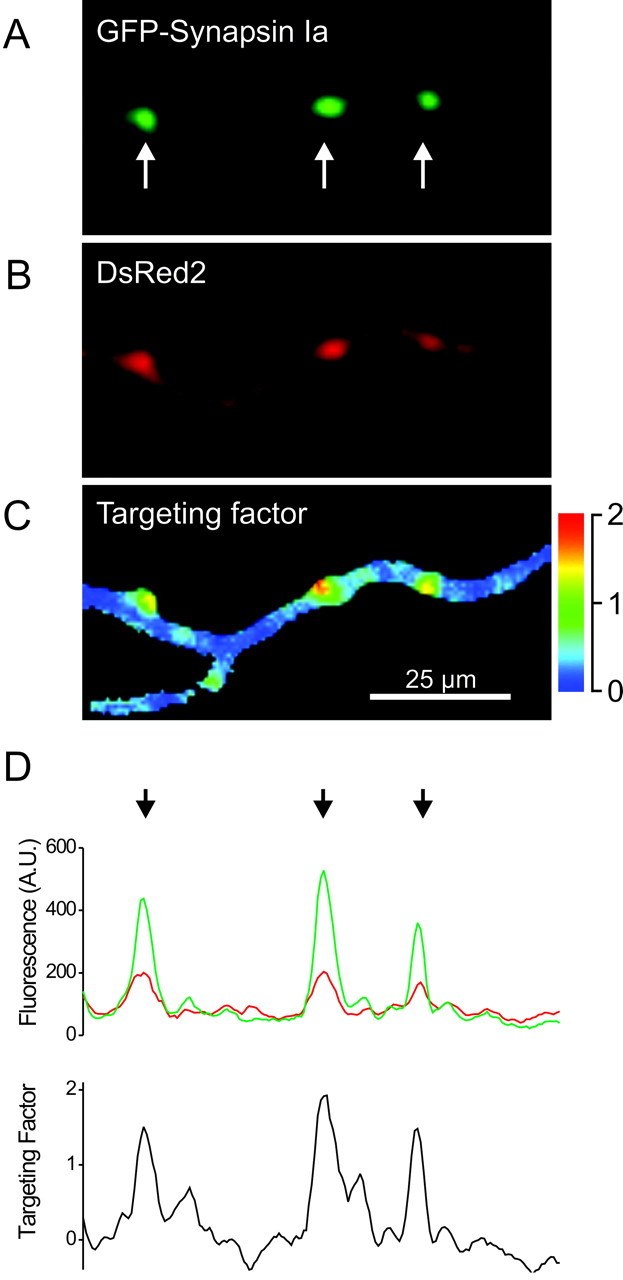
Quantification of synaptic targeting by fluorescence ratio imaging. Images of GFP-tagged synapsin Ia (A), DsRed2 (B), and the synaptic targeting factor ratio (C) are shown. The arrows in A indicate synaptic puncta. D, Calculation of the synaptic targeting factor. Top, Intensities of GFP and DsRed2 fluorescence along the axon shown in A and B. Bottom, Targeting factor was calculated for all points along the line, using the average intensity of axonal fluorescence. Note correspondence of the peaks in the targeting factor (arrows) with the puncta indicated in A.
We found that the targeting of synapsin Ia was higher in varicosities than in the axons of neurons from TKO mice (Fig. 4C,D), with a synaptic targeting factor of 1.76 ± 0.17. Likewise, synapsin Ib had a targeting factor of 0.24 ± 0.05, whereas the targeting factor of GFP was 0.0 ± 0.03, as expected for a protein that is not targeted. The differences between these values are statistically significant (p < 0.05), indicating that synapsin Ib targets to presynaptic terminals better than GFP does. However, the targeting factor for synapsin Ia was severalfold higher than that of synapsin Ib (p < 0.01), as is also evident in the images shown in Figure 3. Thus, synapsin Ia targets dramatically better than syapsin Ib. These results indicate that the ratiometic method quantitatively defines enrichment of synapsins in presynaptic terminals. The targeting factor varied by <2% over a 10-fold range of synapsin Ia expression levels (data not shown), showing that the ratiometric method also has the expected benefit of quantifying targeting independent of the level of synapsin expression. This finding also indicates that saturation of binding sites was not evident at the levels of synapsin expression used in our experiments and, therefore, does not account for differences in the targeting of the two isoforms.
Synapsin domains involved in targeting
To define the mechanisms that enable the synapsins to target specifically to the presynaptic terminals, we took advantage of the similarity of the Ia and Ib synapsin isoforms and asked why these two isoforms differ in their targeting capabilities. Synapsins Ia and Ib are formed by alternative splicing of the synapsin I gene transcript and are identical, except for the substitution of a 45 amino acid long C-terminal portion of domain E (synapsin Ia) by 9 amino acids belonging to domain F (synapsin Ib) (Südhof et al., 1989). To define the portions of synapsin Ia required for targeting, we examined the ability of GFP-tagged fragments of the two synapsin I isoforms, synapsin Ia and synapsin Ib, to target in TKO neurons (Fig. 5).
Figure 5.

Targeting of GFP-tagged synapsin I fragments in TKO neurons. Left, Diagram of the synapsin I fragments that were examined. Right, Mean targeting factor determined for each construct expressed in 2-week-old neurons. The targeting factor of soluble GFP was also measured as a control (bottom). Gray bars correspond to fragments that do not target to synapses, as defined by a targeting factor that is statistically indistinguishable from that of GFP. Error bars represent SEM values.
Given that synapsin Ia targets robustly in TKO neurons, whereas synapsin Ib targets very weakly, the E domain may play a role in the targeting of synapsin Ia. To examine the role of the E domain, we produced a construct lacking this domain (GFP-ABCD); this construct represents the entire sequence shared by synapsins Ia and Ib. The ABCD fragment did not target in TKO neurons, with a targeting factor similar to that of free GFP (p > 0.05). The lack of targeting of the ABCD fragment was not attributable to misfolding, because this construct targeted well in wild-type neurons (see below). This indicates that the E domain does participate in the targeting of synapsin Ia, whereas domain F does not enable targeting.
Although the E domain is important for targeting, this domain cannot be the sole determinant of targeting because synapsin IIb, which lacks the E domain (Fig. 1A), also targets (Fig. 3C). Given that synapsin IIb also lacks the D domain, we next considered the influence of the D domain on targeting. Remarkably, two constructs containing a truncated D domain (ABCD1 and ABCD4) (Onofri et al., 2000) were capable of targeting in TKO neurons. Their targeting factors were similar (p > 0.05) and intermediate between the values measured for the ABCD fragment and for full-length synapsin Ia (p < 0.01 for each comparison) (Fig. 5). Taken together, these results lead to several conclusions. First, part of ABCD is capable of targeting to synaptic terminals, meaning that the E domain is not exclusively responsible for the targeting of synapsin Ia. Second, the C-terminal 42 amino acids of the D domain (residues 618–659), which we will refer to as D′, contain a sequence that inhibits targeting because deletion of this region permitted targeting of ABCD1. Third, because synapsin Ia targets, the inhibition produced by D′ must be overcome by the E domain of synapsin Ia. Fourth, synapsin Ib targets weakly because it contains the inhibitory D′ segment while lacking an E domain. Fifth, the inhibitory effect of D′ cannot be overcome by the F domain of synapsin Ib.
Given that the ABCD1 and ABCD4 fragments of synapsin target in TKO neurons, we next determined the minimal structure within the A-D domains that is required for targeting. A fragment consisting of the A, B, and C domains (ABC) targeted well (Fig. 5). In contrast, a truncated version containing domains A and B and the first half of the C domain (ABC1) did not visibly target. Therefore, the sequence responsible for the targeting of synapsin I includes the C-terminal half of domain C. We next examined whether targeting was prevented by removing domain A, which includes a PKA phosphorylation site that has been reported to control the association of synapsins with membranes (Hosaka et al., 1999). This was not the case; the BC fragment targeted (Fig. 5), although to a lesser extent than the ABC fragment (p < 0.01). Interestingly, removing both domains A and B, leaving only the C domain, eliminated targeting altogether. The lack of targeting of domain C is not attributable to misfolding, because this construct targeted well in wild-type neurons (see below). We, therefore, conclude that domains B and C together are sufficient to permit some targeting in TKO neurons. Because the targeting capabilities of the ABC, ABCD1, and ABCD4 fragments were not significantly different (p > 0.05), we also can conclude that the inhibitory activity of the D domain is confined to D′. Finally, because the ABC fragment targeted better than the BC fragment (p < 0.01), we conclude that the A domain enhances targeting but is not essential for it.
The fact that the BC, ABC, ABCD1, and ABCD4 fragments exhibited only partial targeting in comparison to synapsin Ia (Fig. 5; p < 0.01) suggests that the E domain may enhance targeting beyond its ability to overcome the inhibitory influence of D′.To examine this possibility, we tested the targeting capabilities of GFP-tagged fragments consisting of domains CDE, CDF, CD, DE, and E. All of these constructs lacked the B domain that is necessary for the targeting of the BC combination, so that none of these would be expected to target if domain E played no direct role in targeting. In fact, we found that the CDE construct was capable of targeting as well as full-length synapsin Ia (p > 0.05) when expressed in TKO neurons. In contrast, the CD, CDF, DE, and E fragments did not target to presynaptic terminals (Fig. 5). These results indicate that the E domain plays a role in the targeting of synapsin Ia in addition to overcoming the inhibitory effect of D′, but that this role requires the C domain. The fact that both BC and CDE target independently (although not to the same extent; p < 0.01) suggests that each of these fragments harbors one of two (or more) targeting sequences found in synapsin Ia.
We have shown that the punctate pattern formed by GFP–synapsin Ia in a wild-type background corresponds mostly to presynaptic sites that are associated with postsynaptic densities (Fig. 2D). To determine whether this was true for the synapsin Ia fragments in a TKO background, we also examined PSD-95 localization relative to GFP-tagged synapsin fragments. We found that puncta formed by the BC and CDE fragments either overlap or appose one or more PSD-95-positive puncta (Fig. 6), indicating that these fragments target predominantly to presynaptic terminals.
Figure 6.

Localization of synapsin Ia fragments to synapses in TKO neurons. GFP-tagged BC (left) and CDE (right) constructs (green) apposed postsynaptic structures containing PSD-95 (red) in TKO neurons.
In summary, our results indicate that two domain stretches, BC and CDE, are sufficient for synapsin Ia to target to the presynaptic terminal. Although the contributions of these fragments are not equivalent, each of them targets independently of the other, suggesting that the targeting of full-length synapsin Ia is achieved by two separate mechanisms that require domains B, C, and E. The A domain enhances the targeting conferred by BC, whereas the D′ region apparently inhibits it. Finally, this inhibition is overcome by the E domain but not by the F domain.
Dimerization targets synapsin Ib
Synapsin Ib targets much better in wild-type neurons (Fig. 1C) than in TKO neurons (Fig. 3E,F). This indicates that the TKO neurons lack something, presumably other endogenous synapsin isoforms, that is required for the synaptic localization of synapsin Ib. We next tested the hypothesis that heterodimerization to other synapsins is required for the targeting of synapsin Ib.
Our tests of this hypothesis were based on comparison of synapsin targeting to the heterodimerization properties of synapsins described in vitro (Hosaka and Südhof, 1999). First, dimerization takes place via interactions between C domains (Esser et al., 1998). Although the C domain of synapsin I does not target in TKO neurons (Fig. 7A), our hypothesis predicts that dimerization with endogenous synapsins should allow this construct to target in wild-type neurons. This was indeed the case: the GFP-C domain construct targeted in wild-type neurons (Fig. 7B). To determine whether the targeting of the C domain fragment is mediated by direct interactions with synapsins, we coexpressed the C domain of synapsin I along with synapsin IIa in TKO neurons. The localization of both recombinant proteins was determined simultaneously by tagging the C domain with YFP, whereas synapsin IIa was tagged with CFP. As described above (Fig. 3B), synapsin IIa properly targeted to the presynaptic terminals of TKO neurons. We found that the C domain also targeted under these conditions, as predicted by our hypothesis (Fig. 7C). Similar experiments indicated that other fragments containing the C domain, but which do not target in TKO neurons, such as CD and ABCD, could target properly in wild-type neurons. Taken together, these results indicate that the C domain targets to presynaptic terminals in wild-type neurons by interacting with endogenous synapsins.
Figure 7.
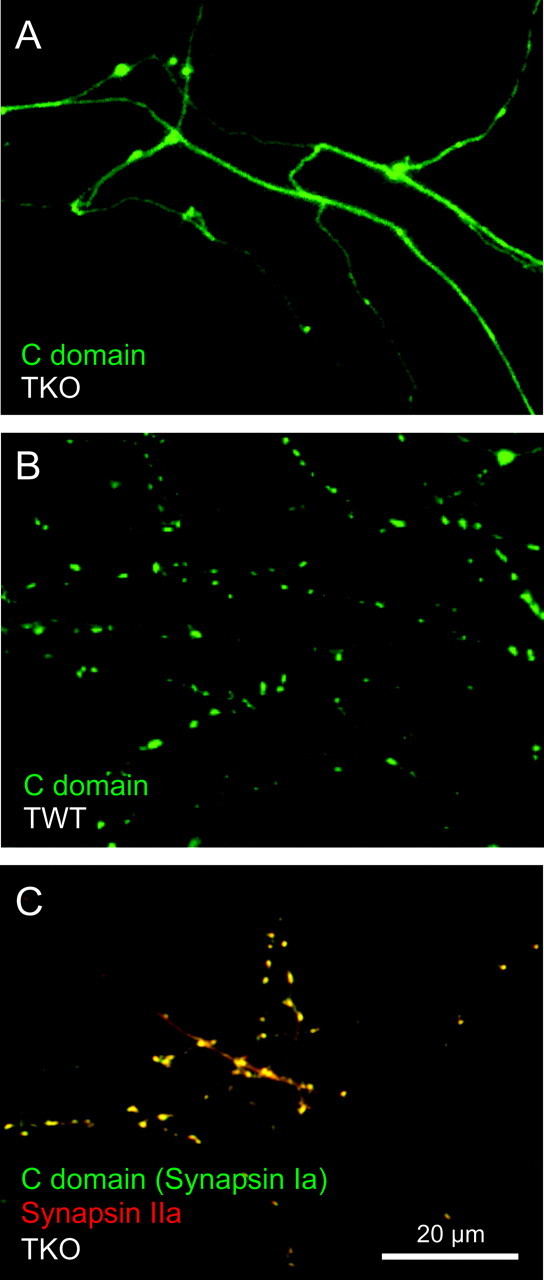
Targeting of domain C of synapsin I in TKO and wild-type neurons. A, GFP-tagged domain C of does not target in 2-week-old cultured TKO neurons. B, The same domain C construct does target in triple wild-type neurons. C, Overlay image of CFP–synapsin IIa (red) and YFP-C domain of synapsin I (green) in a TKO neuron. Synaptic colocalization of these constructs is indicated by yellow regions.
Dimerization of synapsins requires multiple interaction sites that are distributed throughout domain C (Esser et al., 1998). We, therefore, attempted to disrupt dimerization by fragmenting the C domain. Constructs containing either only the first half of the C domain (ABC1), or only the second half of the C domain (C′), did not target in wild-type neurons (data not shown). This illustrates that localization of synapsins requires large regions within the C domain, consistent with the requirements for dimerization. In addition, we found that the DE fragment did not target in wild-type neurons. Because neither DE nor ABC1 target, we conclude that targeting activity, like dimerization, requires the C domain.
We next determined whether dimerization of synapsin Ib with other synapsins is sufficient to target synapsin Ib. For this purpose, we took advantage of the fact that synapsin isoforms differ in their ability to heterodimerize: synapsin I can dimerize with synapsin I or with synapsin II but interacts very weakly with synapsin III (Hosaka and Südhof, 1999). We, therefore, asked whether isoforms that dimerize with synapsin I could also enhance the targeting of synapsin Ib in TKO neurons. For this purpose, we quantified the targeting capability of CFP–synapsin Ib when coexpressed with YFP-tagged versions of either synapsin Ia, synapsin IIa, or synapsin IIIa. Contrary to the exclusively axonal localization of synapsin Ib when expressed alone in TKO neurons (Fig. 8A), synapsin Ib was distributed at synapses when coexpressed with either synapsin Ia or synapsin IIa (Fig. 8B,C). Accordingly, both synapsins Ia and IIa significantly increased the targeting factor of synapsin Ib (p < 0.05) (Fig. 8E). In contrast, synapsin IIIa did not increase the targeting factor of synapsin Ib (p > 0.05) (Fig. 8E). In fact, synapsin Ib and synapsin IIIa remained separate, such that synapsin IIIa was localized to the presynaptic terminals while synapsin Ib remained dispersed throughout the axon (Fig. 8D). The perfect correlation between the ability of various synapsin isoforms to dimerize with synapsin Ib and to enhance the targeting of this isoform indicates that synapsin Ib targets via heterodimerization with other synapsins.
Figure 8.
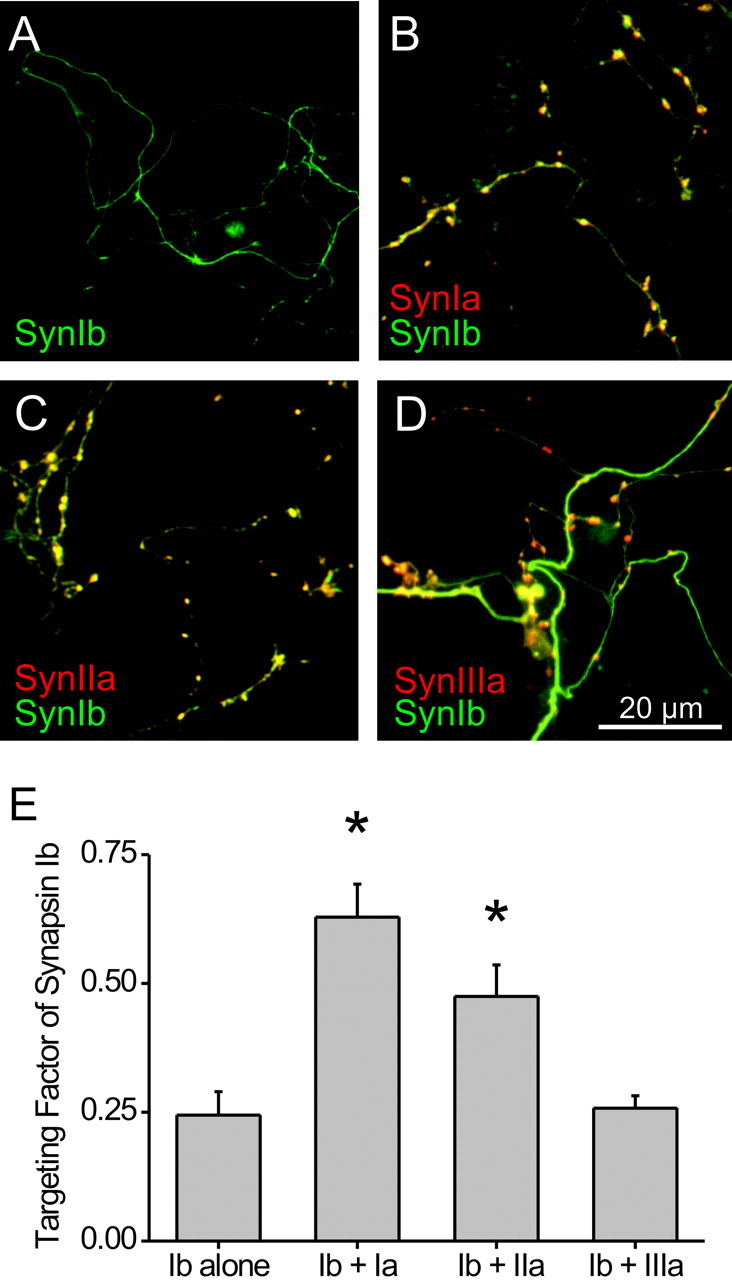
Differential effect of coexpression of various synapsin isoforms on targeting of synapsin Ib. Neurons from TKO mice were transfected with CFP–synapsin Ib singly (A) or together with YFP-tagged versions of synapsin Ia (B), synapsin IIa (C), or synapsin IIIa (D). Synapsin Ib colocalized with synapsins Ia and IIa in presynaptic terminals, as evidenced by the yellow color of the terminals. D, Lack of colocalization of synapsin Ib and of synapsin IIIa is evidenced by the localization of synapsin IIIa to synapses (red) and of synapsin Ib to the axon (green). E, Quantification of the targeting of synapsin Ib in the presence of other synapsin isoforms. The asterisks indicate significant (p < 0.05) differences from controls where the synapsin Ib was expressed alone.
Phosphorylation of sites 1–3 does not affect synapsin targeting
Binding of synapsins to purified SVs and liposomes is negatively regulated by phosphorylation at site 1 by protein kinase A or the type I Ca/calmodulin kinase, or at sites 2 and 3 by the type II Ca/calmodulin kinase (Huttner et al., 1983; Greengard et al., 1993; Stefani et al., 1997; Hosaka et al., 1999). To examine whether phosphorylation regulates the targeting of synapsins, we produced constructs in which these phosphorylation sites were mutated. Site 1 (serine 9) is situated in the A domain, whereas sites 2 (serine 566) and 3 (serine 603) are both in the D domain, proximal to the inhibitory D′ region. The phosphorylation sites were mutated either to alanine, to prevent phosphorylation, or to glutamate, to mimic a constitutively phosphorylated state (Thorsness and Koshland, 1987; Chi et al., 2003). The targeting factors of these mutated synapsins were measured in TKO neurons as described above. Mutation of either site 1 or simultaneous mutation of sites 2 and 3 did not change the targeting characteristics of synapsin Ia in TKO neurons (p > 0.05) (Fig. 9). Likewise, mutating sites 2 or 3 singly had no effect on targeting (data not shown). Because TKO neurons do not express endogenous synapsins that could affect the targeting of the mutant synapsins by dimerization, we conclude that the phosphorylation of site 1, 2, or 3 has no significant effect on the targeting of synapsin Ia to presynaptic terminals.
Figure 9.

Targeting of synapsin Ia with phosphorylation site mutations. GFP-tagged synapsin Ia mutants were examined in 2-week-old cultured TKO neurons, and the targeting factor was unaffected by the mutations in the indicated phosphorylation sites.
Discussion
We have uncovered two levels of mechanism involved in the targeting of synapsins. The first involves the roles of specific structural domains, whereas the second concerns the importance of dimerization.
Intramolecular determinants of targeting
Our results indicate that two combinations of synapsin I domains, BC and CDE, are adequate for targeting to presynaptic terminals. The CDE fragment targeted as well as synapsin Ia, whereas targeting of BC was partial and was enhanced by domain A. D′, located at the C-terminal portion of domain D, exerts a negative effect on targeting. The inhibitory effect of D′ was countered by domain E of synapsin Ia but not domain F of synapsin Ib, consistent with previous indications that domain E plays a role in the function of synapsins (Hilfiker et al., 1998). Based on these results, we propose the following structural model for targeting of synapsin I (Fig. 10). This model assumes that binding of synapsins to SVs contributes to targeting, although other factors also are involved (see below): (1) Domain B and D′: D′ may inhibit targeting by interacting with some other part of synapsin, specifically with domains B, C, or both (Fig. 10A). The N and C termini of the C domain of synapsin I are located only 15 Å apart near one of the tips of the C domain (Esser et al., 1998), indicating that D′ and its interacting partner on the N terminus of synapsin I may be positioned for such an interaction. An alternative explanation is that D′ interacts with amphiphysin II (Onofri et al., 2000), which may disrupt the interaction of synapsin Ib with SVs. (2) Domains E and F: Domain E may disrupt the inhibitory interaction between D′ and domain B, allowing domain B to interact with the SV surface (Fig. 10B). This interaction could be with negatively charged phospholipids, which bind to the N-terminal head portion of synapsin I (Benfenati et al., 1989a), or with an SV protein (Thiel et al., 1990). In addition to masking D′, domain E interacts directly with the SV, consistent with the observation that the C-terminal tail portion of synapsin I interacts with the type II Ca/calmodulin kinase on SVs (Benfenati et al., 1989b, 1992). The interactions of the N and C termini of synapsin Ia localize it to the surface of the vesicle and thereby impart specificity to the interaction with the SV. In the case of synapsin Ib, domain F cannot substitute for domain E and thus allows D′ to prevent this initial interaction (Fig. 10A). (3) Domain C: Domain C is required for synapsin Ia targeting and is known to insert into the SV membrane (Cheetham et al., 2001). To account for our observation that synapsin Ib cannot target, although it includes domain C, we postulate that either domain C cannot insert into the bilayer without the initial interaction of domains B and/or E with the SV or that D′ interacts with domain C to directly prevent membrane insertion. In either case, the interaction of domain C with membrane lipids stabilizes the interaction between synapsin Ia and the SV membrane.
Figure 10.

Molecular models of synapsin targeting. Targeting of synapsin Ib (A) and synapsin Ia (B), indicating the role of interactions of protein domains with each other and with SVs. C, Role of dimerization in synapsin targeting and SV cross-linking. Left, Synapsin Ia/Ia homodimers are divalent and can bind two SVs. Center, Synapsin Ia/Ib heterodimers are monovalent and can bind only one SV, allowing synapsin Ib to target in the presence of other synapsins but preventing SV cross-linking. Right, Synapsin Ib/Ib homodimers do not bind to or cross-link SVs.
In summary, we propose that interaction of synapsin Ia with SVs requires at least two steps: the initial localization of synapsin to SVs, mediated by transient interactions with vesicular constituents, and a subsequent stabilization of this interaction attributable to domain C inserting into the SV membrane. This model extends a previous proposal for the interaction of synapsins with SVs (Cheetham et al., 2001, 2003) and provides a rationale for the complex, mosaic domain structure of the synapsins. Because domain C is highly conserved across all synapsin isoforms (Kao et al., 1999), this domain may also stabilize interactions of synapsins II and III with the vesicle membrane. However, these synapsins do not possess D′, and the primary sequence of their B domains is not well conserved (Kao et al., 1999). Thus, the postulated initial interactions of these synapsins with the SV membrane must be regulated in a different way, although the homologous BC combination could be instrumental for the targeting of synapsin IIb, whereas the stronger BC... E combination could contribute to the targeting of synapsins IIa and IIIa.
Intermolecular determinants of targeting
Synapsin Ib targeting largely required the presence of other synapsins. This surprising characteristic of synapsin Ib probably has evaded previous detection because of cross-interactions between the various synapsin isoforms (Hosaka and Südhof, 1999) and because of the fact that synapsins Ia and Ib comigrate during biochemical purification. Indeed, our work is the first to define a clear functional distinction between the a and b splice variants of any synapsin gene. Our findings contrast with a previous report that overexpressed synapsin Ib apparently targets to the ribbon synapses of photoreceptors, cells that do not naturally express synapsins (Geppert et al., 1994). A possible explanation for this difference is that photoreceptor terminals are situated at the tips of axons, a geometry that makes it difficult to distinguish synaptic targeting from axonal transport. Thus, our finding that synapsin Ib concentrates in axons suggests that synapsin Ib is transported into the axons of the photoreceptors, yet is not specifically targeted to their terminals.
Our findings indicate the importance of heterodimerization in the targeting of synapsin Ib. We identified domain C as the site of this interaction, consistent with previous indications that synapsins dimerize through their C domains (Esser et al., 1998; Hosaka and Südhof, 1999). Our conclusion that heterodimerization is required for targeting of synapsin Ib is also the first indication of the functional significance of synapsin dimerization. Given that synapsin Ib dimers target weakly, if at all, whereas dimers of other synapsins target robustly, mixed dimers of synapsin Ib with synapsins Ia, IIa, or IIb may target only to an intermediate degree (Fig. 10C). Hence, synapsin Ib may interfere with the targeting of other synapsins and consequently with their presynaptic function. Therefore, the relative level of expression of the various synapsin isoforms may determine their ability to regulate SV clustering, the size of the reserve pool of SVs, and neurotransmitter release. Such regulatory interactions among isoforms may be one reason why so many different synapsins have evolved.
Whereas binding to SVs must be important for the targeting of synapsins to presynaptic terminals, our results indicate a number of differences between SV binding and targeting. For example, although the N-terminal head fragment and a C-terminal tail fragment of synapsins are each capable of binding to SVs (Benfenati et al., 1989b), binding of head and tail domains to SVs does not require an intact C domain, whereas targeting does. Furthermore, although phosphorylation of sites 1–3 regulates binding of synapsins to SVs (Huttner et al., 1983; Greengard et al., 1993; Stefani et al., 1997; Hosaka et al., 1999; Chi et al., 2001, 2003), we found that phosphorylation of sites 1–3 does not affect targeting of synapsin Ia. It is likely that targeting also involves other important processes, such as transport of synapsins to presynaptic terminals (Ahmari et al., 2000) and association with actin and other components of the presynaptic cytoskeleton (Hilfiker et al., 1999; Sankaranarayanan et al., 2003). The cellular approach that we have used here should allow future studies to clarify the contributions of SV binding, transport, and other processes to the targeting of the various synapsin isoforms.
In conclusion, we have shown that the targeting of synapsins to presynaptic terminals involves complex interactions within and between synapsin molecules. These results lead to the first model of synapsin targeting and a refined, testable model for the interaction of synapsins with SVs. We also discovered a dramatic difference between the targeting properties of synapsin Ia and synapsin Ib, providing the first suggestion of a functional rationale for the production of a- and b-type splice variants of synapsins.
Footnotes
This work was supported by National Institute of Mental Health Grants MH 39327 and MH 67044 and the Theodore and Vada Stanley Foundation Research Award (P.G. and J.F.). D.G. was supported by a European Molecular Biology Organization long-term fellowship and is a Pfizer fellow of the Life Science Research Foundation. We thank members of our laboratories and Y. Pazy for helpful discussion.
Correspondence should be addressed to Dr. G. Augustine, Department of Neurobiology, Duke University Medical Center, Box 3209, Durham, NC 27710. E-mail: georgea@neuro.duke.edu.
DOI:10.1523/JNEUROSCI.5225-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243711-10$15.00/0
References
- Ahmari SE, Buchanan J, Smith SJ (2000) Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci 3: 445–451. [DOI] [PubMed] [Google Scholar]
- Baumert M, Maycox PR, Navone F, De Camilli P, Jahn R (1989) Synaptobrevin: an integral membrane protein of 18,000 daltons present in small synaptic vesicles of rat brain. EMBO J 8: 379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Greengard P, Brunner J, Bahler M (1989a) Electrostatic and hydrophobic interactions of synapsin I and synapsin I fragments with phospholipid bilayers. J Cell Biol 108: 1851–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Bahler M, Jahn R, Greengard P (1989b) Interactions of synapsin I with small synaptic vesicles: distinct sites in synapsin I bind to vesicle phospholipids and vesicle proteins. J Cell Biol 108: 1863–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ (1992) Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature 359: 417–420. [DOI] [PubMed] [Google Scholar]
- Cheetham JJ, Hilfiker S, Benfenati F, Weber T, Greengard P, Czernik AJ (2001) Identification of synapsin I peptides that insert into lipid membranes. Biochem J 354: 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheetham JJ, Murray J, Ruhkalova M, Cuccia L, McAloney R, Ingold KU, Johnston LJ (2003) Interaction of synapsin I with membranes. Biochem Biophys Res Commun 309: 823–829. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2001) Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci 4: 1187–1193. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA (2003) Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron 38: 69–78. [DOI] [PubMed] [Google Scholar]
- Chin LS, Li L, Ferreira A, Kosik KS, Greengard P (1995) Impairment of axonal development and of synaptogenesis in hippocampal neurons of synapsin I-deficient mice. Proc Natl Acad Sci USA 92: 9230–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Malladi CS, Tan TC, Raymond CR, Smillie KJ, Robinson PJ (2003) Synapsin I-associated phosphatidylinositol 3-kinase mediates synaptic vesicle delivery to the readily releasable pool. J Biol Chem 278: 29065–29071. [DOI] [PubMed] [Google Scholar]
- Czernik AJ, Girault JA, Nairn AC, Chen J, Snyder G, Kebabian J, Greengard P (1991) Production of phosphorylation state-specific antibodies. Methods Enzymol 201: 264–283. [DOI] [PubMed] [Google Scholar]
- Dresbach T, Qualmann B, Kessels MM, Garner CC, Gundelfinger ED (2001) The presynaptic cytomatrix of brain synapses. Cell Mol Life Sci 58: 94–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser L, Wang CR, Hosaka M, Smagula CS, Südhof TC, Deisenhofer J (1998) Synapsin I is structurally similar to ATP-utilizing enzymes. EMBO J 17: 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, Takahashi RH, Kao HT, McEwen BS, Ryan TA, Augustine GJ, Greengard P (2002) Regulation of neurotransmitter release by synapsin III. J Neurosci 22: 4372–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Rapoport M (2002) The synapsins: beyond the regulation of neurotransmitter release. Cell Mol Life Sci 59: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Chin LS, Li L, Lanier LM, Kosik KS, Greengard P (1998) Distinct roles of synapsin I and synapsin II during neuronal development. Mol Med 4: 22–28. [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Kao HT, Feng J, Rapoport M, Greengard P (2000) Synapsin III: developmental expression, subcellular localization, and role in axon formation. J Neurosci 20: 3736–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher TL, Cameron P, De Camilli P, Banker G (1991) The distribution of synapsin I and synaptophysin in hippocampal neurons developing in culture. J Neurosci 11: 1617–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried G, Nestler EJ, De Camilli P, Stjarne L, Olson L, Lundberg JM, Hokfelt T, Ouimet CC, Greengard P (1982) Cellular and subcellular localization of protein I in the peripheral nervous system. Proc Natl Acad Sci USA 79: 2717–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Ullrich B, Green DG, Takei K, Daniels L, De Camilli P, Südhof TC, Hammer RE (1994) Synaptic targeting domains of synapsin I revealed by transgenic expression in photoreceptor cells. EMBO J 13: 3720–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goslin K, Schreyer DJ, Skene JH, Banker G (1990) Changes in the distribution of GAP-43 during the development of neuronal polarity. J Neurosci 10: 588–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F (1993) Synaptic vesicle phosphoproteins and regulation of synaptic function. Science 259: 780–785. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450. [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao HT, Czernik AJ, Greengard P, Augustine GJ (1998) Two sites of action for synapsin domain E in regulating neurotransmitter release. Nat Neurosci 1: 29–35. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P (1999) Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci 354: 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC (1998a) Synapsins I and II are ATP-binding proteins with differential Ca2+ regulation. J Biol Chem 273: 1425–1429. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC (1998b) Synapsin III, a novel synapsin with an unusual regulation by Ca2+ J Biol Chem 273: 13371–13374. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC (1999) Homo- and heterodimerization of synapsins. J Biol Chem 274: 16747–16753. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Südhof TC (1999) A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24: 377–387. [DOI] [PubMed] [Google Scholar]
- Hunt C, Schenker L, Kennedy M (1996) PSD-95 is associated with the postsynaptic density and not with the presynaptic membrane at forebrain synapses. J Neurosci 16: 1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P (1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96: 1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Porton B, Czernik AJ, Feng J, Yiu G, Haring M, Benfenati F, Greengard P (1998) A third member of the synapsin gene family. Proc Natl Acad Sci USA 95: 4667–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HT, Porton B, Hilfiker S, Stefani G, Pieribone VA, DeSalle R, Greengard P (1999) Molecular evolution of the synapsin gene family. J Exp Zool 285: 360–377. [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P (2002) A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci 5: 431–437. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Czernik AJ, De Camilli P, Greengard P, Townes-Anderson E (1992) Differential expression of synapsins I and II among rat retinal synapses. J Neurosci 12: 1736–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H (1996) Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem 271: 21108–21113. [DOI] [PubMed] [Google Scholar]
- Matus-Leibovitch N, Nevo I, Vogel Z (1997) Differential distribution of synapsin IIa and IIb mRNAs in various brain structures and the effect of chronic morphine administration on the regional expression of these isoforms. Brain Res Mol Brain Res 45: 301–316. [DOI] [PubMed] [Google Scholar]
- Menegon A, Dunlap DD, Castano F, Benfenati F, Czernik AJ, Greengard P, Valtorta F (2000) Use of phosphosynapsin I-specific antibodies for image analysis of signal transduction in single nerve terminals. J Cell Sci 113: 3573–3582. [DOI] [PubMed] [Google Scholar]
- Onofri F, Giovedi S, Kao HT, Valtorta F, Borbone LB, De Camilli P, Greengard P, Benfenati F (2000) Specificity of the binding of synapsin I to Src homology 3 domains. J Biol Chem 275: 29857–29867. [DOI] [PubMed] [Google Scholar]
- Pennuto M, Bonanomi D, Benfenati F, Valtorta F (2003) Synaptophysin I controls the targeting of VAMP2/synaptobrevin II to synaptic vesicles. Mol Biol Cell 14: 4909–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perin MS, Brose N, Jahn R, Südhof TC (1991) Domain structure of synaptotagmin (p65). J Biol Chem 266: 623–629. [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P (1995) Distinct pools of synaptic vesicles in neurotransmitter release. Nature 375: 493–497. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Porton B, Rendon B, Feng J, Greengard P, Kao HT (2002) Expression of synapsin III in nerve terminals and neurogenic regions of the adult brain. J Comp Neurol 454: 105–114. [DOI] [PubMed] [Google Scholar]
- Porton B, Kao HT, Greengard P (1999) Characterization of transcripts from the synapsin III gene locus. J Neurochem 73: 2266–2271. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM (1998) Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 18: 1217–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA (2003) Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci 6: 127–135. [DOI] [PubMed] [Google Scholar]
- Sheng M (2001) Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA 98: 7058–7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani G, Onofri F, Valtorta F, Vaccaro P, Greengard P, Benfenati F (1997) Kinetic analysis of the phosphorylation-dependent interactions of synapsin I with rat brain synaptic vesicles. J Physiol (Lond) 504: 501–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, et al (1989) Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science 245: 1474–1480. [DOI] [PubMed] [Google Scholar]
- Tanasugarn L, McNeil P, Reynolds GT, Taylor DL (1984) Microspectrofluorometry by digital image processing: measurement of cytoplasmic pH. J Cell Biol 98: 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel G, Südhof TC, Greengard P (1990) Synapsin II. Mapping of a domain in the NH2-terminal region which binds to small synaptic vesicles. J Biol Chem 265: 16527–16533. [PubMed] [Google Scholar]
- Thorsness P, Koshland Jr D (1987) Inactivation of isocitrate dehydrogenase by phosphorylation is mediated by the negative charge of the phosphate. J Biol Chem 262: 10422–10425. [PubMed] [Google Scholar]
- Trimble WS, Cowan DM, Scheller RH (1988) VAMP-1: a synaptic vesicle-associated integral membrane protein. Proc Natl Acad Sci USA 85: 4538–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Y, Nayak SK, Woodson J, Ross EM (2003) Phosphorylation-regulated inhibition of the Gz GAP activity of RGS proteins by synapsin I. J Biol Chem: M309626200. [DOI] [PubMed]
- Wiedenmann B, Franke WW (1985) Identification and localization of synaptophysin, an integral membrane glycoprotein of Mr 38,000 characteristic of presynaptic vesicles. Cell 41: 1017–1028. [DOI] [PubMed] [Google Scholar]