

Abstract
Previous studies have identified multiple transcription initiation sites for the glutamate receptor 2 (GluR2) gene, resulting in a heterogeneous population of GluR2 transcripts in vivo that differ in the length of their 5′-untranslated leaders (5′-UTR). We designed a series of monocistronic and dicistronic GluR2 cDNA constructs that model the natural in vivo transcripts and investigated their translation efficiencies in rabbit reticulocyte lysates, Xenopus oocytes, and primary cultured neurons. Transcripts containing long 5′ leaders (429 and 481 bases) were translated poorly compared with those with shorter leaders (341 or fewer bases). None of the five initiation codons in the 5′-UTR or the leader length per se were responsible for translation regulation. Rather, control of translation was mediated by a sequence containing a 34–42 nucleotide imperfect GU repeat predicted to form secondary structure in vivo. This translation suppression domain is included in some but not all rat and human GluR2 transcripts in vivo, depending on the site of transcription initiation. Rat cortex GluR2 transcripts that lack the translation suppression sequence were preferentially associated with polyribosomes. Furthermore, the GU-repeat cluster was found to be polymorphic in humans, raising the possibility that expansion or contraction of the GU-repeat cluster in certain populations might modify the level of GluR2 protein expression in neurons.
Keywords: GluR2, translation, polymorphism, hippocampus, GU repeat, human, AMPA receptor, untranslated, stem-loop, Gria2
Introduction
The AMPA subtypes of glutamate receptor (GluR) are assembled from combinations of GluR1, 2, 3, and 4 (GluRA, GluRB, GluRC, GluR) subunits. The Ca2+D permeability, rectification, and single-channel conductance of AMPA receptors are all dominantly influenced by inclusion of an edited GluR2 subunit in the receptor complex (Hollmann et al., 1991; Hume et al., 1991; Burnashev et al., 1992; Dingledine et al., 1992; Bowie and Mayer, 1995; Donevan and Rogawski, 1995; Kamboj et al., 1995; Koh et al., 1995; Swanson et al., 1997; Washburn et al., 1997). Receptors that contain a single edited (Seeburg, 1996) GluR2 subunit have maximally reduced Ca2+ permeability (Geiger et al., 1995; Washburn et al., 1997), whereas inward rectification is reduced in a graded manner as the number of GluR2 subunits in a receptor increases (Washburn et al., 1997). Thus, moderate changes in GluR2 expression are expected to have significant physiological consequences on neuronal excitability.
The relative expression of GluR2 changes in certain neuronal populations during development (Pellegrini-Giampietro et al., 1992a), after seizures or ischemic insult (Pellegrini-Giampietro et al., 1992b, 1994; Pollard et al., 1993; Friedman et al., 1994; Kamphuis et al., 1994; Prince et al., 1995; Gorter et al., 1997; Friedman, 1998), administration of antipsychotics and drugs of abuse (Ortiz et al., 1995; Fitzgerald et al., 1995, 1996), or corticosteroids (Nair et al., 1998), and also after tetanic stimulation of Schaffer collateral–commissural fibers in the hippocampus (Nayak et al., 1998). Recent studies aimed at understanding transcriptional control of GluR2 expression in neurons have identified key positive and negative regulatory elements in the GluR2 promoter (Myers et al., 1998), which play a role in molding GluR2 levels in brain after seizures (Huang et al., 1999, 2001), ischemia (Calderone et al., 2003), and treatment with brain-derived neurotrophic factor (Brene et al., 2000). Changes in GluR2 expression in brain have primarily addressed control of mRNA levels (i.e., transcription); however, translation control mechanisms may also be involved. GluR2 translation occurs locally in isolated dendrites of hippocampal neurons (Kacharmina et al., 2000). Understanding how the translation and subcellular distribution of GluR2 transcripts are regulated in neurons could be important for synaptic plasticity mechanisms. The purpose and regulation of dendritic targeting and translation of mRNAs, including GluR2, is receiving increased attention (Steward and Schuman, 2001; Eberwine et al., 2002; Huang et al., 2002b; Wang et al., 2002). However, the mechanisms involved in the control of GluR2 translation are unknown.
The population of GluR2 transcripts in vivo contain 5′ leaders with different lengths, some as “long” as 481 bases from the translation initiation codon (Köhler et al., 1994; Myers et al., 1998). However, the majority of these GluR2 transcripts include 5′-untranslated leaders (5′-UTR) ranging from 340 to 429 bases in length (Myers et al., 1998). Compared with most eukaryotic mRNA leader sequences (Kozak, 1987, 2002), these GluR2 5′ leaders are uncommonly long and contain noteworthy nucleotide sequence features that in other mRNAs are known to reduce translation under defined conditions (Kozak, 1991, 2002). Here, we evaluate the role of specific sequence elements in GluR2 5′-untranslated leaders on translation in cell-free reticulocyte lysates, in Xenopus laevis oocytes, and in transfected neurons, and we examine the association of native cortical GluR2 transcripts with polyribosomes. We conclude that a subpopulation of GluR2 mRNAs expressed in vivo is subject to translation inhibition by a polymorphic repeat motif in the GluR2 5′-UTR.
Materials and Methods
GluR2 5′-UTR constructs. Numerous constructs were designed to evaluate the influence of GluR2 5′-UTR sequences on translation in vitro and in heterologous expressions systems. Deletions in the GluR2 5′-UTR were generated by digestion with existing or introduced restriction sites, and the resulting sequences were placed upstream of the GluR2 coding sequence in pBluescript (Strategene, La Jolla, CA). The 5′-UTR regions were cloned at their 5′-ends into the XbaI site 40 bp downstream from the T3 RNA polymerase initiation site and at their 3′-ends using a natural NsiI site 12 bp into the GluR2 open reading frame (ORF), with the exception of the –302 leader, which was the original GluR2 cDNA cloned into the EcoRI site 72 bp from the T3 promoter (Boulter et al., 1990). Deletion constructs are named according to the length and source of the 5′-UTR (e.g., –429GluR2), with nucleotide designations given relative to the initiating AUG codon. Substitution mutations that abolish upstream initiation codons (AUG) codons residing in the GluR2 5′-UTR were made by a two-round PCR mutagenesis protocol (Cormack, 1997). These constructs are identified by the rank order position of the mutated AUG from the GluR2 AUG [e.g., δ(–5)GluR2 refers to removal of the fifth AUG upstream of the GluR2+1 AUG]. AUG codons were converted to GUU (δ-5), ACG (δ-4), or GCG (δ-3, δ-2, δ-1). Portions of the GluR2 5′-UTR were also inserted upstream of the GluR1 cDNA in pBluescript. The GluR1 5′-UTR, bases 1–139 (Hollmann et al., 1989), was also placed upstream of the GluR2 5′-UTR by similar standard cloning methods.
GluR2 5′-untranslated leaders were also inserted upstream of Photinus (firefly) luciferase in the pGL2 vector (Promega, Madison, WI). A T3 RNA polymerase promoter was inserted into the pGL2 polylinker SmaI site, and an NsiI site was engineered at the luciferase ATG. GluR2 5′-UTRs were restricted with XbaI and NsiI and cloned into pGL2 NheI and NsiI sites. In these pGL2-luciferase constructs, T3 RNA polymerase initiates transcripts 10 bases upstream of the inserted GluR2 leader. In addition, the GluR2 initiating AUG codon and downstream 12 bases were strictly preserved.
A dicistronic vector designed to drive expression of both firefly and Renilla luciferases from a single mRNA transcript was constructed. The purpose of this vector was to assay translation of firefly luciferase by ribosomes engaging the transcript at the 5′-end of a GluR2 5′-UTR sequence but, as an internal control, use the same mRNA transcript to monitor the synthesis of Renilla luciferase via ribosome entry at the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES). Both firefly and Renilla luciferase activities were measured in the same reaction tube using the Dual Firefly/Renilla assay kit (Promega). To generate these constructs, the EMCV IRES was first cloned upstream of Renilla luciferase in the pRL vector backbone by a standard PCR strategy that introduced a unique MscI restriction site at the Renilla AUG. This IRES-Renilla fragment was then excised and inserted into the 3′-untranslated region of firefly luciferase, moving the sv40 polyadenylation signal sequence to the end of the construct. This cloning step retained the T7 RNA polymerase promoter just upstream from the IRES to allow independent, diagnostic Renilla expression from this promoter. Finally, GluR2 5′-UTR fragments were inserted into the NheI-NsiI sites between a T3 RNA polymerase promoter and the firefly luciferase open reading frame, as above. The parent dicistronic construct design was confirmed by restriction and sequence analysis and assayed for effective firefly and Renilla luciferase expression (using T3 and T7 promoters, respectively) in rabbit reticulocyte lysate reactions before introduction of GluR2 sequences (data not shown). The dicistronic constructs were also modified for plasmid DNA transfections by cloning the cytomegalovirus (CMV) promoter into the SmaI-NheI polylinker sites, removing the T3 RNA polymerase promoter, and placing the CMV+1 transcription initiation site at the start of the GluR2 5′-UTR.
Cell-free translation in rabbit reticulocyte lysates. All cell-free rabbit reticulocyte lysate studies were conducted with trace amounts of 35[S]-methionine to label newly synthesized protein using a transcription–translation-coupled expression kit (TNT; Promega). Briefly, 1 μg of circular plasmid DNA was combined with TNT reaction buffer, amino acid mix (40 μm each amino acid except methionine), Rnasin (40 U), and 35[S]-methionine (5–10 μCi; 1000 Ci/mmol), in the absence of 5′-capping analog, plus T3 RNA polymerase in a total volume of 12.5 μlon ice. Reactions were initiated by the addition of 12.5 μl of rabbit reticulocyte lysate and incubation of tubes at 30°C. Assays were incubated from 10 to 45 min and terminated by freezing in a dry-ice ethanol bath. Circular DNA plasmids used in TNT reactions were phenol:chloroform extracted, precipitated, and resuspended in RNase-free water and carefully quantified before adding to the reaction mixture. Multiple plasmid preparations for each construct were tested.
Translation products were analyzed on SDS-PAGE gels or by TCA precipitation onto glass fiber filters. For analysis by TCA precipitation, 3–5 μl aliquots were removed and brought to 100 μl with 1N NaOH and then quick-frozen in a dry-ice ethanol bath. Samples were thawed at 37°C for 10 min followed by addition of 0.9 ml of an ice-cold 25% TCA/2% casamino acid solution and further incubated on ice for 30–45 min. The protein precipitate was collected onto glass fiber filters (glass B fibers; Schleicher & Schuell, Dassel, Germany) and washed three times with 3 ml of ice-cold 5% TCA by vacuum filtration. Filters were dried and counted by liquid scintillation spectroscopy. The background from blank reactions (no added plasmid) was subtracted from all samples. The recovered luciferase enzyme activity and 35S-methionine incorporation into protein in the TNT lysates were colinear over a 1000-fold range of expression (r = 0.995; data not shown). For gel analysis, 3–4 μl of each reaction was solubilized in 4 μl of loading buffer (0.2 m Tris, pH 6.5, 30% glycerol, 3.2% SDS, 15% δ-mercaptoethanol, 0.01% bromophenol blue), incubated at room temperature for 15–20 min, and loaded onto a 4% stacking/7% resolving SDS-polyacrylamide gel. The stacking gel contained 125 mm Tris-HCl, pH 6.8, and 0.1% SDS, and the resolving gel 375 mm Tris-HCl, pH 8.8, and 0.1% SDS. The running buffer consisted of 25 mm Tris base, 192 mm glycine, and 0.1% SDS. After electrophoresis, gels were soaked for >6 hr in isopropranol:acetic acid:H2O (25:10:65) and then in Amplify (Amersham Biosciences, Arlington Heights, IL) for 30 min before drying onto Whatman (Ann Arbor, MI) paper and exposure to Kodak (Rochester, NY) X-Omat film or a Molecular Dynamics (Sunnyvale, CA) phosphoimager plate for visualization of protein bands.
Sucrose gradient analysis of in vivo transcripts. Sucrose gradient fractionation of polysomes derived from rat cortex was based on the procedure by Feng et al. (1997), with the following modifications. Rats were anesthetized deeply with isoflurane, decapitated, and the cortex quickly dissected and weighed. Tissue was homogenized (Dounce type-B tight-fitting pestle; 50 strokes) at 150 mg tissue/ml in either ice-cold polysome-preserving or ice-cold polysome-disrupting buffer. Both buffers contained the following components: 20 mm Tris-HCl, pH 7.5, 100 mm KCl, 350 mm sucrose, 3.3 μl/ml Rnasin (Promega), 100 μg/ml cycloheximide, and 1 μg/ml each of phenylmethylsufonyl fluoride, aprotinin, leupeptin, and pepstatin A. The polysome-preserving buffer, in addition, contained 5 mm MgCl2, whereas the disrupting buffer contained 20 mm EDTA. In both cases, NP-40 detergent was added immediately after homogenization to a final concentration of 1.5%. Cellular debris was pelleted twice in sterile Microfuge tubes at 16,000 × g for 15 min. No more than 800 μgof A260 material was loaded in a volume of 700–900 μl onto the top of a 15–45% sucrose gradient for subsequent fractionation. Both polysome-preserving and polysome-disrupting gradient buffers contained 10 mm Tris-Cl, pH 7.5, and 80 mm NaCl. The preserving buffer, in addition, contained 5 mm MgCl2, whereas the disrupting buffer contained 1 mm EDTA. The gradients were run at 192,000 × g for 90 min at 4°. Twelve 1 ml fractions from each gradient were collected under RNase-free conditions. Each fraction was phenol extracted, and equivalent numbered fractions from four gradients were combined following standard alcohol precipitation.
The RNA pellet was rinsed once with 70% ethanol and repelleted. The entire pellet was the substrate in an RNase-protection assay using a ribonuclease protection assay (RPA) III kit (Ambion, Austin, TX), according to the instructions of the manufacturer, except that hybridization was performed overnight at 63°C. The 32P radiolabeled probe “D” (Myers et al., 1998) was used in all assays at 5–7 × 104 cpm/reaction. This probe is complementary to the region from bases –361 to –136 in the 5′-UTR of the rat GluR2 gene and yields products of 205 bases corresponding to the major “short” 340 base leader transcript and a 225 base product corresponding to all transcripts with 5′-UTRs longer than 361 bases combined into a single band. The radiolabeled products were ethanol precipitated, digested with RNase A and RNAseT1, and loaded directly onto a 5% denaturing sequencing gel. The dried gel was exposed on a phosphoimager screen for subsequent quantitation of band intensities using NIH Image.
RNase protection assay of human cortex mRNA. A human 5′-UTR construct was synthesized by PCR amplification of human genomic DNA between a GluR2 UP primer (TAGATCTAGACGTGAGTGAGAGAGGAG) and a GluR2 DOWN primer (TGAGCTCGAGCTGAAGTGGAGGCAGAAG). These primers amplify the human GluR2 5′-UTR between –136 and –360 bases upstream from the ATG. This PCR product was cloned into pBluescript XbaI (5′-end) and XhoI (3′-end) sites. The construct was linearized with XbaI for synthesis of 32P-radiolabeled probe as described above using T7 RNA polymerase. Six human neocortex samples were obtained from the Emory Alzheimer's Disease Center (Atlanta, GA). Total RNA isolated from each sample was subject to RNase protection with the human probe as described previously (Myers et al., 1998).
Xenopus oocyte expression of synthetic RNA. The GluR2 cDNA was modified in the 5′-UTR to compare the effect of different length 5′ leader sequences on GluR2 translation. All RNAs were synthesized in vitro from cDNA in pBluescript using a Stratagene (La Jolla, CA) RNA transcription kit. All RNAs synthesized for oocyte expression included the m7(5′)ppp(5′)G cap analog to enhance the stability of the RNA. A trace amount of [α-32P]CTP (∼0.5 μCi, 800 Ci/mmol) was included in the reactions to quantify the synthesis of RNA product by a DE81 filter adsorption assay. [α-32P]CTP-labeled RNAs were also resolved on denaturing agarose gels to verify the synthesis of full-length GluR2 and GluR1 RNA product. The original GluR1 cDNA (Hollmann et al., 1989) containing a 196 base leader was used in oocyte expression studies. All synthetic RNAs were stored under ethanol at –80°Cat15–20 ng/μl until use.
Oocytes were harvested from Xenopus laevis and injected with RNA as described by Dingledine et al. (1992). Different amounts of the –481GluR2, –302GluR2, or –7GluR2 RNAs were combined with GluR1 to yield GluR2:GluR1 mRNA mole ratios ranging from 1:1000 to 3:1. Each of the GluR2 RNAs representing the –481, –302, and –7 leader constructs, and the GluR1 mRNA, was derived from a “pool” of four or five independent RNA reactions. The relative RNA concentration of each GluR2 pool was verified by dot-blot hybridization on nylon membranes probed with a GluR2 coding-domain probe. Overall, 37 separate mRNA injections were made into seven different batches of oocytes for voltage-clamp recordings. In separate experiments, oocytes were injected with 35 ng of the 5′-capped dicistronic mRNA, and firefly and Renilla luciferase activities were measured from individual oocytes 24 hr later. In this experiment, oocytes were lysed by trituration in a pipette tip with 150 μl of lysis buffer (25 mm Tris-phosphate, pH 7.8, 2 mm DTT, 2 mm 1,2-diaminocyclo-hexane-N,N,N′,N′-tetraacetic acid, 10% glycerol, 1% Triton X-100) at room temperature. The lysates were then cleared by centrifugation and 10 μl assayed for both firefly luciferase and Renilla activity in the same tube in a Turner TD-20e luminometer using the Dual Firefly/Renilla assay kit (Promega).
Measurement of AMPA receptor currents in oocytes by voltage clamp. Oocytes were perfused with normal frog Ringer's solution (flow rate, 1–2 ml/min) composed of (in mm): 88 NaCl, 1.0 KCl, 24 NaHCO3, 10 HEPES, pH 7.4, 0.4 MgCl2, and 0.1 CaCl2. Recording microelectrodes (1–2.5 MΩ) were filled with a 3 m CsCl, 0.4 m EGTA solution. Once the membrane potential stabilized, oocytes were voltage clamped at –70 mV and subjected to a current–voltage (I–V) ramp from –100 to +50 mV applied immediately before, twice during, and once after bath application of 100 μm kainate. The I–V ramps measured before and after kainate application were averaged and then subtracted from the averaged current response measured from the two ramps conducted during the kainate application. The AMPA receptor I–V relationship was then fit by a least squares analysis to the following equation:
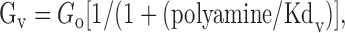 |
where Gv is the voltage-dependent channel conductance, and G0 is the channel conductance in the absence of internal polyamine block adjusted by the following equation to produce a rectification ratio of 2.9 to approximate the most outwardly rectifying currents observed for native and GluR2-saturated recombinant AMPA receptors as described by Washburn et al. (1997):
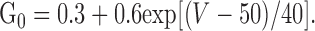 |
The affinity of internal blocking ions for their blocking site (Kdv) is a function of voltage according to the Woodhull equation for internal channel block by impermeable ions:
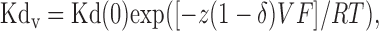 |
where Kd(0) is the blocker dissociation constant at 0 mV, z is the average effective valence of the impermeant blocker, δ is the effective electrical distance of the blocker through the membrane, and RT/F = 25.3 mV at 20°C (Woodhull, 1973). The calculated Kd reflects the combined effects of at least two internal polyamines (spermine and spermidine) and depends on the concentration of free polyamines in the oocyte, which for stage V oocytes has been estimated at ∼300 μm (Osborne et al., 1989). Because the affinity of channel blocker is a function of the relative numbers of GluR2 subunits in the heteromeric AMPA receptor complex (Washburn et al., 1997), we used the Kdv values as a measure of the relative amount of GluR2 protein in the AMPA receptor population.
mRNA stability in Xenopus oocytes. RNA was synthesized in vitro and radiolabeled with trace [α-32P]CTP as described above. mRNA for the –481GluR2, –302GluR2, and –7GluR2 leader constructs was injected (10–15 ng RNA/oocyte) into Stage V Xenopus oocytes followed by incubation in culture Barth's solution at 17°C. At various times after injection, six to seven oocytes from each group were harvested by transfer to an RNase-free tube and quickly frozen in a dry ice:ethanol bath. After all oocytes were harvested, RNA was isolated with the TRIzol reagent (Invitrogen, Gaithersburg, MD) and precipitated. The RNA pellet was dissolved in TE, resolved on an RNA formaldehyde gel, and exposed to Kodak X-Omat film or a Molecular Dynamics phosphoimager plate for visualization.
Culture and transfection of neurons with monocistronic DNA and dicistronic mRNA. Primary rat cortical cultures were prepared from embryonic day 18 rat pups resected from timed-pregnant Sprague Dawley rats and maintained as described previously (Myers et al., 1998). Cultures were grown for 3–7 d until transfection. These cultures are a mixture of neurons and glia with ∼60% of cells expressing the neuron-specific marker MAP-2 at the time of transfection.
Primary neurons were transfected with DNA plasmids using Lipofectamine according to the instructions of the manufacturer as described previously (Myers et al., 1998). Each well received 2.5–400 ng of the –429GluR2 or –341GluR2 luciferase reporter plasmid, 0.3 μg of the pRL control vector (Renilla luciferase driven by a thymidine kinase promoter; Promega), and empty pBluescript to balance all transfections to 1.5 μg/well. Ten to eighteen hours after transfection, cells were rinsed with 2 ml of PBS and harvested with 180 μl of lysis buffer (25 mm Trisphosphate, pH 7.8, 2 mm DTT, 2 mm EDTA, 10% glycerol, 1% Triton X-100) for 15 min at room temperature. The lysates were then cleared by centrifugation and 30 μl assayed for luciferase and Renilla activity as above.
Primary neurons were also transfected with synthetic dicistronic mRNA, essentially the same as for DNA transfections accounting for RNase-free conditions. Here, 0.5 μg of m7(5′)ppp(5′)G-capped dicistronic mRNA was combined with 5 μl of Lipofectamine and 50 μl of 50 mm NaCl in transfection media. Five to eighteen hours after the start of transfections, cells were rinsed with 2 ml of PBS and harvested with 180 μl of lysis buffer, and 30 μl aliquots of the cleared lysates were assayed for firefly luciferase and Renilla activity with a Dual Firefly/Renilla assay kit as above.
Sequence analysis of human GU repeats. Two sets of PCR primers flanking the GluR2 GU-repeat domain were used to amplify 100–200 ng human genomic DNA from 30 individuals drawn from a variety of ethnic backgrounds. The PCR primers were radiolabeled at their 5′-ends, and the products were analyzed on 6% denaturing polyacrylamide sequencing gels. PCR products were also resolved on high-resolution agarose gels and cloned into the TA cloning vector for direct sequence analysis of the resolved products (Sequenase 2.0; United States Biochemicals, Cleveland, OH). The following synthetic primers were used: Up1, TGCCCCTCTCTGTGACTTGC; Up2, GGACTGCCTGCCCCTCTCT, Down 1, TTCAGCACCCTCCCATGCAC; and Down 2, TGGTCCCAGTGTCTCGGAAT.
Results
Effect of 5′-UTR deletions on GluR2 translation
In oocytes, kainate-evoked currents through homomeric GluR1 receptors exhibit strong inward rectification, which is progressively relieved as the ratio of GluR2:GluR1 mRNA injected is increased (Fig. 1A). Interestingly, GluR2 mRNA containing a very short (7 bases) 5′ leader was much more effective in relieving rectification than GluR2 mRNA with a longer (481 bases) leader. For example, the I–V curve produced by a GluR2:GluR1 ratio of 1:100 with the short 7 base GluR2 leader is similar in shape to that produced by a ratio of 1:1.5 with the long 481 base leader (Fig. 1A). Because the GluR2 protein-coding region is identical, regardless of the 5′-UTR length, the shape of the I–V curve can be used to compare the translatability in oocytes of GluR2 RNAs bearing different sized leaders.
Figure 1.
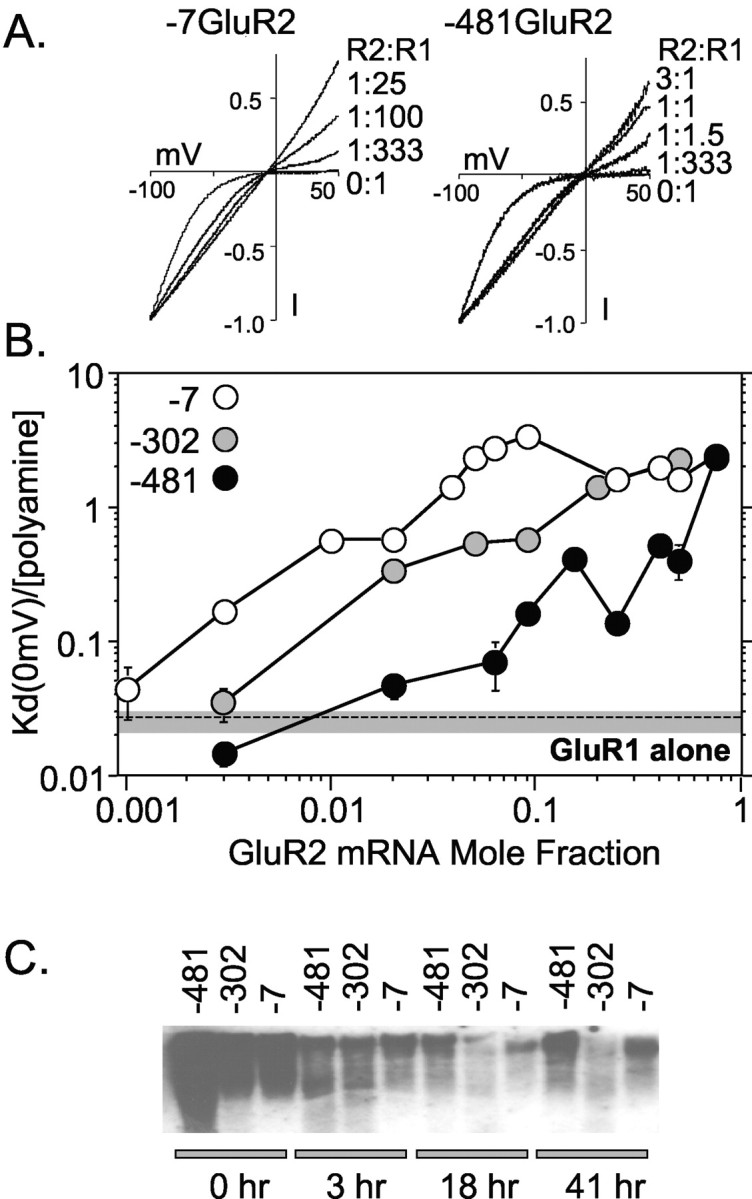
Effect of 5′-UTR truncations on GluR2 expression in Xenopus oocytes. GluR2 mRNAs with a different length 5′-UTR (481, 302, and 7 bases long) were mixed with GluR1 mRNA to achieve different GluR2:GluR1 mRNA ratios and injected into oocytes. After 2–4 d, oocytes were voltage clamped, and kainate I–V curves were generated. A, I–V curves for selected ratios of injected GluR2:GluR1 mRNA. Each curve shown is the average I–V plot from 5–24 oocyte recordings, normalized to the current at –100 mV. Data were fit by a least squares criterion to the Woodhull equation for internal channel block by an impermeable channel blocker. The fitted lines superimpose on the experimental data. B, Comparison of the translation efficiency of GluR2 mRNAs with different 5′ leader sequences. I–V curves were fit to derive the affinity of channel block by the polyamine blocker(s) at 0 mV [Kd(0 mV)], which is related to the number of GluR2 subunits in an heteroligomeric AMPA receptor complex (Washburn et al., 1997). Each data point represents the mean ± SE for 5–24 oocytes. Injections and recordings were made from seven oocyte preparations. For reference, the mean Kd(0 mV)/[polyamine] and 95% confidence limits for GluR1 homomeric receptors are shown as a shaded box with a dashed line. These data suggest that GluR2 mRNA translation in Xenopus oocytes is reduced by the 302 base and the 481 base leaders compared with the no-leader (7 base) control by up to fivefold and 30-fold, respectively. C, Blot of mRNA recovered from oocytes injected with 32[P]-labeled mRNA for each of the GluR2 5′ leaders. Each lane represents RNA recovered from six to seven oocytes for each construct at the indicated time point after injection. A representative blot from one of three experiments is shown.
Inward rectification of kainate-activated AMPA receptors results from voltage-dependent channel block by internal polyamines (Bowie and Mayer, 1995; Donevan and Rogawski, 1995; Kamboj et al., 1995; Koh et al., 1995). The shape of the I–V relationship can be adequately described by the Woodhull equation for internal channel block by impermeable ions to estimate the affinity of the channel blocking site at 0 mV (Kd[0 mV]) for the blocking ion (Bowie and Mayer, 1995; Washburn et al., 1997). The affinity of polyamines for the channel-blocking site is strongly and progressively reduced by the presence of increasing amounts of the GluR2 subunit (Fig. 1B). Approximately 30-fold more –481GluR2 than –7GluR2 mRNA was required to produce macroscopic AMPA receptor currents with equivalent rectification properties. Similarly, approximately fivefold more of the –481GluR2 than the –302GluR2 mRNA was required to produce similar AMPA receptor rectification properties. Importantly, for each of the GluR2 5′ leaders injected, if enough GluR2 mRNA was combined with GluR1, the same maximum increase in the Kd(0 mV) was observed (Fig. 1B). The large differences in the required amount of injected GluR2 mRNA to produce similar receptor properties is unlikely attributable to mRNA degradation, because the three mRNAs exhibited no evidence of differential stability in oocytes (Fig. 1C). Furthermore, dot-blot analysis of the synthetic mRNA pools showed that errors in the amount of mRNA injected per oocyte were very low (data not shown). The conclusion that differences in RNA stability do not underpin the observed differences in translation efficiency is reinforced by findings with dicistronic mRNA and cDNA (see below). Thus, these data are consistent with the interpretation that long GluR2 leaders are poorly translated compared with short leader sequences.
We investigated this finding further by conducting in vitro translation reactions with a transcription–translation-coupled expression system and collection of 35[S]-methionine-labeled protein precipitated onto glass fiber filters. A series of seven GluR2 5′ leader sequences was inserted between the T3 RNA polymerase promoter and the GluR2 protein-coding domain (Fig. 2A). At various times during the incubation, aliquots were removed and analyzed for 35[S]-methionine incorporation. Constructs containing the longest GluR2 leaders of 481 and 429 bases produced little protein accumulation even after 30 min of incubation, whereas constructs with 341 and 302 base leaders showed significant protein synthesis (Fig. 2B). Additional truncation of the GluR2 leader region to 250, 109, or 7 bases from the GluR2 AUG resulted in maximum relief from translation block (Fig. 2B). When these data are presented as rates of protein synthesis over the 10–20 min interval (Fig. 2C), an eightfold increase in mRNA translation efficiency is observed when the leader was truncated from 481 to 341 bases, and approximately fourfold from 429 to 341 bases. Thus, short uncapped GluR2 5′-UTRs appear to be translated more efficiently than long uncapped 5′-UTRs in the TNT assay, a finding consistent with our results in oocytes injected with exogenous 5′-capped mRNA.
Figure 2.

Effect of 5′-UTR truncations and upstream AUG codons on GluR2 expression in rabbit reticulocyte lysates. A, Schematic of the GluR2 5′-UTR deletion series. Top numbers indicate the 5′-ends of a series of deletion constructs relative to the GluR2 AUG. Middle numbers refer to upAUG codons (black bars) and the length of the associated short ORF (arrows). B, Time course of GluR2 protein synthesis in cell-free reticulocyte lysates for a 5′-UTR deletion series. 35[S]-methionine-labeled protein was precipitated with TCA and collected onto glass fiber filters. Blank reactions were subtracted as background. Results shown represent the mean ± SE for four to seven experiments except for the –481 and –7 leaders, which had 16 and 13 experiments, respectively. Each symbol represents results from a different 5′ leader. Inset shows the protein products at 30 min resolved on a denaturing polyacrylamide gel and visualized by exposure to film. C, The average rate of protein synthesis for the 10–20 min interval (B) was calculated for each GluR2 5′-UTR deletion construct. *p < 0.05 by ANOVA and post hoc Dunnett's test, compared with the –481 construct. D, The effect of upAUG codons removed by substitution mutations [i.e., δ(–5)] in two leader contexts on the rate of GluR2 protein synthesis during the 10–20 min reaction interval. Mean ± SE (n = 3–8) for all mutant constructs, except for the –481GluR2 and –302 GluR2 control constructs, which had 13 and 10 experiments, respectively. *p < 0.05 by ANOVA and post hoc Dunnett's test compared with the –481 or –302 construct.
The –481GluR2 and –429GluR2 constructs represent naturally occurring transcripts in vivo. The 481 base leader is a rare transcript, whereas the 429 base leader is abundant and has been designated as the +1 transcriptional start site for both mouse and rat GluR2 genes (Köhler et al., 1994; Myers et al., 1998). The –341GluR2 construct represents another abundant, but short, GluR2 transcript identified in vivo (Myers et al., 1998).
Influence of upstream AUG codons
Depending on the site of transcription initiation for in vivo GluR2 transcripts, the 5′ leader contains up to five AUG codons (Table 1) that could serve as premature translation initiation sites and thereby lure ribosomes away from the GluR2 initiation AUG codon (Kozak, 1991, 2002; Morris and Geballe, 2000). Alternatively, the short peptides derived from the upstream reading frames might themselves inhibit translation machinery engaged with the GluR2 mRNA (Hill and Morris, 1993). To address the potential impact of these upAUG codons, we removed them by substitution mutation and assayed for effects on GluR2 translation in TNT-reticulocyte lysate reactions. These data are summarized in Figure 2D. Essentially, elimination of each of the five upAUGs from the longest GluR2 leader context had no effect on the rate of protein synthesis (Fig. 2D, left). Likewise, elimination of the two most 5′ AUGs [δ(-5/-4)] or all five AUGs together [δ(-all five)] had no significant effect on the rate of protein synthesis. The δ(-3/-2/-1) triple mutation, however, in the short –302GluR2 and long –481GluR2 contexts, showed a substantial yet nonsignificant increase in translation efficiency (Fig. 2D). The –3AUG might influence translation in combination with other AUGs in short transcripts as suggested by the observed increase in translation when the –302GluR2 leader was further shortened to –250GluR2 (Fig. 2B,C). Nonetheless, these data on the whole suggest that upAUG codons in the GluR2 leader have nominal effect on translation in cell-free rabbit reticulocyte lysates.
Table 1.
AUG codons and associated short ORFs in the GluR2 5′-UTR
|
AUGa |
Positiona |
Kozak sequenceb |
ORFa |
Length |
Speciesa |
|---|---|---|---|---|---|
| -5 | -379 | uguguAUGu | MCVYVCARSCERGEREKRARERVIV | 25 | R, M |
| -4 | -369 | uguguAUGu | MCVRARVREERGRRELERG | 19 | H, R, M |
| -3 | -297 | agtgcAUGg | MGGC | 4 | H, R |
| -2 | -98 | tcacaAUGc | MQRI | 4 | H, R, M |
| -1 | -20 | tgtggAUGc | MLYFSWKCKRLCIFLSSFLLFYGD | 24 | H, R, M |
| +1 |
1 |
tggaaAUGc |
MQKIMHISVLLS... GluR2 protein |
883 |
H, R, M |
Rank order AUG number, nucleotide position relative to the rat GluR2 AUG, and uORF for rat sequences. R, Rat; M, mouse; H, human.
The Kozak consensus sequence is CCACCAUGG; underlined bases match the consensus (Kozak, 1987).
Transfer of suppression to another ORF in a dicistronic construct
Use of the TNT assay to assess the rates of translation efficiencies can be problematic if the 5′-UTR itself exerts effects on the rates of transcription from the T3 promoter. For this reason, we designed dicistronic vector constructs to assess the effects of the 5′-UTR on translation in both TNT and Xenopus oocyte translation assays. Because both firefly and Renilla protein-coding regions are contained within a single RNA species, bias in the results because of errors in the amount or stability of RNAs injected in the oocytes is minimized. Furthermore, use of the dicistronic vectors in the TNT assay should help guard against untoward 5′-UTR effects on transcription. We fused a series of GluR2 5′ leaders upstream of firefly luciferase in a pGL2 vector that had been modified to include an EMCV IRES sequence and Renilla luciferase at the 3′-end (Fig. 3A). This dicistronic vector assayed GluR2 5′-UTR-dependent translation of firefly luciferase via scanning ribosomes as well as IRES-dependent translation of Renilla luciferase translation via ribosomes engaging the RNA internally at the IRES. Four GluR2 5′-UTR-dicistronic mRNA constructs containing leader lengths of 481, 429, 341, and 302 bases were compared. The context of the GluR2 AUG codon including the Kozak consensus sequence and the first five GluR2 amino acids was exactly transferred to firefly luciferase in all constructs. The control transcript (Fig. 3B,C, Vect) contained 240 bp of the vector sequence as the 5′-UTR.
Figure 3.

The GluR2 5′-UTR is sufficient for translation inhibition. A, Schematic of the dicistronic constructs used in TNT reticulocyte lysate and mRNA injections into Xenopus oocytes. Several GluR2 5′ leader sequences were cloned downstream of a T3 promoter. Firefly luciferase translation is affected by the inserted GluR2 5′-UTR, whereas the EMCV IRES-driven Renilla luciferase translation is not. B, A series of dicistronic constructs were assayed in TNT-lysate reactions. The average protein synthesis rate for the 10–20 min reaction interval (inferred from enzymatic activity) from two separate reactions is plotted in the left panel. The ratio of firefly (Fire) to Renilla (Ren) activity is shown in the right panel. The control construct (Vect) contains 240 bases of vector sequence as its 5′ leader. Error bars in the right graph represent the range from two experiments. TLU, Turner light units. C, 5′-Capped mRNA made in vitro from the dicistronic constructs were injected into Xenopus oocytes, and luciferase activities were measured at 24 hr. Average raw firefly (Fire) and Renilla (Ren) luciferase activities from a representative experiment (6 oocytes per group) are plotted in left graph. The firefly to Renilla enzyme activity ratios in the right graph are the mean ± SE (n = 18–21 per construct) pooled from three experiments. *p < 0.05 from –429 constructs by ANOVA and post hoc Neuman–Keuls test. These results show that translation of firefly luciferase is attenuated by the 5′-most region of the longest GluR2 5′ leaders, between 341 and 429 bases from the AUG. The attenuation of translation is independent of the GluR2 coding and 3′-untranslated regions.
In both the oocyte mRNA expression and rabbit reticulocyte TNT translation assays, Renilla expression was similar, regardless of the GluR2 5′-UTR inserted into the construct (Fig. 3B,C, black bars, left panel), suggesting that IRES-directed ribosome entry and stability of the dicistronic mRNAs was unaffected by the GluR2 5′-UTR. However, translation of firefly luciferase depended significantly on the GluR2 5′-UTR sequence in the construct (Fig. 3B,C, left panel, open bars). Inspection of the firefly to Renilla activity ratios for the various GluR2 5′-UTRs confirms that a sequence lying between –341 and –429 has substantial translation suppressing activity in both expression assays (Fig. 3B,C, right panels). These data demonstrate that the long GluR2 5′-UTR (429 bases) is capable of suppressing translation not only of GluR2 (Figs. 1, 2) but also heterologous ORFs such as firefly luciferase (Fig. 3). These results also show that the GluR2 5′-UTR is necessary and sufficient to reduce translation in the absence of GluR2 coding and 3′-UTR domains.
To determine whether translation suppression by the longer GluR2 leaders is attributable to the nucleic acid sequence itself or simply an effect of lengthening the leader, we exchanged regions of the GluR2 5′-UTR with regions of the GluR1 5′-UTR. The original GluR1 cDNA (Hollmann et al., 1989) includes a 5′-UTR of 196 bases. Transfer of 139 bases of the GluR1 5′-UTR (lacking internal AUGs) to the 5′-end of –341GluR2 produced a 480 base leader that did not suppress translation of the GluR2 cDNA (Fig. 4A, compare lanes 2 and 3). Conversely, transfer of the candidate GluR2 translation suppression domain, from –341 to –429 bases, to the 5′-end of the GluR1 5′-UTR resulted in a strong reduction in translation (Fig. 4A, compare lane 4 with lanes 5 and 6). The addition of a different region of the GluR2 leader to GluR1, from –250 to –109 bases, was without effect (Fig. 4, lane 7). Finally, placement of the GluR2 (–341 to –429) and the GluR1 (–57 to –196) 5′-UTRs in front of the firefly luciferase 5′-UTR transferred their respective activities to this cDNA as well (Fig. 4, constructs 9–11). Sample bands of protein products are presented in Figure 4B. The results with different reporters, in both monocistronic and dicistronic constructs, reinforce the conclusion that translation suppression is dependent on the sequence but not the length of the GluR2 5′-UTR.
Figure 4.

Transfer of the GluR2 translation inhibition domain to other mRNAs. A, Portions of the GluR2 and GluR1 5′-UTRs were shuffled to make 5′-UTR chimeras indicated by shaded or hatched bars. Open bars represent sequence regions unchanged in the construct design. The graphs in the right panels show translation rates in TNT reactions, normalized to the respective wild-type (wt) 5′-UTR and ORF (i.e., GluR2, GluR1, and Firefly luciferase). Mean ± SE (n = 10–15) from eight experiments is shown. *p < 0.05, from construct 7 or construct 11 by ANOVA and post hoc Neuman–Keuls test. Note the different scales on bar graphs. B, Representative 35[S]-methionine-labeled protein products from TNT-lysate reactions. Lane numbers indicate different constructs as shown in A. np, No plasmid control.
Analysis of GluR2 transcripts in rat brain regions
Previously, it had been shown in rodent brain that GluR2 transcription initiated at multiple sites between –341 and –481 bases from the AUG (Köhler et al., 1994; Myers et al., 1998). We were interested to determine whether all GluR2 transcripts were equally associated with brain polyribosomes. Ribosome-associated mRNA from rat brain was fractionated by sucrose gradient in the presence or absence of Mg2+. RNA near the bottom of the gradient is dominated by the denser polyribosome fractions and considered to be actively translated, whereas mRNA sedimenting near the top of the gradient (associated with monosomes and small polyribosomes) is stalled or poorly translated. Indeed, disruption of translating polyribosomes by chelation of Mg2+ caused the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript, normally translated efficiently, to shift from the bottom to near the top of the gradient (Fig. 5A). Parallel mRNA fractions prepared with Mg2+ were probed with a GluR2 cRNA designed to identify both short (341 bases) and long (360–481 bases) GluR2 leaders in an RNase protection assay. Short GluR2 transcripts were present predominantly in the heavy polyribosome fractions (Fig. 5B), suggesting these transcripts were loaded with ribosomes and actively transcribed. In contrast, long GluR2 transcripts with leaders from 360 to 481 bases were distributed over most of the gradient, including the lightest fraction at the top (Fig. 5B). These results are consistent with the notion that cortical GluR2 mRNA bearing long leaders are found in a variety of states of translation, whereas transcripts with short leaders are actively translated. The results from eight experiments are summarized in Figure 5C. The relative abundance of long and short GluR2 transcripts is different between cortex, hippocampus, and cerebellum (Fig. 5D,E), reinforcing the possibility that different neurons use different transcriptional start sites and thus translate GluR2 mRNA with different degrees of efficiency. In hippocampus, the ratio of long and short GluR2 transcripts approaches unity, whereas in adult cerebellum and cortex, this ratio favors long transcripts by threefold (Fig. 5D,E).
Figure 5.

Differential association of long and short GluR2 transcripts with polyribosomes. A, Sucrose gradient fractions of rat cortex mRNA probed by RNase protection assay with a GAPDH cDNA. When tissue was homogenized in the presence of Mg2+, the majority of transcripts are recovered in the heavy polyribosome fraction, whereas disrupting polysomes with EDTA causes GAPDH to migrate to the lighter regions of the gradient. B, Sucrose gradient fractions prepared in the presence of Mg2+ were analyzed in an RNase protection assay with a GluR2 probe to detect short and long GluR2 transcripts. Short transcripts include leaders 341 bases long, and long transcript leaders are >360 bases from the AUG (see Materials and Methods for details) (Myers et al., 1998). Lanes on gel are designated as follows: P, probe; D, digested probe; T, unfractionated cortical mRNA; lanes 1–11, sucrose gradient fractions (11 = heaviest). C, Quantified ratio of long/short GluR2 transcript band intensities in each sucrose gradient fraction from eight experiments (mean ± SE). The average ratio in the four topmost fractions of the gradient is significantly larger than that at the bottom of the gradient (p < 0.05). D, RNase protection assays with the GluR2 RPA probe across three brain regions. Each lane represents a separate reaction against total poly A + mRNA of a different rat. L, Long; S, short. E, The mean ± SE (n = 7) of the ratio of long/short GluR2 transcripts in cortex, hippocampus (Hippoc.), and cerebellum (Cereb.). Asterisk, Each region is different from others; *p < 0.05 by ANOVA with post hoc Neuman–Keuls test.
Translation suppression in transfected neurons
Finally, we determined whether the region between –429 and –341 bases in the GluR2 5′-UTR could suppress translation in cultured neurons by transfections of 5′-capped mRNA synthesized from the dicistronic constructs. When the ratio of firefly and the Renilla luciferase activity was measured in the cell lysates, the long GluR2 leader (429 bases) significantly reduced translation compared with two short GluR2 leaders (341 and 302 bases) as well as the vector leader (Fig. 6A). This effect was not attributable to differences in the amount of Renilla luciferase produced by the –429GluR2, –341GluR2, and –302GluR2 dicistronic constructs (35 ± 10, 27 ± 10, and 30 ± 11 Turner light units, respectively; n = 6). Transfections were done on 4-, 5-, and 7-d-old neuronal cultures, and the lysates were prepared 5 and 15 hr after transfection with no difference in the results. Thus, results from all experimental conditions are combined in Figure 6A. In addition to RNA transfections, the 429 and 341 base GluR2 leaders were fused to firefly luciferase downstream of a CMV promoter, and the plasmid DNA was transfected into cultured neurons. For these experiments, the cultures were cotransfected with the pRL Renilla vector to normalize for well-to-well variability. Over a range of 10 to 400 ng DNA per well, the plasmid bearing the 341 base GluR2 leader consistently showed increased expression compared with the 429 base GluR2 leader. The results are summarized in Figure 6B. Together, these results demonstrate that the long GluR2 leader (429 bases) is less active than the short GluR2 transcript (341 bases or less) in transfected cultured neurons, consistent with translation attenuation by long GluR2 5′-UTRs.
Figure 6.

Translation suppression in cultured neurons. A, mRNA from dicistronic constructs was synthesized in vitro and transfected into cultured neurons as described in Materials and Methods. These mRNAs drive expression of both firefly and Renilla luciferases as depicted in Figure 3A. The ratio of firefly to Renilla luciferase activity recovered in the cell lysates is shown; mean ± SE for 10–11 transfections for each construct except vector control (V; n = 6) in five cortical cultures. *p < 0.05 from –429 construct by ANOVA and post hoc Neuman–Keuls test. The mean Renilla activity recovered per well per construct was 35.1, 26.8, 29.8, and 15.5 light units for the –429, –341, –302, and the vector control, respectively. B, Plasmid DNA transfections into cultured neurons. Both constructs include a CMV promoter just upstream from the GluR2 5′-UTR and firefly luciferase. Neurons were cotransfected with pRL-Renilla to normalize for well-to-well variation and then normalized to the 400 ng –429GluR2 construct to compare across experiments. Results are mean ± SE (n = 3). These results show that in mixed neuronal–glial cultures from rat cortex, translation is suppressed by the –341 to –429 region of the GluR2 5′-UTR.
Polymorphic human GluR2 GU-repeat domain
The most striking feature of the minimally effective translation suppression domain (Figs. 2, 3, 4) is the presence of a 40-base imperfect GU-repeat sequence. This sequence is also present in the human GluR2 gene. To determine whether this domain is polymorphic, we analyzed 30 human genomic DNA samples by PCR amplification across the GU-repeat region using two sets of primers (see Materials and Methods). PCR products were resolved on polyacrylamide sequencing and high-resolution agarose gels to identify changes in length of the GU-repeat region. Several unique PCR product sizes were observed on the gels. To confirm that these human allele variants were indeed caused by changes in the GU-repeat size, PCR products for each allele size were cloned and sequenced. In sum, 62 alleles from a variety of ethnic groups were analyzed, including the GluR2 sequence deposited in the human genome database (Lander et al., 2001) plus a human genomic clone obtained from the United Kingdom Medical Research Council Human Genome Resource Center (GenBank accession number HS115G5R). Our results are summarized in Figure 7A. The most common GluR2 allele (74% of all sequences) contained a GU-repeat length of 36 nucleotides. Additional GU variants ranged from 34 to 42 nucleotides long.
Figure 7.
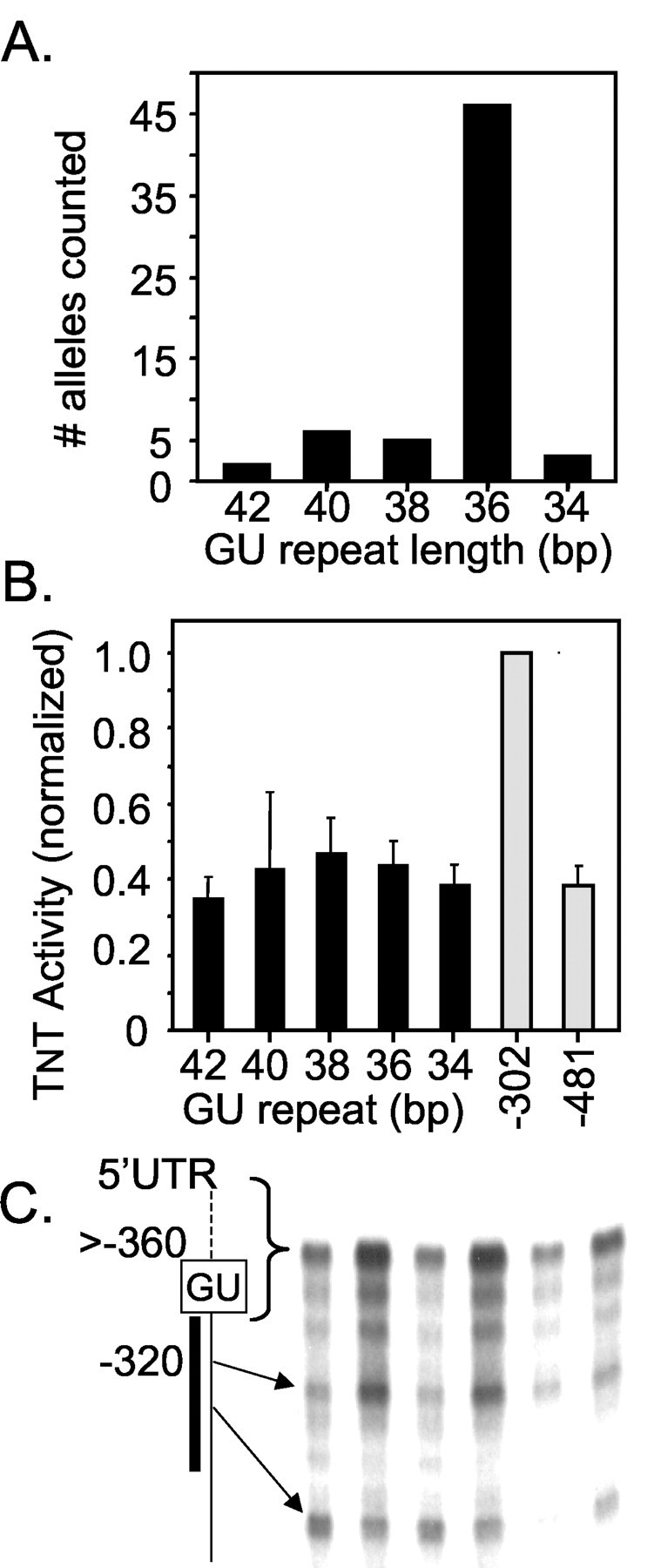
Human GluR2 polymorphisms. A, PCR amplification and sequencing of human genomic DNA was conducted to identify the length of the GU repeat for 62 human alleles. The graph summarizes the frequency of each allele containing GU-repeat lengths of 34, 36, 38, 40, and 42 nucleotides. B, Effect of the human GU-repeat lengths on translation in a TNT reticulocyte lysate assay. Human GU-repeat lengths were fused to the 5′-end of the rat –302GluR2 leader. The translation rate for each construct was then normalized to the –302GluR2 control (mean ± SE; n = 5). The activity for rat –481GluR2 is also shown (light gray bars). C, RNase protection against six human cortex mRNA samples shows the presence of both shorter and long GluR2 transcripts (compare Myers et al., 1998). The vertical diagram to the left represents the 5′-ends of human GluR2 mRNAs showing the position of the GU repeat and the position of the RNase probe (thick line). All transcripts longer than the probe length are summed into the slowest band. The prominent transcriptional start site near –320 bases is shown.
The five human GU-repeat variants were subcloned to the 5′-end of the rat –302GluR2 construct, and the plasmid DNA was added to TNT reticulocyte lysate assays to measure the rate of 35[S]-methionine-labeled protein production, as in previous experiments. Each of the variant human GU repeats tested resulted in a 2.5- to 3-fold reduction in translation efficiency compared with the –302GluR2 control leader. The activity of these human GU constructs is similar to the activity seen for the long rat –481GluR2 construct (Fig. 7B). No correlation between the length of the GU repeat and rates of protein translation was observed, suggesting that a GU-repeat length of 34 nucleotides is sufficient to maximally reduce translation of GluR2 under the conditions tested.
To confirm that multiple transcription start sites are used in the human GluR2 gene to produce transcripts that either lack or contain the translation–suppression GU motif, we performed an RNase protection experiment on total RNA isolated from neocortex of six individuals. The 32P-labeled probe was designed to sum all natural transcripts that contain the GU repeat and longer (more than –360) (Fig. 7C, in brackets) into a single band as described by Myers et al. (1998). In addition to the long band indicative of transcripts containing the GU repeat, two prominent shorter protected bands were observed (Fig. 7C), the longer of which represents a transcriptional start site at approximately –320 bases from the initiating AUG. This site corresponds to a cluster of 5′ rapid amplification of cDNA ends sites identified in rat GluR2 mRNA located near –300 from the AUG (Myers et al., 1998). These results suggest conserved transcriptional start sites in rat and human and confirm that not all human GluR2 transcripts contain the GU repeat.
Discussion
The major conclusion from these studies is that different populations of rat and human GluR2 transcripts exhibit different translation efficiencies resulting from the presence or absence of a short polymorphic sequence in their 5′-untranslated leaders (Fig. 8A). This conclusion is supported by the following findings. First, short but not long GluR2 transcripts were preferentially associated with polyribosomes in rat brain, indicating that short transcripts are more favorably translated in vivo. Second, the rate of GluR2 translation was reduced more than fourfold for GluR2 mRNAs with a long 5′-UTR (>429 bases long) compared with those with a short (341 or fewer bases) 5′-UTR in multiple assays involving both monocistronic and dicistronic cDNAs and mRNAs expressed in Xenopus oocytes, rabbit reticulocyte lysates, and transfected rat cortical neurons. Third, the major GluR2 transcription initiation sites identified in rat and human brain flank the translation suppression region, which resides within a sequence 341–429 bases upstream of the AUG. The expression of both short and long GluR2 transcripts with different translation efficiencies in vivo, combined with the polymorphic nature of the GU repeat, enriches the ability of neurons to shape synaptic responses through control of GluR2 expression.
Figure 8.

Translation control domain in the GluR2 5′-UTR. A, GluR2 transcription is regulated by an RE1 silencer (black box) and by Sp1 and NRF-1 transcriptional elements (stippled boxes) near a zone of transcription initiation sites (diagonal bars). Translation of GluR2 mRNA is suppressed by a region from –341 to –429 that includes a GU-repeat sequence, which is present only in the subset of long native GluR2 mRNAs transcribed from upstream start sites. B, Secondary structure prediction for the 5′-end of rat GluR2 mRNA using the FoldRNA algorithm from Wisconsin GCG. The input sequence for the analysis was from –429 to the GluR2+1 AUG, but only the structure for the 5′-end of the predicted folded segment is shown. The stem-loop structure that includes the GU-repeat sequence (enclosed in the dashed box) has a predicted free energy, ΔG, of –40 kcal/mol.
Potential mechanisms of GluR2 translation suppression
Ribosome scanning is a well supported model that explains how translation is initiated in eukaryotes. In this model, the 40S subunit, primed with initiation factors and Met-tRNAi, engages the mRNA at the 5′-cap and “scans” the transcript in a 5′ to 3′ direction, seeking an AUG with favorable surrounding sequence to initiate translation. Initiation is generally the rate-limiting step in eukaryotic translation (Mathews et al., 1996), thus factors diminishing ribosome access to or scanning of the 5′-UTR, such as the absence of a 5′-cap, a long 5′-UTR with complex secondary structure, the presence of upAUGs with their associated ORFs, or a poor Kozak context of the initiator AUG, can reduce overall translation efficiency (Morris and Geballe, 2000; Kozak, 2002). The GluR2 5′-UTR includes rare features, such as several upAUG codons, a region of high GC content including a dinucleotide repeat sequence, and an overall length of 341–481 bases, each of which by itself might reduce translation (Kozak, 1991, 2002). Our results demonstrate that translation suppression was not mediated by the upAUG codons, an interaction between the 5′-UTR and the GluR2 coding or 3′-UTR, changes in mRNA stability, or the leader length per se. Lack of effects by the upAUGs was somewhat unexpected, because the first AUG encountered by a scanning ribosome is almost always recognized for translation initiation. However, the influence of upAUGs can be averted if scanning ribosomes bypass the upAUG altogether or reinitiate translation at the downstream AUG (Kozak, 1987, 2002; Morris and Geballe, 2000). In the case of GluR2, none of the upAUGs, except the –2AUG, bears a strong Kozak signature sequence (Table 1). Given a poor context, ribosomes might simply traverse the GluR2 upAUG codons without initiating translation, a process termed “leaky scanning” (Kozak, 1991; Lin et al., 1993). Another mechanism by which ribosomes can bypass the upAUGs is by engaging the mRNA internally, downstream of the upAUGs directed by an IRES-like structure in the 5′-UTR of long transcripts. IRES-dependent translation of several viral and cellular mRNAs has been shown, but as yet no consensus IRES sequence has emerged and the extent of IRES-dependent translation in eukaryotes, although growing in number, is still debated (Hellen and Sarnow, 2001; Kozak, 2001).
A more likely mechanism for the GluR2 translation suppression, similar to that proposed for the NR2A 5′-UTR (Wood et al., 1996), involves interference of ribosome scanning through mRNA secondary structure. A stable stem-loop structure would reduce translation by forcing the scanning ribosome to slow down as it melts the double-stranded RNA. Modeling of a long GluR2 construct (–429GluR2) by an RNA-folding algorithm (FoldRNA; Genetics Computer Group, Madison, WI) predicted a structured hairpin loop at the extreme 5′-end of the mRNA (Fig. 8B). This stem-loop structure has a calculated free energy of –33 kcal/mol, if just the region between –429 and –341 is considered. Typically, a free energy of approximatley –30 kcal/mol is not sufficient to impede ribosome scanning, unless the loop is near the 5′-cap site; thus, the position of the structural motif is important for translation control (Kozak, 1989; Goossen and Hentze, 1992). If additional downstream GluR2 sequence is included in the analysis, however (Fig. 8B), the free energy of the entire loop that includes the GU repeat increases to approximately –40 kcal/mol. Thus, the predicted structure in the longer GluR2 mRNAs appears stable enough and is positioned in a manner to impair ribosome scanning.
Functional implications for translation regulation of GluR2
The preferential association of short GluR2 transcripts with heavy polyribosomes indicates that these transcripts are actively translated in vivo, whereas the dispersal of GluR2 transcripts with long 5′-UTRs (>360 bases) throughout the sucrose gradient is indicative of transcripts in various stages of translation (Fig. 5B). A large fraction of GluR2 transcripts must be poorly translated in vivo, if at all, because long transcripts are more abundant than short GluR2 transcripts in the lightest sucrose fractions (Fig. 5B,C), and the bulk level of long transcripts is greater than that of short ones (Fig. 5D,E). However, the association of polyribosomes with some GluR2 mRNAs bearing long 5′-UTRs suggests that a mechanism might exist for overcoming translation suppression, such as the binding of a regulatory protein to the 5′-UTR that can melt the hairpin loop. Alternatively, the long transcripts in Figure 5 could include some natural transcripts that are 368 or 399 nucleotides long (Myers et al., 1998), because the RNase protection probe used in Figure 5 summates all GluR2 transcripts with 5′-UTRs >360 bases long. Shortening the –429GluR2 transcript by even 30 bases could partially disrupt the hairpin loop structure predicted in Figure 8B, relieving translation inhibition. Additional work is needed to choose between these alternatives.
The finding that the GU-repeat sequence in the GluR2 translation suppression domain is polymorphic in humans raises the possibility that contraction or expansion of this sequence could lead to constitutive differences in GluR2 expression in different people. Allele-length dependent translation rates were not observed in reticulocyte lysates, but effects on translation in neurons are not ruled out and, perhaps more importantly, our small sample of only 62 human alleles may have been insufficient to identify rare repeat lengths that influence translation. A GU-repeat length of 34 nucleotides was sufficient to reduce translation of GluR2 under the conditions of our experiments (Fig. 7B). The 5′ leader is highly conserved between rat and human, including the Sp1 and NRF-1 transcriptional control elements located upstream of the GU repeat (Myers et al., 1998). Importantly, RNase protection experiments show that human GluR2 transcripts exist that lack the GU-repeat segment entirely, similar to those in mouse and rat transcripts (Köhler et al., 1994; Myers et al., 1998), suggesting that in human brain, GluR2 transcripts with different translation efficiencies are present.
The relative amount of GluR2 transcripts with long versus short 5′-UTRs varies across brain regions (Fig. 5C,D) (Myers et al., 1998), suggesting that favored transcription start sites are neuron specific. As a consequence, different neuron populations express pools of GluR2 transcripts with different translation efficiencies. Indeed, a significant proportion of CA3 stratum radiatum interneurons express GluR2 mRNA that is edited at the Q/R site but is apparently not translated, as judged by the properties of functional AMPA receptors in these interneurons (Washburn et al., 1997). Together, these observations point to the intriguing possibility that neurons have devised a regulatory system to increase or decrease GluR2 protein levels via changes in transcription initiation, which consequently enables or disables the translation control system. Another intriguing possibility is that long GluR2 transcripts could bind an endogenous protein that targets the mRNA to subcellular compartments ready for local GluR2 translation. The subcellular targeting and local translation of mRNAs in neurons (Steward and Schuman, 2001; Eberwine et al., 2002), including synthesis of GluR2 protein in hippocampal pyramidal cell dendrites (Kacharmina et al., 2000), increases the ability of neurons to regulate synaptic transmission and plasticity in a more spatial and temporal manner. This study shows that endogenous GluR2 transcripts possess variable potentials for translation into protein. However, additional work is needed to determine whether GluR2 transcripts containing the GU-repeat domain are preferentially targeted to dendrites and to uncover the mechanisms by which translation control is itself regulated.
Footnotes
This work was supported by the National Institutes of Health (NIH) National Institute of Neurological Disorders and Stroke (R.D.) and an individual NIH National Research Service Award (S.J.M.). We thank Sunan Zhang for providing neuronal cultures.
Correspondence should be addressed to Ray Dingledine, Department of Pharmacology, Emory University School of Medicine, Atlanta, GA 30322. E-mail: rdingledine@pharm.emory.edu.
S. J. Myers's present address: Department of Pharmacology, University of Washington, Seattle, WA 98195.
Y. Huang's present address: Department of Neuroscience, The Johns Hopkins University, Baltimore, MD 21205.
T. Genetta's present address: Department of Pediatrics, Emory University, Atlanta, GA 30322.
DOI:10.1523/JNEUROSCI.4127-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243489-11$15.00/0
References
- Boulter J, Hollmann M, O'Shea-Greenfield A, Hartley M, Deneris E, Maron C, Heinemann S (1990) Molecular cloning and functional expression of glutamate receptor subunit genes. Science 249: 1033–1037. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15: 453–462. [DOI] [PubMed] [Google Scholar]
- Brene S, Messer C, Okado H, Hartley M, Heinemann SF, Nestler EJ (2000) Regulation of GluR2 promoter activity by neurotrophic factors via a neuron-restrictive silencer element. Eur J Neurosci 12: 1525–1533. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Khodorova A, Jonas P, Helm PJ, Wisden W, Monyer H, Seeburg PH, Sakmann B (1992) Calcium-permeable AMPA-kainate receptors in fusiform cerebellar glial cells. Science 256: 1566–1570. [DOI] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh K-M, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MVL, Zukin RS (2003) Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci 23: 2112–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack B (1997) Directed mutagenesis using the polymerase chain reaction. In: Current protocols in molecular biology (Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, eds), pp 8.5.1–8.5.10. New York: Wiley. [DOI] [PubMed]
- Dingledine R, Hume RI, Heinemann SF (1992) Structural determinants of barium permeation and rectification in non-NMDA glutamate receptor channels. J Neurosci 12: 4080–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA (1995) Intracellular polyamines mediate inward rectification of Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptors. Proc Natl Acad Sci USA 92: 9298–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberwine J, Belt B, Karcharmina JE, Miyashiro K (2002) Analysis of subcellularly localized mRNAs using in situ hybridization, mRNA amplification, and expression profiling. Neurochem Res 27: 1065–1077. [DOI] [PubMed] [Google Scholar]
- Feng Y, Absher D, Eberhart DE, Brown V, Malter HE, Warren ST (1997) FMRP associates with polyribosomes as an mRNP, and the I304N mutation of severe Fragile X Syndrome abolishes this association. Mol Cell 1: 109–118. [DOI] [PubMed] [Google Scholar]
- Fitzgerald LW, Deutch AY, Gasic G, Heinemann SF, Nestler EJ (1995) Regulation of cortical and subcortical glutamate receptor subunit expression by antipsychotic drugs. J Neurosci 15: 2453–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ (1996) Drugs of abuse and stress increase the expression of GluR1 and NMDAR1 glutamate receptor subunits in the rat ventral tegmental area: common adaptations among cross-desensitizing agents. J Neurosci 16: 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LK (1998) Selective reduction of GluR2 protein in adult hippocampal CA3 neurons following status epilepticus but prior to cell loss. Hippocampus 8: 511–525. [DOI] [PubMed] [Google Scholar]
- Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS (1994) Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci 14: 2697–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JRP, Melcher T, Koh D-S, Sakmann B, Seeburg PH, Jonas P, Monyer H (1995) Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principle neurons and interneurons in rat CNS. Neuron 15: 193–204. [DOI] [PubMed] [Google Scholar]
- Goossen B, Hentze MW (1992) Position is the critical determinant for function of iron-responsive elements as translational regulators. Mol Cell Biol 12: 1959–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MVL, Conner JA, Zukin RS (1997) Global ischemia induces downregulation of GluR2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci 17: 6179–6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellen CUT, Sarnow P (2001) Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev 15: 1593–1612. [DOI] [PubMed] [Google Scholar]
- Hill JR, Morris DR (1993) Cell-specific translation translational regulation of S-adenosyl-methionine decarboxylase messenger RNA-dependence on translation and coding capacity of the cis-acting upstream open reading frame. J Biol Chem 268: 726–731. [PubMed] [Google Scholar]
- Hollmann M, O'Shea-Greenfield A, Rogers SW, Heinemann SF (1989) Cloning by functional expression a member of the glutamate receptor family. Nature 342: 643–648. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S (1991) Ca2+ permeability of KA-AMPA gated glutamate receptor channels depends on subunit composition. Science 252: 851–853. [DOI] [PubMed] [Google Scholar]
- Huang Y, Myers SJ, Dingledine R (1999) Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci 10: 867–872. [DOI] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R (2002a) Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22: 8422–8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-S, Jung MJ, Sarkissian M, Richter JD (2002b) N-methyl-d-aspartate receptor signaling results in Aurora kinase–catalyzed CPEB phosphorylation and αCAMKII mRNA polyadenylation at synapses. EMBO J 21: 2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF (1991) Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 253: 1028–1031. [DOI] [PubMed] [Google Scholar]
- Kacharmina JE, Job C, Crino P, Eberwine J (2000) Stimulation of glutamate receptor protein synthesis and membrane insertion within isolated neuronal dendrites. Proc Natl Acad Sci USA 97: 11545–11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG (1995) Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. J Physiol (Lond) 486: 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphuis W, De Rijk TC, Talamini LM, Lopes da Silva FH (1994) Rat hippocampal kindling induces changes in the glutamate receptor mRNA expresssion patterns in dentate granule neurons. Eur J Neurosci 6: 1119–1127. [DOI] [PubMed] [Google Scholar]
- Koh D-S, Burnashev N, Jonas P (1995) Block of native Ca2+-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. J Physiol (Lond) 486: 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler M, Kornau H-C, Seeburg PH (1994) The organization of the gene for the functionally dominant α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit GluR-B. J Biol Chem 269: 17367–17370. [PubMed] [Google Scholar]
- Kozak M (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger mRNAs. Nucleic Acids Res 15: 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1989) Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol 9: 5134–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1991) An analysis of vertebrate mRNA sequences: intimations of translational control. J Cell Biol 115: 887–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (2001) New ways of initiating translation in eukaryotes? Mol Cell Biol 21: 1899–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (2002) Pushing the limits of the scanning mechanism for initiation of translation. Gene 299: 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, et al. (2001) Initial sequence and analysis of the human genome. Nature 409: 860–921. [DOI] [PubMed] [Google Scholar]
- Lin F, MacDougald OA, Diehl AM, Lane MD (1993) A 30 kDa alternative translation product of the CCATT/enhancer binding protein α message: transcriptional activator lacking antimitotic activity. Proc Natl Acad Sci USA 90: 9606–9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Sonenberg N, Hershey JWB (1996) Origins and targets of translational control. In: Translational control (Hershey JWB, Mathews MB, Sonenberg N, eds), pp 1–30. New York: Cold Spring Harbor, NY.
- Morris DR, Geballe AP (2000) Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol 20: 8635–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers SJ, Peters J, Huang Y, Comer MB, Barthel FD, Dingledine R (1998) Transcriptional regulation of the GluR2 gene: neural-specific expression, multiple promoters, and regulatory elements. J Neurosci 18: 6723–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SM, Werkman TR, Craig J, Finnell R, Joels M, Eberwine JH (1998) Corticosteroid regulation of ion channel conductances and mRNA levels in individual hippocampal CA1 neurons. J Neurosci 18: 2685–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak A, Zastrow DJ, Lickteig R, Zahniser NR, Browning MD (1998) Maintenance of late phase LTP is accompanied by PKA-dependent increase in AMPA receptor synthesis. Nature 394: 680–683. [DOI] [PubMed] [Google Scholar]
- Ortiz J, Fitzgerald LW, Charlton M, Lane S, Trevisan L, Guitart X, Shoemaker W, Duman RS, Nestler EJ (1995) Biochemical actions of chronic ethanol exposure in the mesolimbic dopamine system. Synapse 21: 289–298. [DOI] [PubMed] [Google Scholar]
- Osborne HB, Mulner-Lorillon O, Marot J, Belle R (1989) Polyamine levels during Xenopus laevis oogenesis: a role in oocyte competence to meiotic resumption. Biochem Biophys Acta 158: 520–526. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Bennett MVL, Zukin RS (1992a) Are Ca2+-permeable kainate/AMPA receptors more abundant in immature brain? Neurosci Lett 144: 65–69. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Zukin RS, Bennett MVL, Cho S, Pulsinelli WA (1992b) Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA 89: 10499–10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Pulsinelli WA, Zukin RS (1994) NMDA and non-NMDA receptor gene expression following global brain ischemia in rats: effect of NMDA and non-NMDA receptor antagonists. J Neurochem 67: 1067–1073. [DOI] [PubMed] [Google Scholar]
- Pollard H, Hèron A, Moreau J, Ben-Ari Y, Krestchatisky M (1993) Alterations of the GluR-B AMPA receptor subunit flip/flop expression in kainate-induced epilepsy and ischemia. Neuroscience 57: 545–554. [DOI] [PubMed] [Google Scholar]
- Prince HK, Conn PJ, Blackstone CD, Huganir RL, Levey AI (1995) Down regulation of AMPA receptor subunit GluR2 in amygdaloid kindling. J Neurochem 64: 462–465. [DOI] [PubMed] [Google Scholar]
- Seeburg PH (1996) The role of RNA editing in controlling glutamate receptor channel properties. J Neurochem 66: 1–5. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299–25. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG (1997) Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci 17: 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Iacoangeli A, Popp S, Muslimov IA, Imataka H, Sonenberg N, Lomakin IB, Tiedge H (2002) Dendritic BC1 RNA: Functional role in regulation of translation initiation. J Neurosci 22: 10232–10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Numberger M, Zhang S, Dingledine R (1997) Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci 17: 9393–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MW, VanDongen HMA, VanDongen AMJ (1996) The 5′-untranslated region of the N-methyl-d-aspartate receptor NR2A subunit controls efficiency of translation. J Biol Chem 271: 8115–8120. [DOI] [PubMed] [Google Scholar]
- Woodhull A (1973) Ionic blockage of sodium channels in nerve. J Gen Physiol 61: 687–708. [DOI] [PMC free article] [PubMed] [Google Scholar]