

Abstract
The present experiments examined the role of spinal proinflammatory cytokines [interleukin-1β (IL-1)] and chemokines (fractalkine) in acute analgesia and in the development of analgesic tolerance, thermal hyperalgesia, and tactile allodynia in response to chronic intrathecal morphine. Chronic (5 d), but not acute (1 d), intrathecal morphine was associated with a rapid increase in proinflammatory cytokine protein and/or mRNA in dorsal spinal cord and lumbosacral CSF. To determine whether IL-1 release modulates the effects of morphine, intrathecal morphine was coadministered with intrathecal IL-1 receptor antagonist (IL-1ra). This regimen potentiated acute morphine analgesia and inhibited the development of hyperalgesia, allodynia, and analgesic tolerance. Similarly, intrathecal IL-1ra administered after the establishment of morphine tolerance reversed hyperalgesia and prevented the additional development of tolerance and allodynia. Fractalkine also appears to modulate the effects of intrathecal morphine because coadministration of morphine with intrathecal neutralizing antibody against the fractalkine receptor (CX3CR1) potentiated acute morphine analgesia and attenuated the development of tolerance, hyperalgesia, and allodynia. Fractalkine may be exerting these effects via IL-1 because fractalkine (CX3CL1) induced the release of IL-1 from acutely isolated dorsal spinal cord in vitro. Finally, gene therapy with an adenoviral vector encoding for the release of the anti-inflammatory cytokine IL-10 also potentiated acute morphine analgesia and attenuated the development of tolerance, hyperalgesia, and allodynia. Taken together, these results suggest that IL-1 and fractalkine are endogenous regulators of morphine analgesia and are involved in the increases in pain sensitivity that occur after chronic opiates.
Keywords: proinflammatory cytokines, anti-inflammatory cytokines, hyperalgesia, allodynia, rats, interleukin-1, interleukin-10
Introduction
Opioids remain a drug of choice for alleviation of acute and chronic pain. However, the clinical efficacy of these drugs is reduced by the fact that opioids also rapidly stimulate pain facilitatory systems within the brain and spinal cord that not only offset or inhibit opioid analgesia (analgesic tolerance) but also increase basal sensitivity to heat (hyperalgesia) and touch (allodynia) when opioid administration is discontinued (Mao, 2002). These pain facilitatory systems normally regulate opioid analgesia, and the analgesic efficacy of opioids can be prolonged by antagonizing neurochemical systems important for the development of hyperalgesia. For example, antagonizing cholecystokinin receptors can potentiate the acute analgesic effects of morphine and prevent the establishment of tolerance (Watkins et al., 1984), and preventing the development of hyperalgesia after opiate administration by blocking NMDA receptors also prevents the development of analgesic tolerance (Celerier et al., 1999). Thus, it is thought that analgesic tolerance arises when pain facilitatory systems become sensitized or hyperactive after repeated opioid use.
In addition to these classic pain facilitatory systems, the importance of spinal glial activation in a wide range of acute and chronic enhanced pain states is a topic currently receiving intense study. Many forms of exaggerated pain are associated with the activation of glia, the resident immune cells of the CNS, and the release of proinflammatory cytokines such as interleukin-1β (IL-1) within the spinal cord (Watkins and Maier, 2003).
Given this profile, recent studies have investigated whether spinal cord glia may also contribute to the development of analgesic tolerance and opioid-induced hyperalgesia. Chronic administration of systemic morphine activates spinal glia and upregulates proinflammatory cytokines in spinal cord, and administration of glial metabolic inhibitors or cytokine antagonists attenuates the development of analgesic tolerance and prevents the development of hyperalgesia and allodynia in response to systemic morphine (Song and Zhao, 2001; Raghavendra et al., 2002). Similarly, acute tolerance after a single infusion of opioids can be attenuated by pretreatment with the anti-inflammatory cytokine IL-10 or an IL-1 receptor antagonist (IL-1ra) (Fairbanks and Wilcox, 2000). In addition, the analgesic effects of opioids are inhibited by the central administration of proinflammatory cytokines (Gul et al., 2000; Szabo et al., 2002).
Whether glial activation and proinflammatory cytokines regulate analgesic tolerance, thermal hyperalgesia, and/or mechanical allodynia when morphine is delivered to the spinal cord, rather than systemically, is unknown. Therefore, the aim of the present study was threefold: (1) to determine whether acute or chronic intrathecal morphine alters spinal cytokine expression; (2) to examine the effects of antagonizing putative pain facilitatory proinflammatory cytokines (IL-1) and chemokines (fractalkine) on acute intrathecal morphine analgesia and the development of tolerance, hyperalgesia, and allodynia; and (3) to examine whether gene therapy with the anti-inflammatory cytokine IL-10 would affect acute intrathecal morphine analgesia and/or the development of tolerance, hyperalgesia, or allodynia.
Materials and Methods
Subjects
Pathogen-free adult male Sprague Dawley rats (350-450 gm; Harlan Laboratories, Madison, WI) were used in all experiments. On arrival to the colony, the rats were housed in pairs in temperature-controlled (23 ± 3°C) and light-controlled (12 hr light/dark cycle; lights on at 7:00 A.M.) rooms with standard rodent chow and water available ad libitum. After surgery, rats were housed individually to maintain catheter integrity. Behavioral testing was performed between 7:00 A.M. and 4:00 P.M.. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Drugs
Morphine hydrochloride (5 mg/ml; National Institute on Drug Abuse, Research Triangle Park, NC) was dissolved in sterile endotoxin-free isotonic saline (0.9% w/v). Endotoxin-free solutions of recombinant met-human IL-1ra (100 mg/ml; lot number 25005E7; Amgen, Thousand Oaks, CA) and IL-1ra placebo solution (lot number 0210306L6; Amgen) were stored at 4°C. Neutralizing rabbit antibodies against the rat fractalkine receptor (CX3CR1; anti-FKR; 1 mg/ml; lot number 110101; Torrey Pines Biolabs, Houston, TX) and placebo rabbit IgG antibodies (normal IgG; 1 mg/ml; lot number 32K9057; Sigma, St. Louis, MO) were suspended in PBS and stored at -70°C.
Replication-defective adenovirus expression vector containing the cDNA encoding for the expression and extracellular release of human interleukin-10 (AD-IL10), driven by the Rous sarcoma virus (RSV) promoter, was purchased from Dr. Beverly Davidson (Gene Transfer Vector Core, University of Iowa, Iowa City, IA). The control adenovirus (AD-control) was an analogous adenovirus expression vector in which the RSV promoter directed the intracellular expression of Escherichia coli β-galactosidase (a generous gift from Jerome Schaack, University of Colorado Health Sciences Center, Denver, CO). Recombinant adenoviruses were grown by infecting 36 100 mm plates of HEK293 (human embryonic kidney 293) cells (5 × 106 cells/plate) at a multiplicity of 25 pfu/cell. The infected cells were collected after 48 hr, concentrated by low-speed centrifugation, and resuspended in 20 ml of growth media (DMEM and 10% calf serum). After four freeze-thaw cycles, the cell lysates were layered on cesium chloride step gradients (1 ml of 1.4 gm/ml cesium chloride/PBS cushion, 1.5 ml of 1.25 gm/ml cesium chloride/PBS step) and centrifuged in a Beckman SW40 rotor (Beckman Instruments, Fullerton, CA) for 1 hr at 36,000 rpm. Viral bands were harvested and further purified in isopycnic gradients consisting of 1.35 gm/ml cesium chloride/PBS in a Beckman VTi65 rotor (Beckman Instruments) centrifuged for 2 hr at 65,000 rpm. Mature viral particles were isolated and dialyzed for 1 hr against Dulbecco's PBS (DPBS) (1×) and twice each for 2 hr against DPBS-3% sucrose. Dialysed virus preparations were stored as 10 μl aliquots at -80°C. Viral titers were determined by viral plaque assay as described previously (Schaack et al., 1995).
Behavioral measures
Tail flick test for thermal hyperalgesia. The tail flick test, which measures withdrawal latencies of the tail from a noxious radiant heat source (Yaksh et al., 1977), was used to measure thermal nociceptive sensitivity. Rats were habituated to the testing apparatus and procedures across 4 d before the experiment. During testing, rats were transported to the laboratory and placed in individual plastic buckets (45 × 26 cm) for 20 min to minimize any stress-induced alterations in tail flick latencies. Each rat was removed from its bucket, the distal 5 cm portion of its tail was placed over an infrared light source, and the tail flick withdrawal latencies were recorded. Voltage to the light source was adjusted to achieve baseline latencies between 2 and 3 sec (high intensity) or 8-11 sec (low intensity). A cutoff time of 10 and 14 sec, respectively, were imposed to avoid tissue damage. This procedure was repeated four times every 5 min for each rat. The average of the last three of these measurements constituted the baseline pain sensitivity. After intrathecal infusions, tail flick latencies were recorded once every 20 min for 2 hr, and the average response over this time was calculated. von Frey test for mechanical allodynia. The von Frey test measures response thresholds to calibrated light pressure stimuli applied to the ventral surface of the paws (Chaplan et al., 1994). The test was performed using 0.406-15.136 gm calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL) as described in detail previously (Milligan et al., 2000). Briefly, rats were first assessed for baseline response thresholds (average of two to three consecutive withdrawal assessments at 20 min intervals) from each paw at 15 min intervals, and the average response threshold from both feet was calculated. The behavioral responses were used to calculate the log pressure that would have resulted in the 50% paw withdrawal threshold, by fitting a Gaussian integral psychometric function using a maximum-likelihood fitting method (Harvey, 1986; Treutwein and Strasburger, 1999), as described in detail previously (Milligan et al., 2000).
Surgery and microinjections
Chronic intrathecal catheters. Chronic lumbrosacral indwelling catheters were constructed and implanted using a lumbar approach, as described previously (McNally and Westbrook, 1998). Briefly, intrathecal catheters were implanted under anesthesia (isoflurane; Phoenix Pharmaceuticals, St. Joseph, MO) by threading sterile polyethylene-10 tubing (PE-10 Intramedic Tubing; Becton Dickinson Primary Care Diagnostics, Sparks, MD) between the L5 and L6 vertebrae, suturing to the superficial musculature of the lower back and tunneling the exterior end subcutaneously to exit through a small incision at the back of the neck. The rats were allowed 5-9 d to recover before experiments began and received at least one additional habituation session during this time. Infusions of drugs were followed by an additional 20 μl of saline to flush through the catheter. Catheter placement was verified immediately before tissue dissection by visualization of the catheter tip at the level of the lumbosacral spinal cord, and data from animals with misplaced catheters were excluded from analyses.
Lumbar puncture. Acute administrations of viral vectors and drugs to the spinal cord were performed by direct injection. The rats were anesthetized lightly with isoflurane, and the lumbar region of their backs were shaved and cleaned. Viral vectors were administered by inserting an 18 gauge guide catheter between the L5 and L6 vertebrae, and sterile PE-10 tubing was inserted rostrally to extend 3.5 cm from the guide catheter so as to terminate at the lumbosacral level of the spinal cord. Viral vectors were injected (5 μl, followed by 10 μl of saline), the catheters were removed, and the animal was allowed to recover. Morphine (10 μl at 1 μg/μl, followed by 10 μl of saline) was acutely administered under light anesthesia by direct injection between the L5 and L6 vertebrae.
Cytokine measures: ELISA
Tissue and CSF collection. Under anesthesia [50 mg/kg pentobarbital (Abbott Laboratories, North Chicago, IL) and, if required, methoxyflurane (Pitman-Moore, Mundelein, IL)], the lumbosacral enlargement of the spinal cord was exposed by laminectomy. A nick was made in the lumbar dura, and PE-10 tubing attached at one end to a syringe was inserted caudally into the CSF. Approximately 10 μl of CSF was withdrawn and immediately flash-frozen in liquid nitrogen. After verifying intrathecal catheter placement, the lumbosacral spinal cords were then dissected free and placed on an ice-chilled glass plate. The dorsal aspects of these tissues were dissected, divided into left and right halves, and separately flash-frozen in liquid nitrogen. The entire procedure required a maximum of 20-25 min per animal, and each animal was killed by cervical dislocation immediately on completion of sample collection. Samples were stored at -75°C until the time of assay.
Sample preparation and assay. Procedures for tissue processing and ELISAs were identical to those described in detail previously (Hansen et al., 2000a,b; O'Connor et al., 2004). CSF samples were prepared for assay by being slowly thawed and then quick spun (Quick Spin Mini-centrifuge; National Labnet, Woodbridge, NY). Supernatants were removed for immediate use in ELISAs. For spinal tissues, protein was mechanically dissociated by sonication, and the total protein per milliliter was determined by the Bradford protein assay (Bio-Rad, Hercules, CA). Sonicated samples were centrifuged, and supernatants were removed and stored at 4°C until the time ELISAs were performed. IL-1 protein in the dorsal spinal cord and CSF was assayed using commercially available rat-specific ELISA kits (R & D Systems, Minneapolis, MN) in accordance with the manufacturer's instructions.
Cytokine measures: real-time reverse transcription-PCR
RNA isolation and enrichment. Total RNA was isolated based on the method of Chomczynski and Sacchi (1987). Briefly, tissue was homogenized in TRIzol reagent (Invitrogen, Carlsbad, CA). After incubation, homogenates were centrifuged (12,000 × g) at 4° C. After chloroform and isopropyl alcohol isolation steps, samples were vortexed, incubated, and centrifuged (12,000 × g) at 4°C. Nucleic acid precipitates were washed twice in 75% ethanol and centrifuged (7500 × g) at 4° C. UV spectrophotometry was used to assess purity and concentration. Samples were DNase treated (DNA-free kit; Ambion, Austin, TX), followed by requantitation before cDNA synthesis.
cDNA synthesis. Total RNA was reverse transcribed using the SuperScript II First Strand Synthesis system for reverse transcription (RT)PCR (Invitrogen). RNA (2.5 μg) was incubated for 10 min at 70°C in a total reaction volume of 10 μl containing random hexamer primers (5 ng/μl) and dNTPs (1 mm). Samples were chilled and then added to a cDNA synthesis mixture (25 mm MgCl2,5× reverse transcriptase buffer, and 0.1 m dithiothreitol) and incubated at 37°C for 2 min. Reverse transcriptase (1 μl; 200 U of SuperScript II) was added to the reaction and incubated at 42°C for 70 min. Reaction was terminated by heating to 95°C for 10 min. Control reactions lacking either reverse transcriptase or RNA template were included to assess carryover of genomic DNA and contamination of RNA, respectively. The no reverse transcriptase control consisted of pooled RNA equally representing all experimental samples. cDNA was diluted twofold in DNase-free water and stored at -20°C.
Primer specifications. cDNA sequences for rat tumor necrosis factor-α (TNFα) (accession number D00475), IL-1 (accession number M98820), IL-6 (accession number NM_012589), and rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH; accession number M17701) were obtained from GenBank at the National Center for Biotechnology Information (NCBI; www.ncbi.nlm.nih.gov). Primer sequences for rat TNFα (forward, 5′-CTTCAAGGGACAAGGCTG-3′; reverse, 5′-GAGGCTGACTTTCTCCTG; 87 bp product), IL-1β (forward, 5′-CATTGTGGCTGTGGAGAAG-3′; reverse, 5′-ATCATCCCACGAGTCACAGA-3′; 130 bp product), IL-6 (forward, 5′-ACTTCACAGAGGATACCA-3′; reverse, 5′-GCATCATCGCTGTTCATAC-3′; 143 bp product), and GAPDH (forward, 5′-GTTTGTGATGGGTGTGAACC-3′; reverse, 5′-TCTTCTGAGTGGCAGTGATG-3′; 162 bp product) were designed using the Oligo Analysis and Plotting Tool (Qiagen, Valencia, CA; oligos.qiagen.com/oligos/toolkit.php?) and tested for sequence specificity using the Basic Local Alignment Search Tool (Altschul et al., 1997) at NCBI. Primer specificity was further verified by melt curve analysis (see below).
Quantitative real-time PCR. PCR amplification of cDNA was performed using the Quantitect SYBR Green PCR kit (Qiagen). cDNA (1 μl) was added to a reaction master mix (25 μl) containing 2.5 mm MgCl2, HotStar TaqDNA polymerase, SYBR Green I, dNTPs, fluorescein (10 nm), and gene-specific primers (500 nm each of forward and reverse primer). Reactions were conducted in triplicate in 200 μl thin wall tubes (Bio-Rad). PCR cycling conditions consisted of a hot-start activation of HotStar TaqDNA polymerase (95°C, 15 min) and 40 cycles of denaturation (95°C, 15 sec), annealing (58°C, 30 sec), and extension (72°C, 30 sec). A melt curve analysis was conducted to confirm uniformity of product formation, primer dimer formation, and amplification of nonspecific products.
Real-time detection and quantitation of PCR product. Formation of PCR product was monitored in real time using the MyiQ Single-Color Real-Time PCR Detection system (Bio-Rad). Fluorescence of SYBR Green I was captured at 72°C. Threshold for detection of PCR product above background was set at 10 times the SD of mean background fluorescence for all reactions. Background fluorescence was determined from cycles 2-5 before exponential amplification of product and subtracted from raw fluorescence of each reaction per cycle. Threshold for detection of PCR product fell within the log-linear phase of amplification for each reaction. Threshold cycle (CT; number of cycles to reach threshold of detection) was determined for each reaction.
Relative quantitation of gene expression. Relative gene expression was determined using the 2-ΔΔCt method (Livak and Schmittgen, 2001; Pfaffl, 2001). The mean CT of triplicate measures was computed for each sample. The sample mean CT of GAPDH (internal control) was subtracted from the sample mean CT of TNF, IL-1, and/or IL-6 (ΔCT). The sample with the absolute highest mean ΔCT was selected as a calibrator and subtracted from the mean ΔCT of each experimental sample (ΔΔCT). 2-ΔΔCt yields a relative fold change in gene expression of the target gene normalized to the internal control gene expression and relative to the calibrator sample.
In vitro lumbar dorsal spinal cord preparation and IL-1 release assays
An in vitro assay was used to test whether fractalkine induces the release of IL-1. All media products and supplements were from Invitrogen, unless specified otherwise. The lumbar dorsal spinal cord was rapidly exposed using aseptic technique from 70% ethanol-rinsed vertebral columns of unanesthetized decapitated rats. This method was used to avoid the detrimental effects of anesthetics on glial function (Mantz et al., 1993). The dorsal spinal cord was the focus because it, rather than the ventral spinal cord, is implicated in pain facilitation. The lumbar spinal cord was tested to parallel the in vivo studies of intrathecally injected fractalkine. After sterile removal of dorsal vertebrae, the exposed spinal cord was rinsed with sterile endotoxin-free DPBS. The dorsal spinal cord was isolated and rinsed with sterile HBSS, and an ∼1.75 cm section of the lumbar enlargement was isolated. Tissue was placed in 25 μl of incubation medium (DMEM, supplemented with 2 mm l-glutamine, 100 U penicillin, 100 μg streptomycin, 10 mm HEPES buffer (Sigma), and 10% heat-inactivated fetal bovine serum), inside a sterile modified 500 μl Eppendorf centrifuge tube. The tube was modified by removal of the snap cap and creation of a 2 mm wide slit down one side to within 7 mm of the base. Once the tissue was inserted into the tube, dorsal side up, media and drug were added such that the total volume was 200 μl. Tissues were exposed to either 30 ng of r-fractalkine or equivolume vehicle and incubated for 2 hr at 37°C, 5% CO2-95% air. After incubation, supernatants were collected and assayed for rat IL-1 protein by ELISA.
Data analysis
Data from the tail flick tests were analyzed as the withdrawal latency in seconds, and the data from the von Frey tests were analyzed as the interpolated 50% threshold in log10 of stimulus intensity (monofilament stiffness in milligrams × 104). Baseline tail flick and von Frey test results were analyzed by ANOVAs, and postdrug measures were analyzed by repeated-measures ANOVAs to partition variance into the main effects and their interactions with α set at 0.05. Statistical analyses conducted for all tissue, and CSF ELISAs were by ANOVA. Where appropriate, ANOVAs were followed by post hoc contrast tests using Scheffé's adjusted critical value to control α to 0.05.
Experiment 1: effect of intrathecal morphine on lumbar dorsal spinal cord and CSF IL-1 protein and/or mRNA
This experiment examined the effects of single or repeated (chronic) intrathecal morphine infusions on lumbar spinal tissue and CSF IL-1 protein and/or mRNA. On day 1 of the experiment, the rats' baseline von Frey and tail flick responses (with low- and high-intensity light sources) were measured. The rats then received an intrathecal infusion of morphine (10 μg in 2 μl of saline) or equivolume saline via chronic indwelling intrathecal catheters, and postdrug tail flick latencies were measured as described above for 2 hr. In the acute treatment condition, the rats were then anesthetized with pentobarbital, and lumbar dorsal spinal cord and lumbosacral CSF were collected for ELISA and RT-PCR. Tissue and CSF collections were performed either 2 hr (saline-2 hr, n = 8; morphine-2 hr, n = 7) or 24 hr (saline-24 hr, n = 8; morphine-24 hr, n = 7) after infusion to determine the time course of changes in spinal IL-1 protein and mRNA in response to acute morphine. On days 2-4, chronically treated rats received intrathecal infusions of morphine or saline once a day but were not behaviorally tested. On day 5, the rats were tested for evidence of mechanical allodynia (von Frey test) and thermal hyperalgesia (tail flick test). The rats then received a final infusion of morphine or saline, and postdrug tail flick latencies were measured for 2 hr to determine whether animals had developed analgesic tolerance to morphine. The animals were then anesthetized with pentobarbital, and lumbar dorsal spinal cord and lumbosacral CSF were collected as above. That is, tissue and CSF collections were performed either 2 hr (saline-2 hr, n = 8; morphine-2 hr, n = 7) or 24 hr (saline-24 hr, n = 8; morphine-24 hr, n = 8) later. Lumbar spinal CSF and one half of the dissected dorsal horn tissue were analyzed by ELISA for IL-1 protein content, and the other half of the lumbar spinal dorsal horn tissue was analyzed by RT-PCR for relative cytokine mRNA expression. As a first insight into whether morphine may regulate proinflammatory cytokines in addition to IL-1, relative expressions of IL-6 and TNF mRNA were also determined.
Experiment 2: effect of intrathecal IL-1ra on intrathecal morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
On day 1 of the experiment, after assessing baseline von Frey and tail flick measurements (with low- and high-intensity light sources), the rats were given an intrathecal infusion, via chronic indwelling intrathecal catheters, of IL-1ra (100 μg in 1 μl) or equivolume vehicle, followed by morphine (10 μg in 2 μl of saline) or equivolume saline. Group sizes were vehicle-vehicle (n = 7), vehicle-morphine (n = 7), IL-1ra-vehicle (n = 6), and IL-1ra-morphine (n = 7). Postdrug tail flick latencies were measured as described for 2 hr. At 24 hr intervals on days 2-5, the rats were infused with the same drugs as on day 1. On days 2 and 4, no behavioral testing occurred. On days 3 and 5, the rats were tested for predrug and postdrug tail flick latencies on the high-intensity light source only. On day 6, baseline von Frey and tail flick measurements (with low- and high-intensity light sources) were recorded to measure mechanical allodynia and thermal hyperalgesia, respectively. All rats then received an intrathecal infusion of morphine (5 μg in 1 μl of saline) to determine their response to morphine in the absence of IL-1ra to clearly test whether IL-1ra treatment on previous days had attenuated analgesic tolerance. Tail flick latencies were recorded 2 hr after infusion as described previously.
Experiment 3: effect of intrathecal IL-1ra on established morphine-induced analgesic tolerance, hyperalgesia, and allodynia
On day 1 of the experiment, after assessing baseline von Frey and tail flick measurements (with low- and high-intensity light sources), the rats were given an intrathecal infusion, via chronic indwelling intrathecal catheters, of morphine (10 μgin2 μl of saline) or equivolume saline. Postdrug tail flick latencies were measured as described for 2 hr. At 24 hr intervals on days 2-5, the rats were infused with the same drugs as on day 1. On days 2-4, no behavioral testing occurred. On days 5 and 6, after baseline tail flick measurements were recorded, all rats then received another infusion of morphine or saline, and postdrug latencies were again measured for 2 hr to determine whether animals had developed analgesic tolerance to morphine. Half the animals in each group then received an infusion of IL-1ra (100 μg in 1 μl) or equivolume vehicle. Group sizes were saline-vehicle (n = 7), morphine-vehicle (n = 7), saline-IL-1ra (n = 8), and morphine-IL-1ra (n = 8). On days 7-9, the rats received a single infusion containing IL-1ra or vehicle and morphine or saline, as described previously. However, tail flick latencies were measured before and for 2 hr after infusion on day 9 only. On day 10, baseline von Frey and tail flick latencies were measured to measure tactile allodynia and thermal hyperalgesia, respectively. All rats then received an intrathecal infusion of morphine (5 μg in 1 μl of saline) to determine their response to morphine in the absence of IL-1ra to clearly test whether IL-1ra treatment on previous days had reversed analgesic tolerance. Tail flick latencies were recorded 2 hr after infusion, as described previously.
Experiment 4: effect of blocking fractalkine (CX3CR1) receptors on morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
Baseline von Frey and tail flick responses (on low-intensity light source) were measured on the day before the first infusion of morphine or vehicle. Between 5:00 P.M. and 6:00 P.M. the same day, the rats then received an intrathecal infusion of anti-FKR (10 μgin10 μl of PBS) or IgG (10 μg in 10 μl of PBS) via a chronic indwelling catheter. The following morning, the rats' baseline responses in the von Frey and tail flick tests (with low- and high-intensity light sources) were measured again. The rats then received an intrathecal infusion containing anti-FKR or IgG, followed by morphine (10 μgin2 μl of saline) or equivolume saline. Group sizes were IgG-saline (n = 7), IgG-morphine (n = 8), anti-FKR-saline (n = 8), and anti-FKR-morphine (n = 8). Postdrug tail flick latencies were measured as described for 2 hr. On days 2-4, the rats were infused with the same drugs as on day 1 but were not behaviorally tested. On day 5, the rats were tested for predrug and postdrug tail flick latencies on the high-intensity light source only. On day 6, baseline von Frey and tail flick measurements (with low- and high-intensity light sources) were recorded to measure mechanical allodynia and thermal hyperalgesia, respectively.
Experiment 5: effect of fractalkine (CX3CL1) on in vitro release of IL1 from acutely isolated dorsal spinal cord
Given the key role of glial IL-1 in pain facilitation (Watkins et al., 2001), we tested whether fractalkine may potentially exert its effects via release of IL-1 from dorsal spinal cord. Experimentally naive rats were decapitated, and the lumbar dorsal spinal cords were aseptically isolated. Once the lumbar enlargements were placed in incubation medium, tissues were exposed to either 30 ng of fractalkine (a behaviorally active dose) (Milligan et al., 2002) or vehicle (n = 6/group). The supernatants were collected after a 2 hr incubation for assay by ELISA for rat IL-1. This time point was chosen because IL-1 is a large protein and so is expected to slowly diffuse from its site of release within the dorsal horn and to require considerable time before it can accumulate to a sufficient concentration to be detectable in the 200 μl of incubation media.
Experiment 6: effect of intrathecal AD-IL10 on intrathecal morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
Five days before the start of behavioral testing, the rats received an acute intrathecal infusion of AD vector (5 μg) encoding for either IL-10 (ADIL10; n = 8) or β-galactosidase (AD-control; n = 7) via lumbar puncture. These doses of AD-IL10 and AD-control have previously been shown to have no effect on mechanical or thermal response thresholds yet inhibit pain facilitation by diverse procedures for approximately 1 week (Milligan et al., 2003; Watkins and Maier, 2003). On the first, third, and fifth days of the experiment, the rats' pre-morphine von Frey and tail flick responses (with low- and high-intensity light sources) were measured. The rats then received an intrathecal infusion of morphine (10 μg in 10 μl of saline) via lumbar puncture, and post-morphine tail flick latencies were measured as described above for 6 hr. On the second and fourth days, the rats received a single intrathecal infusion of morphine but were not tested.
Results
Experiment 1: effect of intrathecal administration of morphine on lumbar dorsal spinal cord and CSF IL-1 protein and/or mRNA
The spinal release of the proinflammatory cytokine IL-1 is an important factor in the development of hyperalgesia and allodynia in a wide range of exaggerated pain states (Watkins et al., 2003). Similarly, abstinence-induced hyperalgesia and allodynia after chronic administration of systemic morphine are also associated with an increase in the spinal levels of this cytokine (Raghavendra et al., 2002). Although abstinence-induced hyperalgesia and allodynia are only evident after the analgesic effects of morphine have abated, there is evidence that hyperalgesic mechanisms are rapidly activated by morphine because antagonizing opioid receptors shortly after opiate administration not only eliminates analgesia but also reduces pain thresholds below pre-opiate baselines (Celerier et al., 1999). Therefore this experiment aimed to determine the time course of the production and release of IL-1 after intrathecal infusion with morphine, by measuring levels of IL-1 protein and/or mRNA in lumbar dorsal spinal cord tissue and CSF at 2 and 24 hr after morphine administration. Moreover, tolerance and hyperalgesia can result from a single dose of an opiate (Fairbanks and Wilcox, 1997; Larcher et al., 1998) as well as from chronic schedules. Therefore, this experiment also examined whether spinal levels of these cytokines are changed by a single dose of morphine.
Acute morphine
A single lumbrosacral intrathecal infusion of morphine did not significantly affect lumbar dorsal spinal cord (Fig. 1, top) or CSF IL-1 (Fig. 1, bottom) protein levels (all F(1,26) < 2; p > 0.05) or dorsal spinal cord gene expression of either IL-1 (Fig. 2,top), IL-6 (Fig. 2, middle), or TNF (Fig. 2, bottom) (F(1,26) < 1).
Figure 1.
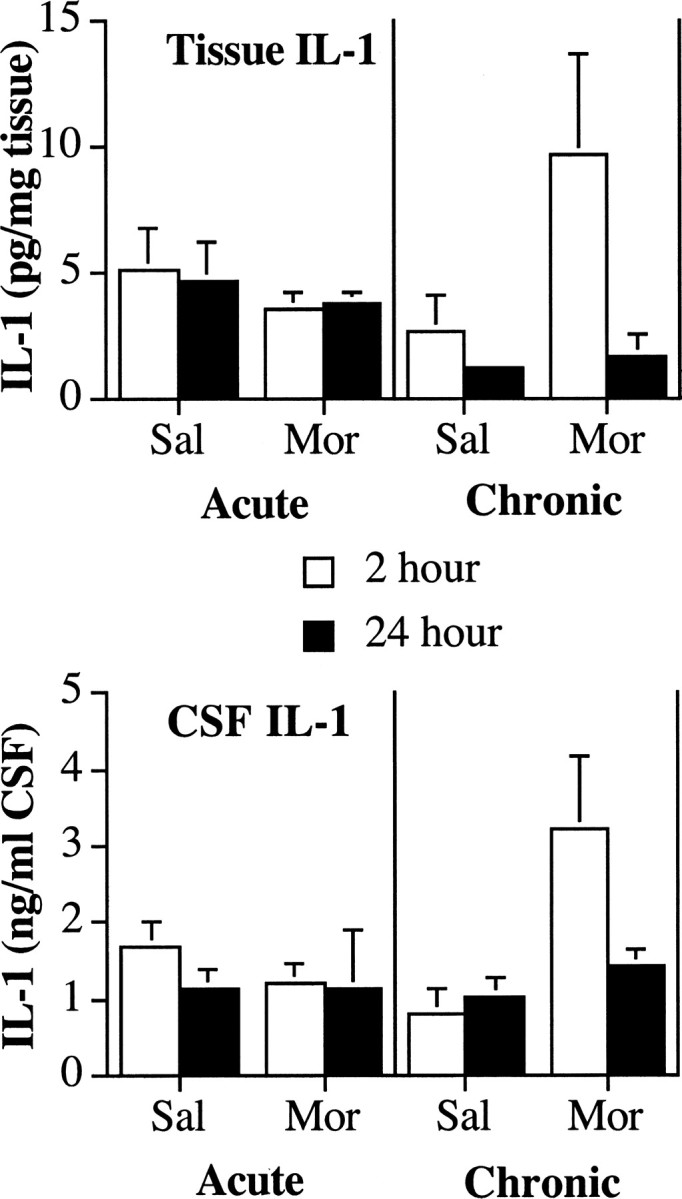
Chronic, but not acute, intrathecal infusion with morphine (Mor) induces the rapid release of IL-1 protein from the lumbar spinal cord determined by ELISA. Top, After five once-daily (chronic) intrathecal infusions of morphine (10 μg) or saline (Sal), there was an increase in IL-1 protein in lumbar spinal dorsal horn tissue 2 hr, but not 24 hr, after the last infusion of morphine. There was no change in IL-1 levels after a single ′infusion of morphine. Bottom, After five once-daily (chronic) intrathecal infusions of morphine (10 μg) or saline, there was an increase in IL-1 protein in lumbar spinal CSF 2 hr, but not 24 hr, after the last infusion of morphine. There was no change in IL-1 levels after a single infusion of morphine.
Figure 2.
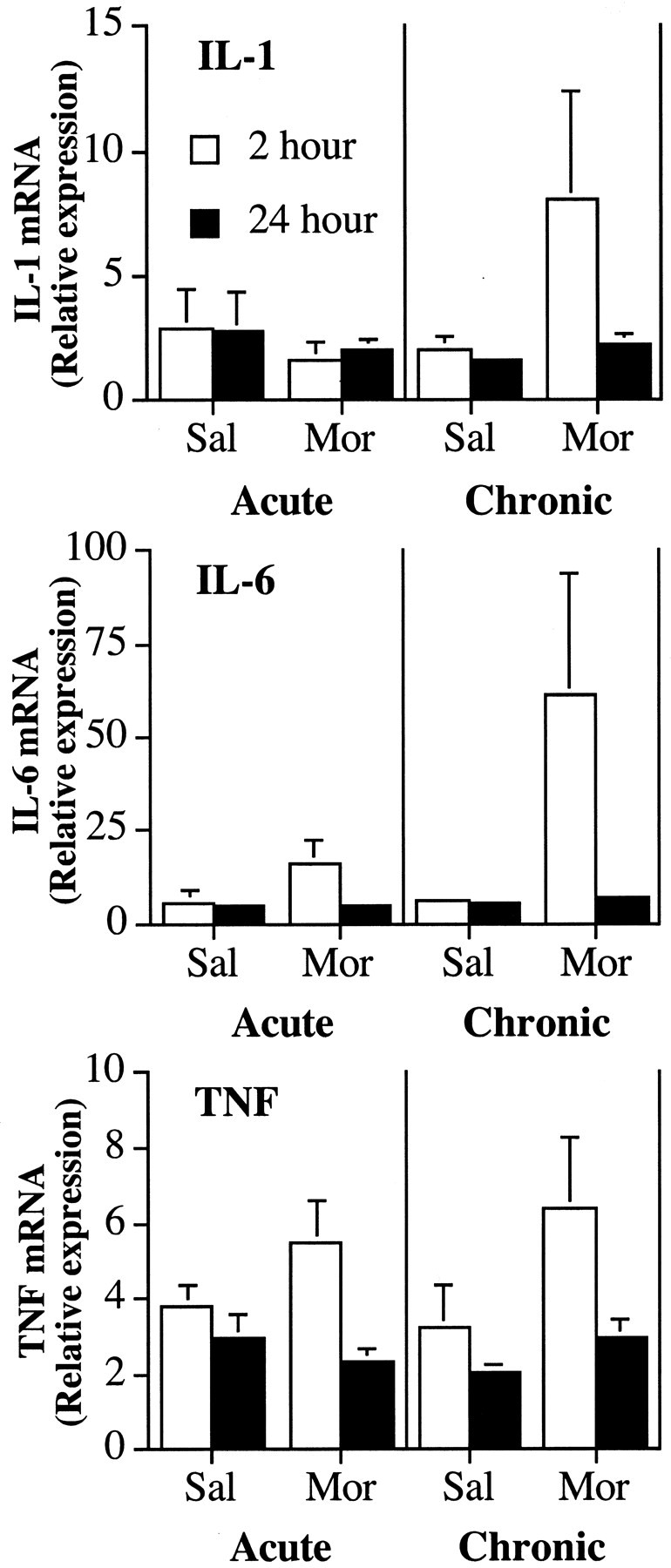
Chronic, but not acute, intrathecal infusion with morphine (Mor) increases the expression of IL-6 and TNF-α mRNA in the lumbar spinal cord determined by RT-PCR. Top, There were no significant differences in relative levels of IL-1 mRNA expression after a single or five once-daily (chronic) infusions of morphine (10 μg) or saline (Sal) in lumbar spinal dorsal horn tissue. Middle, After five once-daily (chronic) intrathecal infusions of morphine (10 μg) or saline, there was an increase in IL-6 mRNA expression in lumbar spinal dorsal horn tissue 2 hr, but not 24 hr, after the last infusion of morphine. There was no change in IL-6 mRNA expression after a single infusion of morphine. Bottom, After five once-daily (chronic) intrathecal infusions of morphine (10 μg) or saline, there was an increase in TNF mRNA expression in lumbar spinal dorsal horn tissue 2 hr, but not 24 hr, after the last infusion of morphine. There was no significant change in TNF mRNA expression after a single infusion of morphine.
Chronic morphine
Lumbosacral intrathecal infusion of morphine once per day for 5 d produced time-dependent increases in lumbar dorsal spinal cord IL-1 protein compared with saline-infused controls. In dorsal spinal cord tissues collected 2 hr after the last infusion of morphine, there was an increase in IL-1 protein (Fig. 1, top, morphine-2 hr) but there were no similar elevations in tissue IL-1 24 hr after the last infusion of morphine (Fig. 1, top, morphine-24 hr). This resulted in reliable main effects of intrathecal morphine (ANOVA; F(1,27) = 5.3; p < 0.03) and time since last infusion (F(1,27) = 8.0; p < 0.01) and a reliable interaction between intrathecal morphine and time (F(1,27) = 4.3; p < 0.05).
Similar results were observed with CSF collected from the lumbosacral spinal cord. There was an elevation in CSF IL-1 protein when collected 2 hr (Fig. 1, bottom, morphine-2 hr) but not 24 hr (Fig. 1, bottom, morphine-24 hr) after the last infusion of morphine. This resulted in a main effect of intrathecal morphine (F(1,27) = 10.0; p < 0.004) and interaction between intrathecal morphine and time since last infusion (F(1,27) = 5.1; p < 0.04) but not a reliable main effect of time (F(1,27) = 3.1; p > 0.05).
The results suggested an elevation of IL-6 and TNF gene expression in lumbar spinal tissue when collected 2 hr after the last infusion with morphine (Fig. 2, middle and bottom, morphine-2 hr). There were main effects of morphine on IL-6 mRNA (F(1,27) = 4.3; p < 0.05), TNF mRNA (F(1,27) = 4.2; p = 0.051), and time of tissue collection for both IL-6 mRNA (F(1,27) = 4.1; p = 0.054) and TNF mRNA (F(1,27) = 5.5; p < 0.03).
Experiment 2: effect of intrathecal IL-1ra on intrathecal morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
The results of experiment 1 suggest that release of IL-1 in the spinal cord is potentiated in rats given five daily intrathecal infusions of morphine. Therefore, this experiment tested whether IL-1 is a key mediator of the tolerance, hyperalgesia, and/or allodynia that results from chronic morphine administration.
Analgesia and tolerance
On the first day of testing, all groups exhibited comparable baseline tail flick latencies on the high-intensity bulb used for testing analgesia before intrathecal infusions (all F < 1; p > 0.05). Infusion of morphine produced a robust analgesic effect (Fig. 3, top, Veh-Mor), and coadministration of IL-1ra enhanced this effect (Fig. 3, top, IL1ra-Mor) compared with saline-injected controls. This resulted in reliable main effects of intrathecal morphine (F(1,23) = 597.7; p < 0.0001) and intrathecal IL-1ra (F(1,23) = 14.6; p < 0.001) and a significant interaction of intrathecal morphine and intrathecal IL-1ra (F(1,23) = 4.8; p < 0.04). Across the first 5 d of the experiment, there were no reliable changes in the groups' baseline tail flick latencies under the high-intensity bulb (repeated-measures ANOVA; all F < 3; p > 0.05). Repeated infusion of morphine across this time resulted in a reduction of analgesic efficacy of morphine or tolerance. However, coadministration of IL-1ra reduced the development of tolerance to morphine across these days. This resulted in reliable interactions between intrathecal morphine and time (F(2,46) = 51.3; p < 0.0001), between intrathecal IL-1ra and time (F(2,46) = 3.9; p < 0.03), and a three-way interaction between intrathecal morphine, intrathecal IL-1ra, and time (F(2,46) = 5.1; p < 0.01).
Figure 3.
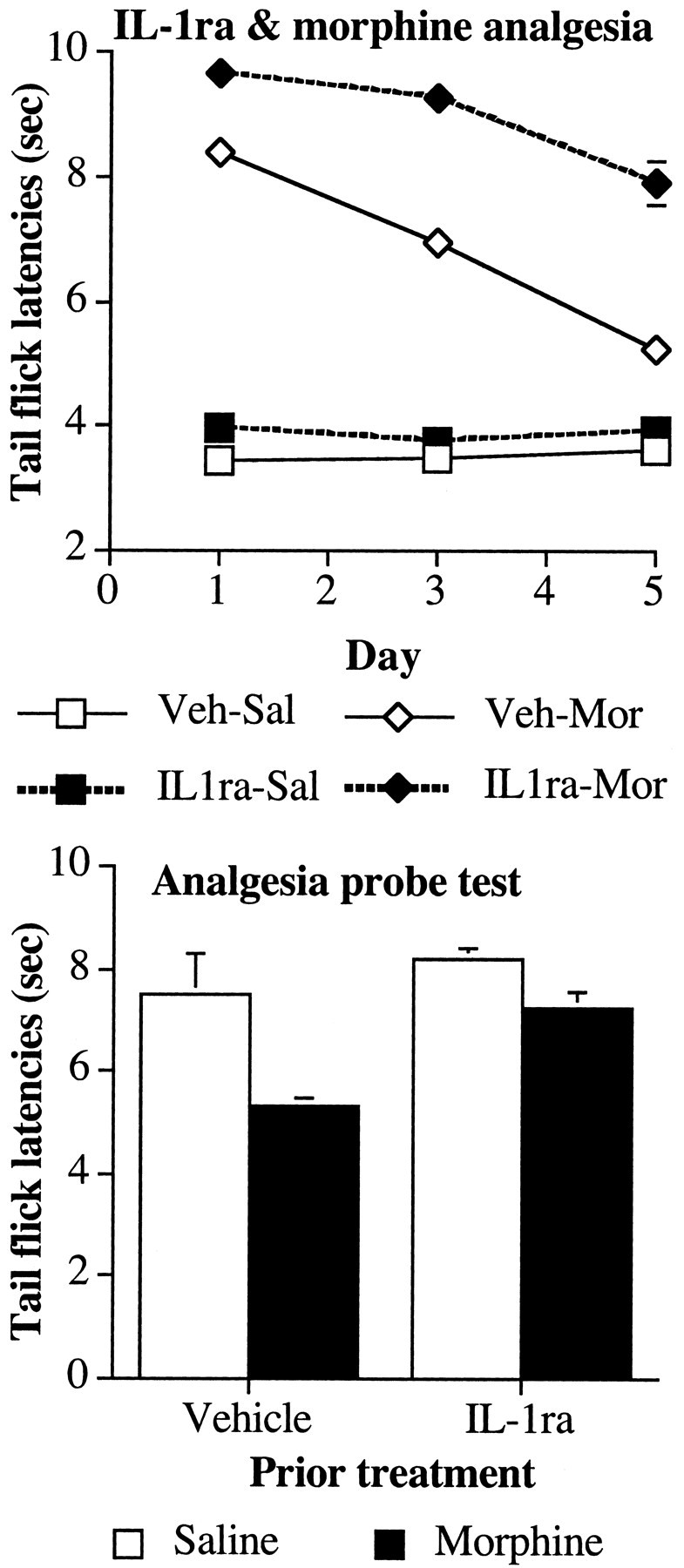
Coadministration of an IL-1ra with intrathecal morphine potentiates morphine analgesia. Top, The analgesic effect of intrathecal morphine (10 μg) was reduced by five once-daily infusions of morphine in the tail flick withdrawal reflex from an infrared light source. Coadministration of IL-1ra with intrathecal morphine potentiates acute morphine analgesia on day 1 and attenuates the development of analgesic tolerance. Bottom, Previous treatment with IL-1ra reduced the expression of tolerance in rats previously given five once-daily infusions of morphine. A probe trial for analgesia was conducted on day 6 to determine the extent of tolerance in the absence of IL-1ra. All subjects received an intrathecal infusion of morphine (5 μg). Veh, Vehicle; Sal, saline; Mor, morphine.
Allodynia
On the sixth day, the rats were tested for tactile sensitivity to von Frey pressure test hairs before morphine administration that day. Treatment with morphine on days 1-5 resulted in an increase in tactile sensitivity compared with saline-infused controls on day 6 (Fig. 4, bottom). Moreover, coadministration of IL-1ra with morphine on days 1-5 reduced the allodynic effect of repeated morphine administration measured on day 6. This was revealed by a significant main effect of morphine on withdrawal thresholds (F(1,23) = 24.6; p < 0.0001) and intrathecal IL-1ra (F(1,23) = 5.6; p < 0.03) and a reliable interaction between intrathecal morphine and intrathecal IL-1ra (F(1,23) = 11.6; p < 0.002).
Figure 4.
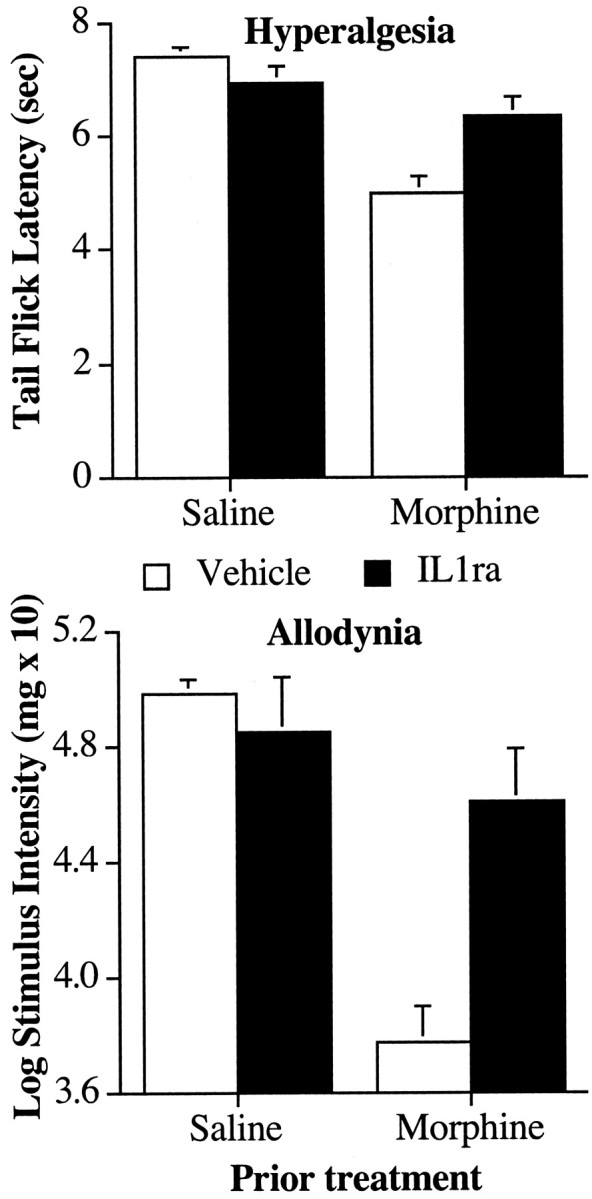
Coadministration of an IL-1ra with an intrathecal infusion of morphine attenuates the development of exaggerated pain sensitivity after morphine. Top, Animals given five once-daily infusions of morphine (10 μg) developed thermal hyperalgesia in the tail flick test using an infrared light source when tested 24 hr after the last infusion of morphine. Coadministration of IL-1ra (100 μg) reduced the hyperalgesic effect of morphine. Bottom, Animals given five once-daily infusions of morphine (10 μg) developed tactile allodynia in the von Frey test when tested 24 hr after the last infusion of morphine. Coadministration of IL-1ra (100 μg) reduced the allodynic effect of morphine.
Thermal hyperalgesia
On the sixth day, the rats were tested for thermal hyperalgesia in the tail flick test on a low-intensity infrared bulb. The tail flick latencies were reduced in rats administered morphine for the previous 5 d, indicating an increased sensitivity to painful thermal stimuli (Fig. 4, top), but coadministration with IL-1ra reduced this effect. These differences resulted in reliable main effects of intrathecal morphine (F(1,23) = 34.0; p < 0.0001) and a reliable interaction between intrathecal morphine and intrathecal IL-1ra (F(1,23) = 12.0; p < 0.02).
Analgesia probe trial without IL-1ra
All rats received an intrathecal infusion of morphine without IL-1ra (or vehicle) on the sixth day to determine whether previous treatment with IL-1ra had altered the response of control rats to morphine as well as reducing the extent of analgesic tolerance in morphine-treated rats. Before intrathecal infusion of morphine, all groups exhibited comparable baseline tail flick latencies (all F(1,23) < 3.8; p > 0.05). Infusion with morphine had an analgesic effect in all rats (Fig. 3, bottom), however there was a reduced analgesic effect of morphine in those rats treated with morphine on previous days. Previous treatment with IL-1ra reduced analgesic tolerance. These differences resulted in reliable main effects of intrathecal morphine treatment (F(1,23) = 79.7; p < 0.0001) and intrathecal IL-1ra (F(1,23) = 17.4; p < 0.0001) and a reliable interaction of intrathecal morphine with intrathecal IL-1ra (F(1,23) = 22.1; p < 0.0001). Post hoc mean comparisons revealed there was no effect of treatment with IL-1ra in the preceding days in the analgesic effect of morphine in control animals (Fig. 3, bottom, Veh-Sal and IL1ra-Sal) (F(1,23) = 0.1; critical, F(3,23) = 9.1).
Experiment 3: effect of intrathecal IL-1ra on established morphine-induced analgesic tolerance, hyperalgesia, and allodynia
The results of experiment 2 suggest that blocking spinal IL-1 receptors potentiates the acute analgesic effect of intrathecal morphine and attenuates the development of analgesic tolerance, hyperalgesia, and allodynia. The aim of the current experiment was to determine whether antagonizing spinal IL-1 receptors would reverse established tolerance, hyperalgesia, or allodynia after rats had received five once-daily intrathecal infusions of morphine.
Analgesia and tolerance
Before the first injection of morphine, there were no reliable differences in the groups' baseline tail flick latencies (all F < 1; p > 0.05). Infusion of morphine produced a robust analgesic effect on day 1 (Fig. 5, top, Mor-Veh and Mor-IL1ra), but the analgesic effect of morphine diminished across the first 5 d of infusion. This resulted in a reliable main effect of morphine on tail flick latencies (F(1,26) = 169.6; p < 0.0001), a reliable effect of time (F(1,26) = 47.1; p < 0.0001), and a significant interaction between morphine and time (F(1,26) = 36.1; p < 0.0001). At the end of the fifth day of testing, the animals received their first intrathecal infusions of IL-1ra or vehicle. Before receiving this first infusion, there were no reliable differences in morphine analgesia between the groups receiving IL-1ra or saline (all F < 1; p > 0.05). However, when the animals were tested on days 6 and 9, there was a significant increase in the analgesic effect of morphine in animals receiving treatment with IL-1ra. This resulted in reliable main effects of morphine (F(1,26) = 49.6; p < 0.0001), IL-1ra (F(1,26) = 4.2; p < 0.05), and an interaction between morphine and IL-1ra (F(1,26) = 5.4; p < 0.03). Moreover, there was a reliable decrease in the analgesic effect of morphine between days 6 and 9 in rats receiving vehicle and IL-1ra. However, treatment with IL-1ra did not affect the rate of decline of morphine analgesia across this time. This was reflected by a reliable main effect of time (Fig. 5, top, day 6 vs day 9) (F(1,26) = 7.5; p < 0.02) and an interaction between time and morphine (F(1,26) = 6.2; p < 0.02) but no interactions between IL-1ra and time or IL-1ra, time, and morphine (F < 1; p > 0.05).
Figure 5.
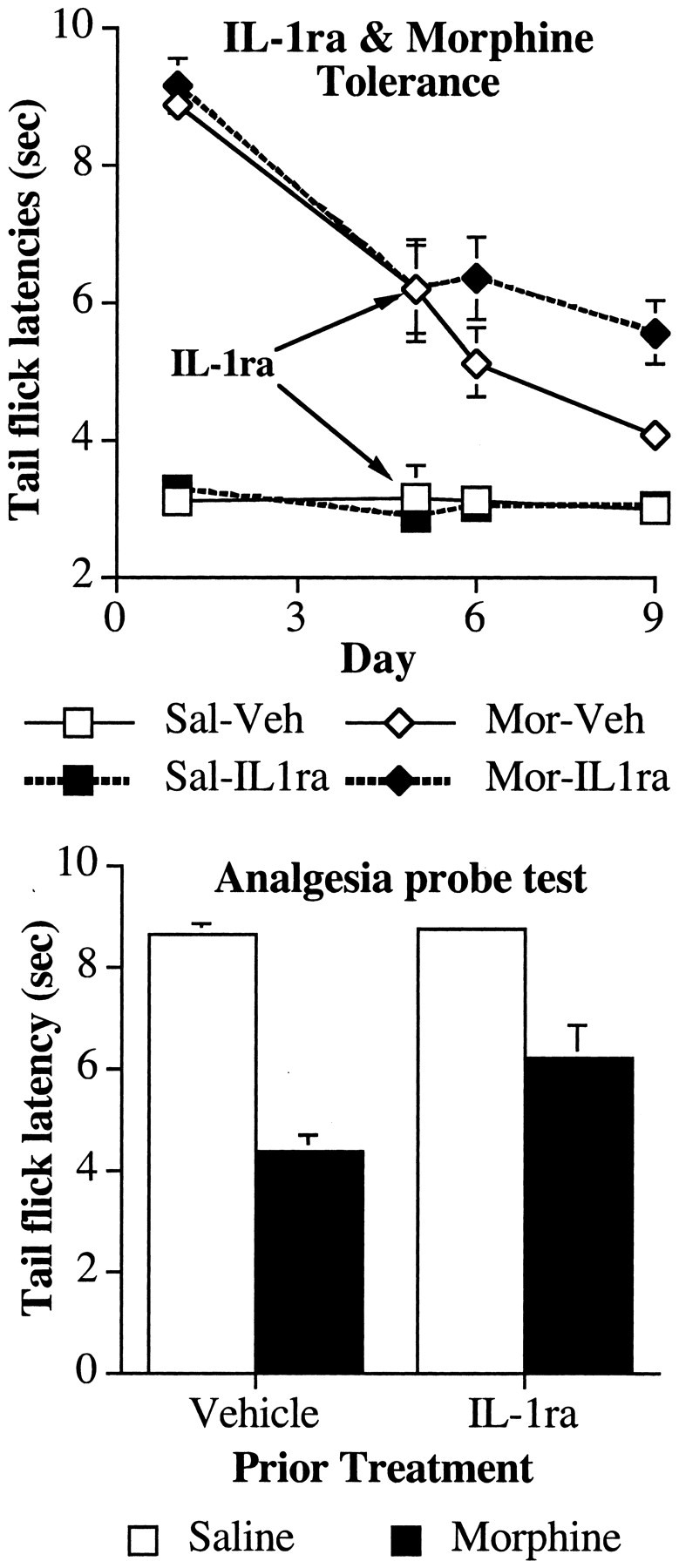
Coadministration of an IL-1ra with intrathecal morphine in morphine-tolerantrats attenuates the additional development of analgesic tolerance. Top, Rats received five once-daily intrathecal infusions of morphine (Mor; 10 μg) or saline to induce analgesic tolerance in the tail flick test. On days 6-9, the rats were coadministered IL-1ra (100 μg) or its vehicle (Veh) with morphine (10 μg) or saline (Sal). Treatment with IL-1ra attenuated the additional development of analgesic tolerance. Bottom, A probe trial for analgesia was conducted on day 10 to determine the extent of tolerance in the absence of IL-1ra in which all subjects received an intrathecal infusion of morphine (5 μg). Treatment with IL-1ra across days 6-9 increased the analgesic effect of morphine in morphine-treated rats.
Allodynia
On the 10th day, the rats were tested for tactile sensitivity to von Frey pressure test hairs in the absence of morphine. Previous treatment with intrathecal morphine resulted in an increase in tactile sensitivity compared with saline-infused controls (Fig. 6, bottom). Moreover, administration of IL-1ra diminished the allodynic effect of repeated morphine administration. This was revealed by a significant main effect of morphine on withdrawal thresholds (F(1,26) = 313.1; p < 0.0001) and intrathecal IL-1ra (F(1,26) = 6.5; p < 0.02) and a reliable interaction between intrathecal morphine and intrathecal IL-1ra (F(1,26) = 25.2; p < 0.0001).
Figure 6.
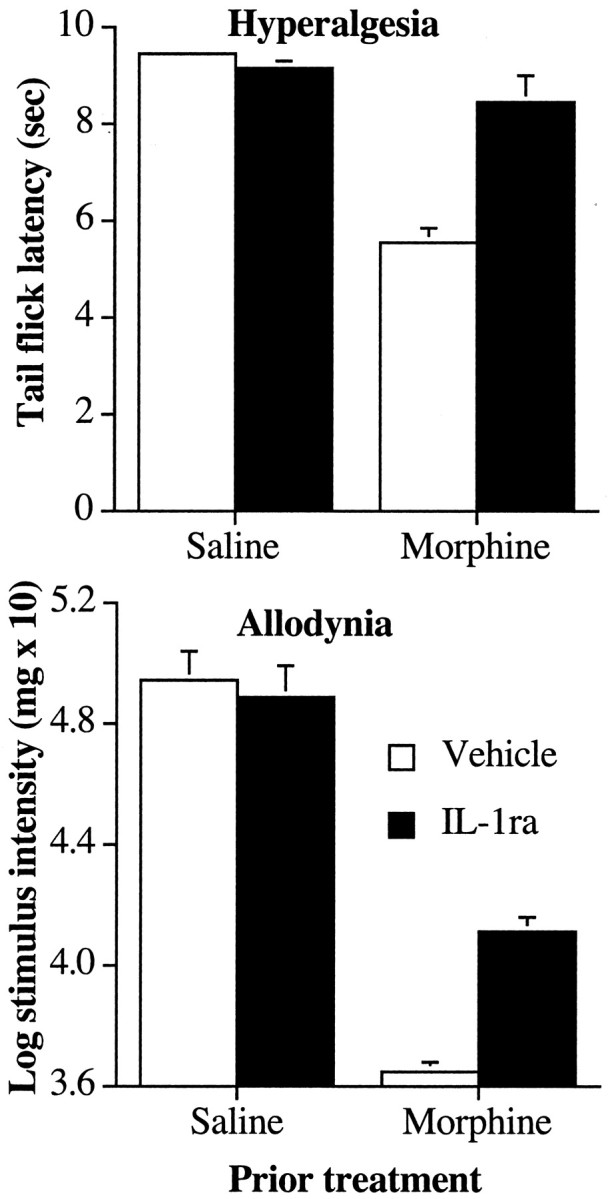
Coadministration of an IL-1ra with an intrathecal infusion of morphine in rats previously made tolerant to morphine attenuates the development of exaggerated pain sensitivity after morphine. Top, Rats received five once-daily intrathecal infusions of morphine (10 μg) or saline to induce analgesic tolerance in the tail flick test. On days 6-9, the rats were coadministered IL-1ra (100 μg) or its vehicle with morphine (10 μg) or saline. Treatment with IL-1ra attenuated the development of thermal hyperalgesia in the tail flick test when tested on day 10, 24 hr after the last drug infusion. Bottom, Treatment with IL-1ra on days 6-9 also attenuated the development of tactile allodynia in the von Frey test when tested on day 10, 24 hr after the last drug infusion.
Thermal hyperalgesia
On the 10th day, the rats were tested for thermal hyperalgesia in the tail flick test on a low-intensity infrared bulb. The tail flick latencies were reduced in rats administered morphine for the previous 9 d, indicating an increased sensitivity to painful thermal stimuli (Fig. 6, top), but administration with IL-1ra reduced this effect. These differences resulted in reliable main effects of intrathecal morphine (F(1,26) = 48.9; p < 0.0001) and IL-1ra (F(1,26) = 17.4; p < 0.0003) and a reliable interaction between intrathecal morphine and intrathecal IL-1ra (F(1,26) = 24.5; p < 0.0001).
Analgesia probe trial without IL-1ra
All rats received an intrathecal infusion of morphine without IL-1ra (or vehicle) on the sixth day to determine whether previous treatment with IL-1ra had altered the response of control rats to morphine as well as reduce the extent of analgesic tolerance in morphine-treated rats. Before intrathecal infusion of morphine, all groups exhibited comparable baseline tail flick latencies (all F(1,23) < 2; p > 0.05). Infusion with morphine had an analgesic effect in all rats (Fig. 5, bottom). However, there was a reduced analgesic effect of morphine in those rats treated with morphine on previous days. Treatment with IL-1ra diminished the extent of analgesic tolerance. These differences resulted in reliable main effects of intrathecal morphine treatment (F(1,26) = 85.2; p < 0.0001) and intrathecal IL-1ra (F(1,26) = 6.9; p < 0.02) and a reliable interaction of intrathecal morphine with intrathecal IL-1ra (F(1,26) = 5.7; p < 0.03). Post hoc mean comparisons revealed there was no effect of treatment with IL-1ra in the preceding days in the analgesic effect of morphine in control animals (Fig. 5, bottom, Veh-Sal and IL1ra-Sal) (F(1,26) = 0.3; critical, F(3,26) = 8.9).
Experiment 4: effect of blocking fractalkine receptors on morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
In addition to the proinflammatory cytokines, recent evidence demonstrates that pain sensitivity can also be modulated by the release of chemokines in the spinal cord (Boddeke, 2001; Milligan et al., 2002). In addition, chemokines affect opioid analgesia. Activity at CCR5, CCR2, CCR7, and CXCR4 but not of the CXCR1 or CXCR2 receptors desensitizes μ- and δ-opioid receptors in vitro, and infusion of ligands for CCR1, CCR5, or CXCR4 receptors in the periaqueductal gray reduces opioid analgesia (Szabo et al., 2002). As yet unpublished work in this laboratory suggests that spinal fractalkine (CX3CL1) plays an important role in pain facilitation and glial activation after inflammatory and traumatic neuropathies. Here, we aimed to determine whether coadministration of a fractalkine receptor neutralizing (anti-FKR) antibody would affect the development of opioid analgesic tolerance, hyperalgesia, and allodynia after chronic morphine.
Analgesia and analgesic tolerance
At the start of the experiment, there were no differences between the groups in baseline tail flick responses (all F < 1; p > 0.05). Infusion of intrathecal morphine induced a large analgesic effect (Fig. 7, left, IgG-Morphine), and administration of anti-FKR antibodies enhanced this effect on day 1 (Fig. 7, left, anti-FKR-Morphine). This resulted in a reliable main effect of intrathecal morphine (F(1,27) = 1361.2; p < 0.0001) and anti-FKR-antibody (F(1,27) = 5.1; p < 0.04) and an interaction between morphine and anti-FKR antibody treatment (F(1,27) = 6.3; p > 0.02). The analgesic effect of intrathecal morphine was drastically reduced after 5 d of injection (Fig. 7, left, IgG-Morphine). However, the anti-FKR antibody reduced the development of tolerance over the first 5 d of the experiment (Fig. 7, left, anti-FKR-Morphine). This was reflected by significant main effects of morphine (F(1,27) = 1058.0; p < 0.0001) and antibody (F(1,27) = 15.8; p < 0.0001) and an interaction between antibody and morphine (F(1,27) = 19.6; p < 0.0001). Moreover, there was a reliable main effect of time (F(1,27) = 25.0; p < 0.0001) with interactions between time and morphine (F(1,27) = 34.8; p < 0.0001) and between time, morphine, and antibody (F(1,27) = 4.8; p < 0.04) but not between time and antibody (F(1,27) = 3.8; p > 0.05).
Figure 7.

Coadministration of a fractalkine receptor (CX3CR1) neutralizing antibody (anti-FKR) with intrathecal morphine potentiates morphine analgesia. Rats received an intrathecal infusion of anti-FKR (10 μg) or its vehicle on day 0. On days 1-5, rats received an intrathecal infusion of anti-FKR or its vehicle and morphine (10 μg) or saline. Left, The analgesic effect of intrathecal morphine was reduced by five once-daily infusions of morphine in the tail flick withdrawal reflex from an infrared light source. Coadministration of anti-FKR with intrathecal morphine potentiates acute morphine analgesia on day 1 and attenuates the development of analgesic tolerance apparent on day 5. Middle, Five once-daily infusions of morphine increased pain sensitivity in the tail flick test. This thermal hyperalgesia was reduced by coadministration of anti-FKR with intrathecal morphine when tested on day 6, 24 hr after the last drug infusion. Right, Five once-daily infusions of morphine increased tactile sensitivity in the von Frey test. This tactile allodynia was reduced by coadministration of anti-FKR with intrathecal morphine when tested on day 6, 24 hr after the last drug infusion.
Allodynia
There was no effect of antibody injection on tactile sensitivity on day 1 of the experiment (all F < 2; p > 0.05). However, tactile sensitivity increased markedly in rats receiving intrathecal infusions of morphine (Fig. 7, right, IgG-Morphine) by day 6 of the experiment. Concurrent infusion with the anti-FKR antibody reduced this allodynic effect of morphine (Fig. 7, right, anti-FKR-Morphine). This was reflected by significant main effects of intrathecal morphine (F(1,27) = 280.2; p < 0.0001) and intrathecal anti-FKR antibody (F(1,27) = 53.9; p < 0.0001) and an interaction between this antibody and intrathecal morphine (F(1,27) = 52.8; p < 0.0001). Moreover, there was a reliable main effect of time (F(1,27) = 258.2; p < 0.0001) with interactions between time and morphine (F(1,27) = 264.9; p < 0.0001), time and antibody (F(1,27) = 52.7; p < 0.0001), and between time, morphine, and antibody (F(1,27) = 41.7; p < 0.0001).
Hyperalgesia
There was no effect of antibody injection on thermal pain sensitivity on day 1 of the experiment (all F < 2; p > 0.05). However, thermal pain sensitivity increased markedly in rats receiving intrathecal infusions of morphine (Fig. 7, middle, IgG-Morphine) by day 6 of the experiment. Concurrent infusion with the anti-FKR antibody reduced this allodynic effect of morphine (Fig. 7, middle, anti-FKR-Morphine). This was reflected by significant main effects of morphine (F(1,27) = 5.9; p < 0.03) and antibody (F(1,27) = 6.7; p < 0.02) and an interaction between antibody and morphine (F(1,27) = 8.1; p < 0.01). Although there was not a reliable main effect of time (F(1,27) < 1; p > 0.05), there were interactions between time and morphine (F(1,27) = 25.9; p < 0.0001), time and antibody (F(1,27) = 39.7; p < 0.0001), and between time, morphine, and antibody (F(1,27) = 36.9; p < 0.0001).
Experiment 5: effect of fractalkine (CX3CL1) on in vitro release of IL-1 from acutely isolated dorsal spinal cord
One potential explanation for the finding that both IL-1ra and fractalkine receptor antagonists enhanced acute morphine analgesia is that IL-1 is released downstream from fractalkine. If this were true, this would predict that fractalkine should cause the release of IL-1 from spinal cord. Thus, this experiment examined whether IL-1 would indeed be released by fractalkine from isolated dorsal spinal cord tissue. Incubating acutely isolated dorsal spinal cord tissue with fractalkine induced the release of IL-1 protein into the incubation media. This resulted in a reliable difference in IL-1 protein between cultures incubated with fractalkine (20.3 ± 3.1 pg IL-1/ml) and those incubated with PBS (9.5 ± 4.3 pg IL-1/ml; p < 0.05).
Experiment 6: effect of intrathecal AD-IL10 on intrathecal morphine analgesia and subsequent development of analgesic tolerance, hyperalgesia, and allodynia
Just as proinflammatory cytokines are important for the genesis of hyperalgesia and allodynia, anti-inflammatory cytokines, such as IL-10, have been shown to prevent hyperalgesic responses in a wide range of exaggerated pain states (Kanaan et al., 1998; Brewer et al., 1999; Laughlin et al., 2000). Previous work in this laboratory has demonstrated that lumbosacral intrathecal injection of adenovirus vector encoding for IL-10 at this dose site-specifically induces high levels of human IL-10 protein in lumbosacral CSF and reduces hyperalgesia and allodynia in diverse pain facilitation states (Milligan et al., 2003; Watkins and Maier, 2003). Therefore, this experiment examined whether this adenoviral vector would also reduce hyperalgesia and allodynia associated with chronic morphine administration. In addition, this experiment investigated whether IL-10 would affect morphine analgesia and the development of analgesic tolerance.
Allodynia
Before the first injection of morphine, there was no difference between the groups' baseline withdrawal thresholds in the von Frey test (F(1,13) = 0.2; p > 0.05). However, there was a decrease in response threshold to tactile stimuli after treatment with morphine (Fig. 8, right, Control). This effect was attenuated in rats previously treated with AD-IL10 (Fig. 8, right, IL-10). This was evident statistically by a significant main effect of adenovirus treatment (F(1,13) = 185.2; p < 0.0001), a reliable main effect of time (repeated-measures ANOVA; F(2,26) = 74.0; p < 0.0001), and a reliable interaction between treatment and time (F(2,26) = 74.1; p < 0.0001).
Figure 8.

Lumbosacral intrathecal injection with an adenovirus expressing interleukin-10 (ADIL-10) potentiates intrathecal morphine analgesia. Rats received an intrathecal infusion with adenoviral vectors expressing IL-10 (5 μg) or β-galactosidase (Control) 5 d before the start of testing. Rats then received five once-daily infusions of morphine (10 μg). Left, The analgesic effect of intrathecal morphine was reduced by five once-daily infusions of morphine in the tail flick withdrawal reflex from an infrared light source. Treatment with ADIL-10 with intrathecal morphine potentiated acute morphine analgesia on day 1 and attenuated the development of analgesic tolerance apparent on day 5. Middle, Five once-daily infusions of morphine increased pain sensitivity in the tail flick test. Prevoius treatment with ADIL-10 reduced this thermal hyperalgesia. Right, Five once-daily infusions of morphine increased tactile sensitivity in the von Frey test. This tactile allodynia was reduced by previous treatment with ADIL-10.
Hyperalgesia
Before the first infusion of morphine, there was no difference in the groups' baseline tail withdrawal latency on the low-intensity infrared beam (F(1,13) < 0.1; p > 0.05). However, there was a decrease in tail flick latencies after treatment to morphine (Fig. 8, middle, Control). This effect was attenuated in rats previously treated with the IL-10 expressing adenovirus (Fig. 8, middle, IL-10). This was evident statistically by a significant main effect of adenovirus treatment (F(1,13) = 71.5; p < 0.0001), a reliable main effect of time (repeated-measures ANOVA; F(2,26) = 7.6; p < 0.003), and a reliable interaction between treatment and time (F(2,26) = 33.8; p < 0.0001).
Analgesia and tolerance
On the first day of testing, all groups exhibited comparable baseline tail flick latencies on the high-intensity bulb used for testing analgesia before intrathecal infusions (F(1,13) < 0.1; p > 0.05). The first infusion of morphine produced a robust analgesic effect (Fig. 8, left, Control), and treatment with AD-IL10 enhanced this effect (Fig. 8, left, IL-10). This resulted in a significant effect of treatment of IL-10 on analgesia on the first day (F(1,13) = 4.7; p < 0.05). Across the 5 d of the experiment, there were no reliable changes in the groups' baseline tail flick latencies under the high-intensity bulb (repeated-measures ANOVA; all F < 1; p > 0.05). Repeated infusion of morphine across this time resulted in a reduction of analgesic efficacy of morphine, or tolerance, in the control animals. However, treatment with AD-IL10 reduced the development of tolerance to morphine across these days. This resulted in a reliable main effect of treatment (F(1,13) = 26.7; p < 0.0002), a main effect of time (F(2,26) = 13.7; p < 0.0001), and an interaction between treatment and time (F(2,26) = 5.6; p < 0.01).
Discussion
The present studies provide evidence that spinal proinflammatory cytokines and chemokines modulate the development of tolerance to the analgesic effects of intrathecal morphine and consequent development of mechanical allodynia and thermal hyperalgesia. Spinal IL-1 protein and spinal IL-6 and TNF mRNAs increased after chronic, but not acute, intrathecal morphine. These studies also demonstrate that modulation of spinal cytokine and chemokine activity with IL-1ra, a fractalkine receptor neutralizing (anti-FKR) antibody, or gene therapy encoding for the anti-inflammatory cytokine IL-10 (AD-IL10) not only attenuated the development of tolerance, hyperalgesia, and allodynia but also potentiated acute morphine analgesia. Moreover, in rats with established morphine tolerance, IL1ra reduced hyperalgesia and attenuated the additional development of tolerance and allodynia. Taken together, the present study shows that intrathecal morphine is associated with the activation of spinal proinflammatory systems.
This study indicates that activation of spinal proinflammatory systems in opioid analgesia can occur via the local action of morphine within the spinal cord. Problems arise in studies using systemic morphine in determining how morphine may be activating these spinal proinflammatory systems. Morphine has profound immunomodulatory effects, influencing immune cells directly via opioid receptors and indirectly by modulating neuroendocrine systems that regulate immune function (e.g., glucocorticoids, catecholamines) (Mellon and Bayer, 1998; McCarthy et al., 2001). Moreover, brain-to-spinal cord projections contribute significantly to systemic opioid tolerance (Vanderah et al., 2001). Thus, activation of spinal proinflammatory systems after systemic morphine may be indirect. The present results indicate that spinal proinflammatory processes involved in analgesic tolerance and opioid-induced hyperalgesia and allodynia can be stimulated directly by morphine. These studies are the first to demonstrate the in vivo release of IL-1 from spinal cord tissue and the role of spinal IL-1 and fractalkine receptors in analgesic tolerance, hyperalgesia, and allodynia from intrathecal morphine.
These results are also consistent with recent evidence that cytokines, chemokines, and glial activation contribute to the development of analgesic tolerance. Central infusion of cytokines (Gul et al., 2000) or chemokines (Szabo et al., 2002) reduces opioid analgesia, and simultaneously inhibiting IL-1, IL-6 and TNF, or inhibiting glial metabolism attenuates development of opioid tolerance after chronic morphine (Song and Zhao, 2001; Raghavendra et al., 2002). A novel and important finding from the present study is that central immune processes also modulate acute opioid analgesia. That is, the analgesic effect of morphine was potentiated and prolonged by treatments that antagonized receptors of IL-1 or fractalkine or induced the expression of the anti-inflammatory cytokine IL-10.
Moreover, the increased sensitivity to heat (hyperalgesia) and tactile (allodynia) stimuli after chronic intrathecal morphine could be attributed to actions of spinal proinflammatory cytokines and chemokines. We show for the first time that (1) activity at IL-1 receptors is sufficient for the upregulation of pain sensitivity in opioid-tolerant rats, (2) administration of IL-1ra to morphine-tolerant rats reduces hyperalgesia and attenuates the additional development of allodynia, (3) activity at fractalkine receptors is also important for opioid-induced hyperalgesia and allodynia, and (4) treatment with the anti-inflammatory cytokine IL-10 can attenuate these changes in pain sensitivity. These results support and extend the well documented role of cytokines and chemokines in modulating pain sensitivity (Boddeke, 2001; Watkins et al., 2003). Hyperalgesia and allodynia can be induced by central IL-1 or fractalkine (Oka et al., 1993; Reeve et al., 2000; Falchi et al., 2001; Milligan et al., 2002) and reduced by IL-10 or antagonists against IL-1 or fractalkine receptors (Poole et al., 1995; Tadano et al., 1999; Laughlin et al., 2000; Ji et al., 2002; Milligan et al., 2002).
This suggests that morphine induces proinflammatory cytokine and chemokine release and that their actions contribute to the termination and inhibition of morphine analgesia. However, ELISA analysis of IL-1 protein in CSF or lumbar spinal tissue failed to find a corresponding increase in IL-1 after one morphine injection. This may be because changes in spinal IL-1 were below the sensitivity of the ELISA and that IL-1 produces physiological responses via its receptor at low concentrations and/or via localized release. In support, although previous studies in this laboratory found evidence that blocking IL-1 receptors reduces hyperalgesia or allodynia in models of central inflammation (intrathecal infusion of gp120), peripheral nerve inflammation (sciatic inflammatory neuropathy), and peripheral nerve injury (chronic constriction injury), we have only detected increases in IL-1 protein by ELISA when spinal glia are directly activated by intrathecal gp120 (Milligan et al., 2001, 2003). Taken together, this suggests that small local changes in IL-1 are sufficient to induce changes in opioid efficacy.
Nevertheless, development of tolerance to chronic intrathecal morphine is paralleled by an increased release of IL-1 within 2 hr after morphine. This suggests that chronic morphine exposure sensitizes the release of spinal proinflammatory cytokines. It is notable that the expression of glial activation markers (e.g., GFAP) increases in the spinal cord and brain only after chronic exposure to morphine (Beitner-Johnson et al., 1993; Song and Zhao, 2001; Raghavendra et al., 2002). Moreover, in vitro chronic opioid treatment potentiates the activation of microglia and macrophages and the release of IL-1, TNF, and nitric oxide (NO) when these cells are stimulated with classic immune activators (Apte et al., 1990; Chao et al., 1994; Das et al., 1995; Kowalski et al., 2000; Peng et al., 2000; Gomez-Flores et al., 2001; Azuma and Ohura, 2002). In fact, morphine has the potential to induce the release of these proinflammatory cytokines directly: glial cells express opioid receptors (Maderspach and Solomonia, 1988) and chronic opioid treatment can induce the naloxone-reversible release of IL-1 from mixed brain cell cultures (Das et al., 1995). Thus, intriguing parallels may be found to recent clinical reports that long-term and/or high-dose intrathecal morphine administration is associated with the invasion of spinal tissue with immune cells and development of inflammatory masses at the catheter tip (for review, see Yaksh et al., 2002). Thus, in addition to evidence for glial inflammatory processes induced by shorter-term and lower-dose intrathecal morphine provided by the present study, intrathecal morphine has the capability of altering the function of immune cells as well.
These results also show a role for spinal fractalkine in regulating morphine analgesia. Fractalkine (CX3CL1) is a recently identified chemokine that acts exclusively at the CX3CR1 receptor and that exists in two forms on neurons: an inactive membrane-bound form and, after cleavage from the membrane, an active soluble form. Fractalkine cleavage can be induced by glutamate (Chapman et al., 2000). This suggests that the rapid sensitization of glutamatergic NMDA receptors by opioids (Mao and Mayer, 2001) may induce fractalkine cleavage. Also, spinal fractalkine is expressed on neurons and sensory afferents, whereas its receptor is predominately expressed on microglia, indicating that soluble fractalkine may play a role in neuron-to-glia communication. Given that fractalkine can induce the release of IL-1 from spinal cord tissue, the present results also suggest that fractalkine cleavage may play an important part in the activation of glia and the release of cytokines by opioids in vivo.
These proinflammatory substances may reduce opioid analgesia by directly affecting opioid receptor activity or they may indirectly offset opioid antinociception by inducing hyperalgesia. For instance, there are several potential mechanisms by which these inflammatory elements may rapidly and directly modulate opioid receptor function. First, there is the potential for heterologous desensitization between receptors for opioids and those for inflammatory substances via shared G-protein-coupled systems (Szabo et al., 2002) or other secondary messenger systems within neurons and glia, such as MAP kinases, nuclear factor-κB, and calcium (Roy et al., 1998; Law et al., 2000; Haddad, 2002). Second, glia regulate glutamatergic NMDA receptor-mediated neuroplastic changes known to contribute to opioid-induced tolerance and hyperalgesia (Fairbanks and Wilcox, 1997; Celerier et al., 1999; Ma and Zhao, 2002). Chronic opioid treatment reduces glial glutamate transporter expression (Mao et al., 2002), and the decreased glial uptake of synaptic glutamate may contribute to NMDA receptor activation. Third, activated glia release other substances that contribute to analgesic tolerance and hyperalgesia and allodynia in diverse exaggerated pain states (e.g., NO, ATP, excitatory amino acids, prostaglandins, dynorphin) (Watkins et al., 2003). Indeed, such mechanisms may contribute to the hyperalgesia and allodynia apparent 24 hr after the last injection of morphine in morphine-tolerant rats, even when there was no detectable increase in IL-1 protein levels in the spinal cord. That is, the rapid but transient increase in spinal IL-1 after morphine may activate downstream mechanisms that induce and maintain hyperalgesic and allodynic states.
Together, these results demonstrate that daily intrathecal morphine activates spinal proinflammatory responses. Moreover, these responses contribute to the inhibition of acute morphine analgesia and development of tolerance, hyperalgesia, and allodynia after chronic morphine. In particular, IL-1 and fractalkine are early signals in the recruitment of endogenous pain facilitatory mechanisms that modulate opioid analgesia. Finally, these results demonstrate that gene therapy with anti-inflammatory cytokines such as IL-10 are promising analgesic adjuvants in opioid-tolerant conditions and are effective at preventing increases in pain sensitivity after treatment with opioids.
Footnotes
This work was supported by National Institutes of Health Grants DA015632, DA015656, NS38020, NS40696, AI051093, and HL56510. We thank Amgen for the gift of IL1ra and vehicle and GlaxoSmithKline for the gift of anti-CX3CR1.
Correspondence should be addressed to Dr. Linda R. Watkins, Department of Psychology, University of Colorado, Campus Box 345, Boulder, CO 80309. E-mail: lwatkins@psych.colorado.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249353-13$15.00/0
References
- Apte RN, Durum SK, Oppenheim JJ (1990) Opioids modulate interleukin-1 production and secretion by bone-marrow macrophages. Immunol Lett 24: 141-148. [DOI] [PubMed] [Google Scholar]
- Azuma Y, Ohura K (2002) Endomorphin-2 modulates productions of TNF-alpha, IL-1beta, IL-10, and IL-12, and alters functions related to innate immune of macrophages. Inflammation 26: 223-232. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Guitart X, Nestler EJ (1993) Glial fibrillary acidic protein and the mesolimbic dopamine system: regulation by chronic morphine and Lewis-Fischer strain differences in the rat ventral tegmental area. J Neurochem 61: 1766-1773. [DOI] [PubMed] [Google Scholar]
- Boddeke EW (2001) Involvement of chemokines in pain. Eur J Pharmacol 429: 115-119. [DOI] [PubMed] [Google Scholar]
- Brewer KL, Bethea JR, Yezierski RP (1999) Neuroprotective effects of interleukin-10 following excitotoxic spinal cord injury. Exp Neurol 159: 484-493. [DOI] [PubMed] [Google Scholar]
- Celerier E, Laulin J, Larcher A, Le Moal M, Simonnet G (1999) Evidence for opiate-activated NMDA processes masking opiate analgesia in rats. Brain Res 847: 18-25. [DOI] [PubMed] [Google Scholar]
- Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK (1994) Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther 269: 198-203. [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55-63. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ (2000) Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci 20: RC87(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Das KP, McMillian MK, Bing G, Hong JS (1995) Modulatory effects of [Met5]-enkephalin on interleukin-1 beta secretion from microglia in mixed brain cell cultures. J Neuroimmunol 62: 9-17. [DOI] [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL (1997) Acute tolerance to spinally administered morphine compares mechanistically with chronically induced morphine tolerance. J Pharmacol Exp Ther 282: 1408-1417. [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL (2000) Spinal plasticity of acute opioid tolerance. J Biomed Sci 7: 200-212. [DOI] [PubMed] [Google Scholar]
- Falchi M, Ferrara F, Gharib C, Dib B (2001) Hyperalgesic effect of intrathecally administered interleukin-1 in rats. Drugs Exp Clin Res 27: 97-101. [PubMed] [Google Scholar]
- Gomez-Flores R, Rice KC, Zhang X, Weber RJ (2001) Increased tumor necrosis factor-alpha and nitric oxide production by rat macrophages following in vitro stimulation and intravenous administration of the delta-opioid agonist SNC 80. Life Sci 68: 2675-2684. [DOI] [PubMed] [Google Scholar]
- Gul H, Yildiz O, Dogrul A, Yesilyurt O, Isimer A (2000) The interaction between IL-1beta and morphine: possible mechanism of the deficiency of morphine-induced analgesia in diabetic mice. Pain 89: 39-45. [DOI] [PubMed] [Google Scholar]
- Haddad JJ (2002) Cytokines and related receptor-mediated signaling pathways. Biochem Biophys Res Commun 297: 700-713. [DOI] [PubMed] [Google Scholar]
- Hansen MK, Nguyen KT, Fleshner M, Goehler LE, Gaykema RP, Maier SF, Watkins LR (2000a) Effects of vagotomy on serum endotoxin, cytokines, and corticosterone after intraperitoneal lipopolysaccharide. Am J Physiol Regul Integr Comp Physiol 278: R331-R336. [DOI] [PubMed] [Google Scholar]
- Hansen MK, Nguyen KT, Goehler LE, Gaykema RP, Fleshner M, Maier SF, Watkins LR (2000b) Effects of vagotomy on lipopolysaccharide-induced brain interleukin-1beta protein in rats. Auton Neurosci 85: 119-126. [DOI] [PubMed] [Google Scholar]
- Harvey LO (1986) Efficient estimation of sensory thresholds. Behav Res Methods Instr Comp 18: 623-632. [Google Scholar]
- Ji G-C, Ma F, Zhang Y-Q, Wu G-C (2002) Thermal hyperalgesic effects induced by intracerebroventricular injection of interleukin-1beta in rats. Sheng Li Xue Bao 54: 325-328. [PubMed] [Google Scholar]
- Kanaan SA, Poole S, Saade NE, Jabbur S, Safieh-Garabedian B (1998) Interleukin-10 reduces the endotoxin-induced hyperalgesia in mice. J Neuroimmunol 86: 142-150. [DOI] [PubMed] [Google Scholar]
- Kowalski J, Makowiecka K, Belowski D, Herman ZS (2000) Augmenting effect of methionine-enkephalin on interleukin-6 production by cytokine-stimulated murine macrophages. Neuropeptides 34: 187-192. [DOI] [PubMed] [Google Scholar]
- Larcher A, Laulin JP, Celerier E, Le Moal M, Simonnet G (1998) Acute tolerance associated with a single opiate administration: involvement of N-methyl-d-aspartate-dependent pain facilitatory systems. Neuroscience 84: 583-589. [DOI] [PubMed] [Google Scholar]
- Laughlin TM, Bethea JR, Yezierski RP, Wilcox GL (2000) Cytokine involvement in dynorphin-induced allodynia. Pain 84: 159-167. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Ann Rev Pharmacol Toxicol 40: 389-430. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402-408. [DOI] [PubMed] [Google Scholar]
- Ma J-Y, Zhao Z-Q (2002) The involvement of glia in long-term plasticity in the spinal dorsal horn of the rat. NeuroReport 13: 1781-1784. [DOI] [PubMed] [Google Scholar]
- Maderspach K, Solomonia R (1988) Glial and neuronal opioid receptors: apparent positive cooperativity observed in intact cultured cells. Brain Res 441: 41-47. [DOI] [PubMed] [Google Scholar]
- Mantz J, Cordier J, Giaume C (1993) Effects of general anesthetics on inter-cellular communications mediated by gap junctions between astrocytes in primary culture. Anesthesiology 78: 892-901. [DOI] [PubMed] [Google Scholar]
- Mao J (2002) Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain 100: 213-217. [DOI] [PubMed] [Google Scholar]
- Mao J, Mayer DJ (2001) Spinal cord neuroplasticity following repeated opioid exposure and its relation to pathological pain. Ann NY Acad Sci 933: 175-184. [DOI] [PubMed] [Google Scholar]
- Mao J, Sung B, Ji RR, Lim G (2002) Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 22: 8312-8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy L, Wetzel M, Sliker JK, Eisenstein TK, Rogers TJ (2001) Opioids, opioid receptors, and the immune response. Drug Alcohol Depend 62: 111-123. [DOI] [PubMed] [Google Scholar]
- McNally GP, Westbrook RF (1998) Effects of systemic, intracerebral, or intrathecal administration of an N-methyl-d-aspartate receptor antagonist on associative morphine analgesic tolerance and hyperalgesia in rats. Behav Neurosci 112: 966-978. [DOI] [PubMed] [Google Scholar]
- Mellon RD, Bayer BM (1998) Evidence for central opioid receptors in the immunomodulatory effects of morphine: review of potential mechanism(s) of action. J Neuroimmunol 83: 19-28. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR (2000) Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res 861: 105-116. [DOI] [PubMed] [Google Scholar]
- Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR (2001) Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci 21: 2808-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chapman GA, O'Connor K, Maier SF, Watkins LR (2002) Spinal cord fractalkine, a chemokine, is involved in exaggerated pain states. Brain Behav Immunity 16: 201. [Google Scholar]
- Milligan ED, Langer SJ, Sloane EM, Chacur M, Weiseler-Frank J, Frank MG, Flotte TR, Cruz P, Leinwand L, Maier SF, Watkins LR (2003) Inflammatory and chronic constriction injury-induced pain states are controlled by spinal delivery of viral and non-viral vectors encoding the anti-inflammatory gene, interleukin-10 (IL10). Soc Neurosci Abstr 29: 587.9. [Google Scholar]
- O'Connor K, Holguin A, Hansen MK, Maier SF, Watkins LR (2004) A method for measuring multiple cytokines from small samples. Brain Behav Immunity 18: 274-280. [DOI] [PubMed] [Google Scholar]
- Oka T, Aou S, Hori T (1993) Intracerebroventricular injection of interleukin-1 beta induces hyperalgesia in rats. Brain Res 624: 61-68. [DOI] [PubMed] [Google Scholar]
- Peng X, Mosser DM, Adler MW, Rogers TJ, Meissler Jr JJ, Eisenstein TK (2000) Morphine enhances interleukin-12 and the production of other pro-inflammatory cytokines in mouse peritoneal macrophages. J Leukocyte Biol 68: 723-728. [PubMed] [Google Scholar]
- Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole S, Cunha FQ, Selkirk S, Lorenzetti BB, Ferreira SH (1995) Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10. Br J Pharmacol 115: 684-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA (2002) The role of spinal neuro-immune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci 22: 9980-9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 4: 247-257. [DOI] [PubMed] [Google Scholar]
- Roy S, Cain KJ, Chapin RB, Charboneau RG, Barke RA (1998) Morphine modulates NF kappa B activation in macrophages. Biochem Biophys Res Commun 245: 392-396. [DOI] [PubMed] [Google Scholar]
- Schaack J, Langer S, Guo X (1995) Efficient selection of recombinant adenoviruses by vectors that express beta-galactosidase. J Virol 69: 3920-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Zhao ZQ (2001) The involvement of glial cells in the development of morphine tolerance. Neurosci Res [Suppl] 39: 281-286. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen X-H, Xin L, Adler MW, Howard OMZ, Oppenheim JJ, Rogers TJ (2002) Heterologous desensitization of opioid receptors by chemokines inhibits fchemotaxis and enhances the perception of pain. Proc Natl Acad Sci USA 99: 10276-10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadano T, Namioka M, Nakagawasai O, Tan-No K, Matsushima K, Endo Y, Kisara K (1999) Induction of nociceptive responses by intrathecal injection of interleukin-1 in mice. Life Sci 65: 255-261. [DOI] [PubMed] [Google Scholar]
- Treutwein B, Strasburger H (1999) Fitting the psychometric function. Percept Psychophys 61: 87-106. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan Jr TP, Porreca F (2001) Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain 92: 5-9. [DOI] [PubMed] [Google Scholar]
- Watkins L, Kinscheck I, Miller J, Coghill R, Frank H, Mayer D (1984) CCK antagonists enhance opiate analgesia and appear to reverse morphine tolerance. Pain 18: S353. [Google Scholar]
- Watkins LR, Maier SF (2003) Glia: a novel drug discovery target for clinical pain. Nat Rev 2: 973-985. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF (2001) Spinal cord glia: new players in pain. Pain 93: 201-205. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF (2003) Glial proinflammatory cytokines mediate exaggerated pain states: implications for clinical pain. Adv Exp Med Biol 521: 1-21. [PubMed] [Google Scholar]
- Yaksh TL, Kohl RL, Rudy TA (1977) Induction of tolerance and withdrawal in rats receiving morphine in the spinal subarachnoid space. Eur J Pharmacol 42: 275-284. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Hassenbusch S, Burchiel K, Hildebrand KR, Page LM, Coffey RJ (2002) Inflammatory masses associated with intrathecal drug infusion: a review of preclinical evidence and human data. Pain Med 3: 300-312. [DOI] [PubMed] [Google Scholar]