

Abstract
Glutathione, a major endogenous antioxidant, is found in two intracellular pools in the cytoplasm and the mitochondria. To investigate the importance of the smaller mitochondrial pool, we developed conditions based on treatment with ethacrynic acid that produced near-complete and highly selective depletion of mitochondrial glutathione in cultured astrocytes. Recovery of mitochondrial glutathione was only partial over several hours, suggesting slow net uptake from the cytoplasm. Glutathione depletion alone did not significantly affect mitochondrial membrane potential, ATP content, or cell viability when assessed after 24 hr, although the activities of respiratory chain complexes were altered. However, these astrocytes showed a greatly enhanced sensitivity to 3-morpholinosydnonimine, a peroxynitrite generator. Treatment with 200 μm 3-morpholinosydnonimine produced decreases within 3 hr in mitochondrial membrane potential and ATP content and caused the release of lactate dehydrogenase, contrasting with preservation of these properties in control cells. These properties deteriorated further by 24 hr in the glutathione-depleted cells and were associated with morphological changes indicative of necrotic cell death. This treatment enhanced the alterations in activities of the respiratory chain complexes observed with glutathione depletion alone. Cell viability was markedly improved by cyclosporin A, suggesting a role for the mitochondrial permeability transition in the astrocytic death. These studies provide the most direct evidence available for any cell type on the roles of mitochondrial glutathione. They demonstrate the critical importance of this metabolite pool in protecting against peroxynitrite-induced damage in astrocytes and indicate a key contribution in determining the activities of respiratory chain components.
Keywords: glutathione, SIN-1, astrocytes, mitochondria, cyclosporin A, peroxynitrite
Introduction
Glutathione is a major endogenous antioxidant in cells, acting both to directly detoxify reactive oxygen species and as a substrate for several peroxidases (Dringen and Hamprecht, 1997; Dringen, 2000). This tripeptide is found in both the cytoplasm and the mitochondria but is synthesized only in the cytoplasm (Fernandez-Checa et al., 1998). Mitochondrial glutathione is maintained by uptake from the cytoplasmic pool. Despite the interdependence of the two glutathione pools, they apparently interact slowly and can be differentially affected by some treatments (Meredith and Reed, 1983; Colell et al., 1998; Wullner et al., 1999).
Diseases or treatments that deplete cellular glutathione commonly promote cell death. The loss of viability appears to relate more closely to the extent of depletion of the mitochondrial glutathione rather than the changes in the cytoplasmic pool (Meredith and Reed, 1983; Garcia-Ruiz et al., 1994; Colell et al., 1998; Fernandez-Checa et al., 1998; Wullner et al., 1999). Thus, alterations of mitochondrial glutathione could underlie tissue damage in some diseases. Consistent with this possibility, selective depletion of mitochondrial glutathione has been observed after exposure of the liver to ethanol and other toxins (Fernandez-Checa et al., 1991; Garcia-Ruiz et al., 1994; Colell et al., 1998). Furthermore, the affected liver cells showed an increased susceptibility to oxidative stress, which was ameliorated by treatments that prevented the glutathione depletion.
The possible contribution of changes in mitochondrial glutathione in neurological disorders has received little attention. However, we recently obtained evidence that glutathione is partially lost from brain mitochondria in an animal model of stroke (Anderson and Sims, 2002). Furthermore, a close apparent association between the regional distribution of mitochondrial glutathione loss and the subsequent development of tissue infarction was observed, suggesting a role for this change in promoting cellular vulnerability to cerebral ischemia.
Accumulations of nitric oxide and its derivatives are likely to contribute to oxidative stress in nervous system disorders, with particularly strong evidence for a role in promoting tissue damage in stroke (Eliasson et al., 1999; Gursoy-Ozdemir et al., 2000; Sims and Anderson, 2002). Nitric oxide can directly modulate cell function via inhibition of mitochondrial respiration but conversion to peroxynitrite in the presence of superoxide also appears to underlie many of its deleterious effects (Beckman et al., 1990; Brown and Borutaite, 2001). Peroxynitrite and its degradation products can modify many cellular components (Goldstein et al., 1996). The inhibition of mitochondrial activity is again a prominent consequence (Brown and Borutaite, 2002; Radi et al., 2002; Szabo, 2003).
A direct test of the role of mitochondrial glutathione is lacking. In previous studies, treatments that depleted mitochondrial glutathione also produced substantial changes in the cytoplasmic pool. To address this issue more directly and to investigate the importance of mitochondrial glutathione in a major brain cell population, we established conditions that produce a highly selective loss of this metabolite in the mitochondria of astrocytes in culture. We investigated the consequences of this change for mitochondrial function and cell viability and examined the susceptibility of these cells to the peroxynitrite-generating agent 3-morpholinosydnonimine (SIN-1).
Materials and Methods
Materials. All of the chemicals were of analytical grade and were obtained from Sigma (St. Louis, MO) unless stated otherwise. Tissue culture re-agents were obtained from Invitrogen (San Diego, CA). Tacrolimus (FK506) was from Alexis (San Diego, CA). 5,5,6,6-Tetrachloro-1,1,3,3-tetraethylbenzi-midazolocarbo-cyanine iodide (JC-1), propidium iodide (PI), rhodamine 123, and the Annexin-V-Fluos kit were from Molecular Probes (Leiden, The Netherlands), and Hoechst 33258 was from Calbiochem (La Jolla, CA).
Cell cultures. Primary astroglial cell cultures were prepared from neonatal [postnatal day 1 (P1)-P2] Sprague Dawley rat pups [bred in-house at the School of Medicine, Flinders University, or obtained from Charles River (Uppsala, Sweden)] as previously described (Muyderman et al., 2001). In brief, the brains were rapidly removed from the skulls, and the meninges were removed. The cerebral cortices were dissected from the brains and passed through an 80 μm mesh nylon net into sera-containing modified Eagle's MEM supplied with Earl's salts (Invitrogen, Paisley, UK). The medium was changed after 3 d of cultivation, and thereafter three times a week. Cultures were grown to confluence in a humidified atmosphere of 5% CO2 in air at 37°C and were used for experiments between 14 and 16 d.
Separation of mitochondrial and cytosolic components. Cells were harvested by mild trypsinization [0.05% (w/v) trypsin and 0.02% EDTA; 3-5 min] to minimize any microglial contamination. Trypsinization was halted by addition of HEPES-buffered salt solution (HBSS) (in mm: 137 NaCl, 5.4 KCl, 0.41 MgSO4, 0.49 MgCl2, 1.26 CaCl2, 0.64 KH2PO4, 3.0 NaHCO3, 5.5 glucose, and 20 HEPES, pH 7.4), containing 10% calf sera. The cell suspension was washed twice in HBSS (without sera), and samples for determining total glutathione content were removed. The pellet was resuspended in “isolation medium” (10 mm Tris, 1 mm EDTA, 0.32 m sucrose, pH 7.4) containing 0.2 mg/ml digitonin and centrifuged (15,000 × g) for 5 min. The supernatant was removed and used as the “cytoplasmic fraction” in subsequent studies. To produce a crude “mitochondrial fraction,” the pellet was washed twice with isolation medium with centrifugation after each wash. In initial studies, to establish the subfractionation conditions, 93 ± 2% of total cellular citrate synthase activity (a mitochondrial marker) and 2.5 ± 0.3% of the total lactate dehydrogenase (LDH) activity (a cytoplasmic marker) were recovered in the mitochondrial fraction (n = 8). The cytoplasmic fraction contained 98 ± 2% of the LDH activity and no detectable citrate synthase activity (n = 8). The small residual LDH activity in the mitochondrial fraction was not decreased by additional washing or by increasing the concentration of digitonin used initially to disrupt the plasma membranes (n = 5). This suggests that either the LDH was bound to noncytoplasmic components in this fraction or this apparent activity was attributable to the reduction of NAD+ by a reaction catalyzed by an enzyme other than LDH.
Determination of glutathione content. Samples were treated with 2 m perchloric acid containing 4 mm EDTA for 3 min, centrifuged, and neutralized with 2 m potassium bicarbonate and 0.3 m 3-(N-morpholino)propanesulfonic acid. Total (reduced plus oxidized) glutathione was determined spectrophotometrically using the procedure of Akerboom and Sies (1981), adapted for use on a Cobas Fara (Roche, Basel, Switzerland) spectrophotometric analyzer. The assay measured the rate of formation of 5-thio-2-nitrobenzoate from 5,5′-dithio-bis(2-nitrobenzoate) in the presence of NADPH and glutathione reductase. Glutathione monoethylester at concentrations equivalent to those of the glutathione standards did not produce a measurable response in this assay.
Treatment with ethacrynic acid. A stock solution of 1 m ethacrynic acid in ethanol was prepared and further diluted into fresh HBSS, pH 7.4. Cultures were rinsed twice in HBSS and incubated with ethacrynic acid at 37°C for desired times and concentrations.
Enzyme assays: respiratory chain complexes, citrate synthase, and lactate dehydrogenase. The activity of nicotinamide adenine dinucleotide (NADH)-ubiquinone oxidoreductase (complex I) was determined spectrophotometrically at 340 nm by following the rotenone-sensitive oxidation of NADH in the presence of ubiquinone-1 (Ragan et al., 1987). Succinate-cytochrome c oxidoreductase (complexes II-III) activity was determined by measuring the reduction of cytochrome c in the presence of sodium succinate (King, 1967), and cytochrome c oxidase (complex IV) activity was assessed from the oxidation of reduced cytochrome c (Wharton and Tzagoloff, 1967; Zaidan and Sims, 1997). All of the measurements were made using a Shimadzu UV-3000 spectrophotometer (Shimadzu Scientific Instruments, Rydalmere, New South Wales, Australia). Enzyme activities were calculated as nanomoles per minute per milligram of protein, except for cytochrome c oxidase, which was expressed as the first-order rate constant (k) per milligram of protein. All of the results are presented as relative changes compared with untreated control cultures.
Citrate synthase activity was measured at 410 nm using a Shimadzu UV-3000 spectrophotometer at 30°C as described by Shepherd and Garland (1969). The reaction buffer contained 100 mm Tris-HCl, pH 8.1, 0.1 mm 5,5′-dithio-bis(2-nitrobenzoic acid), 0.3 mm acetyl-CoA, and 0.1% (v/v) Triton X-100. The assay was initiated by the addition of oxaloacetic acid (final concentration, 0.05 mm).
Lactate dehydrogenase activity was determined spectrophotometrically from the oxidation of NADH in the presence of pyruvate (Vassault, 1983). The method was adapted for use in the Cobas Fara spectrophotometric analyzer and also on a Wallac 1420 Victor multilabel reader. Comparable results were obtained from both systems. Release into the supernatant was determined as a percentage of the total lactate dehydrogenase activity, which was measured after treating cells for 10 min with 0.25% Triton X-100.
ATP measurements. Cultured cells were rinsed twice in ice-cold PBS and then lysed in 5% trichloroacetic acid for 15 min on ice. The content of ATP in the lysate was measured using the ATP Bioluminescence Assay kit CLS II (Roche, Basel, Switzerland) in accordance with the manufacturer's instructions.
Estimation of mitochondrial transmembrane potential in intact cultured astrocytes. The ratiometric fluorescent dye JC-1 was used to provide relative measurements of mitochondrial membrane potential (Reers et al., 1991, 1995; Salvioli et al., 1997). At depolarized potentials, JC-1 forms monomers producing a green fluorescence detectable at 530 nm. At higher membrane potentials, JC-1 forms multimers (J-aggregates) producing a red fluorescence that is detectable at 590 nm. Both the monomer and the J-aggregate were excited simultaneously at 488 nm. The ratio of fluorescence emission at 590:530 nm was determined as an indicator of the mitochondrial membrane potential (Reers et al., 1995).
For the measurements, cultured cells were washed twice and incubated in HBSS containing 0.125 μm JC-1 for 30 min. The cultures were then rinsed and left in dye-free HBSS for 20 min at 37°C before being viewed in a Nikon Optiphot-2. Images were captured using a Hamamatsu C5810 color-intensified 3-CCD camera and corrected for autofluorescence. Of nine predefined areas on each coverslip, three were randomly chosen for analysis. In untreated cultures, the mean value of the JC-1 fluorescence ratio was 1.4 ± 0.4 (mean ± SD; n = 10). Maximum depolarization of the mitochondria was obtained by treatment with the potassium ionophore valinomycin (500 nm; 10 min), which rapidly reduced the ratio by 62% to 0.53 ± 0.09 (mean ± SD; n = 10). All of the estimations of alterations in mitochondrial membrane potential have been presented as changes in the JC-1 fluorescence ratio expressed relative to the response to valinomycin. No changes in JC-1 fluorescence ratio were observed after depolarization of the plasma membrane for 10 min using high K+ (56 mm), indicating that the response of JC-1 after loading was not influenced by changes in the plasma membrane potential.
Estimation of mitochondrial transmembrane potential in isolated mitochondria. Rhodamine 123 was used to evaluate the membrane potential in mitochondria isolated from the astrocytic cultures as described above. In short, the mitochondrial fraction was transferred to analysis buffer containing 100 mm KCl, 10 mm potassium phosphate buffer, 10 mm Tris-HCl, 10 mm MgCl2, 1 mg/ml fatty acid-free BSA, 5 mm pyruvate, 2.5 mm malate, and 20 nm rhodamine 123. (In some initial studies, 40 nm rhodamine 123 was used. This increased the fluorescence signal from the mitochondria but comparable effects on the fluorescence profiles were produced by the experimental treatments investigated.) After 2 min, the incubation mixture was analyzed by flow cytometry in a FACScan (BD Biosciences, San Jose, CA) equipped with a single argon ion laser. Samples were excited at 488 nm, and the emission was collected at 529 nm. A minimum of 10,000 events per sample were analyzed and stored. ADP (100 nmol) was added, and the analysis was repeated after an additional 2 min incubation and again 2 min after addition of carbonyl cyanide m-chlorophenylhydrazone (CCCP) (final concentration, 5 μm). Data analysis was performed using the standard CellQuest software (BD Biosciences).
Assessment of mitochondrial respiratory activity. Oxygen consumption in the mitochondrial fractions from astrocytic cultures was determined polarographically using a Clark-type oxygen electrode (Instech Laboratories, Plymouth Meeting, PA) and a Yellow Springs 5300 oxygen monitor (Yellow Springs Instruments, Yellow Springs, OH). Mitochondria (usually 100-250 μg of protein in 30 μl) were added to “respiration buffer” in a total volume of 300 μl in a magnetically stirred water-jacketed Perspex chamber at 30°C. The respiration buffer contained 100 mm KCl, 25 mm sucrose, 75 mm mannitol, 5 mm phosphoric acid (adjusted to pH 7.4 with Tris), 50 μm potassium EDTA, and 10 mm Tris-HCl, pH 7.4. Substrates were added before the mitochondria to give the following concentrations: 5 mm pyruvate plus 2.5 mm malate or 10 mm succinate plus 5 μm rotenone. State 3 respiration rate (substrates plus ADP present) was determined after the addition of 25-100 nmol of ADP, and the state 4 respiration rate (substrate only) was determined after the depletion of this ADP (Chance and Williams, 1956). The ADP/O ratio was calculated from the oxygen use required to fully metabolize the added ADP. The maximal uncoupled rate was determined after several small additions of the mitochondrial uncoupling agent, CCCP, typically producing a final concentration of 0.4 μm.
Staining of cells with annexin V, PI, and Hoechst 33258. Early apoptotic cell death was evaluated using an Annexin-V-Fluos kit, which assesses the translocation of phosphatidylserine from the inner to the outer leaflet of the plasma membrane lipid bilayer. PI (25 μm; 20 min) was used to visualize dead cells and Hoechst 33258 (1:500) was used to visualize chromatin condensation and apoptotic bodies if present. Staining procedures were performed in accordance with the manufacturer's instructions with minor modifications.
Annexin V-positive/PI-negative cells were considered to be apoptotic, whereas cells that were only PI positive were seen as necrotic. The small proportion of cells that were both annexin-V positive and PI positive were considered apoptotic only if there was also evidence of apoptotic bodies or condensed chromatin. Images were taken from nine predefined areas per coverslip. Three of these were randomly chosen and analyzed. Staurosporine (0.5-10 μm; 4 hr) was used to induce apoptosis in normal astrocytes as a positive control (n = 4 for each concentration). Total and apoptotic nuclei were counted, and the data are presented as percentage changes compared with controls.
Protein determination. Cell suspensions and subcellular fractions were solubilized in 2 m NaOH, and the protein content was determined by the method of Lowry et al. (1951) using bovine serum albumin as standard.
Statistical analyses. Results are presented as mean ± SD. Each experimental condition was tested on cultures obtained from at least three separate seedings of astrocytes. Individual values were determined as the average of the results from at least two (and usually three or more) identically treated wells on individual culture plates. Statistical analyses were performed on raw data by one-way ANOVA followed by Student-Newman-Keuls test.
Results
Glutathione content in astrocyte cultures
The total glutathione content of primary astrocyte cultures was 31 ± 4 nmol of glutathione per milligram of protein. Most of the glutathione in these cells were found in the cytoplasmic compartment, which contained 27 ± 3 nmol of glutathione per milligram of total cell protein. The mitochondrial fraction contained 0.49 ± 0.07 nmol of glutathione per milligram of cell protein (equivalent to 1.1 nmol per milligram of mitochondrial protein), which was <2% of the total pool (n = 15-20). These findings are similar to those reported from a previous investigation of glutathione distribution of cultured astrocytes (Huang and Philbert, 1995). Although the mitochondrial glutathione pool was only small, it could be manipulated independently of the cytoplasmic glutathione in the present study, as detailed below. Thus, the glutathione detected in the mitochondrial fraction apparently did not primarily arise from cytoplasmic contamination.
Selective modulation of mitochondrial glutathione
Experiments to modify glutathione content were initially performed in both serum-containing MEM and in HBSS (without serum). The latter does not contain extracellular precursors for glutathione synthesis or known antioxidants. The initial content of glutathione in the cytoplasm and mitochondria of the astrocytes was very similar in the two media. Furthermore, in both media, ethacrynic acid produced a pronounced depletion of glutathione, which was initially selective for the mitochondrial pool (Fig. 1). However, the depletion of glutathione in both the mitochondria and cytoplasm was severalfold faster in HBSS than in MEM, perhaps reflecting a lower effective concentration of ethacrynic acid in MEM as a result of interaction with other components in this more complex medium. HBSS was used as incubation medium in all of the subsequent studies. Incubation in this medium alone for 24 hr did not affect the mitochondrial glutathione content (96 ± 3% at 24 hr; n = 5). However, the cytoplasmic pool showed a small loss of glutathione after this time (71 ± 8% of initial values; n = 5).
Figure 1.

Depletion of mitochondrial (•/○) and cytoplasmic (▪/□) glutathione by 50 μm (•/▪) and 100 μm (○/□) ethacrynic acid in HBSS. Note the earlier and more pronounced depletion of the mitochondrial glutathione pool. Values are shown as mean ± SD. *p < 0.05 and ***p < 0.001, compared with untreated cells (one-way ANOVA with Student-Newman-Keuls test; n = 15-20). Control (100%) values were 1.3 ± 0.3 nmol of glutathione per milligram of mitochondrial protein in the mitochondria and 26 ± 6 nmol of glutathione per milligram of cell protein in the cytoplasm.
Because the mitochondrial pool was much more sensitive to ethacrynic acid, it was possible to identify conditions in which there was a highly selective and prolonged depletion of this pool (Fig. 1). After treatment with 100 μm ethacrynic acid for 15 min in HBSS, no mitochondrial glutathione was detectable, whereas cytoplasmic glutathione remained essentially unaffected (96 ± 4% of untreated cells). The mitochondrial fraction prepared after treatment with ethacrynic acid showed no difference in the proportion of the citrate synthase activity (92 ± 3% of total cellular activity compared with 93 ± 2% in untreated cells; n = 5) or of the LDH activity (2.5 ± 0.2% of total LDH activity compared with 2.5 ± 0.3% in untreated cells; n = 5) that was recovered when compared with the mitochondrial fraction from untreated cells. In addition, no citrate synthase activity was detectable in the cytosolic preparation from cells treated with ethacrynic acid (n = 5). More importantly, the mitochondria prepared from ethacrynic acid-treated and control cells showed similar membrane potentials, assessed using rhodamine 123, in the presence of pyruvate plus malate as respiratory substrates and after subsequent additions of ADP and the uncoupling agent CCCP (Fig. 2). Thus, treatment with ethacrynic acid did not seem to be associated with any major disruption of mitochondrial structure or substantial changes in the function of these organelles. Treatment with 100 μm ethacrynic acid for 15 min was used in subsequent investigations.
Figure 2.

Fluorescence responses of rhodamine 123 (40 nm final concentration) in mitochondria isolated from normal astroglial cultures (A) and from cultures treated with 100 μm ethacrynic acid for 15 min (B). Traces show the initial reading with pyruvate and malate as respiratory substances (a), a small shift to the left in response to 100 μm ADP (b), and a large change in transmembrane potential in response to the mitochondrial uncoupler CCCP (5 μm) (c). The traces show one typical experiment of five.
Restoration of mitochondrial glutathione
The spontaneous restoration of mitochondrial glutathione during subsequent incubation in HBSS medium was slow and reached only 26 ± 4% of pretreatment values 3 hr after removal of ethacrynic acid (Fig. 3) Thus, there was apparently only limited exchange between the cytoplasmic and mitochondrial pools after the treatment with ethacrynic acid. Nonetheless, the mitochondrial glutathione could be readily replenished by incubation with glutathione monoethylester. Exposure to 5 mm glutathione monoethylester for 30 min completely restored the mitochondrial glutathione pool, while having little effect on glutathione content in the cytoplasm (Fig. 3). Longer incubations resulted in a substantially increased glutathione content in both pools, although the proportional change was much larger in the mitochondria (Fig. 3).
Figure 3.

Recovery of mitochondrial glutathione after treatment with 100 μm ethacrynic acid for 15 min. The slow spontaneous restoration of mitochondrial glutathione during incubation in HBSS alone is shown (•) and compared with rapid recovery in the presence of 5 mm glutathione monoethylester ( ). Treatment with the glutathione ester also increased cytoplasmic glutathione content (
). Treatment with the glutathione ester also increased cytoplasmic glutathione content ( ), but the changes were slower to develop and less pronounced than those in the mitochondria. Values are shown as mean ± SD. **p < 0.01 and ***p < 0.001, compared with glutathione content measured after 15 min of treatment with 100 μm ethacrynic acid (one-way ANOVA with Student-Newman-Keuls test; n = 15). Control (100%) values were 1.0 ± 0.1 nmol of glutathione per milligram of mitochrondrial protein in the mitochondria and 27 ± 2 nmol of glutathione per milligram of cell protein in the cytoplasm.
), but the changes were slower to develop and less pronounced than those in the mitochondria. Values are shown as mean ± SD. **p < 0.01 and ***p < 0.001, compared with glutathione content measured after 15 min of treatment with 100 μm ethacrynic acid (one-way ANOVA with Student-Newman-Keuls test; n = 15). Control (100%) values were 1.0 ± 0.1 nmol of glutathione per milligram of mitochrondrial protein in the mitochondria and 27 ± 2 nmol of glutathione per milligram of cell protein in the cytoplasm.
Effects of mitochondrial glutathione depletion on astrocytic cell viability, ATP content, and mitochondrial membrane potential
Despite the marked and persisting decrease in mitochondrial glutathione produced by ethacrynic acid, this treatment did not significantly increase extracellular LDH activity (11.5 ± 1.8% of total compared with 8.5 ± 6.5% without ethacrynic acid treatment; n = 10) or the number of PI-positive cells (3.5 ± 3.0 vs 2.2 ± 1.8%; n = 5) when assessed 24 hr after the onset of the treatment. The content of ATP was unaltered at 6, 12, and 24 hr after the 15 min exposure to ethacrynic acid (98 ± 4, 103 ± 7, and 97 ± 2% of control; n = 5). Mitochondrial transmembrane potential also remained essentially unaffected (94 ± 13% of control at 24 hr; n = 5), although isolated cells with decreases in the JC-1 fluorescence ratio were observed 24 hr after the onset of the treatment.
Effects of SIN-1 on astrocytes with intact and depleted mitochondrial glutathione
Exposure of normal astrocytes to 1 mm SIN-1 led to a progressive loss of both mitochondrial and cytoplasmic glutathione (Fig. 4). Furthermore, exposure to 1 or 2 mm SIN-1 resulted in cell disruption by 6 hr as indicated by significant increases in LDH release with additional deterioration as the incubation time increased (Fig. 5). In addition, both the ATP content and the JC-1 fluorescence ratio dropped rapidly after 6 hr exposure to 1 mm SIN-1. After 24 hr exposure, PI staining confirmed a large proportion of disrupted cells (46 ± 8%; n = 5). No significant changes in annexin V labeling or number of apoptotic bodies were observed. In contrast, treatment with staurosporine (10 μm; 4 hr), as a positive control, induced apoptosis in >80% of the astrocytes (n = 4).
Figure 4.

Changes in glutathione content in the cytoplasm (▪/) and mitochondria (•/) in response to 1 mm (▪/•) and 200 μm (/) SIN-1. Values are shown as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with untreated controls (one-way ANOVA with Student-Newman-Keuls test; n = 10). Control (100%) values were 1.3 ± 0.5 nmol of glutathione per milligram of mitochondrial protein in the mitochondria and 27 ± 3 nmol of glutathione per milligram of cell protein in the cytoplasm.
Figure 5.

Extracellular increase of LDH activity in response to treatment with 2 mm (), 1 mm (), and 200 μm ( ) SIN-1 in HBSS. Control values (•) were obtained after incubation in HBSS only. Values are shown as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with HBSS alone (one-way ANOVA with Student-Newman-Keuls test; n = 10).
) SIN-1 in HBSS. Control values (•) were obtained after incubation in HBSS only. Values are shown as mean ± SD. *p < 0.05, **p < 0.01, and ***p < 0.001, compared with HBSS alone (one-way ANOVA with Student-Newman-Keuls test; n = 10).
In contrast to the higher SIN-1 concentrations, treatment with 200 μm SIN-1 had no effect on mitochondrial and cytoplasmic glutathione in normal astrocytes for at least 3 hr (Fig. 4). In addition, no significant change in extracellular LDH activity was seen until beyond 12 hr of exposure (Fig. 5). Thus, most cells apparently remained intact after treatment with 200 μm SIN-1. Treatment with this concentration of SIN-1 for 3 hr was selected for subsequent comparisons of the effects on glutathione-depleted and normal astrocytes.
The effects of this treatment with SIN-1 (in HBSS) on the properties of normal and glutathione-treated astrocytes are summarized in Figure 6 and compared with the responses of cells incubated in HBSS alone. SIN-1 again produced a small but significant increase in LDH release in astrocytes with intact glutathione by 24 hr after initiation of the 3 hr treatment. The number of PI-positive cells was 18 ± 6% compared with untreated cultures at 2.2 ± 2.0% (n = 5-10). These changes were accompanied by a significant loss of ATP and JC-1 fluorescence ratios at 24 hr, although both measures remained above 65% of the response of cells treated with HBSS alone.
Figure 6.

The effects of treatment with SIN-1 (in HBSS) on LDH release, ATP content, and JC-1 fluorescence in normal astrocytes and astrocytes that were treated with ethacrynic acid (+EA) to deplete mitochondrial glutathione. The responses of these cells to incubation in HBSS without SIN-1 are also presented for comparison. Results for JC-1 fluorescence ratio are expressed relative to values in untreated control cultures (1.4 ± 0.4). The results are shown as mean ± SD. Values significantly different in astrocytes with depleted glutathione exposed to SIN-1 compared with normal cells also treated with SIN-1 are indicated as follows: **p < 0.01 and ***p < 0.001 (one-way ANOVA with Student-Newman-Keuls test). All of the values for the glutathione-depleted astrocytes exposed to SIN-1 were significantly different (p < 0.001) compared with these same cells incubated in HBSS without SIN-1. Significant differences (p < 0.001) were also seen at 24 hr in LDH release, ATP content, and JC-1 fluorescence ratio for normal astrocytes exposed to SIN-1 compared with these cells incubated in HBSS alone. At the earlier time points examined, only the JC-1 fluorescence ratio measured at 3 hr showed a statistically significant difference (p < 0.05) after SIN-1 treatment of these normal astrocytes.
The depletion of mitochondrial glutathione markedly increased the susceptibility to SIN-1. In these cells, exposure to 200 μm SIN-1 for 3 hr increased extracellular LDH activity threefold compared with normal cells and produced large decreases in both ATP content and the mitochondrial membrane potential as assessed from JC-1 fluorescence ratios (Fig. 6). Furthermore, these effects deteriorated substantially after the removal of SIN-1. Thus, at 24 hr after the onset of the SIN-1 exposure, extracellular LDH activity had reached >70% of the total. Both the mitochondrial membrane potential and ATP content were also much more affected compared with either normal astrocytes exposed to SIN-1 or glutathione-depleted astrocytes incubated in HBSS alone (Fig. 6).
Mitochondrial fractions prepared from these cells showed changes in membrane potential measured using rhodamine 123 and flow cytometry that were consistent with the JC-1 fluorescence ratio measured in the intact astrocytes (Table 1). The rhodamine fluorescence in mitochondria prepared from astrocytes after glutathione depletion and SIN-1 treatment was markedly lower than fractions from untreated control cells or cells exposed to either ethacrynic acid or SIN-1 alone when assessed in the presence of the respiratory substrates, pyruvate plus malate. Similar differences were seen after addition of ADP to the mitochondria, although there was not a statistically significant difference between the glutathione-depleted cells exposed to SIN-1 and SIN-1 treatment alone. The rhodamine fluorescence showed comparably low values in all of the preparations after addition of the mitochondrial uncoupler CCCP.
Table 1.
Rhodamine 123 fluorescence in mitochondrial fractions prepared from astrocytes after mitochondrial glutathione depletion and exposure to SIN-1
|
|
Mean fluorescence (arbitrary units) |
||||
|---|---|---|---|---|---|
| Astrocyte treatment |
Respiratory substrates only |
Respiratory substrates plus ADP |
Respiratory substrates plus ADP plus CCCP |
||
| Untreated cells | 84 ± 19 | 48 ± 11 | 16 ± 5 | ||
| Ethacrynic acid | 77 ± 21 | 48 ± 14 | 15 ± 8 | ||
| SIN-1 | 67 ± 17 | 41 ± 8 | 15 ± 2 | ||
| Ethacrynic acid plus SIN-1 |
42 ± 5*
|
30 ± 8*
|
15 ± 5 |
||
Mitochondrial fractions were isolated from astrocytes at 24 hr after exposure to ethacrynic acid to deplete mitochondrial glutathione and/or treatment with 200 μm SIN-1 for 3 hr. The fractions were incubated with 20 nm rhodamine 123 in the presence of 5 mm pyruvate and 2.5 mm malate as respiratory substrates and fluorescence analyzed by flow cytometry. Additional measurements were obtained after additions of ADP and CCCP. Values represent the geometric mean fluorescence shown as mean ± SD (n = 5-10). Values significantly different compared with mitochondrial fractions from the untreated cells (incubated in HBSS) or cells treated with ethacrynic acid are indicated: *p<0.05 (one-way ANOVA with Student-Newman-Keuls test). For the incubations with respiratory substrates alone, the fluorescence in mitochondria from cells exposed to ethacrynic acid plus SIN-1 also differed significantly to the mitochondria from cells treated with SIN-1.
Treatment with SIN-1 also increased the number of PI-positive cells in the glutathione-depleted cells to 62 ± 7%. However, no significant increase in annexin-positive cells was observed (n = 5). Thus, in both normal cultures and cultures with depleted mitochondrial glutathione, the cell death induced by SIN-1 appeared to be necrotic rather than apoptotic based on both the lack of annexin-V staining and morphological examination of the cells.
Protection against SIN-1-induced cell damage
The decrease in cell viability observed after SIN-1 treatment of cells lacking mitochondrial glutathione could be prevented by restoring the mitochondrial glutathione pool with glutathione monoethylester. Thirty minute exposure to 5 mm glutathione monoethylester after the ethacrynic acid treatment completely abolished the increase in extracellular LDH activity on subsequent exposure to SIN-1 (Fig. 7). Under these conditions, the glutathione ester restored the mitochondrial pool without significantly affecting cytoplasmic glutathione (Fig. 3). Thus, the findings provide good evidence that the increased susceptibility of cells to SIN-1 treatment after ethacrynic acid treatment was attributable to the selective loss of mitochondrial glutathione and not some other unintended consequence of the ethacrynic acid treatment.
Figure 7.

LDH release at 24 hr after the onset of a 3 hr exposure to 200 μm SIN-1 in cells with mitochondrial glutathione either intact (first bar) or depleted (remaining five bars). Significant protective effects were seen after inclusion of superoxide dismutase (SOD) (500 U/ml; 3 hr), glutathione monoethylester (GSH-E) (5 mm; 30 min), and cyclosporin A (CsA) (2 μm; 3 hr) in the incubation media. Treatment with FK506 (200 nm) was not protective. Values are shown as mean ± SD. ***p < 0.001, compared with SIN-1 exposure in cells with depleted mitochondrial glutathione (one-way ANOVA with Student-Newman-Keuls test; n = 5-10).
Cyclosporin A (2 μm), an inhibitor of the mitochondrial permeability transition, did not protect against SIN-1-induced damage in cells with an intact mitochondrial glutathione pool (data not shown). However, it abolished the additional increase in extracellular LDH activity (24 ± 2% compared with 73 ± 7%) (Fig. 7) and the drop in mitochondrial membrane potential produced by SIN-1 in cells with depleted mitochondrial glutathione (Fig. 8). The effects of FK506 on LDH release were also examined (Fig. 7). This compound does not block induction of the permeability transition but does mimic other effects of cyclosporin A, including inhibition of the protein phosphatase calcineurin (Griffiths and Halestrap, 1991). In contrast to the findings with cyclosporin A, FK506 did not decrease LDH release in the glutathione-depleted cells exposed to SIN-1. This compound also had no effect on LDH release in normal cells (data not shown). Superoxide dismutase (500 U/ml), which removes superoxide and inhibits the generation of peroxynitrite, prevented all of the SIN-1-mediated effects in both normal and ethacrynic acid-treated cultures (Fig. 7), demonstrating that the effects elicited by the SIN-1 treatment resulted from peroxynitrite or from superoxide itself.
Figure 8.
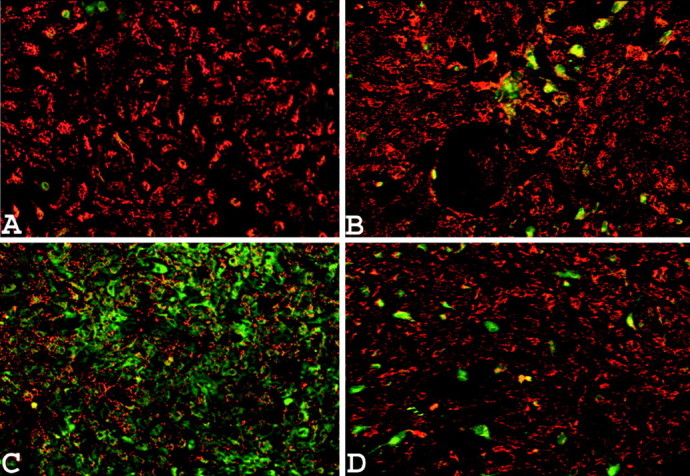
Astrocytes loaded with the mitochondrial membrane potential-sensitive dye JC-1. A, Nontreated cells. B, Normal cells exposed to 200 μm SIN-1 for 3 hr. C, Cells with depleted mitochondria exposed to 200 μm SIN-1 for 3 hr. D, Cells with depleted mitochondria exposed to 200 μm SIN-1 for 3 hr in the presence of 2 μm cyclosporin A. Each picture was taken 24 hr after the onset of the treatment and shows the results of one typical experiment of five. Quantitative analysis of the JC-1 fluorescence revealed that the effect of SIN-1 on cells with depleted mitochondrial glutathione was significantly different when tested in the absence of cyclosporin A (39 ± 9% of control) compared with the presence of this compound (74 ± 6%) (p < 0.01; one-way ANOVA with Student-Newman-Keuls test; n = 5).
Effect of glutathione depletion and SIN-1 treatment on activity of electron transport chain complexes
In contrast to the apparent preservation of most of the measured cell properties after depletion of mitochondrial glutathione alone, there was a substantial (approximately fourfold) increase in the activity of complex I of the electron transport chain, when assessed in mitochondrial fractions 24 hr later (Fig. 9a). This was accompanied by small but significant decreases in the activities of complexes II-III and complex IV (Fig. 9b). The mitochondrial fractions from these treated cells showed an approximately twofold increase compared with fractions from untreated astrocytes in respiratory rates assessed under state 3, state 4, and uncoupled conditions in the presence of the NAD-linked substrate pyruvate plus malate (Table 2). The ADP/O ratio was similar under the two conditions. No differences in respiratory rates were seen with the flavin adenine dinucleotide-linked substrate succinate (in the presence of rotenone). These findings are consistent with increased flux through the electron transport chain because of increased activity of complex I at 24 hr after selective glutathione depletion.
Figure 9.

Changes in the activities of complex I (a) and complexes II-III and complex IV (b) after depletion of mitochondrial glutathione by 100 μm ethacrynic acid for 15 min (A), 3 hr exposure to 200 μm SIN-1 (B), the combination of the two (C), and cells treated with ethacrynic acid and exposed to 200 μm SIN-1 in the presence of 500 U/ml superoxide dismutase (D). Samples were analyzed 24 hr after the onset of the treatments. Values are shown as mean + SD (n = 5). Activities in untreated control cultures (100% values) were 53 ± 8 nmol · min-1 · mg of protein -1 for complex I, 13.2 ± 1.3 nmol · min-1 · mg of protein -1 for complexes II-III, and 1.14 ± 0.14 k·mg·protein-1 for complex IV. Statistically significant differences in the activities of each of the complexes was produced by depletion of mitochondrial glutathione as a result of treatment with ethacrynic acid or by SIN-1 alone (p < 0.001; one-way ANOVA with Student-Newman-Keuls test). SIN-1 treatment of cells with depleted mitochondrial glutathione significantly exacerbated the effects of glutathione depletion alone (p < 0.001). Superoxide dismutase significantly decreased these effects on the activity of complex I (p < 0.001), complexes II-III (p < 0.05), and complex IV (p < 0.001).
Table 2.
Respiratory properties of mitochondrial fractions prepared from astrocytes after mitochondrial glutathione depletion and exposure to SIN-1
|
|
Oxygen uptake rate (ng-atom 0·min−1·mg of protein−1) |
|
||||
|---|---|---|---|---|---|---|
| Conditions |
State 3 respiration |
State 4 respiration |
Uncoupled respiration |
ADP/0 |
||
| Respiratory substrate: pyruvate plus malate | ||||||
| Untreated cells | 25.1 ± 4.1 | 8.9 ± 3.0 | 25.8 ± 2.0 | 2.4 ± 0.6 | ||
| Ethacrynic acid | 54.0 ± 5.9** | 19.0 ± 7.3* | 59.1 ± 6.0** | 2.0 ± 0.5 | ||
| Ethacrynic acid plus SIN-1 | 15.8 ± 0.5* | 17.9 ± 6.4 | ||||
| Respiratory substrate: succinate | ||||||
| Untreated cells | 43.6 ± 8.9 | 14.2 ± 3.7 | 48.5 ± 9.1 | 1.3 ± 0.3 | ||
| Ethacrynic acid | 42.1 ± 3.3 | 20.5 ± 6.7 | 47.3 ± 3.4 | 1.5 ± 0.2 | ||
| Ethacrynic acid plus SIN-1 |
24.2 ± 2.0*
|
|
24.3 ± 5.7**
|
|
||
Mitochondrial fractions were isolated from astrocytes after 24 hrs in HBSS. Treated cells were exposed to ethacrynic acid to deplete mitochondrial glutathione, and some preparations were further treated for 3 hr with 200 μm SIN-1. Respiratory properties were measured using an oxygen-sensitive electrode. State 3 respiration rate was determined in the presence of ADP plus respiratory substrates. State 4 respiration was measured following depletion of the ADP, and the uncoupled rate was determined after the addition of CCCP. Samples from cells treated with ethacrynic acid plus SIN-1 were poorly coupled, and state 4 values could not be reliably determined. Values are shown as mean ± SD (n = 3-5). Values significantly different from those for mitochondrial fractions from the untreated cells are indicated as follows: *p < 0.05, **p < 0.01 (one-way ANOVA with Student-Newman-Keuls test). All of the values for mitochondria from cells treated with ethacrynic were also significantly different from those for cells exposed to ethacrynic acid plus SIN-1 (p < 0.01).
Treatment of normal cells with 200 μm SIN-1 for 3 hr also resulted in losses of activity of complexes II-III and complex IV at 24 hr and an increase in complex I activity (Fig. 9). However, complex I activity was increased to only a minor extent (Fig. 9), which was substantially less than the response to mitochondrial glutathione depletion. The effects of SIN-1 were again greatly exacerbated in glutathione-depleted astrocytes. At 24 hr after initiation of 3 hr SIN-1 treatment, complex I activity was increased to about ninefold that of normal untreated cells, whereas the activities of complexes II-III and complex IV were decreased to <15% of untreated values (Fig. 9). Mitochondrial fractions prepared from these cells showed markedly decreased respiration with little or no evidence of coupling (Table 2), suggesting that possible effects from the large increase in complex I activity were overridden by the marked decreases in activity of the other respiratory chain complexes. These results are again consistent with the measurements of mitochondrial membrane potential obtained using JC-1 in intact cells. Changes in activity of the electron transport chain complexes produced by glutathione depletion alone were not blocked by superoxide dismutase (data not shown). However, a concentration of 500 U/ml of this enzyme did block much of the exacerbation of these effects by exposure to SIN-1 (Fig. 9).
Discussion
The present study has identified conditions in which glutathione in the mitochondria of astrocytes was essentially completely depleted, whereas the cytoplasmic pool of this metabolite remained intact. There have been previous reports of selective partial decreases in the mitochondrial pool in response to experimental manipulations in vitro (Shan et al., 1993; Raza et al., 2002) or in animal models of disease (Fernandez-Checa et al., 1991; Garcia-Ruiz et al., 1994; Colell et al., 1998; Anderson and Sims, 2002). However, this is the first time for any cell type that such highly selective depletion of mitochondrial glutathione has been achieved. Thus, the astrocytes treated with ethacrynic acid provide a unique opportunity to directly investigate the role of the smaller glutathione pool in the function and survival of a major brain cell population.
The characterization of these glutathione-depleted cells in the present study has generated several interesting new insights into the properties of this key mitochondrial antioxidant. Although astrocytes in culture were able to tolerate the loss of mitochondrial glutathione, at least for 24 hr, they were much more susceptible to stress as a result of exposure to peroxynitrite. Thus, despite glutathione in the mitochondria being only a very small part of the total in these astrocytes in culture, this pool seems to be critically important in the overall defenses against at least some forms of oxidative damage. The findings also provide evidence for a strong influence of the glutathione pool in the mitochondria on the activity of the respiratory chain complexes, particularly complex I. Finally, they demonstrate very slow recovery of the depleted mitochondrial glutathione despite the preservation of the cytoplasmic pool. This suggests that there may be limited net uptake of glutathione into mitochondria in these intact astrocytes either because of an intrinsic property of these cells or as a response to the conditions used to produce the selective glutathione depletion.
Cyclosporin A substantially ameliorated the mitochondrial changes and cell death produced by SIN-1 in astrocytes with depleted mitochondrial glutathione but not the milder responses to SIN-1 in cells with intact glutathione. These findings suggest that different mechanisms contributed to the response to SIN-1 in the presence and absence of mitochondrial glutathione and that the mitochondrial permeability transition was an important contributor to the outcome when glutathione was depleted. Key sulfhydryl groups on the adenine nucleotide translocator are known to be readily oxidized by peroxynitrite, and this modification promotes formation and opening of the permeability transition pore (Vasquez et al., 2001; Radi et al., 2002). Thus, the loss of protection for these protein sulfhydryls probably contributed to increased vulnerability of the cells to peroxynitrite after the depletion of mitochondrial glutathione. The development of the permeability transition could potentially lead to apoptosis because of the release of cytochrome c and other mitochondrial inducers of this process. However, the morphological findings suggest a necrotic form of cell death. The increase in release of lactate dehydrogenase was associated with a loss of ATP, but whether this metabolite change was a contributor to cellular degeneration or a consequence of this process cannot be resolved from the present study. Cell death arising solely because of disrupted mitochondrial ATP generation seems unlikely in these cells in culture that are normally heavily dependent on glycolysis. However, other cellular changes including an increased generation of reactive oxygen species by the damaged mitochondria could also increase susceptibility.
The major alteration detected after mitochondrial glutathione depletion alone was a substantial (fourfold) increase in the activity of complex I of the electron transport chain with small but significant decreases in the activities of complexes II-III and IV when assessed at 24 hr. These mitochondria also showed an increase in NAD-linked respiratory capacity, consistent with previous findings indicating that complex I is a major determinant of respiratory rate in intact mitochondria (Davey et al., 1998; Bianchi et al., 2004).
A similar but smaller increase in complex I activity has recently been reported in astrocytes in culture after a partial depletion of glutathione as a result of exposure to buthionine sulfoxamine (Vasquez et al., 2001), a treatment that primarily alters the cytoplasmic pool of glutathione. The complex I change in this previous study was associated with increased expression of the mitochondrially encoded ND6 subunit, which is involved in the assembly and respiratory activity of complex I (Vasquez et al., 2001). Additional stimulation of complex I subunit synthesis could underlie the changes observed in our study and also the even larger increases resulting from SIN-1 treatment of the astrocytes with depleted mitochondrial glutathione.
Studies in mitochondria and cells from non-neural tissues have indicated that complex I can be a target of inactivation by peroxynitrite (Brown and Borutaite, 2002; Radi et al., 2002). However, such damage was clearly not a major consequence of peroxynitrite exposure in the astrocytes with either intact or depleted mitochondrial glutathione. This resistance of complex I to peroxynitrite-induced inactivation is consistent with previous findings of preserved complex I activity in normal neurons and astrocytes exposed to a single burst of peroxynitrite, although this treatment did lead to loss of activity of other respiratory complexes in the neurons (Bolanos et al., 1995). Complex II can also be directly modified and inactivated by peroxynitrite (Radi et al., 2002). Thus, the large decreases in activity of complexes II-III after SIN-1 treatment of the astrocytes with depleted glutathione could be a primary effect of the peroxynitrite. However, similar changes were also seen in complex IV activity, which is usually less susceptible to direct modifications by this substance (Bolanos et al., 1995; Radi et al., 2002). Therefore, losses of activity in these complexes may result from other changes that were associated with the advanced deterioration of cellular structure at 24 hr after the exposure of glutathione-depleted astrocytes to peroxynitrite.
The selective depletion of glutathione in the present study was achieved using ethacrynic acid. This compound acts to deplete glutathione by glutathione S-transferase conjugation and has been widely used to produce loss of glutathione from both the cytoplasm and mitochondria in various cell types (Meredith and Reed, 1982, 1983; Huang and Philbert, 1995, 1996; Wullner et al., 1999). An initially greater effect of ethacrynic acid on the mitochondrial pool in cultured astrocytes is consistent with previous reports (Huang and Philbert, 1995, 1996). However, the present investigation showed a much more pronounced selectivity in the response of the two glutathione pools during the initial phase of treatment. This could reflect differences in the incubation medium during exposure to ethacrynic acid as seen in our initial comparisons of treatments in HBSS or in serum-containing MEM. Differences in the origin of the astrocytes in these studies could also contribute. Cerebellum was used in the studies of Huang and Philbert (1995, 1996), whereas cerebral cortex was the source of cells for our investigations. Other studies indicate that the response to ethacrynic acid is cell type specific, probably reflecting differences in the activity, subcellular distribution, and substrate specificity of isoforms of the glutathione S-transferases. For example, the mitochondrial pool in cerebellar granule cells has been reported to also show a greater susceptibility to ethacrynic acid (Wullner et al., 1999), whereas liver cells show more pronounced initial changes in the cytoplasmic pool (Meredith and Reed, 1982).
The small size of the glutathione pool detected in the mitochondrial fraction prepared from the cultured astrocytes (<2% of total glutathione) is consistent with previous reports for these cells (Huang and Philbert, 1995). Mitochondrial glutathione has similarly been reported to be low in cultured neurons, accounting for only ∼4% of the total (Wullner et al., 1999). These results contrast with findings in various tissues in vivo, in which ∼15% of the glutathione is found in the mitochondria (Meredith and Reed, 1982; Meister, 1995). The much lower proportion of mitochondrial glutathione in the cultured cells perhaps relates in part to their high dependence on glycolysis for ATP generation and therefore the presence of fewer mitochondria. However, the glutathione content of the mitochondrial fraction (1.1 nmol/mg protein) prepared from the cultures was severalfold lower than values reported for mitochondria isolated from adult brain (Anderson and Sims, 2002). Although this difference is almost certainly exaggerated by contaminants in the crude mitochondrial fraction prepared in the present study, the results do suggest real differences between the content of glutathione in the organelles from the astrocytes in culture and those from adult brain. Nonetheless, modulation of this small proportion of total cellular glutathione had major effects on the cellular defense against peroxynitrite, indicating a pronounced protective function for the mitochondrial glutathione pool.
Restoration of the mitochondrial pool after depletion occurred only slowly, suggesting a very limited capacity for net glutathione uptake under these conditions despite the ongoing availability of glutathione in the cytoplasm. This could possibly be attributable to an intrinsic lack of rapid transport into mitochondria in these cells or to modulation of the transport process by the products of the interaction of glutathione with ethacrynic acid. To our knowledge, there are no studies of the properties of glutathione uptake into mitochondria from any brain cell populations. Investigations using mitochondria isolated from liver and other tissues have provided evidence for several different mechanisms of glutathione uptake that would be expected to rapidly replenish the mitochondria from an intact cytoplasmic pool (Chen and Lash, 1998; Coll et al., 2003). However, slow recovery of mitochondrial glutathione has also recently been reported for COS cells in which this pool was partially depleted after exposure to 4-hydroxynonenal (Raza et al., 2002). This compound, like ethacrynic acid, is conjugated by glutathione S-transferases. Together, these results suggest that there is a limited capacity to restore mitochondrial glutathione under some conditions, which could contribute to long-term compromise of this antioxidant defense.
Additional studies are needed to determine whether the roles demonstrated for mitochondrial glutathione in astrocytes are also a feature in neurons and other brain cell populations and whether alterations in this antioxidant pool could contribute to cell loss in degenerative neurological diseases. Our finding of partial losses of mitochondrial glutathione during focal cerebral ischemia (Anderson and Sims, 2002) suggests that this change could be an important factor in the development of tissue infarction in stroke. The critical role of glutathione in the cellular defense against peroxynitrite, as demonstrated in the present study, and recent evidence that infarct volume can be reduced by treatment with glutathione monoethylester (Anderson et al., 2004) provides additional support for this possibility. Thus, additional understanding of the factors influencing mitochondrial glutathione content in brain cells could contribute to the development of new therapeutic interventions for stroke and perhaps other neurological diseases.
Footnotes
This work was supported by the National Health and Medical Research Council (Australia), The Royal Swedish Academy of Sciences, The Swedish Research Council, The Swedish Foundation for International Cooperation in Research and Higher Education, Swedish Society for Medical Research, The Helmuth Hertz Foundation, Edit Jacobssons Stiftelse, Hjalmar Svenssons Stiftelse, and Ulla och Rune Amlövs Stiftelse. The protocol for the measurements of rhodamine-labeled mitochondria and advice on these measurements were kindly provided by Dr. Diane Lee. We also thank Sheila Engdahl for general technical assistance.
Correspondence should be addressed to Dr. Håkan Muyderman, Department of Medical Biochemistry, School of Medicine, Flinders University, G.P.O. Box 2100, 5001 Adelaide, SA, Australia. E-mail: hakan.muyderman@flinders.edu.au.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248019-10$15.00/0
References
- Akerboom TP, Sies H (1981) Assay of glutathione, glutathione disulfide, and glutathione mixed disulfides in biological samples. Methods Enzymol 77: 373-382. [DOI] [PubMed] [Google Scholar]
- Anderson MF, Sims NR (2002) The effects of focal ischemia and reperfusion on the glutathione content of mitochondria from rat brain subregions. J Neurochem 81: 541-549. [DOI] [PubMed] [Google Scholar]
- Anderson MF, Nilsson M, Eriksson PS, Sims NR (2004) Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci Lett 354: 163-165. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 87: 1620-1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi C, Genova ML, Castelli GP, Lenaz G (2004) The mitochondrial respiratory chain is partially organized in a supercomplex assembly: kinetic evidence using flux control analysis. J Biol Chem 279: 36562-36569. [DOI] [PubMed] [Google Scholar]
- Bolanos JP, Heales SJ, Land JM, Clark JB (1995) Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem 64: 1965-1972. [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V (2001) Nitric oxide, mitochondria, and cell death. IUBMB Life 52: 189-195. [DOI] [PubMed] [Google Scholar]
- Brown GC, Borutaite V (2002) Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic Biol Med 33: 1440-1450. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR (1956) The respiratory chain and oxidative phosphorylation. Adv Enzymol 17: 65-137. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lash LH (1998) Evidence for mitochondrial uptake of glutathione by dicarboxylate and 2-oxoglutarate carriers. J Pharmacol Exp Ther 285: 608-618. [PubMed] [Google Scholar]
- Colell A, Garcia-Ruiz C, Miranda M, Ardite E, Mari M, Morales A, Corrales F, Kaplowitz N, Fernandez-Checa JC (1998) Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 115: 1541-1551. [DOI] [PubMed] [Google Scholar]
- Coll O, Colell A, Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC (2003) Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 38: 692-702. [DOI] [PubMed] [Google Scholar]
- Davey GP, Peuchen S, Clark JB (1998) Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem 273: 12753-12757. [DOI] [PubMed] [Google Scholar]
- Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62: 649-671. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hamprecht B (1997) Involvement of glutathione peroxidase and catalase in the disposal of exogenous hydrogen peroxide by cultured astroglial cells. Brain Res 759: 67-75. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Huang Z, Ferrante RJ, Sasamata M, Molliver ME, Snyder SH, Moskowitz MA (1999) Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci 19: 5910-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Garcia-Ruiz C, Ookhtens M, Kaplowitz N (1991) Impaired uptake of glutathione by hepatic mitochondria from chronic ethanol-fed rats. Tracer kinetic studies in vitro and in vivo and susceptibility to oxidant stress. J Clin Invest 87: 397-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A (1998) Mitochondrial glutathione: importance and transport. Semin Liver Dis 18: 389-401. [DOI] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Morales A, Ballesta A, Rodes J, Kaplowitz N, Fernandez-Checa JC (1994) Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest 94: 193-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S, Squadrito GL, Pryor WA, Czapski G (1996) Direct and indirect oxidations by peroxynitrite, neither involving the hydroxyl radical. Free Radic Biol Med 21: 965-974. [DOI] [PubMed] [Google Scholar]
- Griffiths EJ, Halestrap AP (1991) Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Biochem J 274: 611-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gursoy-Ozdemir Y, Bolay H, Saribas O, Dalkara T (2000) Role of endothelial nitric oxide generation and peroxynitrite formation in reperfusion injury after focal cerebral ischemia. Stroke 31: 1974-1980. [DOI] [PubMed] [Google Scholar]
- Huang J, Philbert MA (1995) Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res 680: 16-22. [DOI] [PubMed] [Google Scholar]
- Huang J, Philbert MA (1996) Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res 711: 184-192. [DOI] [PubMed] [Google Scholar]
- King TE (1967) Preparation of succinate cytochrome c reductase and the cytochrome b-c1 particle, and reconstitution of succinate cytochrome c reductase. Methods Enzymol 10: 216-225. [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275. [PubMed] [Google Scholar]
- Meister A (1995) Mitochondrial changes associated with glutathione deficiency. Biochim Biophys Acta 1271: 35-42. [DOI] [PubMed] [Google Scholar]
- Meredith MJ, Reed DJ (1982) Status of the mitochondrial pool of glutathione in the isolated hepatocyte. J Biol Chem 257: 3747-3753. [PubMed] [Google Scholar]
- Meredith MJ, Reed DJ (1983) Depletion in vitro of mitochondrial glutathione in rat hepatocytes and enhancement of lipid peroxidation by adriamycin and 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU). Biochem Pharmacol 32: 1383-1388. [DOI] [PubMed] [Google Scholar]
- Muyderman H, Angehagen M, Sandberg M, Bjorklund U, Olsson T, Hansson E, Nilsson M (2001) Alpha 1-adrenergic modulation of metabotropic glutamate receptor-induced calcium oscillations and glutamate release in astrocytes. J Biol Chem 276: 46504-46514. [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L (2002) Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33: 1451-1464. [DOI] [PubMed] [Google Scholar]
- Ragan CI, Wilson MT, Darley-Usmar VM, Lowe PN (1987) Subfraction of mitochondria and isolation of the proteins of oxidative phosphorylation. In: Mitochondria: a practical approach (Darley-Usmar VM, Rickwood D, Wilson MT, eds), pp 79-112. London: IRL.
- Raza H, Robin MA, Fang JK, Avadhani NG (2002) Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. Biochem J 366: 45-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reers M, Smith TW, Chen LB (1991) J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30: 4480-4486. [DOI] [PubMed] [Google Scholar]
- Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB (1995) Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol 260: 406-417. [DOI] [PubMed] [Google Scholar]
- Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A (1997) JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess Δψ changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 411: 77-82. [DOI] [PubMed] [Google Scholar]
- Shan X, Jones DP, Hashmi M, Anders MW (1993) Selective depletion of mitochondrial glutathione concentrations by (R, S)-3-hydroxy-4-pentenoate potentiates oxidative cell death. Chem Res Toxicol 6: 75-81. [DOI] [PubMed] [Google Scholar]
- Shepherd D, Garland PB (1969) The kinetic properties of citrate synthase from rat liver mitochondria. Biochem J 114: 597-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR, Anderson MF (2002) Mitochondrial contributions to tissue damage in stroke. Neurochem Int 40: 511-526. [DOI] [PubMed] [Google Scholar]
- Szabo C (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140-141: 105-112. [DOI] [PubMed] [Google Scholar]
- Vasquez OL, Almeida A, Bolanos JP (2001) Depletion of glutathione up-regulates mitochondrial complex I expression in glial cells. J Neurochem 76: 1593-1596. [DOI] [PubMed] [Google Scholar]
- Vassault A (1983) l-Lactate dehydrogenase. UV method with pyruvate and NADH. In: Methods of enzymatic analysis (Bergmeyer J, Grassl M, eds), pp 118-126. Weinheim, Germany: Verlag Chemie.
- Wharton A, Tzagoloff A (1967) Cytochrome oxidase from beef heart mitochondria. Methods Enzymol 10: 245-251. [Google Scholar]
- Wullner U, Seyfried J, Groscurth P, Beinroth S, Winter S, Gleichmann M, Heneka M, Loschmann P, Schulz JB, Weller M, Klockgether T (1999) Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Res 826: 53-62. [DOI] [PubMed] [Google Scholar]
- Zaidan E, Sims NR (1997) Reduced activity of the pyruvate dehydrogenase complex but not cytochrome c oxidase is associated with neuronal loss in the striatum following short-term forebrain ischemia. Brain Res 772: 23-28. [DOI] [PubMed] [Google Scholar]