

Abstract
In hippocampal slice models of epilepsy, two behaviors are seen: short bursts of electrical activity lasting 100 msec and seizure-like electrical activity lasting seconds. The bursts originate from the CA3 region, where there is a high degree of recurrent excitatory connections. Seizures originate from the CA1, where there are fewer recurrent connections. In attempting to explain this behavior, we simulated model networks of excitatory neurons using several types of model neurons. The model neurons were connected in a ring containing predominantly local connections and some long-distance random connections, resulting in a small-world network connectivity pattern. By changing parameters such as the synaptic strengths, number of synapses per neuron, proportion of local versus long-distance connections, we induced “normal,” “seizing,” and “bursting” behaviors. Based on these simulations, we made a simple mathematical description of these networks under well-defined assumptions. This mathematical description explains how specific changes in the topology or synaptic strength in the model cause transitions from normal to seizing and then to bursting. These behaviors appear to be general properties of excitatory networks.
Keywords: epilepsy, networks, small-world networks, seizures, computational modeling, interictal burst
Introduction
Epilepsy is characterized by two electrographic behaviors: interictal bursts of activity that last ∼100 msec and “seizures” that last from seconds to minutes (Steriade, 2003). In slice models of epilepsy, bursts and seizures can be elicited in different regions of the hippocampus bathed in 4-aminopyridine (4-AP). Bursts originate in region CA3 of hippocampus (Chesnut and Swann, 1988), whereas seizures originate in region CA1 (Netoff and Schiff, 2002). 4-AP increases excitability and effective synaptic strength (Perreault and Avoli, 1989); however, epileptiform behavior can be induced in slices through a variety of methods (Traub and Miles, 1991), suggesting that the cause of these behaviors is a general property of the network.
Traditionally, epilepsy is viewed as a disease of “hypersynchronous” neuronal activity (Penfield and Jasper, 1954; Steriade, 2003). Evidence from hippocampal slices shows that bursts in CA3 are caused by neuronal activity that is synchronous on a fine time scale (<10 msec); however, neuronal activity during slice seizures in CA1 is not synchronous (Netoff and Schiff, 2002; Van Drongelen et al., 2003). The most notable difference between the hippocampal regions is that CA3 has more recurrent synaptic connections than CA1. Staley et al. (1998) hypothesized that bursts originate in region CA3 because the network activates quickly, via recurrent excitation, depleting the primary glutamate stores of the neurons and thus shutting down the network.
Our goal in this study was to use computational models to explore how epileptiform behaviors relate to the connectivity of the underlying networks. Our operating hypothesis for CA3 bursts is similar to that of Staley et al. (1998), except that our models rely on generally defined neuronal “refractoriness” to terminate burst activity. Refractoriness may arise via a number of mechanisms, including synaptic depletion, inhibition, or voltage-dependent properties in postsynaptic cells. In the CA1, which has less recurrent excitation, the activity spreads slower. Thus, an excitable pool of CA1 neurons is always available, leading to sustainable seizure-like activity. To test these hypotheses, we simulated networks intended to mimic regions CA3 and CA1. We used “small-world” network topologies, in which the majority of connections between cells are “local,” but a few cells have “long-distance” connections (Watts and Strogatz, 1998; Watts, 1999). Small-world networks were used because they are simple, flexible, and reminiscent of the connectivity patterns of networks in the brain. As connectivity in the networks was changed, we observed activity resembling epileptiform behaviors seen in slice models. Networks with large numbers of long-distance connections were more prone to generating self-terminating bursts. Additionally, our results suggest that the greater level of interconnectivity in CA3 may be responsible for its tendency to burst rather than seize. Results were independent of the specific neuronal model (Poisson, integrate-and-fire, Hodgkin-Huxley) used in the simulations. We derived a reduced mathematical description of the networks that helped us to describe the conditions under which networks transition from “normal” to “seizing” to “bursting.” Although this model provides only a heuristic description of the slice behavior, it demonstrates how epileptiform behaviors may depend on specific physical parameters.
Materials and Methods
Structure of the network and connectivity. We generated simple network models of excitatory neurons in hippocampus. To keep the number of free parameters manageable, to more easily constrain activity to spread in a controlled manner, and to eliminate the effects of boundary conditions, we restricted our analyses to one-dimensional “rings” of neurons. Because organization of the synaptic connections within cortical regions is neither a lattice of nearest-neighbor connections nor completely randomly connected (Mountcastle, 1997; Gonzalez-Burgos et al., 2000; McCormick and Contreras, 2001), we used a simple method to construct networks that lie between these two extremes. We began with a model in which each neuron is connected to a specific number k of its nearest neighbors and then randomly disconnected a proportion ρ of the synaptic connections and reconnected these synapses to a randomly chosen postsynaptic cell. This method of network construction leads to small-world networks (Watts and Strogatz, 1998; Watts, 1999), in which most connections made by a given presynaptic neuron are local, but an important few can spread activity over long distances. An illustration of networks with varying amount of long-distance connections is given in Figure 1. In the network in Figure 1a, each “node” represents a neuron. The ring of neurons is connected in a perfect “lattice” (ρ = 0), with each cell connected to its four nearest neighbors. Figure 1, b and c, represents networks increasing values of ρ. These are small-world networks because they include a preponderance of local, regular connections but a small number of long-distance connections, which greatly reduce the number of synaptic steps required to connect any pair of neurons in the network.
Figure 1.

Small-world network. a, Networks of neurons are generated in which all cells are only coupled to their nearest neighbors (4 in this case). b, To generate small-world networks, small numbers of connections are broken and rewired to make long-distance connections at random locations. Long-distance connections reduce the number of synapses between any pair of neurons in the network. c, As more long-distance connections are added, the network loses the property that most connections are local, and the network looks much more random. We find a range of normal and epileptiform behaviors in the small-world network regimen, where few connections are necessary to connect any pair of the neurons, but local connections still predominate.
The anatomy of our small-world networks is characterized by three free parameters: network size (N), the number of synaptic connections per neuron (k), and the proportion of randomly made long-distance connections (ρ). For most simulations, we used N = 3000, approximately corresponding to the smallest population size within which epileptiform activity is seen in the hippocampus (Fox et al., 2001). In some simulations, we used N = 24,000 to examine the generality of our results. We examined many values of k but focused our attention on networks with 1% total connectivity (k = 30 for N = 3000) to represent region CA1 and networks with 3% total connectivity (k = 90 for N = 3000) to represent region CA3. Both k values are overestimated as follows: excitatory to excitatory coupling in region CA3 is closer to 2% (MacVicar and Dudek, 1980) and for region CA1, it is <1%. Because the numbers of local versus long-distance connections are unknown in hippocampus, we treated ρ (the proportion of random connections in the network) as an explicit free parameter that we varied from 0 to 1. ρ is a particularly important parameter for these models, because it controls the rate at which local “waves” of activity give rise to new waves at distant locations in the network.
Model neurons and synapses. We ran simulations using three different types of model neurons: noisy and leaky integrate-and-fire neurons, stochastic Hodgkin-Huxley cells (Chow and White, 1996), and a Poisson spike-train cell model that is equivalent to the other models in terms of first-order interspike interval statistics. The integrate-and-fire model is a regular leaky integrate-and-fire model with a stochastic component, given by
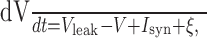 |
where V indicates the membrane potential, Vleak is the resting potential of the neuron, Isyn is the synaptic input (described in a subsequent paragraph), and ξ is the stochastic component (white noise of sufficient variance to generate spontaneous activity at a target rate). The neuron fires when V reaches a threshold, resulting from noise or when a synaptic current is injected into postsynaptic neurons. The neuron is then reset to zero, and all synaptic inputs are blocked for an absolute “refractory” period of time τR (see below for additional information on refractoriness).
The stochastic Hodgkin-Huxley model is a conductance-based model, which is described in detail by Chow and White (1996). Parameters were as in the original 1952 study, except that sodium channels were modeled as discrete, stochastic elements. The Langevin method was used to describe the effects of channel noise (Chow and White, 1996). In this model, membrane noise causes the membrane potential to fluctuate and occasionally causes the neuron to fire spontaneously. The number of sodium channels was “tuned” to 3375 to match a target average spontaneous firing rate (see below).
Synaptic currents for the integrate-and-fire and stochastic Hodgkin-Huxley models were calculated using a double exponential function Isyn = A(e-t/τs - e-t/τf) (Vsyn - V), where A is the synaptic amplitude, t is the time since synaptic input occurred, τs and τf are the slow and fast decay rates, respectively, and Vsyn is the reversal potential of the synapse (Bower and Beeman, 1995).
To construct the Poisson model, we used Matlab (MathWorks, Natick, MA) to select spike times from a Poisson process. Synaptic inputs for this model were simulated in the following way: in response to the arrival of a presynaptic spike, the postsynaptic cell has a probability of immediately firing, where the probability for a single synaptic input was set to 2.5%. We set the probability of firing after two or more simultaneous synaptic inputs to 1. If the cell does not immediately fire in response to an input, then it uses the spike time previously drawn from the Poisson distribution.
All models were adjusted to have an absolute refractory period, τR (after spiking), 10 times longer than the synaptic delay, τd. This allows the active population to travel 10 steps before the neurons recover. For the Poisson model, the synaptic delay was set at 3.7 msec and a refractory time of 36 msec, whereas in the integrate-and-fire model, the synaptic delay was 2.8 msec and the refractory time was 28 msec. The Hodgkin-Huxley model had a synaptic delay dependent on the distance between the neurons with a 1 msec delay between the local cells and up to 5 msec for long-distance connections. An absolute refractory time was 36 msec and was set to behave qualitatively similarly to the other models. The one exception is the network simulation with 24,000 neurons where the refractory time was 28 msec and the synaptic delay was 2 msec, which was necessary to allow enough time for the entire network to be activated before the first cells recovered. The refractory period can be generated by many mechanisms, either presynaptic or postsynaptic, and we leave this undetermined. The important factor is that the refractory time is on the order of the time it takes activity to spread throughout the network in the bursting regimen; otherwise, the activity will re-enter and a clean transition to bursting will not occur. We also matched all models so that they had an approximately exponential interspike interval distribution and firing probability of 0.0315 spikes/sec. In most simulations, synaptic efficacies were set in all models such that single inputs caused postsynaptic action potentials 2.5% of the time, and two simultaneous inputs led to postsynaptic firing with probability of firing approximately equal to one (assumed to be exactly one in our later theoretical analysis). For the 24,000 cell simulations (see Fig. 3), we compensated for the eightfold increase in network size with a 20-fold decrease in synaptic weight. As successfully predicted by our reduced theoretical model, this change kept the same overall rate of wave generation, leading to results (see Fig. 3).
Figure 3.
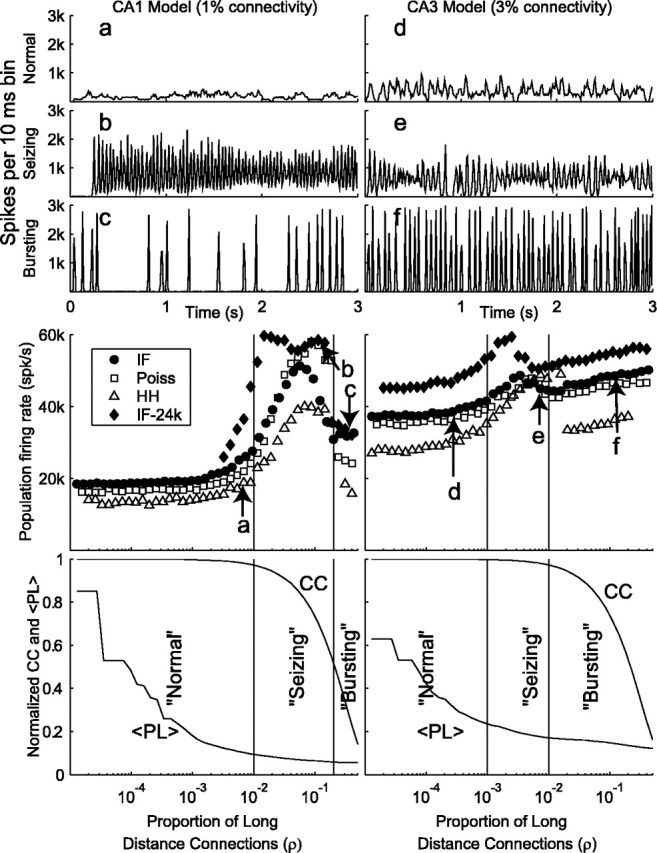
Transition from normal → seizing → bursting behavior as a function of the number of long-distance connections (ρ). The left column shows the results from the CA1 model with N = 3000 and k = 30, whereas the right column shows the results from a CA3 model with N = 3000 and k = 90. At the top are three examples of data (taken from a Poisson-simulated network) for normal, seizing, and bursting, showing the count of neurons that fired in a 10 msec time bin. Middle panels illustrate the total population activity for Poisson (Poiss), noisy leaky integrate-and-fire (IF), and stochastic Hodgkin-Huxley (HH) simulations with the examples from above indicated by a-f. Vertical bars indicate boundaries between normal, seizing, and bursting as identified by eye from the time traces of population activity from the Poisson model. Simulations of IF network with 24,000 neurons and reduced synaptic strength are displayed as well (IF-24k). These networks show qualitative behaviors similar to the 3000 neurons. The bottom panels illustrate the normalized clustering coefficients and mean path length between neurons in the network as the proportion of long-distance connections in the network (ρ) is increased. The left side of these graphs indicates a network topology in which the ring of neurons has only local connections; the right side indicates a nearly randomly connected network.
Results
Basic properties of propagating activity
In all three types of networks (Poisson, integrate-and-fire, and stochastic Hodgkin-Huxley), we observed qualitatively similar behaviors. Networks are quiescent at first, but eventually a spontaneous action potential in one neuron initiates activity in two neurons with common local postsynaptic targets. Because convergence of two simultaneous inputs fires postsynaptic cells in these networks with probability near one, this event generates two waves that travel in opposite directions around the ring. With few long-distance connections, a small number of waves sweeps across the network. This results in a small, stable amount of activity. With more long-distance connections, existing waves frequently give rise to new waves in distant locations, and network activity transitions to sustained high activity, which we liken to seizures. Figure 2c illustrates still frames from a movie of an ongoing seizure (see Fig. 2, a and b, for an explanation of the display method). With a large enough number of long-distance connections (Fig. 2d), we observed bursts in which the majority of the network fired synchronously and then became refractory. Movies of seizures and bursts can be seen at http://www.bu.edu/ndl/people/netoff/SWN/JNeurosciSupplement.html. As we show, transition points between normal, bursting, and seizing vary according to the number of long-distance connections, network size, synaptic strength, and number of synapses per neuron.
Figure 2.
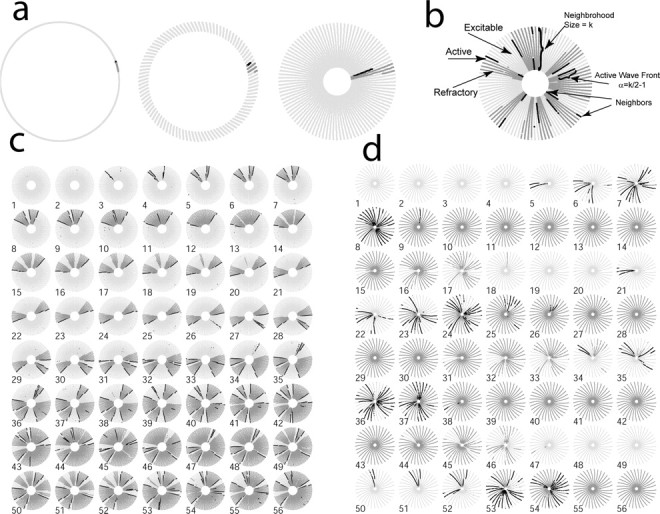
Bursting and seizing behaviors as the number of long-distance connections are changed. a, The ring contains N neurons, each of which are connected to k, mostly local neighbors (left). To visualize the activity of this large network, we color coded each point according to the state of the neuron and pulled every kth point in the ring toward the center to make a spoke. Therefore, a neuron in the center of a spoke is connected to all the neurons in the spoke, assuming that all synaptic connections are local. A neuron at the end of the spoke is connected to half of the neurons on the spoke and half of the neurons on the opposite end of the next spoke. This results in a plot of the ring that resembles a slinky. b, An illustrative temporal snapshot of network activity, with N = 3000, k = 30 synapses per neuron (i.e., 1% network connectivity), and ρ = 0.1. Light gray dots represent excitable neurons, black dots are firing neurons, and dark gray dots are refractory neurons. The wave front size stabilizes to approximately half the size of the local neighborhood k and is followed by a refractory tail. This tail is determined by how many steps the wave front can travel before the neurons begin to recover. c, Successive frames from a movie of seizing activity, with N = 3000, k = 30, and ρ = 0.1 (i.e., that 10% of synapses have been rewired). The frame rate is 250 Hz, corresponding to approximately two synaptic time delays; therefore, the active waves appear twice as large (in space) as their actual size. Spontaneous background activity generates a cascade of activity, which stabilizes into two traveling waves (frames 5-25). These traveling waves generate other waves in the network through the long-distance connections (e.g., frames 26, 31, 34). Eventually, waves start to meet and annihilate each other (e.g., frames 4, 33, 43). This network attains equilibrium when the new waves are generated at the same rate that the waves annihilate each other. d, Still frames from a movie of bursting activity (N = 3000; k = 90; ρ = 0.1). In this network, the number of long-distance connections causes waves to generate new waves faster than the waves annihilate each other. This results in all of the neurons firing in the network, all of the neurons becoming refractory, and the activity in the network shutting off. Movies of network activity can be seen at: http://www.bu.edu/ndl/people/netoff/SWN/JNeurosciSupplement.html.
Dependence on model parameters
Figure 3 illustrates how network activity depends on the proportion of long-distance connections (ρ), which controls the spread from local activity to distant portions of the network. Each neuron is coupled, predominantly locally, to 1% (Fig. 3, left panels) (approximately corresponding to connectivity in region CA1) or 3% (right panels) (approximately corresponding to region CA3) of the rest of the network. The top six panels (Fig. 3a-f) show traces of population activity versus time for specific cases. Normal activity is characterized by a low sustained population firing rate. Seizing activity is characterized by significantly higher, sustained firing rates with some evidence of coherence. Bursting activity is characterized by network activity that rises and falls rapidly and coherently.
The middle set of panels in Figure 3 show time-averaged population firing rates as a function of ρ, the proportion of long-distance connections. a-f in Figure 3 correspond to the examples from the top six traces. For the CA1 model, the slope of the population firing rate versus ρ becomes steep at approximately ρ = 0.01. We define this point as the transition from normal firing to seizing in the network. Population oscillations, with a period close to the neuronal refractory period, are seen in parts of the seizing region (Fig. 2b,e). For the CA3 model, the transition to seizing occurs atρ = 4 × 10-4. As more long-distance connections are added to the network, coherent bursting begins. Interestingly, the onset of bursting, which occurs at ρ = 0.01 for the CA3 model and ρ = 0.2 for the CA1 model, leads to a marked decrease in population activity. The period of the bursting is erratic, because after a burst, there is no residual activity in the network, implying that the next burst is triggered only at the rate of the random spontaneous background activity. These results suggest that the observed phenomena are independent of the individual neuron models used in the network (Poisson, integrate-and-fire, stochastic Hodgkin-Huxley).
The integrate-and-fire network was also scaled up eightfold, to 24,000 neurons, while decreasing synaptic strength 20-fold to balance the excitation in the network. (The formula for determining this decrease in synaptic strength came from our reduced model, which is discussed below.) This manipulation results in very similar transitions from normal to seizing to bursting behavior (Fig. 3, filled diamonds), indicating that these results do not depend on network size. Very similar results (data not shown) were also seen in 24,000 member Poisson-process networks with downsized synapses, further indicating that these results depend critically on connectivity but not on other details.
The bottom two panels in Figure 3 show the normalized clustering coefficient (CC) and mean path length (<PL>) plotted versus ρ. The clustering coefficient is a measure of how likely it is that two interconnected neurons both make connections to the same neighbor. In particular, it is the average probability that the number of observed overlap in neighbors would occur by chance if the network had been connected randomly. Mean path length is the average number of “degrees of separation” between two randomly chosen neurons. It is calculated by averaging the measured distance between all pairs in the network. Both measures decrease with increasing proportion of long-distance connections ρ, but an intermediate value of ρ corresponds to the so-called small-world regimen where the clustering coefficient is high and the mean path length is low. The regimen of seizing activity in the CA1 model corresponds with this small-world regimen. The beginning of the bursting regimen corresponds with the drop in clustering coefficient, which signals the transition of the network from a small world to a random graph. As indicated by comparing the right and left columns in Figure 3, having a larger number of connections leads to bursting at lower values of ρ.
Scaling relationships and the reduced model of propagating activity
Propagating activity in simulated networks has a number of stereotypical characteristics that allow us to represent it accurately using a reduced model. First, because the time needed for supra-threshold inputs to activate the model neurons is small compared with the synaptic delay time, the synaptic delay sets the amount of time required for wave fronts to propagate from one group of active neurons to another. Consequently, the network evolves in approximately discrete time steps, corresponding to the synaptic delay. Second, assuming a nearly regular lattice (i.e., that the number of long-distance connections is small relative to N), the wave front size, α, is just less than half the neighborhood size: α = k/2 - 1. The division by 2 comes from the fact that the wave spreads in both directions (unless one set of cells is refractory); subtracting 1 from the total removes the neuron that only receives a single synaptic input from the active neurons. Third, following each wave is a wake of refractory neurons, approximately the size αR = α × τR/τd, where τR denotes the refractory time of each neuron, and τd denotes the synaptic delay. This equation defines the number of time steps that a neuron remains refractory, which we have denoted R. Finally, knowing the number of waves, their size, and their wake size, it is straightforward to calculate the characteristics of propagation and the probability of emergence of a new wave per time step, as well as the probability per unit time that two waves will collide and thus annihilate. Thus, we should be able to construct a discrete time, birth-and-death process describing activity in the ring network.
Let wi denote the number of active waves at time step i. We define the number of new waves that will begin spontaneously in one time step to be Si = seip2. Here, s is the probability that any given neuron will fire in that time, which we set at s = 0.0315 × τd, where 0.0315 is the spontaneous spike rate per second. ei is the number of excitable neurons present in the network at time i. p2 is the probability that two or more neurons fire in response to the firing of the same presynaptic neuron. The dependent variable p2 can be calculated from the binomial theorem using synaptic strength p1 (which we define as the probability that firing of a single presynaptic cell induces an action potential in a given postsynaptic cell) and the number of synapses per neuron k: p2 = 1 - (1 - p1)k - kp1(1 - p1)k-1. In our reduced model, if two such neurons in a neighborhood fire, we will assume that a traveling wave will be initiated with probability of exactly one (the relative frequency of such an event was observed to be ∼1 in the simulations), with wave front size exactly equal to α. This assumption holds true for our 3000 cell simulations but not for the 24,000 cell simulations. Interestingly, behavior of the network is not sensitive to violations of this assumption (Fig. 3).
Once a wave has been initiated, it can start new waves of activity in other regions of the network through long-distance connections. The rate at which new waves are generated depends on the current number of waves, wi, which determines the number of active neurons, αwi. These active neurons have αwikρ long-distance connections on average. The probability that synapses at the end of these connections start a wave at a postsynaptic neuron is p1p2ei/N, where ei is the number of excitable cells (see below). The approximate symmetry in the connectivity of the ring (because ρ is assumed to be much less than 1) means that a new wave will propagate in two directions as two separate wave fronts. This is reflected in the formula for the new wave rate by multiplying by a factor of two. This approximation is valid, provided that the network is far from being saturated with activity (i.e., when the network size N is much larger than the number of active and refractory neurons) and the resulting wave fronts develop into full waves with refractory wakes, an assumption that is true as long as the waves emerge in a region of nonrefractory cells. The number of excitable cells, ei, is the total number of neurons minus the number that are active or refractory:
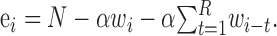 |
The last term in this equation for ei accounts for the recent history of activity, by summing that past activity up to the refractory time. This sum gives the total number of refractory neurons at time i. Initially, we will further simplify this equation by assuming that the number of refractory cells is simply proportional to the number of active waves: ei = N - αwi(1 + R). This approximation is also valid only when the network activity is far from saturation. According to this model, the number of new waves born at time step i is approximated by ni = (2αwikρ) (p1p2ei/N) + Si. This function, a quadratic function of wi, is plotted in the top panels of Figure 4 (dashed lines) for three different values of ρ, the proportion of long-distance connections.
Figure 4.

The birth- death process (1-dimensional map) model of wave generation and annihilation. The top panels show how many new waves are generated (ni; dashed lines) and annihilated (di; solid lines) per time step as functions of the number of currently active waves, with k = 90. The y-intercept of the dashed lines indicates the spontaneous background rate of wave generation. An equilibrium point exists where the new wave rate is equal to the dying wave rate (indicated by the arrows). The bottom panels map the number of waves on one time step to the average number expected on the next time step (solid lines). Equilibria occur when the number of waves on the next time step is equal to that on the current time step [i.e., at the intersection of the solid line of f(wi) and the dotted line of identity (also indicated by arrows)]. Forρ = 0.0001, the equilibrium point at wi ≈ 1.82 is only weakly attracting (the slope of the solid line is approximately + 1), and the number of waves changes only slightly at each time step. Forρ = 0.01, the system has a strongly attracting equilibrium (where the slope of the solid line is approximately zero), corresponding to ongoing seizures characterized by an average of 3.87 existing simultaneous waves. For ρ = 0.05, the equilibrium is unstable, and the dynamics has entered a chaotic regimen. The onset of chaos indicates that the entire network will repeatedly fire brief synchronous bursts.
The average number of wave collisions per time step depends on how many waves are present in the network. The more waves in the network, the more likely collisions will occur. The expected number of dying waves in a time step can be approximated by the time it takes the currently active waves, evenly distributed around the ring, to collide. We estimate the death rate to be di = 2αwi/ei. In this equation, the term 2α reflects the fact that two wave fronts propagate through an excitable region toward each other at a rate of 2α neurons per time step; the term wi reflects the fact that evenly distributed waves grow, on average, closer as their number grows; and the term ei reflects the fact that the average distance between two waves is proportional to the number of remaining excitable cells. As for ei, the approximation in di is most valid when the network activity is far from saturation. The death rate di is plotted (solid lines) in the top panels in Figure 4. Its value increases without bound as the number of active and refractory neurons approaches N, when the denominator approaches zero (at the point where the network reaches saturation). The net growth in the number of waves at time i is ni - di, and thus the wave birth-death process can be written as a one-dimensional map, wi+1 = f(wi) = wi + ni - di, in which we have defined the function f(wi) for notational convenience. This one-dimensional map is only valid under the assumption that the number of refractory cells is directly proportional to the current number of active wave (see above) (Table 1).
Table 1.
Principal definitions, symbols, and default parameter values used in equations
|
Symbol |
Identification |
Definition/values used |
|---|---|---|
| N | Number of neurons in network | 3000 (24,000 in some network simulations) |
| k | Number of synapses per neuron | 30 (for CA1), 90 (for CA3) |
| s | Spontaneous firing rate of a single neuron per time step of size τd | 0.0315 × τd |
| τR | Absolute refractory time of neuron | 28 msec (IF), 36 msec (Poiss, HH) |
| τd | Synaptic time delay | 2.8 msec (IF), 3.7 msec (Poiss), 1-5 msec (HH) |
| ρ | Proportion of long-distance connections generated by breaking a synapse and rewiring it to a randomly chosen postsynaptic cell | Varied from 1.0 × 10−5 to 0.4 |
| p1 | Synaptic strength (i.e., probability that postsynaptic neuron will fire given that a particular presynaptic neuron fired) | 0.025 |
| p2 | Probability that two postsynaptic neurons fire given the presynaptic neuron fired (dependent on k and p1) | p2 = 1 - (1 - p1)k - kp1(1 - p1)k−1 |
| α | Approximate number of neurons in wave front | k/2 - 1 |
| R | Number of time steps that a neuron remains refractory | R = τR/τd ≈ 10 |
| Wi | Number of waves present in network at time i | Wi + 1 = f(Wi) |
| ei | Number of excitable neurons in the network at time i |
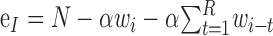 or ei = N − αwi(1+R) (for one-dimensional map) or ei = N − αwi(1+R) (for one-dimensional map) |
| ni | Number of new waves generated at time i resulting from long-distance connections | ni = (2αWi kρ)(p1p2ei/N) + Si |
| di | Number of waves that die in time step i resulting from wave collision | di = 2αWi/ei |
| Si | Spontaneous wave generation resulting from spontaneous cellular activity | Si = seip2 |
| W* | Number of waves in network where new wave rate and dying wave rate are equal (equilibrium point) | f(W*) = W* |
|
f(Wi) |
Function describing number of waves on next time step given number of waves on time step i
|
f(Wi) = Wi + ni − di
|
Activity in the network reaches an equilibrium state when new waves are generated at the same rate that they annihilate each other, i.e., when there is a steady state number of waves w* that solves the equation f(w*) = w*. A stable equilibrium corresponds to the slope of the function f(wi) at the equilibrium between 1 and -1; otherwise, it is unstable and the new wave rate is faster than the dying wave rate. The strength of attraction of this equilibrium affects the spread of the distribution in the number of neurons firing at any given time in a stochastic network.
The equilibrium level of activity and its stability depend on the parameters in the new wave rate ni and dying wave rate di.As we vary parameters such as the proportion of long-distance connections (ρ), we can investigate how changes in the equilibrium cause the network to switch from normal to seizing to bursting. In the bottom panels in Figure 4, we plot the number of waves on the next time step wi+1 versus the number of waves on this time step wi (solid line) as well as the line of identity (y = x; dotted line). In these plots, equilibrium points f(w*) are indicated by intersections of the solid and dotted lines. Stability of equilibrium points is given by the slope of f(w*). At low long-distance connectivity (ρ = 0.001), both ni and di are small, resulting in a weakly attracting equilibrium (with a slope just less than one). As indicated by the fact that wi+1 ≈ wi, waves are rarely born or die, implying that the network is dominated by the low rate of spontaneous wave generation. As more long-distance connections are introduced (ρ = 0.01), the rate of emergence of new waves increases. This trend increases the number of waves that we expect to see at the equilibrium and also makes the equilibrium more strongly attracting (the slope at the equilibrium point is near zero). As even more long-distance connections are introduced (ρ = 0.05), the slope of f at the equilibrium point is less than -1, implying that the equilibrium is unstable. [This is known as a “flip bifurcation,” or sometimes as a “pitchfork bifurcation” for maps (Baker and Gollub, 1996).] At this point in the full network model, new waves are generated rapidly, giving rise to a burst (i.e., a large, synchronous increase in the amount of activity). The burst (when wi > 6) would lead to a system that is dominated by wave death on subsequent time steps. Then, almost the entire network will become refractory, implying that the system will be quiescent until enough cells have returned to the excitable state and the network can burst again. This detail of bursting behavior is not captured by our reduced model, which was derived under the assumption that the number of active and refractory cells at any given time is small compared with the network size N. Instead, as ρ is increased above a critical point at ∼0.05, the reduced system continues to oscillate between increasingly higher and lower states (around the unstable equilibrium) until the state fluctuates chaotically with a very large amplitude. This describes a well-understood “period-doubling route to chaos” for discrete-time maps such as that described by f (which resembles the classic “logistic” map) (Baker and Gollub, 1996). The amplitudes are large enough that the activity of a full network model would reach saturation at this point and therefore enter our “bursting” regimen. Because the large-amplitude chaotic behavior is reached for ρ values very close to that which first caused the equilibrium to become unstable, the onset of instability is a reasonable indicator for the onset of bursting in the full network model. Results from this reduced model and those from simulations are compared in Figure 5 (discussed below).
Figure 5.

Change in network behavior as a function of number of synaptic connections per neuron and proportion of long-distance connections using the reduced model (for network size of 3000 neurons). The left panel shows curves delineating the normal, seizing, and bursting regimens as the number of synapses per neuron, and the proportion of long-distance connections are changed. The solid black curve is calculated from analysis of the one-dimensional map and the dotted black curve from the (1 + R)-dimensional map. Tick marks indicate the boundaries of normal, seizing, and bursting behavior in network simulations from Figure 3. Horizontal lines indicate specific parameter choices for the CA3 and the CA1 models. Points labeled a-f correspond to the conditions simulated in Figure 3a-f. These plots imply that the CA3 network will transition from normal to bursting at a much smaller proportion of long-distance connections or smaller synaptic strength than the CA1. The right panel illustrates the boundaries between the behavioral regimens as the number of synapses per neuron and the synaptic strength are varied (with the proportion of long-distance connections fixed at ρ = 0.01). The curves were calculated in a similar way to that in the left panel. The tick mark in the line for the CA3 indicates the boundary between bursting and seizing observed in the integrate-and-fire model, where the population firing rate as a function of synaptic strength are plotted in the inset. The results of the simulations correlate well with the analyses of the reduced map models. No clear transition from seizing to bursting was seen in the full CA1 model, as predicted by the one-dimensional map model.
For both CA1 and CA3 network simulations, the transition from normal activity to seizures corresponds to an increase in the percentage of active cells and the onset of a sustained oscillation in the population activity. Because analysis of the one-dimensional map does not provide us with an explicit condition for this transition, we used the full refractory dynamics in the definition of ei, making the map (1 + R)-dimensional, where R is the length of the refractory period. In a similar way to the one-dimensional map, the (1 + R)-dimensional map can be analyzed to predict qualitative changes in the dynamics. For few long-distance connections, both maps exhibit the same equilibria and stability properties. As more long-distance connections are added, the (1 + R)-dimensional map exhibits oscillations not present in the one-dimensional map, after a “Hopf bifurcation for maps” occurs as ρ is increased (Agarwal et al., 2000). These small-amplitude oscillations resemble those in the full network simulations during seizures (data not shown) and also have a period approximately equal to the refractory time of the neurons. The transition to oscillation occurs at a value of ρ that can be computed analytically and compared with results from computational simulations (see below). Numerical implementations of the two maps both transition to bursting for similar values of model parameters. However, the (1 + R)-dimensional map does not provide an explicit analytic condition for the burst onset. This is because the transition occurs in the full map when the oscillations grow so large that the activity saturates. The saturation of activity breaks the assumption of the derivation of the map and is not a precisely definable transition. Thus, it cannot be captured by an analytical expression involving a local change in stability. In contrast, the one-dimensional map represents an “averaged” view of the full dynamics (through its simplified treatment of the refractory wake), for which transition to bursting occurs while the low-activity assumption still holds.
Figure 5a shows the boundaries of the different behavior regimens predicted by both of the analytical models as the number of synapses per neuron (k) and proportion of long-distance connections (ρ) is varied. The seizing-bursting border (solid black lines) was calculated from the simplified one-dimensional map. The normal-seizing border (dotted black lines) was derived from the (1 + R)-dimensional map. The two horizontal gray lines in each panel correspond to the CA1 and CA3 models from Figure 3. a-f in Figure 5a correspond to the examples from Figure 3, again as the proportion of long-distance connections ρ is changed over several orders of magnitude. Vertical tick marks in Figure 5a correspond to the borders between the normal, bursting, and seizing regimens for the simulated CA1 and CA3 models from Figure 3. Predicted transitions from simplified maps occur near the transition points observed in simulations for a wide range of parameter values.
In the brain and in slice models, epilepsy is probably not caused by an increase in long-distance connections but rather a change in the synaptic strength resulting from drugs or changes in the ionic concentrations of the fluid bathing the slice. Using our reduced map models, we can explore the effects of changing the synaptic efficacy p1 (Fig. 5b), which we define as the probability that a single postsynaptic input gives rise to a postsynaptic spike on the next time step. We look for the regimen transitions as p1 is varied, keeping ρ at 0.01 (the same value used for panels a and e in Fig. 3). The transitions predicted by the models are plotted in Figure 5b. The total network activity measured from the network simulations is plotted in the insets and shows a drop in activity coinciding with the burst transition for CA3 as predicted (indicated by the line in the inset and the dash in the full panel). The CA1 network did not transition to bursts and had a smooth transition to seizures with an onset that is harder to define but approximately coincides with the predicted values. Thus, the reduced model predicts that as synaptic strengths are enhanced, CA3 is more likely to transition to bursting, but CA1 is more likely to transition to a seizing regimen, agreeing with experimental results.
Discussion
Here, we have introduced a simple small-world network representation of excitatory neurons in the hippocampal slice. The network was implemented using three different neuron models: noisy and leaky integrate-and-fire, stochastic Hodgkin-Huxley, and a Poisson spike-train model. We used these models to explain why seizures may not be hypersynchronous, but bursts are. In these simulations, seizure activity in the CA1-like networks is not fully synchronous, allowing the activity to be sustained. The same model supports that bursts in CA3 are caused by synchronous rapid recruitment of neuronal activity. Simulation results were independent of particular cellular models used, indicating that the proportion of long-distance connections is more important than the details of individual neurons in determining the epileptiform properties of the network. We derived a reduced mathematical model of the average activity in the network as a birth-death process (map) in discrete time. This model avoids a purely mean-field approximation (which would ignore the small-world properties of the network) and instead retains parameters directly related to the physiology and the connectivity of the network. The one-dimensional map predicted the transition from seizing to bursting found in the network simulations as the number of synapses per neuron, proportion of long-distance connections, and synaptic strength were varied. These transitions were described as a loss of stability of an equilibrium in the map. A more detailed description of the refractory dynamics gave rise to a higher-dimensional map. This map could be analyzed to predict the transition from normal activity to seizing. These reduced models highlight the roles of physical parameters that could underlie the different epileptiform behaviors observed in CA1 versus CA3.
Relation to previous modeling studies
A number of recent modeling studies have examined problems similar to those studied here. For example, Tsodyks et al. (2000) studied coherent activity in randomly connected networks with depressing synapses. They observed coherent activity, somewhat similar to the bursting seen here. In their work and in related work (Tabak et al., 2000; Wiedemann and Luthi, 2003), networks of neurons with depressing synapses initiate bursts from excitatory neurons that had not fired for some time and thus gave rise to large postsynaptic effects. Synaptic depression shut down bursts in these studies. In contrast to these models, our model has explicit small-world connectivity that plays an important role in the behavior of the network. We focused on how changes in the number of long-distance connections, synaptic strength, and overall connectivity led to dramatic changes in network activity.
Nishikawa et al. (2003) studied synchronization in small-world networks that ranged from our Watts-Strogatz style to networks in which a few hyperconnected “hub” neurons served to reduced the mean path length. They found that hubbed networks were less likely to synchronize than Watts-Strogatz networks, although the hubbed networks have smaller mean path lengths. Superficially, this result seems to contrast with our result that the greatest synchronization was seen with small path lengths. But this apparent discrepancy is an artifact of the way the networks were constructed in these two studies. Our results are compatible with the general argument of Nishikawa and colleagues, in that network synchronization decreases with increasing heterogeneity in the number of connections per neuron. Our bursting networks with large ρ have both small mean path and low heterogeneity, because in a randomly connected network, each cell receives approximately the same number of connections. Our seizing networks with small ρ are more heterogeneous and thus are expected to be less synchronized.
Two additional computational efforts have shown that small-world networks similar to ours can synchronize. Networks of oscillatory elements synchronize when the network contains enough long-distance connections of sufficient synaptic strength (Hong et al., 2002). Roxin and colleagues (2004) have showed that adding long-distance connections makes small-world networks of integrate-and-fire neurons transition from sustained activity to synchronous bursts of finite duration. Oscillation of population activity has been studied by Curtu and Ermentrout (2001) and Wilson and Cowan (1973) using differential equations similar in form to our discrete-time maps. Our work utilizes these types of models to study epilepsy. First, we relate our models to epilepsy in a specific brain region (hippocampus) and attempt to explain why regions CA1 and CA3 exhibit different epileptiform behaviors in slice models. Second, we show that the relationship between the number of long-distance connections and seizing or bursting is remarkably independent of the neuronal model used. Third, our derivation of the reduced map models retains important physiological parameters of which the effect on epileptiform behavior can be studied directly.
Relation to experimental results
This model helps to explain how a stable network may become unstable and prone to epileptiform behaviors. Many drugs and disturbances to the cell equilibria can cause this. For example, 4-AP induces epileptiform behaviors in slice models by blocking voltage-gated K+ channels and thus indirectly enhancing EPSP amplitudes (Chesnut and Swann, 1988). It is shown in Figure 5 that enhancing synaptic strength can transition a stable network into bursting or seizing. Epilepsy can also be induced following cell death. Although decreasing the number of cells alone cannot induce epilepsy in our networks, a concomitant increase in synaptic strength to compensate for the reduced synaptic activity might. Although our simulations only included excitatory cells, normal network activity depends on a balance of excitation and inhibition for stability. The parameter p1 could be interpreted as representing the ratio of excitation to inhibition. In hippocampal slices, epileptiform activity can be induced by pharmacologically blocking inhibitory synaptic activity (Amitai et al., 1993). These disinhibited slice models of epilepsy show three stages during the ictal event (Borck and Jefferys, 1999). After the first stage of depolarization, the second consists of high-frequency oscillations similar to our seizures. The third stage consists of postictal bursts that are similar to our bursts. Our model suggests that the seizing-bursting transition in disinhibited slices may correspond to increasing synaptic strength, connectivity, or cellular excitability during the ictal event.
We predict from our model that networks with fewer recurrent connections are more likely to seize than networks with more recurrent connections. Our model is consistent with the theory that epileptiform behaviors are generated by positive feedback in excitatory network activity (Schwartzkroin, 1994), resulting in runaway excitation. This may help to explain why some regions of the brain produce seizures, whereas others produce epileptiform bursts. The range of synaptic strengths for which the network will produce seizing behavior is smaller for networks with more recurrent excitatory synapses. These networks are more likely to transition from normal directly to bursting without observing seizures. Our model suggests that epileptiform bursts in CA1 may occur only when it receives strong synchronous synaptic input from the burst-prone CA3 region. If bursting input to a seizure-producing region is stopped, the region may be released to produce its own seizing behavior, as observed experimentally by Barbarosie and Avoli (1997) and Bragdon et al. (1992).
Organotypic cultures of neocortex generate waves of activity that are reminiscent of our normal and bursting activity (Beggs and Plenz, 2003). Normal activity in the organotypic cultures is scale-free, in that the probability distribution of sizes of waves and the distribution of wave lifetimes both obey a power law, whereas picrotoxin-induced bursts were not. Beggs and Plenz (2003) replicated their scale-free behavior in a multilayer, feed-forward model. In our simulations, distributions of activity do not resemble power laws for normal, seizing, or bursting activity. We believe that our simulated activity is not scale-free primarily because waves of activity, once initiated, almost always propagate over long distances and thus have long lives. This behavior stands in contrast to that observed and modeled by Beggs and Plenz (2003), in which the most common events are small in spatial scale and short in duration.
The time scales that we observe for the bursting and seizing do not necessarily correspond to actual time scales observed experimentally in vitro. Furthermore, the seizures in the model do not end as seizures do in the in vitro models, because the small-world network model and our mathematical description of the model are highly reduced compared with the hippocampal slice. We expect that fuller models, including inhibition and more spatial realism, may address these discrepancies. The purpose of this model is to develop an understanding of how changes in the physiology change the behavior from normal to bursts to seizures. Therefore, we simulated the networks with only excitatory activity and overestimated the number of recurrent excitatory connections in the CA1 and CA3. This model could be expanded to include inhibitory neurons and more realistic connection schemes, such as a two-dimensional lattice of neurons. The purpose of this model was not to calculate the exact transition of the network behavior or to derive physiological values for these transitions but to give an intuitive feel for why these transitions occur. The exact parameter values where the networks transition between behaviors may differ as we change the form and constituents of the network, but we expect that these transitions will remain qualitatively the same. Nevertheless, we feel that our simple network is useful as one approximation to the qualitative properties of collective behavior in the hippocampus.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS34425 and R01-MH43510 and National Science Foundation Grant DMS-0211505. We thank Nancy Kopell, Sid Redner, Steven J. Schiff, Corey Acker, Alan Dorval, and G. Bard Ermentrout for valuable discussions in the development of this work. The reviewers of this paper made a number of suggestions that improved it dramatically.
Correspondence should be addressed to Theoden I. Netoff, Boston University, Department of Biomedical Engineering, 44 Cummington Street, Boston, MA 02215. E-mail: tnetoff@bu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248075-09$15.00/0
References
- Agarwal R, Grace S, O'Regan D (2000) Oscillation theory for difference and functional differential equations, Sec 9, Chap 1. Dordrecht: Kluwer.
- Amitai Y, Friedman A, Connors BW, Gutnick MJ (1993) Regenerative activity in apical dendrites of pyramidal cells in neocortex. Cereb Cortex 3: 26-38. [DOI] [PubMed] [Google Scholar]
- Baker GL, Gollub JP (1996) Chaotic dynamics: an introduction, Chap 4, Ed 2, p 81. New York: Cambridge UP.
- Barbarosie M, Avoli M (1997) CA3-driven hippocampal-entorhinal loop controls rather than sustains in vitro limbic seizures. J Neurosci 17: 9308-9314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs JM, Plenz D (2003) Neuronal avalanches in neocortical circuits. J Neurosci 23: 11167-11177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck C, Jefferys JG (1999) Seizure-like events in disinhibited ventral slices of adult rat hippocampus. J Neurophysiol 82: 2130-2142. [DOI] [PubMed] [Google Scholar]
- Bower JM, Beeman D (1995) The book of GENESIS: exploring realistic neural models with the GEneral NEural SImulation System, Chap 15, Ed 2, p 245. Santa Clara, CA: Telos.
- Bragdon AC, Kojima H, Wilson WA (1992) Suppression of interictal bursting in hippocampus unleashes seizures in entorhinal cortex: a proepileptic effect of lowering [K+]o and raising [Ca2+]o. Brain Res 590: 128-135. [DOI] [PubMed] [Google Scholar]
- Chesnut TJ, Swann JW (1988) Epileptiform activity induced by 4-aminopyridine in immature hippocampus. Epilepsy Res 2: 187-195. [DOI] [PubMed] [Google Scholar]
- Chow CC, White JA (1996) Spontaneous action potentials due to channel fluctuations. Biophys J 71: 3013-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtu R, Ermentrout G (2001) Oscillations in a refractory neural net. J Math Biol 43: 81-100. [DOI] [PubMed] [Google Scholar]
- Fox JE, Bikson M, Jefferys JGR (2001) Minimum neuronal aggregate necessary for the generation of epileptiform discharges in the hippocampal slice exposed to low calcium ACSF. Soc Neurosci Abstr 27: 969.2. [Google Scholar]
- Gonzalez-Burgos G, Barrionuevo G, Lewis D (2000) Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex 10: 82-92. [DOI] [PubMed] [Google Scholar]
- Hong H, Choi MY, Kim BJ (2002) Synchronization on small-world networks. Phys Rev E Stat Nonlin Soft Matter Phys 65: 026139. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Dudek FE (1980) Local synaptic circuits in rat hippocampus: interactions between pyramidal cells. Brain Res 184: 220-223. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63: 815-846. [DOI] [PubMed] [Google Scholar]
- Mountcastle VB (1997) The columnar organization of the neocortex. Brain 120: 701-722. [DOI] [PubMed] [Google Scholar]
- Netoff TI, Schiff SJ (2002) Decreased neuronal synchronization during experimental seizures. J Neurosci 22: 7297-7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Motter AE, Lai YC, Hoppensteadt FC (2003) Heterogeneity in oscillator networks: are smaller worlds easier to synchronize? Phys Rev Lett 91: 014101. [DOI] [PubMed] [Google Scholar]
- Penfield W, Jasper HH (1954) Epilepsy and the functional anatomy of the human brain, Ed 1. Boston: Little Brown.
- Perreault P, Avoli M (1989) Effects of low concentrations of 4-aminopyridine on CA1 pyramidal cells of the hippocampus. J Neurophysiol 61: 953-970. [DOI] [PubMed] [Google Scholar]
- Roxin A, Riecke H, Solla SA (2004) Self-sustained activity in a small-world network of excitable neurons. Phys Rev Lett 92: 198101. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA (1994) Cellular electrophysiology of human epilepsy. Epilepsy Res 17: 185-192. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A (1998) Presynaptic modulation of CA3 network activity. Nat Neurosci 1: 201-209. [DOI] [PubMed] [Google Scholar]
- Steriade M (2003) Neuronal substrates of sleep and epilepsy. New York: Cambridge UP.
- Tabak J, Senn W, O'Donovan MJ, Rinzel J (2000) Modeling of spontaneous activity in developing spinal cord using activity-dependent depression in an excitatory network. J Neurosci 20: 3041-3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Miles R (1991) Neuronal networks of the hippocampus, Chap 3, p 62. New York: Cambridge UP.
- Tsodyks M, Uziel A, Markram H (2000) Synchrony generation in recurrent networks with frequency-dependent synapses. J Neurosci 20: RC50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Drongelen W, Koch H, Marcuccilli C, Pena F, Ramirez JM (2003) Synchrony levels during evoked seizure-like bursts in mouse neocortical slices. J Neurophysiol 90: 1571-1580. [DOI] [PubMed] [Google Scholar]
- Watts DJ (1999) Small worlds, Chap 3, p 58. Princeton: Princeton UP.
- Watts DJ, Strogatz SH (1998) Collective dynamics of “small-world” networks. Nature 393: 440-442. [DOI] [PubMed] [Google Scholar]
- Wiedemann UA, Luthi A (2003) Timing of network synchronization by refractory mechanisms. J Neurophysiol 90: 3902-3911. [DOI] [PubMed] [Google Scholar]
- Wilson H, Cowan J (1973) A mathematical theory of the functional dynamics of cortical and thalamic nervous tissue. Kybernetik 13: 55-80. [DOI] [PubMed] [Google Scholar]