

Abstract
Molecular diffusion in the brain extracellular space (ECS) is an important determinant of neural function. We developed a brain surface photobleaching method to measure the diffusion of fluorescently labeled macromolecules in the ECS of the cerebral cortex. The ECS in mouse brain was labeled by exposure of the intact dura to fluorescein-dextrans (Mr 4, 70, and 500 kDa). Fluorescein-dextran diffusion, detected by fluorescence recovery after laser-induced cortical photobleaching using confocal optics, was slowed approximately threefold in the brain ECS relative to solution. Cytotoxic brain edema (produced by water intoxication) or seizure activity (produced by convulsants) slowed diffusion by >10-fold and created dead-space microdomains in which free diffusion was prevented. The hindrance to diffusion was greater for the larger fluorescein-dextrans. Interestingly, slowed ECS diffusion preceded electroencephalographic seizure activity. In contrast to the slowed diffusion produced by brain edema and seizure activity, diffusion in the ECS was faster in mice lacking aquaporin-4 (AQP4), an astroglial water channel that facilitates fluid movement between cells and the ECS. Our results establish a minimally invasive method to quantify diffusion in the brain ECS in vivo, revealing stimulus-induced changes in molecular diffusion in the ECS with unprecedented spatial and temporal resolution. The in vivo mouse data provide evidence for: (1) dead-space ECS microdomains after brain swelling; (2) slowed molecular diffusion in the ECS as an early predictor of impending seizure activity; and (3) a novel role for AQP4 as a regulator of brain ECS.
Keywords: aquaporin, extracellular space, fluorescence recovery after photobleaching, seizure, brain edema, water intoxication
Introduction
The extracellular space (ECS) in brain comprises ∼20% of brain tissue volume, consisting of a jelly-like matrix in which neurons, glia, and blood vessels are embedded (Sykova, 1997). The ECS contains ions, neurotransmitters, metabolites, peptides, and extracellular matrix molecules, forming the microenvironment for all cells in the brain and mediating glia-neuron communication via diffusible messengers, metabolites, and ions (Fields and Stevens-Graham, 2002). Neuronal activity is associated with depolarization of neurons and adjacent glial cells and with increased extracellular glutamate and K+, which can synchronize neuronal activity and activate glial metabolism and signaling (Grosche et al., 1999; Newman, 2001; Fields and Stevens-Graham, 2002; Nedergaard et al., 2003). High-affinity binding sites for many extracellular messengers as well as ligand- and voltage-gated ion channels and transporters exist on neuronal somata, axons, and glial cells, mediating what has been termed “nonsynaptic communication” or “volume transmission” (Sykova, 1997; Nicholson, 2000; Piet et al., 2004). Defining the structure and function of the ECS is thus critical in determining its role in brain physiology and in pathological conditions such as brain edema and seizure activity.
Various techniques have been applied for measuring ECS parameters in brain slices involving diffusion of labeled molecules including radiotracers, tetramethylammonium+ (TMA+), and fluorescently labeled macromolecules (Nicholson and Sykova, 1998). Using primarily the TMA+ method developed by Nicholson (Nicholson and Phillips, 1981; Nicholson, 1993), it has been shown that cellular swelling is associated with reduced ECS volume and increased ECS tortuosity (a measure of hindrance to diffusion). Although the TMA+ method has provided an important body of information about ECS properties, it has several limitations, including the need for direct micropipette invasion of the measurement site. Also, because the method is limited to measurement of TMA+ diffusion, the diffusion of macromolecules cannot be measured, nor can questions be addressed about the size, shape, or charge dependence of molecular diffusion in the ECS.
We report here a minimally invasive brain surface photobleaching method to quantify the diffusion of fluorescently labeled macromolecules in the ECS in vivo that does not require micropipettes or breach of dural integrity. Diffusion is measured by fluorescence recovery after photobleaching (FRAP) after transdural loading of the ECS with fluorescein-dextrans. FRAP has been used extensively to characterize solute and macromolecular diffusion in cell and membrane biology (Verkman, 2002). The surface photobleaching method was used to characterize size-dependent macromolecule diffusion in the ECS and to demonstrate remarkable changes in diffusion in response to cytotoxic brain swelling, neural activity, and deletion of aquaporin-4 (AQP4), a glial water channel known to modulate brain swelling and neural excitability (Manley et al., 2000; Binder et al., 2004; Papadopoulos et al., 2004).
Materials and Methods
Fluorescent dye loading of ECS in mouse brain in vivo. All experiments were conducted under an approved protocol from the University of California, San Francisco Committee on Animal Research. Twenty-five to 35 gm male mice in a CD1 genetic background (wild type and AQP4 deficient) (Ma et al., 1997) were anesthetized using Avertin (2,2,2-tribromoethanol; 125 mg/kg, i.p.) and immobilized in a stereotactic frame. Additional Avertin was given as needed to maintain an even depth of anesthesia. Rectal temperature was monitored and strictly maintained between 35 and 38°C using a heating lamp. After exposing the skull by midline skin incision, an atraumatic craniectomy was made with a Foredom high-speed drill (45,000 rpm, 1 mm stainless steel burr; Blackstone Industries, Bethel, CT) to expose the intact dura and underlying brain surface. The craniectomy margins were the coronal suture anteriorly, the lambdoid suture posteriorly, and the attachment of each temporalis muscle laterally. In preliminary experiments, exposing the olfactory bulbs more rostrally resulted in excessive venous bleeding. After exposure and craniectomy (see Fig. 1 A), the skin flaps were held open with a cylindrical plastic dam to create a pocket for dye loading (see Fig. 1 B).
Figure 1.
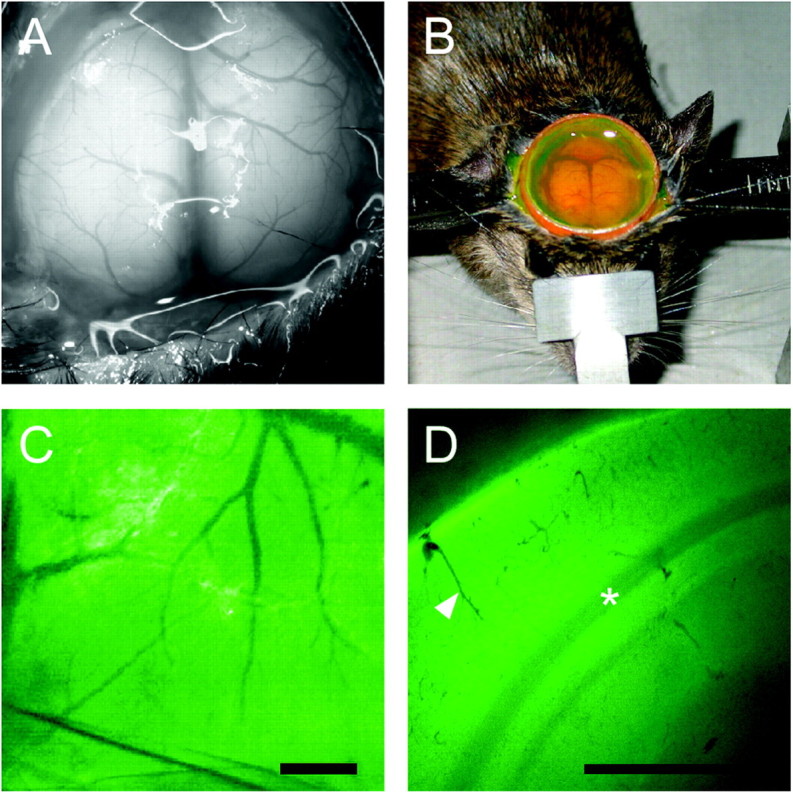
Invivoloading of brain ECS by fluorescein-dextrans. A, Brain surface exposure after craniectomy showing cortical blood vessels and intact dura. B, Transdural loading of brain ECS showing a cylindrical dam containing aCSF solution of fluorescein-dextran. C, Fluorescence image of cortical surface after dye loading. Scale bar, 1 mm. D, Coronal 300 μm brain slice obtained ex vivo after loading demonstrates fluorescence loading of the cortex. Scale bar, 1 mm. A gradient of fluorescence signal is observed from the cortical surface (top left) to the dorsal hippocampus (bottom right). Arrowhead, Cortical blood vessel; asterisk, white matter.
Fluorescein isothiocyanate (FITC)-labeled dextrans (4, 70, and 500 kDa; Sigma, St. Louis, MO) were dissolved in artificial cerebrospinal fluid (aCSF; in mm: 124 NaCl, 4 KCl, 1 MgCl2, 2.5 CaCl2, 1 KH2PO4, and 10 glucose, pH 7.4). The osmolarity of all aCSF solutions applied directly to the dura was 300 mOsm, as measured by a freezing-point depression osmometer (Osmette; Precision Systems, Natick, MA). Thin-layer chromatography confirmed the absence of unconjugated fluorescein. Fresh solutions of fluorescein-dextrans (4 kDa, 50 mg/ml; 70 kDa, 30 mg/ml; 500 kDa, 25 mg/ml) were applied to cover the exposed dura completely (300-400 μl). Loading times to give adequate brain fluorescence signals were established in initial measurements (4 kDa, 1 hr; 10 kDa, 1.5 hr; 500 kDa, 2 hr). Fluorescence labeling of the ECS was verified by imaging of the brain surface and ex vivo coronal brain slices (see Results). After loading, residual dye-containing aCSF was removed from the dural surface by a microsuction catheter constructed to remove fluid and blood from the surgical field, and the dural surface was washed three times via superfusion of ∼3 ml of dye-free aCSF. The skin flaps were then removed, and the mouse in the stereotactic frame was transferred to the stage of an upright epifluorescence microscope for cortical FRAP measurements. Mice with cortical damage or inadequate fluorescence labeling were excluded from FRAP measurements.
Fluorescence recovery after photobleaching. Photobleaching measurements were done on an apparatus described previously (Jayaraman et al., 2001) after modification for confocal detection. Briefly, the first-order beam of an argon ion laser (2 W at 488 nm) diffracted by an acoustooptic modulator was focused onto the surface of the mouse brain through a dichroic mirror (510 nm) and 50× objective lens (numerical aperture, 0.55; working distance, 8 mm; Nikon air). In some experiments, larger bleached spots were obtained using a 20× objective lens (numerical aperture, 0.35; working distance, 2.2 mm; Nikon). Emitted fluorescence was filtered (510 nm long-pass), detected by a photomultiplier, amplified, and digitized by a 14-bit analog-to-digital converter. Confocal detection was achieved by positioning a 400 μm diameter pinhole (with xy micropositioners) at the back focal plane. Axial resolution was ∼20 μm (width at half-maximum of z-point spread function) as measured using fluorescent beads. Fluorescence was sampled continuously over 200 msec before the bleach pulse, at rates of up to 1 MHz for 500 msec after bleaching and then at 1 Hz (shutter opened for 20 msec per acquisition) for longer times (10-60 sec). Fluorescence recovery curves, F(t), were analyzed by nonlinear least-squares regression using the semi-empirical equation: F(t) = (F0 + [R(F - F0) + F0](t/t1/2))[1 + (t/t1/2)]-1, where F is prebleach fluorescence, F0 is fluorescence immediately after bleaching, R is the mobile fraction, and t1/2 is the recovery half-time (Feder et al., 1996).
For in vitro (solution diffusion) experiments, 10 μl of freshly prepared solutions of fluorescein-dextrans (4, 70, or 500 kDa as above) were sandwiched between two coverslips. The laser spot was focused on the solution, and bleaching was accomplished by increasing laser illumination intensity ∼4000-fold for 1-5 msec to bleach fluorescence in the illuminated circular spot (∼5 μm for 50× lens, ∼12 μm for 20× lens) to ∼60% of prebleach fluorescence. Fluorescence recovery curves were obtained from three to five spots and averaged for each condition. In control experiments, droplets of fluorescein-dextran solution (without coverslip) yielded identical fluorescence recovery, indicating insensitivity of measured diffusion to solution geometry.
For in vivo measurements, the laser spot was focused just beneath the brain surface, although fluorescence recovery curves were insensitive to small changes in focus position. An adjustable mechanical arm fixed to the stereotactic frame was used to position a small (3 mm diameter, 150 μm thick) glass window on the cortical surface to dampen cardiorespiratory oscillations. Bleaching was accomplished as above. F(t) from three to seven different cortical spots was averaged per condition for each mouse.
Calibration curves relating t1/2 to the relative diffusion coefficient (D/Do) were generated by measuring t1/2 for aCSF solutions containing fluorescein-dextrans (10-20 mg/ml) and glycerol (0-60 wt%) having different macroscopic viscosities (measured using a Cannon-Fenske viscometer; Fisher Scientific, Pittsburgh, PA). Potential corrections of the t1/2 versus D/Do relationship for light scattering by dura and brain parenchyma were considered, both of which could increase effective spot size and hence increase t1/2. The effect of light scattering by dura was determined by measuring t1/2 for different fluorescein-dextrans in aCSF under a coverglass in which a freshly excised dural fragment was immobilized. Also, in some studies, FRAP measurements were performed just before and after removal of the dura. In each of these two types of control experiments, the dura was removed atraumatically by midline durotomy near the superior sagittal sinus and careful lateral reflection of the dura with microforceps under a dissecting microscope. The effect of light scattering by brain parenchyma was determined using phantoms consisting of aCSF solutions of fluorescein-dextrans containing different concentrations of milk powder (0-10 wt%) prepared to mimic the light scattering properties of brain parenchyma (see Results).
Water intoxication. Cytotoxic brain edema from water intoxication was produced as described previously (Manley et al., 2000). After collection of baseline FRAP data, mice received injections of distilled water (20% body weight, i.p.) prewarmed to 37°C. FRAP measurements were then taken at 10 and 20 min after water administration.
Glutamate application and seizure induction. Glutamate (1 mm in aCSF) was applied epidurally to cover the brain surface for 5 min. FRAP recovery data were collected before and 5 min after glutamate application. In several experiments (n = 3), mannitol (40 gm/kg, i.p.) was administered 10 min after glutamate application in an attempt to reverse glutamate-induced diffusion restriction. To induce generalized seizures, mice received injections of the convulsant pentylenetetrazol (PTZ; 100 mg/kg, i.p.), a GABAA antagonist used widely to model generalized epilepsy (Macdonald and Barker, 1977; Medina et al., 2001; Binder et al., 2004). Seizure activity was monitored by EEG recording using a steel bipolar electrode implanted under the surface of the exposed cortex. Real-time cortical EEG was recorded by a digital acquisition system (Biopac Systems, Goleta, CA) and time-locked to PTZ administration to obtain EEG and photobleaching data before PTZ administration, after PTZ administration but before seizure activity, and after seizure activity.
Results
After surgical exposure of the cerebral cortex in anesthetized mice (Fig. 1A), transdural loading of the fluorescein-dextrans was performed (Fig. 1B). After extensive washing with dye-free aCSF, fluorescence was observed in the cerebral cortex throughout the brain parenchyma but not in blood vessels (Fig. 1C). Freshly cut ex vivo coronal slices taken from dye-loaded brains demonstrated fluorescence loading of cortical cell layers (Fig. 1D). Bright green fluorescence was observed from the pial surface extending throughout the cortical layers, with lower fluorescence in deeper tissues (white matter and dorsal hippocampus). These findings indicate successful transdural dye penetration and loading of the ECS in the cortex of living mice.
After dye loading and washing, the mouse was transferred to the photobleaching apparatus (Fig. 2A), and a small glass window was applied gently to the dura to dampen cardiorespiratory oscillations (Fig. 2B). The window was maintained in contact with the dura only during the FRAP measurement and then removed before a subsequent measurement. To ensure that there was no effect of glass window apposition on ECS diffusion, two types of control studies were performed. First, prolonged application of the glass window (10 min) did not alter FRAP recovery curves. Second, fluorescence recovery was unchanged when measured immediately after anesthetic overdose to eliminate cardiorespiratory oscillations.
Figure 2.

Brain ECS diffusion measured by cortical surface photobleaching. A, Schematic of apparatus for cortical surface photobleaching measurements in vivo. B, Photograph showing glass window positioned at the dural surface to dampen cardiorespiratory brain oscillations. C, Representative fluorescence recovery curves for 4 kDa fluorescein-dextran in aCSF (red), brain cortex (black), and aCSF containing 30% glycerol having viscosity ∼2.7 centipoise (blue; spot size, ∼5 μm). D, Left, Relationship between t1/2 (half-time) for fluorescence recovery and relative brain versus aCSF diffusion coefficient (D/Do; top x-axis) deduced from photobleaching measurements on fluorescein-dextran-containing solutions made viscous with glycerol(bottomx-axis). See Results for details. Right, Fluorescencere covery curves for a CSF containing 4kDa fluorescein-dextran and indicated concentrations of glycerol.
Baseline in vivo fluorescence recovery curves were acquired after briefly increasing laser beam intensity to reduce fluorescence to ∼60% of original fluorescence. Confocal detection was crucial in these studies to reduce the effects of light scattering by brain parenchyma; in initial experiments without confocal detection, qualitatively similar results were obtained but required much longer bleach durations (>20 msec) because of detected fluorescence arising from well beneath the brain surface. Fluorescence recovery in the brain ECS (Fig. 2C, black curve) was slowed compared with recovery for the same sized 4 kDa fluorescein-dextran in aCSF in vitro (Fig. 2C, red curve). In vivo brain recovery curves were insensitive to slight changes in z-axis focus (25-50 μm; accomplished with fine focus) and did not show a rapid recovery phase, indicating no signal contamination by subarachnoid space diffusion. Fluorescence recoveries at baseline were similar in different wild-type mice and stable for at least 30 min, the maximum duration of experiments in any mouse. After 30 min, the fluorescence was generally reduced because of dye diffusion out of the loaded region, resulting in unacceptably poor signal/noise ratios in recovery curves. The fluorescence recovery for fluorescein-dextran in the brain ECS (black curve) was nearly identical to that produced by adding 30% glycerol to the aCSF solution to increase viscosity to ∼2.7 centipoise (Fig. 2C, blue curve). This correspondence in curve shape provides direct evidence that fluorescein-dextran diffusion under baseline conditions is a simple (nonanomalous) one-component process that is describable quantitatively by a single parameter such as the recovery half-time (t1/2).
To quantify relative fluorescein-dextran diffusion in the brain ECS versus aCSF (D/Do), t1/2 values for fluorescence recovery were obtained by numerical regression of recovery curves. A calibration of t1/2 versus D/Do was generated for each size of fluorescein-dextran, in which D/Do could be deduced from measured t1/2. The standards for the calibration consisted of aCSF solutions of fluorescein-dextrans at different viscosities (set by adding glycerol) measured under similar conditions as in the brain (solution layers under glass window). Figure 2D shows the deduced t1/2 versus D/Do calibration from the solution glycerol measurements, which was used to convert t1/2 measured in vivo to D/Do. Possible effects of light scattering from brain parenchyma and dura on the validity of the t1/2 versus D/Do calibration were evaluated. Scattering by brain parenchyma was evaluated using solution glycerol standards made turbid by adding milk powder (up to 10%) to mimic the light-scattering properties of the brain at fluorescence excitation and emission wavelengths. As anticipated from the restricted detection volume in confocal optics, measured t1/2 changed by <4% in solution standards with twofold greater turbidity (at 488 nm) than brain parenchyma, indicating that no correction was needed for scattering by parenchyma. However, scattering by dura produced a consistent ∼28% increase in t1/2 at all viscosities, as determined by photobleaching of standards in which a freshly isolated fragment of dura was interposed between the glass window and fluorescent solution. This correction arises because of a small increase in the bleached area beneath the dura and was taken into account in the t1/2 versus D/Do calibration plotted in Figure 2D. The quantitative validity of this correction factor was verified in vivo in a limited set of measurements comparing t1/2 before versus just after careful removal of the dura (n = 3; data not shown). In other control studies, FRAP measurements of the 4 kDa fluorescein-dextran were performed using a larger spot size (produced by a 20× objective). As expected from the larger bleached area, the 20× objective gave a slower recovery t1/2 (slowed sevenfold to eightfold) for both solution and in vivo measurements, yet a comparable D/Do of ∼0.3, indicating the adequacy of ECS sampling using the 50× objective.
To determine the dependence of D/Do on molecular size, recovery curves were obtained for 4, 70, and 500 kDa fluorescein-dextrans under baseline conditions. Photobleaching recoveries for each fluorescein-dextran were measured in five to seven mice per condition, and three to seven measurements were averaged within each condition to yield recovery curves. Absolute diffusion was slowed with increasing fluorescein-dextran size (Fig. 3A,B), yet there was no significant change in deduced D/Do (Fig. 3B).
Figure 3.

Diffusion of fluorescein-dextrans in the brain ECS before and after cytotoxic brain edema. A, Fluorescence recovery curves for indicated fluorescein-dextrans before (black) and at 10 min (blue) and 20 min (green) after intraperitoneal water injection producing cytotoxic brain edema. Both short and long time scales are shown. B, Averaged t1/2 (left) and deduced D/Do (right) (mean ± SE) from data as in A. The top axis shows equivalent tortuosity (λ = [Do/D]1/2). C, Percentage of fluorescence recovery (mean ± SE) at 10 sec after photobleaching before and after water intoxication.
We tested the hypothesis that brain edema is associated with changes in ECS diffusive properties using a well established model of cytotoxic brain swelling produced by water intoxication (Wasterlain and Torack, 1968; Manley et al., 2000). Water intoxication produced dramatic slowing of fluorescence recovery for each fluorescein-dextran observed within 10 min after injection (Fig. 3A). In many mice, cortical surface fluorescence did not recover to prebleach levels, indicating an immobile fraction of extracellular dye that presumably represents the creation of dead-space microdomains in which free diffusion in the ECS is prevented. This was seen particularly for the 70 and 500 kDa dextrans compared with the smaller 4 kDa dextran (Fig. 3A). The incomplete fluorescence recovery after swelling was quantified by the percentage of recovery at 10 sec, revealing a time- and size-dependent reduction in fluorescence recovery (Fig. 3C).
To determine the effects of acute increases in neuronal activity on molecular diffusion in the ECS in vivo, we examined the effects of glutamate exposure and generalized seizure induction. Application of 1 mm glutamate to the cortex caused a rapid and dramatic reduction of fluorescein-dextran diffusion within 5 min (Fig. 4A). Mannitol partially reversed the reduction in fluorescein-dextran diffusion (Fig. 4A). Induction of generalized seizures by systemic PTZ produced high-amplitude synchronous spike activity on EEG with a latency of several minutes, accompanied by motor manifestations of seizure and followed by post-seizure electrical depression (Fig. 4B, top). ECS diffusion measurements were taken before PTZ injection, after PTZ injection but before seizure initiation, and after seizure termination. Remarkably, diffusion in the ECS was reduced after PTZ administration but before electroencephalographic seizure activity (Fig. 4B, bottom). After seizure activity, there was an even greater reduction in ECS diffusion, presumably corresponding to seizure-related cell swelling (Fig. 4B, bottom). Averaged percentages of recovery are shown at the bottom.
Figure 4.

Reduced macromolecular diffusion in the brain ECS after glutamate- and seizure-induced neuronal activity. A, Fluorescence recovery curves for 70 kDa fluorescein-dextran before and 5 min after application of glutamate (1 mm) to the cortical surface and 10 min after intraperitoneal injection of mannitol (40 gm/kg). Data are representative of three mice. B, Top, Electroencephalographic recordings before and after intraperitoneal injection of PTZ (100 mg/kg). Bottom, Fluorescence recovery curves for 70 kDa fluorescein-dextran before PTZ administration, after PTZ but before electroencephalographic seizure activity, and after seizure activity. The percentage of fluorescence recovery (mean ± SE; 3 mice) at 10 sec after photobleaching is summarized at the bottom.
Finally, fluorescein-dextran diffusion in the ECS was measured in mice lacking AQP4, a glial membrane water channel that facilitates cell-to-ECS water movement (Solenov et al., 2004) and the deletion of which in mice produces alterations in brain water accumulation after injury (Manley et al., 2000) and seizure threshold (Binder et al., 2004). We hypothesized that altered ECS structure may be, in part, responsible for these phenotypes. Diffusion of the 4, 70, and 500 kDa fluorescein-dextrans in the ECS of AQP4-null mice was significantly faster than that in wild-type control mice (Fig. 5A,B). In addition, the restriction to diffusion at 10 and 20 min after water intoxication was attenuated in the AQP4-null mice (Fig. 5C).
Figure 5.

Enhanced ECS diffusion in mice lacking glial water channel AQP4. A, Fluorescence recovery curves for indicated fluorescein-dextrans in the brain cortex of wild-type (black) and AQP4-/- (gray) mice. B, Averaged t1/2 (left) and deduced D/Do (right) (mean ± SE) from data as in A. The top axis on the right shows equivalent tortuosity (λ = [Do/D]1/2). *p<0.01 compared with wild type. C, Averaged t1/2 (mean ± SE) before and at 10 and 20 min after intraperitoneal water injection.
Discussion
We developed and applied a brain surface photobleaching method to measure diffusion of fluorescently labeled macromolecules in the ECS of the cerebral cortex in vivo. Loading of mouse cortex ECS was accomplished by exposure of the intact dura to fluorescein-dextrans, and diffusion was measured quantitatively by fluorescence recovery after cortical photobleaching. Our results establish a novel approach to quantify diffusion in the ECS in vivo. Several interesting observations were made, including (1) an approximate threefold slowed diffusion of fluorescein-dextrans (4-500 kDa) in the ECS relative to solution, (2) dramatic impairment of fluorescein-dextran diffusion in the ECS after cytotoxic brain edema and neural activity (including changes preceding seizure activity), and (3) significant enhancement of fluorescein-dextran diffusion in mice lacking glial water channel AQP4.
We adapted the well established technique of FRAP to quantify fluorescent probe diffusion in the ECS. FRAP has been used extensively in cell and membrane biology to quantify intracellular, intercellular, and membrane diffusion (Verkman, 2002, 2003), as well as to measure diffusion in solid tumors (Pluen et al., 2001). In neurobiology, FRAP has been used to quantify the extent of gap junctional coupling (Lee et al., 1995; Cotrina et al., 2001) and to study membrane receptor diffusion (Choquet and Triller, 2003). The surface cortical photobleaching method has a number of advantages compared with previous methods of studying ECS parameters, including the TMA+ and integrative optical imaging methods (Nicholson and Phillips, 1981; Tao and Nicholson, 1996; Nicholson and Sykova, 1998; Tao, 1999). As an optical method, FRAP is minimally invasive and avoids damage related to micropipette perturbation of the recording site. The diffusion of many types of fluorescent macromolecules can, in principle, be studied, such as proteins, DNA, and carbohydrates. In contrast to methods involving point-source microinjection (Prokopova-Kubinova et al., 2001), the FRAP method has no intrinsic directionality and does not cause convective fluid transport. The FRAP method has excellent spatial and temporal resolution in that measurements can be done repeatedly (within seconds) and any portion of the cortical surface can be sampled. Finally, the FRAP method is well suited for measurements in vivo in real time. A number of technical challenges were encountered and resolved in applying FRAP to the brain ECS in vivo, including optimization of mouse preparation and fluorescent dye loading conditions, mitigation of cardio-respiratory oscillations, modification of the apparatus for confocal detection, and analysis/correction for light-scattering effects from dura and brain parenchyma. Other in vivo cortical imaging methodologies such as intrinsic optical signal imaging and voltage-sensitive dye imaging have also necessitated dampening of cardiorespiratory brain oscillations and/or gating of signal acquisition to the cardiac cycle (Shoham et al., 1999; Kalatsky and Stryker, 2003).
Fluorescein-dextran diffusion in the brain was approximately threefold slower than solution (D/Do, ∼0.3). This corresponds to a tortuosity (λ = [Do/D]1/2, a measure of hindrance to diffusion) of ∼1.7, which is in agreement with a considerable body of data using the TMA+ technique with reported λ in the range 1.3-1.8 (Sykova, 1997; Nicholson and Sykova, 1998; Sykova et al., 2000). In contrast, in studying fluorescein-dextran diffusion in brain cortical slices, Nicholson and Tao (1993) found greater values of λ for 40 and 70 kDa dextrans (2.16 and 2.25, respectively) than for 3 and 10 kDa dextrans (1.70 and 1.63, respectively), which may be related to cell swelling or other phenomena in the in vitro slices. It is important to recognize that “total” tortuosity λ is made up of both geometric and viscous components; “geometric” tortuosity presumably depends on intercellular gap width and architecture (sensitive to cell swelling), whereas “viscous” tortuosity reflects extracellular composition (Rusakov and Kullmann, 1998a,b). The near-complete fluorescence recoveries observed with these fluorescein-dextrans rules out the possibility of a noncommunicating intracellular reservoir of a significant fraction of dye. Interestingly, D/Do was similar for 4, 70, and 500 kDa fluorescein-dextrans under baseline conditions, suggesting that the characteristic spacing of the ECS is greater than that of the 500 kDa dextran, which has a radius of gyration (RG) of <30 nm (Seksek et al., 1997). This is consistent with the approximate intercellular ECS spacing (∼20-50 nm) (Van Harreveld and Malhotra, 1967; Rusakov and Kullmann, 1998c); by comparison, a synaptic vesicle is ∼40 nm (Rusakov and Kullmann, 1998c).
We found that water intoxication, which results in cytotoxic brain edema with glial cell swelling and reduction of ECS volume (Wasterlain and Torack, 1968; Manley et al., 2000), produced a marked reduction in diffusion of 70 and 500 kDa fluorescein-dextrans early after free water administration. The incomplete fluorescence recovery at long times for these fluorescein-dextrans, or “immobile fraction,” provides evidence for creation of dead-space microdomains after brain swelling in vivo. A previous TMA+ study of the effect of ischemia in brain slices suggested the presence of dead spaces indirectly from changes in ECS volume and effects of added dextrans (Hrabetova et al., 2003). Our demonstration of size-dependent diffusion restriction during brain swelling/edema has important implications for drug and gene delivery in vivo because ECS diffusion is greatly impaired by even modest glial swelling. Future studies using the in vivo FRAP technique to characterize ECS parameters in and around pathological brain lesions such as tumors, traumatic lesions, and stroke may help define conditions for optimal delivery of therapeutic agents.
Rapid and dramatic changes in ECS diffusion parameters were also observed after intense neural activity. Glutamate application to the cortex remarkably reduced fluorescein-dextran diffusion in the ECS within 5 min and produced dead-space microdomains. The impaired fluorescein-dextran diffusion could be reversed partially by mannitol administration, consistent with the expected reciprocal relationship of ECS and cell volume. After generalized seizure activity induced by PTZ, fluorescein-dextran diffusion in the ECS was reduced markedly. Although it is known that osmolarity and ECS volume have powerful effects on neuronal excitability and synchronization in vitro (Andrew et al., 1989; Traynelis and Dingledine, 1989; Dudek et al., 1990; McBain et al., 1990; Roper et al., 1992; Chebabo et al., 1995; Hochman et al., 1995; Pan and Stringer, 1996; Schwartzkroin et al., 1998), few studies have examined the association between ECS changes and seizures in vivo. Previous experiments using the iontophoretic TMA+ method in cat cortex in vivo demonstrated ECS volume contraction associated with seizures induced by intravenous PTZ (Heinemann and Dietzel, 1984; Dietzel and Heinemann, 1986; Lux et al., 1986). However, until now, there has not been direct evidence for ECS changes preceding seizures in vivo. Reduced fluorescein-dextran diffusion in the ECS after convulsant administration but before seizure initiation provides support for a potential role of cell swelling and ECS changes in seizure initiation and could, in principle, be incorporated as part of seizure detection algorithms. Interestingly, Heinemann et al. (1977) detected a preictal fall in extracellular calcium levels in the cerebral cortex of PTZ-treated cats; thus, calcium influx before seizure synchronization may represent a potential mechanism for our observed ECS changes.
Fluorescein-dextran diffusion in the ECS was compared in wild-type mice and mice lacking AQP4, a glial water-transporting protein the deletion of which in mice reduces brain water accumulation after water intoxication and acute stroke (Manley et al., 2000), impairs auditory-evoked (Li and Verkman, 2001) and light-evoked (Li et al., 2002) potentials, and increases seizure threshold (Binder et al., 2004). Because AQP4 provides the principal route for water movement between glial cells and the ECS (Solenov et al., 2004), and probably between retinal Müller and bipolar cells and between cochlear supportive and hair cells, we hypothesized that some or all of the phenotypic findings in mice may relate to altered ECS physiology. FRAP measurements indicated significantly enhanced fluorescein-dextran diffusion in the ECS of AQP4-null mice compared with wild-type control mice under baseline conditions and after water intoxication. Increased ECS diffusion in AQP4 deficiency may be related to reduced ECS-to-glial cell fluid transport, which could result from altered functioning of ion channels that are associated with AQP4. AQP4 colocalizes with the inwardly rectifying K+ channel Kir4.1 (Nagelhus et al., 1998; Connors and Kofuji, 2002; Kofuji and Connors, 2003), which has been proposed to be responsible for rapid K+ clearance after neural activity (Newman et al., 1984; Newman, 1993; Kofuji et al., 2000). Because of the potential functional interaction of water transport through AQP4 channels and K+ influx, we hypothesize that AQP4 deficiency leads to reduced steady-state K+ and H2O influx into glial cells, thereby expanding the ECS and contributing to the observed enhanced ECS diffusion in AQP4 deficiency. Proof of this hypothesis will require clarification of ECS ion composition and measurement of K+ fluxes and clearance mechanisms together with ECS changes in AQP4-null mice. Although unlikely, at present we cannot rule out a role of altered extracellular matrix composition or chemistry in AQP4-null mice that may produce enhanced fluorescein-dextran diffusion. In any case, the increased ECS diffusion in AQP4-deficient mice provides a novel mechanism to account for alterations in neural excitability.
In summary, we have established a minimally invasive method to quantify brain ECS diffusion in vivo with unprecedented spatial and temporal resolution. This method should be broadly applicable to many areas of neuroscience, and in combination with other existing methodologies (e.g., voltage-sensitive dye imaging, intrinsic signal imaging, ion- and pH-sensitive dyes, microelectrodes) should help define real-time ECS physiology in the mammalian CNS in normal and pathological states.
Footnotes
This work was supported by National Institutes of Health Grants EB00415, DK35124, EY13574, HL73856, and HL59198 and Research Development Program Grant R613 from the Cystic Fibrosis Foundation to A.S.V. and a Clinician-Scientist Fellowship from the Wellcome Trust to M.C.P. We thank Liman Qian for mouse breeding and genotype analysis.
Correspondence should be addressed to Dr. Alan S. Verkman, Cardiovascular Research Institute, University of California, San Francisco, 1246 Health Sciences East Tower, San Francisco, CA 94143-0521. E-mail: verkman@itsa.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248049-08$15.00/0
References
- Andrew RD, Fagan M, Ballyk BA, Rosen AS (1989) Seizure susceptibility and the osmotic state. Brain Res 498: 175-180. [DOI] [PubMed] [Google Scholar]
- Binder DK, Oshio K, Ma T, Verkman AS, Manley GT (2004) Increased seizure threshold in mice lacking aquaporin-4 water channels. NeuroReport 15: 259-262. [DOI] [PubMed] [Google Scholar]
- Chebabo SR, Hester MA, Aitken PG, Somjen GG (1995) Hypotonic exposure enhances synaptic transmission and triggers spreading depression in rat hippocampal tissue slices. Brain Res 695: 203-216. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A (2003) The role of receptor diffusion in the organization of the postsynaptic membrane. Nat Rev Neurosci 4: 251-265. [DOI] [PubMed] [Google Scholar]
- Connors NC, Kofuji P (2002) Dystrophin Dp71 is critical for the clustered localization of potassium channels in retinal glial cells. J Neurosci 22: 4321-4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Gao Q, Lin JH, Nedergaard M (2001) Expression and function of astrocytic gap junctions in aging. Brain Res 901: 55-61. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U (1986) Dynamic variations of the brain cell microenvironment in relation to neuronal hyperactivity. Ann NY Acad Sci 481: 72-86. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenhaus A, Tasker JG (1990) Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett 120: 267-270. [DOI] [PubMed] [Google Scholar]
- Feder TJ, Brust-Mascher I, Slattery JP, Baird B, Webb WW (1996) Constrained diffusion or immobile fraction on cell surfaces: a new interpretation. Biophys J 70: 2767-2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B (2002) New insights into neuron-glia communication. Science 298: 556-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H (1999) Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci 2: 139-143. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Dietzel I (1984) Extracellular potassium concentration in chronic alumina cream foci of cats. J Neurophysiol 52: 421-434. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ (1977) Extracellular free calcium and potassium during paroxsmal activity in the cerebral cortex of the cat. Exp Brain Res 27: 237-243. [DOI] [PubMed] [Google Scholar]
- Hochman DW, Baraban SC, Owens JWM, Schwartzkroin PA (1995) Dissociation of synchronization and excitability in furosemide blockade of epileptiform activity. Science 270: 99-102. [DOI] [PubMed] [Google Scholar]
- Hrabetova S, Hrabe J, Nicholson C (2003) Dead-space microdomains hinder extracellular diffusion in rat neocortex during ischemia. J Neurosci 23: 8351-8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS (2001) Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci USA 98: 8119-8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalatsky VA, Stryker MP (2003) New paradigm for optical imaging: temporally encoded maps of intrinsic signal. Neuron 38: 529-545. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Connors NC (2003) Molecular substrates of potassium spatial buffering in glial cells. Mol Neurobiol 28: 195-208. [DOI] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA (2000) Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci 20: 5733-5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Magge S, Spencer DD, Sontheimer H, Cornell-Bell AH (1995) Human epileptic astrocytes exhibit increased gap junction coupling. Glia 15: 195-202. [DOI] [PubMed] [Google Scholar]
- Li J, Verkman AS (2001) Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem 276: 31233-31237. [DOI] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS (2002) Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci 43: 573-579. [PubMed] [Google Scholar]
- Lux HD, Heinemann U, Dietzel I (1986) Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv Neurol 44: 619-639. [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS (1997) Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest 100: 957-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Barker JL (1977) Pentylenetetrazol and penicillin are selective antagonists of GABA-mediated post-synaptic inhibition in cultured mammalian neurones. Nature 267: 720-721. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS (2000) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6: 159-163. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Traynelis SF, Dingledine R (1990) Regional variation of extracellular space in the hippocampus. Science 249: 674-677. [DOI] [PubMed] [Google Scholar]
- Medina AE, Manhaes AC, Schmidt SL (2001) Sex differences in sensitivity to seizures elicited by pentylenetetrazol in mice. Pharmacol Biochem Behav 68: 591-596. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Veruki ML, Torp R, Haug FM, Laake JH, Nielsen S, Agre P, Ottersen OP (1998) Aquaporin-4 water channel protein in the rat retina and optic nerve: polarized expression in Muller cells and fibrous astrocytes. J Neurosci 18: 2506-2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA (2003) New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26: 523-530. [DOI] [PubMed] [Google Scholar]
- Newman EA (1993) Inward-rectifying potassium channels in retinal glial (Muller) cells. J Neurosci 13: 3333-3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA (2001) Calcium signaling in retinal glial cells and its effect on neuronal activity. Prog Brain Res 132: 241-254. [DOI] [PubMed] [Google Scholar]
- Newman EA, Frambach DA, Odette LL (1984) Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science 225: 1174-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C (1993) Ion-selective microelectrodes and diffusion measurements as tools to explore the brain cell microenvironment. J Neurosci Methods 48: 199-213. [DOI] [PubMed] [Google Scholar]
- Nicholson C (2000) Volume transmission in the year 2000. Prog Brain Res 125: 437-446. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Phillips JM (1981) Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol (Lond) 321: 225-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Sykova E (1998) Extracellular space structure revealed by diffusion analysis. Trends Neurosci 21: 207-215. [DOI] [PubMed] [Google Scholar]
- Nicholson C, Tao L (1993) Hindered diffusion of high molecular weight compounds in brain extracellular microenvironment measured with integrative optical imaging. Biophys J 65: 2277-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan E, Stringer JL (1996) Influence of osmolality on seizure amplitude and propagation in the rat dentate gyrus. Neurosci Lett 207: 9-12. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS (2004) Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J 18: 1291-1293. [DOI] [PubMed] [Google Scholar]
- Piet R, Vargova L, Sykova E, Poulain DA, Oliet SH (2004) Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci USA 101: 2151-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, Brown EB, Izumi Y, Campbell RB, Berk DA, Jain RK (2001) Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci USA 98: 4628-4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopova-Kubinova S, Vargova L, Tao L, Ulbrich K, Subr V, Sykova E, Nicholson C (2001) Poly[N-(2-hydroxypropyl)methacrylamide] polymers diffuse in brain extracellular space with same tortuosity as small molecules. Biophys J 80: 542-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper SN, Obenhaus A, Dudek FE (1992) Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann Neurol 31: 81-85. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM (1998a) A tortuous and viscous route to understanding diffusion in the brain. Trends Neurosci 21: 469-470. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM (1998b) Geometric and viscous components of the tortuosity of the extracellular space in the brain. Proc Natl Acad Sci USA 95: 8975-8980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM (1998c) Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci 18: 3158-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzkroin PA, Baraban SC, Hochman DW (1998) Osmolarity, ionic flux, and changes in brain excitability. Epilepsy Res 32: 275-285. [DOI] [PubMed] [Google Scholar]
- Seksek O, Biwersi J, Verkman AS (1997) Translational diffusion of macromolecule sized solutes in cytoplasm and nucleus. J Cell Biol 138: 131-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoham D, Glaser DE, Arieli A, Kenet T, Wijnbergen C, Toledo Y, Hildesheim R, Grinvald A (1999) Imaging cortical dynamics at high spatial and temporal resolution with novel blue voltage-sensitive dyes. Neuron 24: 791-802. [DOI] [PubMed] [Google Scholar]
- Solenov E, Watanabe H, Manley GT, Verkman AS (2004) Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am J Physiol Cell Physiol 286: C426-C432. [DOI] [PubMed] [Google Scholar]
- Sykova E (1997) The extracellular space in the CNS: its regulation, volume and geometry in normal and pathological neuronal function. Neuroscientist 3: 28-41. [Google Scholar]
- Sykova E, Mazel T, Vargova L, Vorisek I, Prokopova-Kubinova S (2000) Extracellular space diffusion and pathological states. Prog Brain Res 125: 155-178. [DOI] [PubMed] [Google Scholar]
- Tao L (1999) Effects of osmotic stress on dextran diffusion in rat neocortex studied with integrative optical imaging. J Neurophysiol 81: 2501-2507. [DOI] [PubMed] [Google Scholar]
- Tao L, Nicholson C (1996) Diffusion of albumins in rat cortical slices and relevance to volume transmission. Neuroscience 75: 839-847. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R (1989) Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol 61: 927-938. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Malhotra SK (1967) Extracellular space in the cerebral cortex of the mouse. J Anat 101: 197-207. [PMC free article] [PubMed] [Google Scholar]
- Verkman AS (2002) Solute and macromolecule diffusion in cellular aqueous compartments. Trends Biochem Sci 27: 27-33. [DOI] [PubMed] [Google Scholar]
- Verkman AS (2003) Diffusion in cells measured by fluorescence recovery after photobleaching. Methods Enzymol 360: 635-648. [DOI] [PubMed] [Google Scholar]
- Wasterlain CG, Torack RM (1968) Cerebral edema in water intoxication. II. An ultrastructural study. Arch Neurol 19: 79-87. [DOI] [PubMed] [Google Scholar]