

Abstract
Ionotropic glutamate receptors (iGluRs) in Caenorhabditis elegans are predicted to have high permeability for Ca2+ because of glutamine (Q) residues in the pore loop. This contrasts to the low Ca2+ permeability of similar iGluRs in principal neurons of mammals, because of an edited arginine (R) at the critical pore position in at least one channel subunit. Here, we introduced the R residue into the pore loop of a glutamate receptor subunit, GLR-2, in C. elegans. GLR-2(R) participated in channel formation, as revealed by decreased rectification of kainate-evoked currents in electrophysiological recordings when GLR-2(R) and the wild-type GLR-2(Q) were coexpressed in worms. Notably, the transgenic worms exhibited, at low penetrance, strong phenotypic impairments including uncoordination, neuronal degeneration, developmental arrest, and lethality. Penetrance of adverse phenotypes could be enhanced by transgenic expression of an optimal GLR-2(Q)/(R) ratio, implicating channel activity as the cause. In direct support, a mutation in eat-4, which prevents glutamatergic transmission, suppressed adverse phenotypes. Suppression was also achieved by mutation in calreticulin, which is necessary for maintainance of intracellular Ca2+ stores in the endoplasmic reticulum. Thus, synaptically activated GLR-2(R)-containing iGluR channels appear to trigger inappropriate, neurotoxic Ca2+ release from intracellular stores.
Keywords: glutamatergic synaptic transmission, necrotic-like cell death, transgenic expression, iGluR, intracellular calcium, phenotypic rescue
Introduction
From worm to man, glutamate activates ionotropic receptors (iGluRs), which mediate fast communication between nerve cells. Whereas invertebrate iGluR subunits carry a glutamine (Q) residue in a pore loop position identified as controlling ion permeability, for most mammalian principal neurons, iGluRs incorporate a subunit with an arginine (R) residue in this critical pore loop position (Burnashev et al., 1992). This R residue is introduced by RNA editing (Seeburg et al., 1998), a posttranscriptional process also operative in invertebrates, but not targeting iGluR transcripts (Palladino et al., 2000; Tonkin et al., 2002; Hoopengardner et al., 2003). Although mammalian iGluRs incorporating the R-form subunit become Ca2+ impermeable, with iGluR-mediated currents that have a linear current-voltage (I-V) relationship, iGluRs assembled from subunits with Q at the critical pore loop position are Ca2+ permeable and rectifying (Burnashev et al., 1992; Washburn et al., 1997). These functional properties of mammalian iGluRs, governed by the R-form subunit, are essential for CNS physiology, as demonstrated by early seizure-related death of mice in which part of the R-form subunit population is expressed in the unedited Q-form (Brusa et al., 1995; Feldmeyer et al., 1999). As indicated by recent studies (H. E. Krestel, personal communication), this R/Q change for the population of iGluR subunits normally only expressed in the R-form leads to pathological signaling, resulting in hyperactive principal neurons.
Because Caenorhabditis elegans serves as a powerful model organism, we used the worm to investigate the functional contribution to glutamate signaling of an iGluR subunit in its R-form. Of the 10 iGluR-related C. elegans genes, seven of which are shown in partial alignment with two mammalian pore-forming domains (see Fig. 1A), glutamate receptor (GLR)-1 and GLR-2 are most closely related to subunits of the mammalian iGluR subtype mediating nearly all fast excitatory neurotransmission (Sprengel et al., 2001) and are frequently coexpressed (Brockie et al., 2001). In the AVA interneurons, fast glutamate-activated whole-cell currents are absent in either a glr-1 or glr-2 mutant background (Mellem et al., 2002), so these subunits are likely to form heteromeric channels. GLR-2 is expressed in ∼25 neurons in C. elegans, and in a subset of these neurons, GLR-2 may be the only iGluR (Brockie et al., 2001), suggesting that it might also be able to form homomeric channels in these cells. We therefore chose GLR-2 as the best candidate to investigate iGluR signaling by expression of GLR-2(R).
Figure 1.
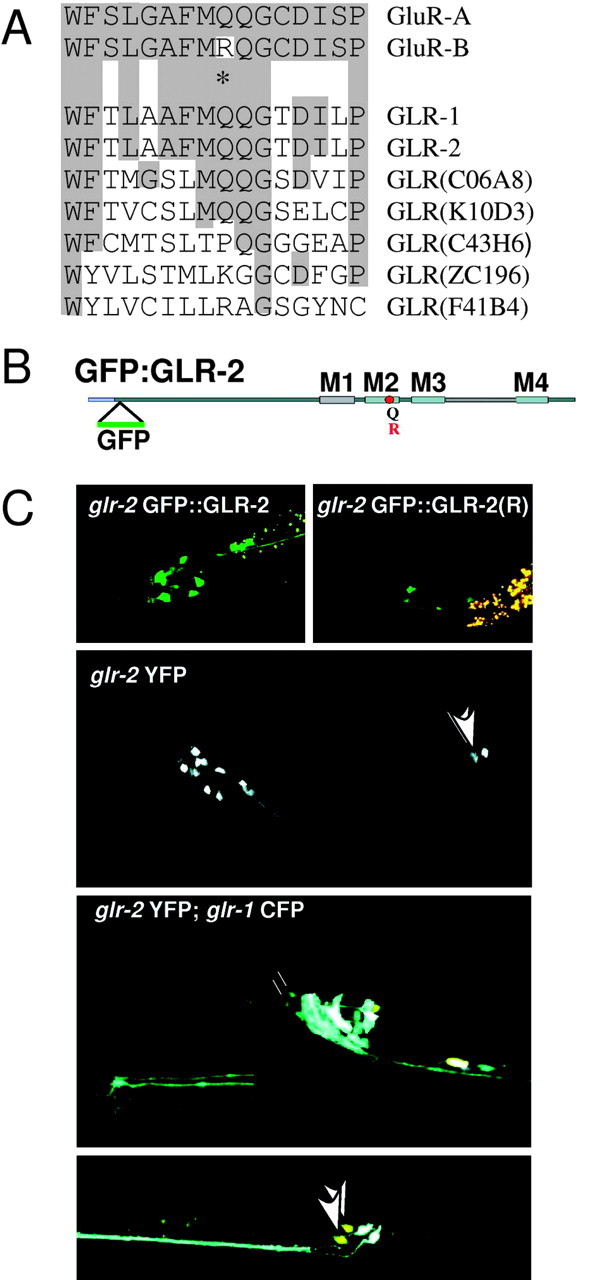
GFP::GLR2(Q)/(R) in C. elegans. A, Alignment of the M2 pore-forming domain from mouse GluR-A and GluR-B subunits and seven C. elegans glr genes, with the Q/R position marked by an asterisk. B, Graphic depiction of GFP::GLR-2, with the Q/R site and membrane domains M1-M4. C, Examples of transgenic worms expressing the glr-2-derived transgenes. GFP::GLR-2 and integrated GFP::GLR-2(R) in the wild-type background are shown above. The middle and bottom panels show expression of soluble YFP driven by the same glr-2 promoter used in the fusion constructs. In the middle panel, it is the only transgene expressed, whereas in the bottom panel, it is in a strain carrying an integrated glr-1::CFP soluble protein fusion. The inset (bottom left) shows URY cell terminal processes, which express glr-1 and not glr-2, in contrast to the M1 pharyngeal neuron process that runs down the middle of the pharynx and only expresses glr-2. Arrowheads point to the DVA neurons.
Although GLR-2(R) expressed as the only GLR-2 form had little, if any, effect on animals, we found that coexpression of GLR-2(R) and GLR-2(Q) elicited deleterious phenotypes that are not caused by glr-2 deletion alone (Mellem et al., 2002), including uncoordination, developmental arrest, and lethality. The classic glutamate-mediated excitotoxicity by excessive Ca2+ influx through glutamate-activated channels cannot explain these phenotypes. Indeed, electrophysiological data showed putative GLR-2(Q/R) channels act similarly to those of mammals containing the edited R-form subunit, exhibiting reduced currents and diminished rectification. Rather, the observed phenotypes were attributable to necrotic-like neuronal cell deaths, which appeared similar to those seen after dominant mutation of other ion channels in C. elegans (Xu et al., 2001). Genetic interference with either glutamate in synaptic vesicules or Ca2+ stores in the endoplasmic reticulum (ER) permitted complete suppression of the adverse phenotypes. Thus, by expression of GLR-2(R) in the worm, we reveal cross talk between iGluR signaling and intracellular Ca2+ stores.
Materials and Methods
Worm culture and transgenic lines. C. elegans was cultured as described (Brenner, 1974). Transgenic worms were generated by standard methods with coinjection markers [including the rol-6 dominant collagen marker (Mello et al., 1991), myo-3::GFP (pPD93_ 97; a gift from A. Fire, Stanford University, Stanford, CA), and unc-47::dsRED2 (a gift from H. Hutter, Max Planck Institute for Medical Research, Heidelberg, Germany)] with similar phenotypes observed in at least six transgenic lines carrying extrachromosomal arrays for each test construct or after UV-mediated integration. The integrated glr-1::CFP strain was kindly provided by H. Hutter. C. elegans strains KP4 glr-1 (n2461), MT6308 eat-4 (ky5), ZB1029 crt-1 (bz30) and ZB1031 crt-1 (bz50) were from the Caenorhabditis Genetics Center.
Test DNAs. The Q/R variants of GFP::glr-2 were generated from expressed-sequence tag yk348g10 (a gift from Y. Kohara, National Institute of Genetics, Mishima, Japan) and thus lacked all glr-2 intronic and noncoding sequences. In-frame MluI-digested green fluorescent protein (GFP) DNA (from the plasmid pPD113.05; gift from A. Fire) was inserted downstream of the predicted signal sequence at an MluI site in the cDNA. This clone, p3.5, was also used for insertion of the R mutation by PCR with mutagenesis primers RA33 and RA38 and outside primers RA1 and RA3. An ∼650 bp Eco32I-Bsu15I fragment carrying the R codon for the Q/R site was substituted into the p3.5 vector. A putative glr-2 promoter comprising sequences upstream of the glr-2 coding region was PCR amplified from genomic DNA with primers RA6 and RA11, and an XhoI-HpaI fragment from this PCR product was blunt-ended and inserted in the sense orientation into XbaI-digested and blunt-ended p3.5 or p3.5(R), to generate GFP::glr-2(Q) and GFP::glr-2(R). The construct for soluble yellow fluorescent protein (YFP) was generated by inserting an Eco32I-BamHI promoter fragment into the HincII/BamHI-digested pVH20.01 promoterless YFP construct (a gift from H. Hutter). Test DNAs were injected at 20-200 ng/μl.
The primers (5′ to 3′) used included the following: RA1, CATGCCAGTTGCGTTTCAACGTGGTCACTG; RA2, TGCAGCCGTTGATCCACCTTCATTGACACC; RA3, TGTCCGACTTTCATTGTATCGCACGGTCTC; RA6, TACGTCGACGCATCACAAAGCAGCCACTGG; RA11, GACCGCGGACAACGCGTTGAATGTCATTTCC; RA33, GGCAGCGTTCATGCGACAAGGAAC; RA38, GTTCCTTGTCGCATGAACGCTGCC.
Nose touch assays. Individual animals were assayed as described previously (Kaplan and Horvitz, 1993). Worms were allowed 10 trials each and scored as responding if they stopped or backed up on collision with a fine hair. Worms expressing the unc-47::dsRed2 coinjection marker were tested blind. See supplemental material (available at www.jneurosci.org) for further details.
Rescue of glr-1/GLR-2(Q/R) lines by mutations in eat-4. Heterozygous eat-4/+ males were crossed with transgenic Roller hermaphrodites from glr-1 lines in strains KP4 glr-1(n2461), KB1 and KNA (with 1:3 Q/R ratio transgenes), and KRA [with only GFP:GLR-2(R)]. F2 progeny from singled F1s were picked based on the weak Dpy phenotype (because of the dpy-19 marker gene, tightly linked to glr-1) and for eat-4-specific nose-up browsing phenotype. Lines from Roller F3s, which bred true for these markers, were scored for incidence of uncoordinated (Unc) and lethal (Let) animals.
Imaging and analysis. Bright-field images were obtained with a camera (Intas, Göttingen, Germany) on a dissecting microscope (Stemi SVII; Zeiss, Oberkochen, Germany). Fluorescence and Nomarski images were from a Zeiss AxioplanII. Confocal images were from an SP2 system (Leica, Bensheim, Germany) with subsequent image processing with Imaris software and Photoshop. Cross progeny data and numerical analyses were in Excel with a standard t test for statistics.
Electrophysiology. Currents were recorded from AVA cells as described (Mellem et al., 2002). The pipette solution contained 1 mm spermine in addition to the usual intracellular solution. The strains used included Q:R 1:1 (VM1836), Q:R 1:3 (VM1826), and R alone (VM1724).
Results
GFP::GLR-2(Q)/(R) subunits form ligand-gated channels in neurons
For functional transgenic expression of GFP::GLR-2 subunits in C. elegans, we generated glr-2 cDNA transgenes for the Q-form (wild type) or R-form of GLR-2 tagged by GFP (Fig. 1B), using 3.5 kb of glr-2 upstream sequence to drive transcription. Transgenic expression of either GFP::GLR-2(Q) or GFP::GLR-2(R) in both the wild-type laboratory strain N2 and in a glr-2 deletion strain (Mellem et al., 2002) resulted in GFP signal in 25 neurons, as reported previously (Brockie et al., 2001), with one additional cell pair (the DVA neurons) consistently seen in the tail (Fig. 1C). Correct cellular expression driven by the glr-2 upstream sequence was confirmed in transgenic worms that expressed from this sequence the soluble YFP (glr-2::YFP) and coexpressed under the glr-1 promoter cyan fluorescent protein (glr-1::CFP).
To demonstrate that GFP::GLR-2 subunits in Q-form and R-form are incorporated into receptor channels and participate in glutamate receptor signaling, we obtained electrophysiological recordings from AVA interneurons of the locomotory control circuit (Mellem et al., 2002) in transgenic lines of glr-2 mutants carrying varying ratios of GFP::GLR-2(Q) and GFP::GLR-2(R) (Fig. 2). Kainate was used as agonist in these experiments to preclude interference from other glutamate-activated channels (Mellem et al., 2002), but results with glutamate were similar (data not shown). The GFP::GLR-2(Q) subunit expressed by itself restored currents in the glr-2 deletion background (Fig. 2A). A 1:1 Q/R ratio of GFP::GLR-2 led to a decrease in current amplitude, accompanied by a significantly reduced current rectification (Fig. 2B,C). By linear regression, the slope from -40 to 0 mV changed from (mean ± SD) 4.0 ± 0.5 (wild type; n = 6) to 1.1 ± 0.6 (1:1 Q/R; n = 7), whereas the slope from 0 to +40 mV was 1.0 ± 0.2 (wild type) and 0.6 ± 0.04 (1:1 Q/R). For the 1:3 Q/R ratio of GFP::GLR-2 and for the GFP::GLR-2(R) subunit alone, currents could be barely detected. In conclusion, channels in either Q or Q/R configuration are functional, but the R-form alone is unlikely to form operative channels.
Figure 2.

Electrophysiology of GLR-2(Q/R) channels in AVA interneurons. A, Representative whole-cell current responses to pressure-applied kainate (100 μm) in strains expressing GLR-2 variants. The holding potential was stepped from -60 to +40 mV. The bar above current traces indicates the period of agonist application. B, Current-voltage relationship for the representative traces. C, Rectification index (I-40/I+40); *p < 0.001.
Transgenic GFP::GLR-2(Q) subunits rescued impaired nose touch response in glr-2 deletion mutants
In vivo function of the GFP-tagged GLR-2 subunits in transgenic lines was tested by nose touch, a simple behavior characterized by the worm stopping or reversing after encountering an obstacle with the tip of its nose (Kaplan and Horvitz, 1993). The ASH sensory neurons, which mediate avoidance of noxious stimuli, signal via iGluRs in this response circuit, and both glr-1 and glr-2 deletion mutants are defective in this response (Hart et al., 1995; Maricq et al., 1995; Mellem et al., 2002). Notably, the behavioral defect of the glr-2 deletion mutant, but not the glr-1 mutant, was rescued by transgenic expression of GFP::GLR-2(Q), whereas GFP::GLR-2(R) failed to rescue (supplemental material, available at www.jneurosci.org). These results indicate that the native pore configuration of GFP::GLR-2 channels defined by the Q residue is functionally required in the worm. The GFP::GLR-2(R) subunit cannot provide wild-type function, consistent with electrophysiological results. The results further demonstrate gene specificity. Although glr-1 mutants exhibit similar nose touch defects, GLR-2 will not compensate for the loss of GLR-1 function in the nose touch response.
GFP::GLR-2(R) expression induced deleterious phenotypes in wild-type and in glr-1, but not glr-2, mutant worms
We observed that in the glr-2 deletion background, expression of neither GFP::GLR-2(Q) nor GFP::GLR-2(R) alone resulted in any apparent phenotype, but adverse phenotypes were seen when GFP::GLR-2(R) was expressed in the wild-type or glr-1 mutant background (Table 1). The worms exhibited Unc, clear (Clr), and Let phenotypes. GLR-2(R) expression in the glr-1 deletion background produced additional posterior defects with aberrant tail morphologies (Fig. 3A; supplemental material, available at www.jneurosci.org) and difficulty in molting. Thus, putative GLR-2(Q/R) channels may substitute for the missing GLR-1-containing channels and exacerbate the phenotypic effects.
Table 1.
Phenotypes from Q/R ratios of GLR-2 in different mutant backgrounds
|
Strain |
wt |
Glr-1 |
glr-2KO |
eat-4 |
crt-1 |
|---|---|---|---|---|---|
| GFP::GLR-2 | + | + | + | + | + |
| GFP::GLR-2(R) | UC | UCL | + | + | + |
| Q/R ratio 1:5 | n.d. | n.d. | −/+ | + | n.d. |
| Q/R ratio 1:3a | UCL | UCL | UCL | + | + |
| Q/R ratio 1:1 | UC | n.d. | +/− | + | n.d. |
| Q/R ratio 5:1 |
n.d. |
n.d. |
+ |
+ |
n.d. |
+, Healthy worms; +/−, very rare sick worms (≤1% of transgenic worms); −/+, rare sick worms (~2-5%); UC, Unc, Clr, and some Let worms (≥5%); UCL, Unc, Clr, Let, and strong developmental arrest (including posterior and molting defects, especially in the glr-1 background); n.d., not determined.
Elevated penetrance of phenotypes was observed in both the wild-type (wt) and knock-out (K0) glr-2 background. Copy numbers within transgene arrays were at least fivefold over the endogenous alleles, as estimated by single-worm PCR, and expression was generally consistent with the input Q/R ratio, as tested by sequence analysis of reverse transcription-PCR products (data not shown).
Figure 3.
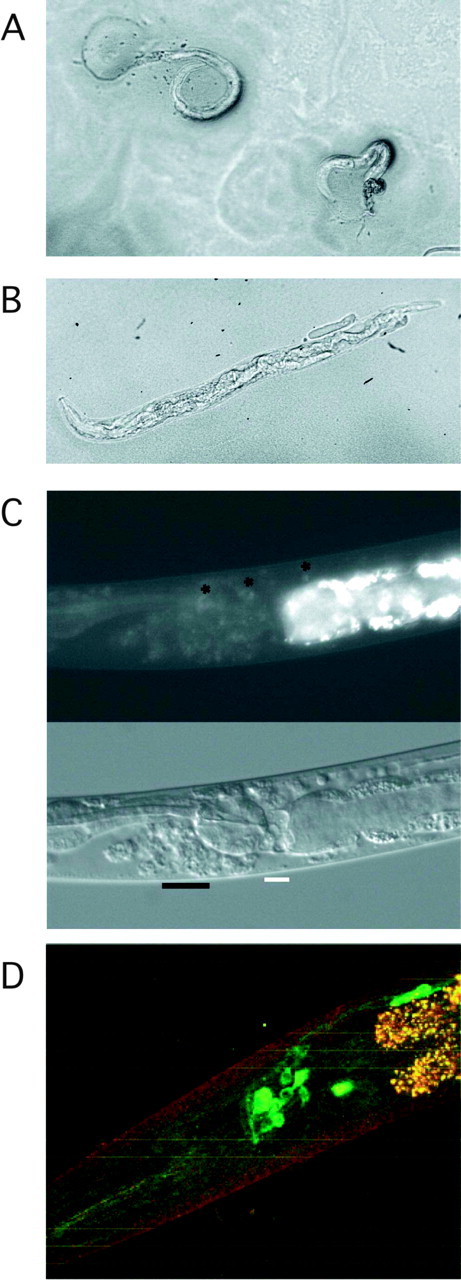
Images of representative animals. A, Young arrested larvae from the glr-1 mutant strain carrying a 1:3 Q/R transgene array. The animal on the left is virtually paralyzed, whereas the one on the right is relatively mobile. The aberrant posterior defect in both is readily apparent. (See also supplemental material, available at www.jneurosci.org.) B, Bright-field image of an Unc cross progeny from mating wild-type males with rescued eat-4 (mutant vGluT) hermaphrodites carrying a 1:3 Q/R transgene array after restoration of the wild-type vGluT. (See also supplemental material, available at www.jneurosci.org.) C, Nomarski and fluorescent images of an Unc glr-2 deletion animal. Degenerating cells and distortion of cell body positions are marked. The asterisks highlight a few relatively healthy cells with residual GFP signal. (See also supplemental material, available at www.jneurosci.org.) D, Confocal image of the head of an animal carrying a 1:3 Q/R transgene array in the mutant calreticulin (crt-1) background.
Deleterious phenotypes induced by GFP::GLR-2(R) require GLR-2(Q) subunits
The penetrance of phenotypes that we obtained by GFP::GLR-2(R) expression in wild-type animals was low. In six independent transgenic lines in the wild-type background, ∼5% of transgenic progeny were Unc, Clr, and/or Let. Notably, however, no phenotypes were observed in nontransgenic siblings, nor in transgenic worms expressing GFP::GLR-2(Q).
We therefore hypothesized that GLR-2(Q/R) channels are directly involved in the development of the phenotypes and that optimizing expression of these channels should enhance the penetrance of the phenotypes. To test this, different ratios of the transgenes GLR-2(Q) and GLR-2(R) were injected directly into the glr-2 deletion strain. Incidence and intensity of the phenotypes in the transgenic worms peaked at a Q/R ratio of 1:3 (Table 1), with phenotypes observed in up to 25% of the animals, along with developmental arrest. Moreover, the number of transgenic lines obtained for this ratio relative to all others was reduced by almost threefold (from, on average, 11 to 4%; n = 40-70 primary transformants). This may reflect the developmental arrest observed. Observable phenotypes could thus be directly correlated with the expression of putative GLR-2(Q/R) channels.
Glutamatergic synaptic transmission is essential for GLR-2(Q/R) effects
To demonstrate that active glutamate receptors are directly involved in the expression of adverse phenotypes, we inhibited glutamatergic transmission. We tested the same Q/R ratios of glr-2 transgenes in a strain with a mutation in the eat-4 gene, which encodes a candidate vesicular glutamate transporter (vGluT) (Bellocchio et al., 2000; Takamori et al., 2000; Otis, 2001; for review, see Rand et al., 2000) required for glutamatergic synaptic function (Lee et al., 1999). We found this mutation fully suppressed the phenotypes caused by coexpression of the Q-form and R-form of GFP::GLR-2 (Table 1). It also allowed transgenic lines to be obtained at the expected frequency of ∼10%. Furthermore, crossing the wild-type eat-4 gene back into these rescued lines reinstated the deleterious effects of GFP::GLR-2(Q/R) expression (Fig. 3B; supplemental material, available at www.jneurosci.org). Strikingly, the eat-4 mutation also suppressed even the most severe phenotypes in the glr-1 mutant. This strongly supports the hypothesis that glutamatergic synaptic transmission, most likely via direct activation of GLR-2(Q/R) channels, is crucial for development of the adverse phenotypes.
GLR-2(Q/R) channels induce neurotoxicity
In worms expressing GLR-2(Q/R) channels, the altered glutamate receptor signaling seems to trigger necrotic-like cell death. Examination with Nomarski optics of affected worms revealed dead and dying neuronal cells that by position and morphology appeared to be cells that normally express glr-2. GFP signal diminished as cells degenerated, starting with cell body swelling and sometimes leaving large holes or vacuoles (Fig. 3C; supplemental material, available at www.jneurosci.org). It seems likely that the Unc phenotype finds its origin in necrosis of the glr-2 expressing command interneurons and the larval arrests in loss of the M1 pharyngeal neuron. Worms are known to be quite tolerant to impairment of neuronal function, and death of other glr-2-expressing cells might not result in overt phenotypes, thus helping to explain the low incidence phenotypes observed from GLR-2(Q/R) expression.
GLR-2(Q/R)-mediated neurotoxicity is induced via intracellular calcium stores
To provide evidence for pathogenic iGluR signaling by GLR-2(Q/R) expression, we again used a genetic approach. In the worm, mutations in calreticulin can be neuroprotective. In case of the hyperactivated MEC-4(d) degenerin ion channel (Driscoll and Chalfie, 1991), elevated Na+ influx leads to the release of Ca2+ from ER stores. The resultant intracellular calcium increase is essential for downstream events that cause cellular demise, which the calreticulin mutant prevents (Xu et al., 2001). When analyzed, calreticulin mutant worms from independent transgenic lines expressing the 1:3 Q/R ratio of GFP::GLR-2 exhibited no neuronal swelling or degeneration, although the expected GFP fluorescence was evident in glr-2-expressing cells (Fig. 3D). Transgenic lines were also obtained at expected frequencies. Thus, as for the mutant Na+ channel MEC-4(d), ligand-activated GLR-2(Q/R) channels generate a signal that provokes release of Ca2+ from ER stores.
Discussion
We generated a glr-2 gene that encodes an R residue for the critical position of the conserved iGluR pore-forming domain. The GFP::GLR-2(R) mutant was expressed normally in the animal and caused no apparent phenotype in the glr-2 deletion strain. In contrast to the results with the wild-type GFP::GLR-2(Q), no phenotypic rescue of glr-2 deletion mutants by the R mutant was observed, as measured by the nose touch assay. However, in the wild-type background, GFP::GLR-2(R) expression caused lethality accompanied by the Unc and Clr phenotypes. Increased penetrance of phenotypes was obtained when GFP::glr-2(Q) and GFP::glr-2(R) transgenes were at an optimal ratio. Because Q/R ratios within a circumscribed range were most efficacious, it seems most likely that both Q and R subunits must be incorporated together into a channel for development of these phenotypes, reminiscent of what is seen after simultaneous expression of the Q-form and R-form by impaired Q/R site editing in the mouse (Brusa et al., 1995; Feldmeyer et al., 1999) (Krestel, personal communication).
The GLR-1 subunit, which is expressed in ∼40 neurons of the worm (Brockie et al., 2001), does not contribute to the Unc, Clr, and Let phenotypes. Moreover, in glr-1 mutant worms, coexpression of the endogenous GLR-2(Q) and transgenic GFP::glr-2(R) produced additional phenotypes, most likely mediated by neurons that usually coexpress GLR-1 and GLR-2. It is possible that GLR-1-containing channels can compensate or override the effects leading to cell death mediated by activated GLR-2(Q/R).
In C. elegans, as in the mammalian system, iGluR channel assemblies contributing R and Q to the channel pore have a strong effect on the electrophysiological properties of the channel. We show GLR-2(Q)/(R)-expressing neurons exhibit a markedly reduced kainate response that, in analogy to the rodent AMPA receptors (Burnashev et al., 1992), may result from reduced single-channel conductance. The participation of GLR-2(R) in channel formation could also be monitored by the diminished rectification seen in the I-V curve and by the significantly decreased rectification index in cells coexpressing the 1:1 ratio of GLR-2(Q) and GLR-2(R). Thus, the R residue in the GLR-2 channel pore affects the same channel parameters described for the rodent subunits (Washburn et al., 1997). Therefore, we expect that in addition to channel conductance and rectification, the Ca2+ permeability of GLR-2(R)-containing channels is strongly reduced.
By using an eat-4 mutation, a loss-of-function mutation of the vGluT, we could show that glutamatergic synaptic transmission is crucial for the development of the adverse phenotypes. The eat-4 mutation not only suppressed the adverse phenotypes caused by GLR-2(Q)/(R) coexpression but also suppressed the most severe phenotypes in the glr-1 mutant background. Interestingly, because eat-4 is not expressed in the PVC neurons (Lee et al., 1999), posterior cells that coexpress both GLR-1 and -2, the observed phenotypic rescue supports the idea that eat-4 works in the presynaptic partner [with possible caveats as discussed in a previous report (Berger et al., 1998)] to supply ligand that activates the postsynaptic GLR-2(Q/R) channel and initiates neurotoxic effects. Furthermore, this result shows that the GLR-2(Q/R) channel activation is necessary to cause neuronal cell death. Nonspecific effects by overexpression of GFP::GLR-2(Q/R) can thus be excluded.
Reduced GLR-2 channel-mediated currents per se can also be excluded as the cause of the phenotypes, because glr-2 deletion mutants, which have very small glutamate-evoked currents, similar to those of the 1:3 Q/R ratio, do not cause adverse effects in the animal. Rather, the phenotypes are explained by the necrotic-like neuronal cell deaths of GLR-2(Q/R)-expressing neurons. As shown by the phenotypic rescue with calreticulin-deficient worms, the activated release of intracellular Ca2+ stores is the ultimate cause for this cellular degeneration, which is in analogy to mammalian models in which neurodegeneration has also been linked to intracellular ER-based Ca2+ stores (Mattson et al., 2000). Thus, ligand-activated GLR-2(Q/R) channels induce ER-driven Ca2+ release, which in turn triggers necrotic cell death. The predicted strongly reduced Ca2+ flux of activated GLR-2(Q/R) channels might induce ER signaling. Alternatively, a PDZ [Postsynaptic density-95/Discs large/Zona occludens-1] consensus binding site (Saras and Heldin, 1996) at the C terminus of GLR-2 (amino acids STLF) could provide an initial physical linkage between glutamate receptor and Ca2+ stores.
These data demonstrate that in C. elegans, Q/R site substitution in the pore loop of GLR-2 triggers adverse effects. These data, furthermore, underscore the importance of appropriate regulation of the activation of iGluR channels and/or ionic flux. In one sense, GLR-2(R) acts as a dominant negative by requiring GLR-2(Q) for effects. Perhaps this too is the case in mammals, particularly if unedited subunits are present. In contrast to worm neurons, however, which appear to require Ca2+ influx by iGluRs, principal neurons in mammals require the edited pore loop residue for correct subunit composition, gating, and conductance properties of their AMPA channels (Seeburg et al., 1998; Greger et al., 2003). Thus, Q/R site editing of iGluRs appeared late in evolution and has become essential for the complex CNS of mammals (Brusa et al., 1995; Higuchi et al., 2000). Collectively, our results indicate that, in addition to depolarizing the membrane after channel activation, iGluR activation can impinge on intracellular Ca2+ signaling.
Footnotes
We thank the Caenorhabditis Genetics Center for strains; Harold Hutter (Max Planck Institute for Medical Research, Heidelberg, Germany) for strains, constructs, reagents, and more; and Nektarios Tavernarakis (Institute of Molecular Biology and Technology, Crete, Greece) for the suggestion to test calreticulin mutants.
Correspondence should be addressed to Dr. Rachel Aronoff, Life Sciences, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: rachel.aronoff@epfl.ch.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248135-06$15.00/0
References
- Bellocchio EE, Reimer RJ, Fremeau Jr RT, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289: 957-960. [DOI] [PubMed] [Google Scholar]
- Berger AJ, Hart AC, Kaplan JM (1998) Gα-induced neurodegeneration in Caenorhabditis elegans J Neurosci 18: 2871-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans Genetics 77: 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockie PJ, Madsen DM, Zheng Y, Mellem J, Maricq AV (2001) Differential expression of glutamate receptor subunits in the nervous system of Caenorhabditis elegans and their regulation by the homeodomain protein UNC-42. J Neurosci 21: 1510-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusa R, Zimmermann F, Koh DS, Feldmeyer D, Gass P, Seeburg PH, Sprengel R (1995) Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270: 1677-1680. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B (1992) Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189-198. [DOI] [PubMed] [Google Scholar]
- Driscoll M, Chalfie M (1991) The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349: 588-593. [DOI] [PubMed] [Google Scholar]
- Feldmeyer D, Kask K, Brusa R, Kornau HC, Kolhekar R, Rozov A, Burnashev N, Jensen V, Hvalby O, Sprengel R, Seeburg PH (1999) Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat Neurosci 2: 57-64. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40: 763-774. [DOI] [PubMed] [Google Scholar]
- Hart AC, Sims S, Kaplan JM (1995) Synaptic code for sensory modalities revealed by C. elegans GLR-1 glutamate receptor. Nature 378: 82-85. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78-81. [DOI] [PubMed] [Google Scholar]
- Hoopengardner B, Bhalla T, Staber C, Reenan R (2003) Nervous system targets of RNA editing identified by comparative genomics. Science 391: 832-836. [DOI] [PubMed] [Google Scholar]
- Kaplan JM, Horvitz HR (1993) A dual mechanosensory and chemosensory neuron in Caenorhabditis elegans Proc Natl Acad Sci USA 90: 2227-2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Sawin ER, Chalfie M, Horvitz HR, Avery L (1999) EAT-4, a homolog of a mammalian sodium-dependent inorganic phosphate cotransporter, is necessary for glutamatergic neurotransmission in Caenorhabditis elegans J Neurosci 19: 159-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maricq AV, Peckol E, Driscoll M, Bargmann CI (1995) Mechanosensory signalling in C. elegans mediated by the GLR-1 glutamate receptor. Nature 378: 78-81. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD (2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23: 222-229. [DOI] [PubMed] [Google Scholar]
- Mellem JE, Brockie PJ, Zheng Y, Madsen DM, Maricq AV (2002) Decoding of polymodal sensory stimuli by postsynaptic glutamate receptors in C. elegans Neuron 36: 933-944. [DOI] [PubMed] [Google Scholar]
- Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis TS (2001) Vesicular glutamate transporters in cognito. Neuron 29: 11-14. [DOI] [PubMed] [Google Scholar]
- Palladino MJ, Keegan LP, O'Connell MA, Reenan RA (2000) A-to-I premRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102: 437-449. [DOI] [PubMed] [Google Scholar]
- Rand JB, Duerr JS, Frisby DL (2000) Neurogenetics of vesicular transporters in C. elegans FASEB J 14: 2414-2422. [DOI] [PubMed] [Google Scholar]
- Saras J, Heldin CH (1996) PDZ domains bind carboxy-terminal sequences of target proteins. Trends Biochem Sci 21: 455-458. [DOI] [PubMed] [Google Scholar]
- Seeburg PH, Higuchi M, Sprengel R (1998) RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res Brain Res Rev 26: 217-229. [DOI] [PubMed] [Google Scholar]
- Sprengel R, Aronoff R, Volkner M, Schmitt B, Mosbach R, Kuner T (2001) Glutamate receptor channel signatures. Trends Pharmacol Sci 22: 7-10. [DOI] [PubMed] [Google Scholar]
- Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407: 189-194. [DOI] [PubMed] [Google Scholar]
- Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL (2002) RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans EMBO J 21: 6025-6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Numberger M, Zhang S, Dingledine R (1997) Differential dependence on GluR2 expression of three characteristic features of AMPA receptors. J Neurosci 17: 9393-9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31: 957-971. [DOI] [PubMed] [Google Scholar]