

Abstract
In vitro studies have provided evidence thatβ1 integrins in motor neurons promote neurite outgrowth, whereasβ1 integrins in myotubes regulate acetylcholine receptor (AChR) clustering. Surprisingly, using genetic studies in mice, we show here that motor axon outgrowth and neuromuscular junction (NMJ) formation in large part are unaffected when the integrin β1 gene (Itgb1) is inactivated in motor neurons. In the absence of Itgb1 expression in skeletal muscle, interactions between motor neurons and muscle are defective, preventing normal presynaptic differentiation. Motor neurons fail to terminate their growth at the muscle midline, branch excessively, and develop abnormal nerve terminals. These defects resemble the phenotype of agrin-null mice, suggesting that signaling molecules such as agrin, which coordinate presynaptic and postsynaptic differentiation, are not presented properly to nerve terminals. We conclude that Itgb1 expression in muscle, but not in motor neurons, is critical for NMJ development.
Keywords: integrin, axon outgrowth, motor neuron, synapse formation, agrin, muscle
Introduction
Motor neurons encounter during their development extracellular matrix (ECM) glycoproteins, including isoforms of laminin (LN) and agrin, and they respond in vitro to ECM glycoproteins in distinct ways. LN1/2 promote neurite outgrowth (Tomaselli and Reichardt, 1988; Varnum-Finney et al., 1995; Weaver et al., 1995). In contrast, a neuron-specific splice variant of agrin and isoforms of LN that contain the β2 chain are deposited into the synaptic basal lamina and are growth inhibitory; these molecules may act as a “stop signal” for motor neurons and induce synapse formation (Campagna et al., 1995; Porter et al., 1995). The effect of LN isoforms on axon outgrowth has not been studied in vivo, but genetic evidence supports a role for specific isoforms of LN and agrin during synaptogenesis. Synaptic defects are observed at the neuromuscular junctions (NMJs) in mice null for the LN β2 and α4 chain genes (Noakes et al., 1995; Patton et al., 2001). In agrin-null mice the motor axons fail to terminate their growth at the muscle midline and exhibit poorly differentiated nerve terminals (Gautam et al., 1996). The receptors that mediate effects of ECM components on motor neurons are not well defined. ECM receptors of the β1 integrin family mediate the growth-promoting effects of LN1/2 in vitro (Tomaselli and Reichardt, 1988; Weaver et al., 1995). However, it is unclear whether β1 integrins are essential for axon outgrowth in vivo and whether they are required in motor neurons for the formation of NMJs.
ECM glycoproteins also regulate the assembly of the postsynaptic apparatus of the NMJ. Postsynaptic differentiation is initiated before innervation by motor neurons and can be visualized by the formation of acetylcholine receptor (AChR) clusters in the muscle membrane (Sanes and Lichtman, 2001). An agrin isoform that is secreted by motor neurons and the muscle-specific kinase (MuSK) that is activated by neural agrin are required to consolidate this AChR prepattern into stable postsynaptic specializations (Reist et al., 1992; DeChiara et al., 1996; Glass et al., 1996). Interestingly, MuSK, but not neural agrin, is essential for the initial formation of the AChR prepattern (Lin et al., 2001; Yang et al., 2001). This suggests that at early stages of skeletal muscle differentiation MuSK is activated by an agrin-independent mechanism. In myotubes in culture AChR clustering is initiated not only by agrin but also by LN1/2/4. Moreover, LN1/2/4 and agrin act synergistically to induce AChRs clustering, a process that is modulated by antibodies that recognize β1 integrins. Because LN, but not agrin, is present in early muscle, a LN/β1-dependent signaling pathway may regulate the development of the AChR prepattern; LN and neural agrin subsequently may cooperate to regulate postsynaptic differentiation (Martin and Sanes, 1997; Sugiyama et al., 1997; Montanaro et al., 1998; Burkin et al., 2000; Marangi et al., 2002; Willmann and Fuhrer, 2002).
To determine the function of β1 integrins in neuromuscular development, we have analyzed mice genetically engineered to lack expression of the Itgb1 gene in spinal motor neurons and in skeletal muscle, respectively. In contrast to expectations, we show that β1 integrins in motor neurons are not essential for axon outgrowth and for the innervation of skeletal muscle fibers. We have shown previously that β1 integrins in myoblasts regulate skeletal muscle fiber development (Schwander et al., 2003). AChR clusters still form in the Itgb1-deficient muscle fibers, but motor axons do not stop in the prospective endplate zone and fail to consolidate this AChR prepattern into stable synaptic contacts. These defects likely are caused by abnormal muscle fiber development that prevents normal adhesive and signaling interactions between motor neurons and muscle.
Materials and Methods
Mouse lines
The mouse lines and protocols for genotyping have been described as follows: Rosa26lacZflox (Mao et al., 1999), Itgb1+/- (Stephens et al., 1995), Itgb1flox (Graus-Porta et al., 2001), HSACre (Leu et al., 2003; Schwander et al., 2003), nestin-CRE (Tronche et al., 1999; Graus-Porta et al., 2001), and HB9-GFP mice (Lee et al., 2004).
Histology and immunohistochemistry
Sections and cells in culture. Nissl and LacZ staining were performed as described (Farinas et al., 1996; Graus-Porta et al., 2001). Cryostat sections and cells in culture were processed for immunohistochemistry as described (Schwander et al., 2003). Primary antibodies were as follows: mouse monoclonal anti-rapsyn (Froehner, 1984), mouse monoclonal anti-utrophin (provided by M. Ruegg, Biozentrum Basel, Basel, Switzerland), rabbit polyclonal anti-β-dystroglycan (also provided by M. Ruegg), rabbit polyclonal anti-integrin α7B subunit (provided by U. Mayer, University of Manchester, Manchester, UK), mouse monoclonal anti-glial fibrillary acidic protein (GFAP; Dako, High Wycombe, UK), and mouse monoclonal anti-synaptophysin (Dako, Glostrup, Denmark). The β1-specific antibody was raised in rabbits against a peptide encompassing the cytoplasmic domain of the β1 integrins. For immunofluorescence, FITC-labeled secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were mixed with TRITC-labeled α-bungarotoxin (Molecular Probes, Eugene, OR). Images were collected on a Deltavision microscope and processed by deconvolution.
Whole-mount neurofilament/α-bungarotoxin staining. Embryos were placed into Ca- and Mg-free PBS and eviscerated; small tail pieces were cut for genotyping. Subsequent steps were performed at 4°C. Rib cages and diaphragm muscle were stained for 2 hr with TRITC-labeled α-bungarotoxin (diluted 1:1000; Molecular Probes) in L-15 medium. Samples were washed four times for 15 min with PBS, fixed for 5 min at -20°C in methanol, washed for 10 min with PBS, blocked, and permeabilized for 2 hr in PBS containing 5% horse serum, 1% BSA, 1% Triton X-100, and 0.1% sodium azide (blocking solution). Samples were incubated for 2 d with neurofilament antibody (diluted 1:200; Chemicon, Temecula, CA) in blocking solution, washed at least four times for 2 hr with PBS containing 0.2% Triton X-100, and incubated overnight with FITC-conjugated secondary antibody (diluted 1:200; Jackson ImmunoResearch) in blocking solution. Samples were washed at least four times for 2 hr with PBS containing 0.2% Triton X-100. Muscles were dissected from rib cages and mounted in Vectashield (Vector Laboratories, Burlingame, CA). The size and number of AChR clusters were determined in three diaphragm muscles each from embryonic day 14.5 (E14.5) wild-type and mutant embryos. To quantify clusters (∼150/sample), we analyzed random fields (0.004 mm2) of muscle fibers in the medial diaphragm.
Whole-mount neurofilament/diaminobenzidine staining. Embryos were recovered, tail pieces were cut for genotyping, and the embryos were eviscerated; heads were sliced open dorsally to allow for antibody penetration. Embryos were fixed overnight at 4°C in methanol/DMSO (4:1), and bleached for 6 hr at 4°C with fixative/30% H2O2 (5:1). Samples were rehydrated by successive 1 hr incubations at room temperature (RT) in 80, 50, 30, and 0% methanol in PBS/0.25% Triton X-100. The samples were incubated for 2 d at 4°C in blocking solution [fetal calf serum (Invitrogen, Gaithersburg, MD)/DMSO (4:1), 0.1% thimerosal] containing a 1:1000 dilution of α-neurofilament antibody (Chemicon). After six 1 hr washes in PBS the embryos were incubated overnight at 4° Cin blocking solution containing a 1:200 dilution of horseradish peroxidaseconjugated goat anti-mouse IgG secondary antibody (Pierce, Rockford, IL). After six 1 hr washes at RT in PBS the embryos were incubated for 1 hr at RT in 0.3 mg/ml diaminobenzidine and 5 mg/ml NiCl2. H2O2 was added to 0.03%. The reaction was stopped after 10-30 min by washing the samples with PBS.
Spinal motor column explants and primary muscle cell cultures
Explant cultures from E10.5 embryos were prepared for neurite outgrowth assays. The ventral third of the spinal cords at forelimb level were dissected, treated for 1 hr on ice with 0.5% trypsin in Ca2+/Mg2+-free Hanks' solution, and transferred into ice-cold DMEM/F-12 (Invitrogen, Carlsbad, CA) supplemented with 25% fetal bovine serum. After complete removal of the floor plate the motor column explants were placed on coverslips double-coated with poly-d-lysine and 10 μg/ml LN1/2 (Invitrogen) in 24-well culture plates (Corning, Acton, MA). Culture conditions were essentially the same as previously described (Shirasaki et al., 1998). After 24 hr in culture the explants were fixed with 4% paraformaldehyde in PBS containing 10% sucrose. Neurites extending from the explants were stained with TRITC-labeled phalloidin (Molecular Probes). The length and number of neurites were determined in four motor column explants each from wild-type and mutant embryos. Statistical significance was evaluated by Student's t test.
Myoblasts were isolated from hindlimb muscle, cultured, and induced to differentiate into myotubes as described (Schwander et al., 2003). AChR clustering was induced by adding a C-terminal fragment of the neuronal isoform of chick agrin (200 pm-2nm) (Ruegg, 1996). Myotubes were analyzed 24 hr later by immunohistochemistry as described above.
Results
Cre mice for the inactivation of the Itgb1 gene in motor neurons and in muscle
To analyze the function of β1 integrins in the development of motor neurons and the formation of the NMJ, we have used three mouse lines that we have described as follows: (1) Itgb1flox mice, which carry an allele of the Itgb1 gene suitable for Cre-mediated gene inactivation (Graus-Porta et al., 2001); (2) HSA-Cre mice, which express Cre in developing skeletal muscle (Schwander et al., 2003); (3) nestin-Cre mice, which express Cre in the developing nervous system (Tronche et al., 1999; Graus-Porta et al., 2001). In HSA-Cre mice Cre is expressed by E9.5 in somites in cells that are committed to the myogenic lineage and stays high throughout skeletal muscle development (Leu et al., 2003; Schwander et al., 2003). In nestin-Cre mice Cre is expressed by E10.5 in the developing cerebral cortex in the precursors of neurons and glia (Graus-Porta et al., 2001). To monitor Cre recombination in the spinal cord also, we crossed nestin-Cre mice with a reporter mouse line that carries a Rosa26lacZflox gene. In this line LacZ expression is activated by Cre recombination (Mao et al., 1999). Stainings of embryos in whole mount revealed that nestin-Cre induced recombination throughout the neural tube as early as E10.5 at a time when the first motor neurons become postmitotic (Fig. 1a). In histological sections of P3 mice the LacZ staining was evident throughout the spinal cord, with pronounced staining in the ventral motor neuron pools (Fig. 1b). LacZ-positive motor neurons were identified by their position and by the morphological appearance of their nuclei in Nissl staining (Fig. 1d, arrowheads). Additional expression was observed in sensory neurons of the dorsal root ganglia (DRGs) (Fig. 1b-d, asterisks). We conclude that the nestin-Cre mouse line used here induces recombination in developing spinal motor neurons and in sensory neurons of the DRGs.
Figure 1.

Analysis of the nestin-Cre mice. Nestin-Cre mice were crossed with Rosa26lacZflox/flox mice. The Cre recombination pattern was analyzed by LacZ staining (blue). a, Whole-mount staining at E10.5. LacZ was expressed throughout the entire neural tube (arrow). b, c, Sections of P3 animals. In transversal sections through spinal cord at forelimb (b) and hindlimb (c) levels LacZ expression was detected in the spinal cord. It was pronounced in motor neuron pools of the ventral horn (b, arrowhead). LacZ expression was also evident in dorsal root ganglia (asterisk). d, Higher magnification view of a transversal section through spinal cord at P3. Sections were stained with Nissl (violet) to label motor neuron nuclei and with LacZ (blue). Essentially all motor neurons (arrowheads) were lacZ-positive. Scale bars: a, 200 μm; b, c, 100 μm; d, 30 μm.
Inactivation of the Itgb1 gene in spinal motor neurons
To analyze the function of the Itgb1 gene in spinal motor neurons, we crossed Itgb1flox/flox mice with Itgb1+/-nestin-Cre mice. As previously described, the Itgb1-deficient offspring, called here nestin-Itgb1 Ko mice, are viable and fertile but display malformations in cortical structures in the CNS. Mutant mice are ataxic and fail to stretch their hindlimbs when picked up by the tail (Graus-Porta et al., 2001) (data not shown). Defects in the cerebellum or the motor cortex or perturbations in neuromuscular transmission could account for motor deficits.
To demonstrate first that the Itgb1 gene was expressed in motor neurons of wild-type embryos, we stained cross sections of E11.5 animals with an antibody to the integrin β1 subunit. The sections were prepared from wild-type embryos expressing an HB9-GFP transgene in motor neurons (Fig. 2a) (Lee et al., 2004). The β1 protein was expressed prominently in the cell bodies and axonal projections of GFP-positive motor neurons (Fig. 2a, thick arrow). Additional staining was observed in sensory neurons of the DRGs (Fig. 2a, thin arrows) and in skin (Fig. 2a, arrowhead). We next stained sections of nestin-Itgb1Ko embryos with antibodies directed against β-tubulin to visualize nerve fibers and with antibodies to the β1 integrin subunit (Fib. 2b). As expected, β1 expression in motor neurons (thick arrows) and sensory neurons (thin arrows) was reduced drastically in the mutants. We also performed Western blots with neural tube tissue from E12.5 embryos. Consistent with the immunohistochemical data, we readily observed β1 protein in extracts from wild-type embryos, but not in extracts from nestin-Itgb1Ko embryos (Fig. 2c).
Figure 2.
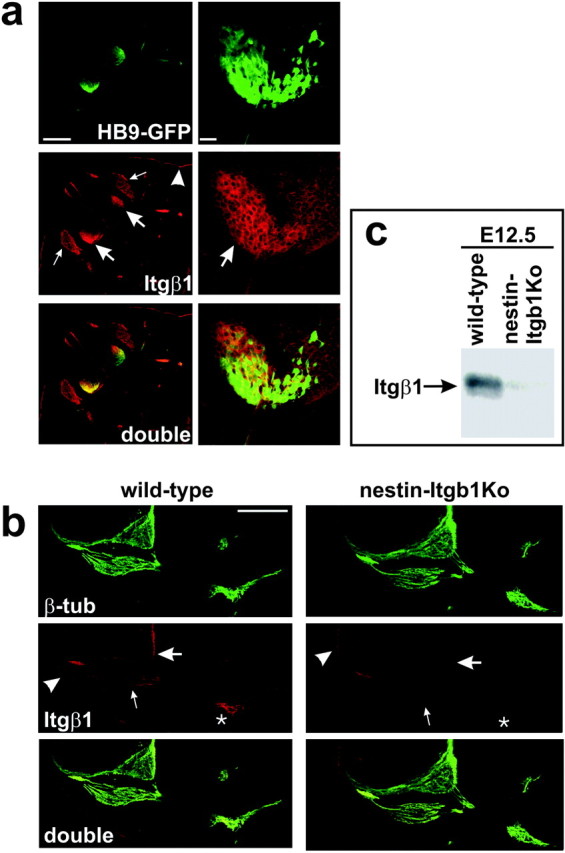
Expression and inactivation ofβ1 integrin expression in motor neurons. a, Transverse sections from E11.5 HB9-GFP embryos were analyzed for GFP fluorescence (green) and β1 expression (red). The GFP transgene is expressed specifically in the cell bodies and projections of spinal motor neurons. Prominent β1 expression was detected in motor neurons (thick arrows), in DRG neurons (thin arrows), and in skin (arrowheads). b, Transverse sections from E11.5 wild-type and nestin-Itgb1Ko animals were stained with antibodies to β-tubulin to visualize neurons and with antibodies to β1 integrin. The β1 expression was diminished drastically in motor neurons (thick arrows), DRG neurons (thin arrows), and peripheral nerve bundles (asterisks), but not in skin (arrowhead). c, Western blot analysis for the β1 protein. Extracts were prepared from the neural tube dissected from E12.5 wild-type and nestin-Itgb1Ko embryos. The β1 protein was readily detectable in wild-type mice, but not in mutant mice. Scale bars: a, left, 200 μm; a, right, 25 μm; b, 200 μm.
To provide additional evidence that β1 integrin expression was abolished in motor neurons of nestin-Itgb1Ko mice, we performed in vitro neurite outgrowth assays with ventral neural tube explants on purified LN1/2 substrates. Cultures obtained from mice expressing the HB9-GFP transgene demonstrated that the neurites that grew in culture were derived from motor neurons (data not shown). Extensive motor neurite outgrowth was observed in explants from wild-type mice, but it was diminished dramatically in explants from the nestin-Itgb1Ko mice (Fig. 3a). The number of neurites and neurite length were strongly reduced (Fig. 3c-e). The few neurites that were present in mutant explants tended to grow in fascicles (Fig. 3a). Occasionally, fibroblasts emigrated from explants onto the surrounding LN substrate. In these cultures extensive neurite outgrowth was observed on top of the fibroblasts layer, and there was no difference in neurite length in cultures derived from wild-type or nestin-Itgb1Ko mice (Fig. 3b,f).
Figure 3.

Neurite outgrowth assay in vitro. a, Phase-contrast and fluorescent images of spinal motor neuron explants prepared from E10.5 wild-type and nestin-Itgb1Ko embryos plated on poly-d-lysine/LN-coated coverslips. Wild-type explants formed numerous processes. In explants derived from nestin-Itgb1Ko embryos the neurite outgrowth was strongly impaired, and neurites formed fascicles (bottompanel). b, The growth of β1-deficient motor neurons was not impaired on cocultured fibroblasts. c, The mean number of neurites per explant on LN substrates was reduced significantly in nestin-Itgb1Ko as compared with wild-type embryos. d, The results of the means of neurite lengths per explant on LN substrates were calculated, demonstrating a strong reduction in neurite length between wild-type and nestin-Itgb1Ko explants; ***p = 0.0012. Error bars indicate SEM. The SEM was determined and a Student t test was performed. e, Cumulative frequency distribution plot of neurite length on LN substrates shows a shift for nestin-Itgb1Ko explants toward shorter neurite lengths. f, Cumulative frequency distribution plot of neurite length on fibroblast substrates shows no difference in neurite length between wild-type and nestin-Itgb1Ko explants. Scale bars: a, b, 120 μm.
We conclude that β1 integrins in spinal motor neurons are required for efficient neurite outgrowth on LN1/2 substrates. Consistent with earlier findings (Tomaselli et al., 1988; Drazba and Lemmon, 1990), neurite outgrowth on top of cells was β1-independent and likely promoted by additional cell surface molecules.
Development of peripheral nerves and motor neurons in the absence of β1 integrins
To examine the role of β1 integrins in the development of axonal projections in peripheral nerve, we performed whole-mount stainings of E11.5 and E12.5 embryos with antibodies to neurofilament (Fig. 4a) (data not shown). Axons that project toward intercostal and limb muscles were present in both wild-type and nestin-Itgb1Ko mice. Axon length and ramification were not affected in the mutants, suggesting that the rate of axon outgrowth was not impaired significantly in the absence of β1 integrins.
Figure 4.
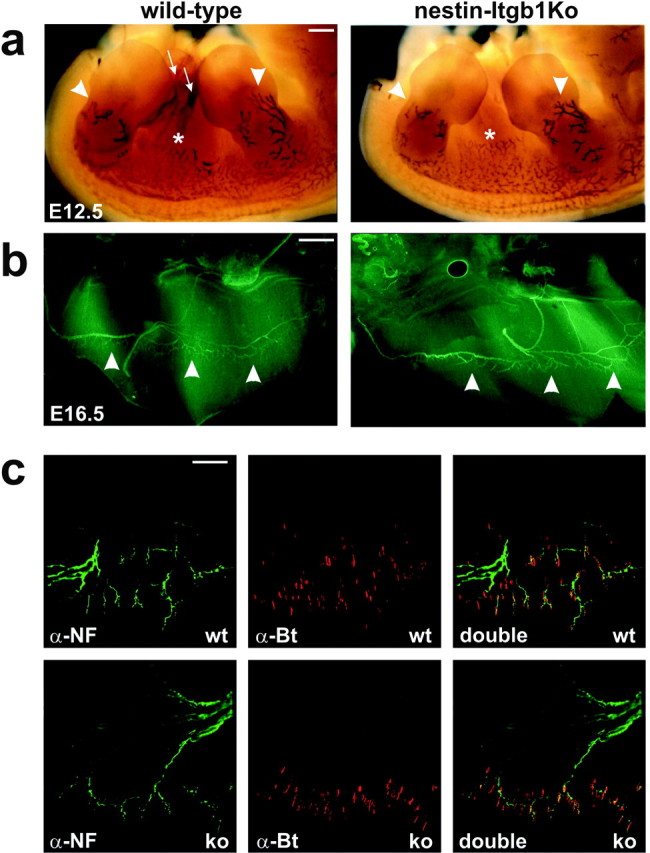
Analysis of axonal projections in vivo. a, Side views of E12.5 wild-type and nestin-Itgb1Ko embryos stained in whole mount with an antibody to neurofilament to visualize axonal projections in limbs (arrowheads) and the intercostal areas (asterisks). Staining occasionally was observed within the body cavity of the eviscerated embryos, likely caused by nonspecific trapping of antibodies (arrows). b, Top view of one-half of a diaphragm from a E16.5 wild-type and nestin-Itgb1Ko embryo stained for neurofilament (green) to reveal motor neurons. The phrenic nerve and its side branches that terminated in the central endplate zone (arrowheads) were present in wild-type and nestin-Itgb1Ko embryos. c, Higher magnification view of the diaphragm shown in b. Neurofilament staining is shown in green and α-bungarotoxin staining in red. AChRs were clustered at synaptic sites in the medial part of the muscle. Scale bars: a, 400 μm; b, 100 μm; c, 50 μm.
We next analyzed in detail the projection pattern of the phrenic nerve, which innervates the diaphragm muscle. Although there are differences in the branching pattern of the phrenic nerve even among wild-type littermates, its overall projection pattern is nevertheless stereotypic. After reaching the diaphragm, the phrenic nerve splits into three primary trunks. The two largest enter the muscle and then turn in opposite directions and run perpendicular to developing muscle fibers; the third runs dorsally to innervate the crus. Small groups of axons extend from the nerve trunks, branch further, and innervate muscle fibers (Greer et al., 1999). To examine the branching of intramuscular nerves and the distribution of synapses, we stained E16.5 whole mounts with neurofilament antibodies to visualize nerve fibers and with α-bungarotoxin to label AChR clusters at developing NMJs. The overall branching pattern was similar in wild-type and nestin-Itgb1Ko mice (Fig. 4b). Innervation was confined to a narrow band in the muscle midline, the endplate band, in which nerve terminals are in close apposition to postsynaptic AChR clusters (Fig. 4c).
We conclude that in the absence of β1 integrins in spinal motor neurons the outgrowth of peripheral nerve fibers was not impaired significantly. No gross defects were observed in the branching and innervation pattern of the phrenic nerve.
NMJ formation in the absence of β1 integrins in spinal motor neurons
Synaptic differentiation depends on interactions of nerve terminals with the synaptic basal lamina of muscle fibers (Sanes et al., 1978; Noakes et al., 1995), suggesting a role for presynaptic β1 integrins in this process. Nascent synapses were evident in the diaphragm muscle midline of wild-type and nestin-Itgb1Ko mice, as demonstrated by the formation of AChR clusters (Fig. 4c). The distribution of postsynaptic antigens was qualitatively normal in muscle from nestin-Itgb1Ko mice (Fig. 5a). The cytoplasmic protein rapsyn (Noakes et al., 1993) colocalized perfectly with AChR clusters at the NMJ. Components of the dystrophin/utrophin-associated glycoprotein complex, such as β-dystroglycan and utrophin (Bewick et al., 1992), but also the integrin α7β1 (Martin et al., 1996) were concentrated in the postsynaptic membrane of NMJs in wild-type and nestin-Itgb1Ko mice (Fig. 5a). In addition, the presence of the laminin α5 chain at mutant NMJs indicated that synaptic laminins were recruited into the synaptic basal lamina (data not shown). Together, these results provide evidence that the molecular assembly of the postsynaptic apparatus was not affected by the loss of β1 integrins in motor neurons.
Figure 5.

Analysis of presynaptic and postsynaptic differentiation. Sections of gastrocnemius muscle from 4-week-old wild-type and nestin-Itgb1Ko mice were stained with TRITC-α-bungarotoxin (red) and antibodies against presynaptic and postsynaptic proteins (green). a, The postsynaptic markers rapsyn (RAP), utrophin (UTP),β-dystroglycan (β-DG), and integrin α7B (Itg-α7B) were colocalized with AChR clusters at synaptic sites in wild-type and nestin-Itgb1Ko mice. b, GFAP-positive terminal Schwann cells apposing AChR clusters were clearly visible in longitudinal sections. c, Synaptophysin (SYN) was enriched in areas that were in register with AChR clusters in the postsynaptic membrane (arrowheads). Scale bars, 5 μm.
Next we analyzed the extent to which presynaptic differentiation was affected in the mutants. Defects in motor neurons often are reflected by changes in gene expression in terminal Schwann cells that cap the nerve terminals (Son et al., 1996). In particular, the expression of GFAP is upregulated in terminal Schwann cells in response to impaired synaptic transmission (Georgiou et al., 1994). In wild-type and nestin-Itgb1Ko mice the expression levels of GFAP were comparable, indicating that capping of nerve terminals and synaptic transmission probably were not affected (Fig. 5b).
Differentiated nerve terminals contain neurotransmitter vesicles near vesicle release sites, which align with AChR clusters in the postsynaptic membrane. No obvious difference in the expression of the synaptic vesicle protein synaptophysin was evident between nerve terminals of wild-type and nestin-Itgb1Ko mice (Fig. 5c). Synaptophysin was concentrated in patches that aligned with AChR aggregates (Fig. 5c, arrowheads). We therefore conclude that, in the absence of β1 integrins in motor neurons, the recruitment of a large number of presynaptic and postsynaptic proteins to NMJs was unaffected. These data provide evidence that β1 integrins in motor neurons are not essential to initiate assembly of the presynaptic and postsynaptic apparatus at the NMJ. However, we cannot exclude that recruitment of some proteins is affected, for example of calcium channels that have been shown to associate with the integrin α3β1 at the frog NMJ (Cohen et al., 2000). Additional studies will be necessary to address this possibility.
Defective nerve branching in the absence of β1 integrins in skeletal muscle
We recently have described the phenotype of mice in which expression of the Itgb1 gene has been inactivated in muscle with the use of a HSA-Cre mouse line (Schwander et al., 2003). The mutant mice, referred here to as HSA-Itgb1Ko mice, die at birth with muscle defects and non-inflated lungs. Overall muscle mass is reduced, and the remaining muscle fibers are short. Mutant embryos never move, do not respond to a tail pinch, and show a bent posture. These are characteristic features of flaccid paralysis. We therefore analyzed the innervation pattern and the development of neuromuscular synapses in the diaphragm muscle. In wild-type mice small-caliber axonal projections left the two major trunks of the phrenic nerve, formed secondary arbors, and established synaptic contacts with muscle fibers at the muscle midline (Fig. 6a,c,e). In HSA-Itgb1Ko mice the main nerve trunks as well as axonal side branches that invaded muscle were apparent. However, axons failed to stop and arborize at the prospective central endplate band and instead projected over long distances toward the borders of muscle (Fig. 6b,d,f). Excessive axonal branches and arbors were evident in those sections of nerve that had projected beyond the prospective central endplate band. Nerve defects were evident by E15.5 and became more pronounced at later embryonic stages (Fig. 6). Remarkably, defects were confined mainly to intramuscular portions of the nerve, supporting the idea that they were a result of defective interactions between nerve and muscle.
Figure 6.
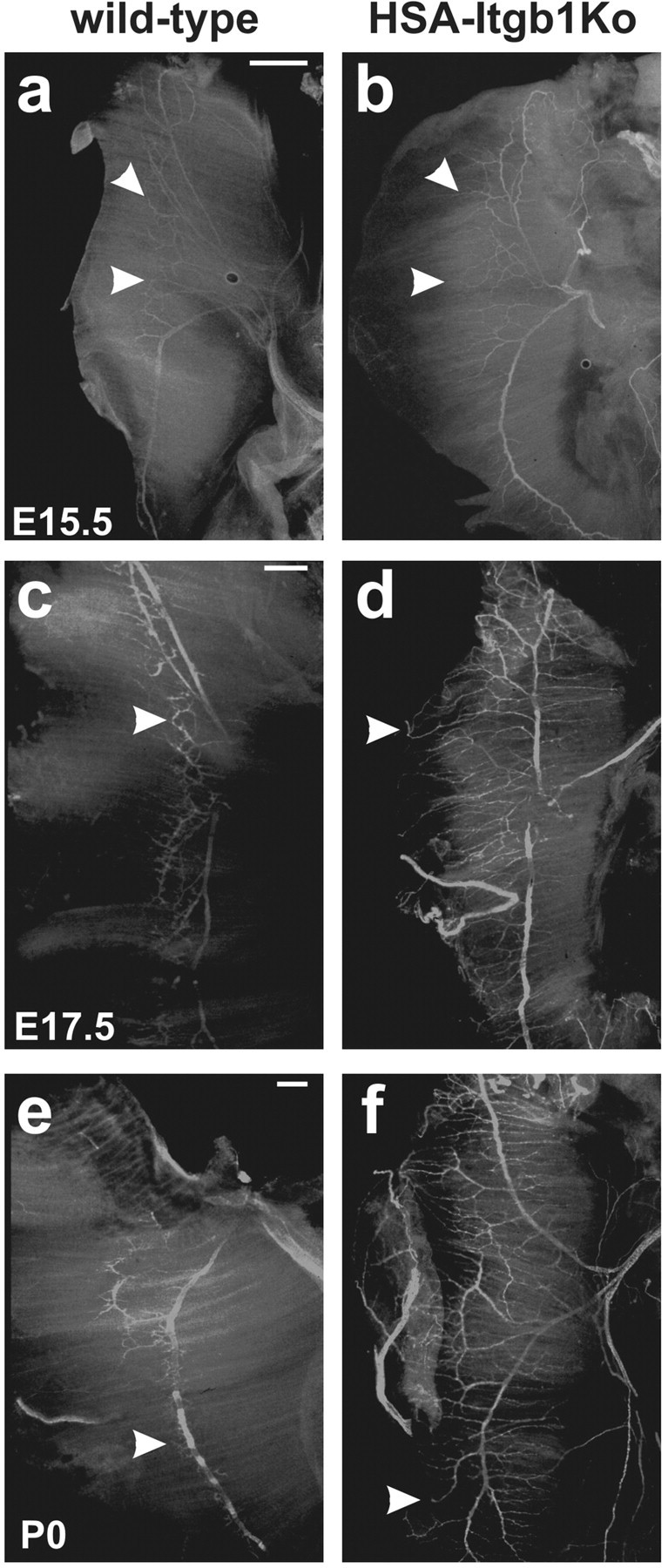
Defective innervation of β1-deficient muscle. a-f, Anti-neurofilament staining of the phrenic nerve innervating diaphragm muscle in wild-type and HSA-Itgb1Ko embryos. a, b, At E15.5 the motor axons extended beyond the medial endplate zone in the mutants(arrowheads). c,d, Severe nerve defasciculation and overgrowth were evident in mutant muscle at E17.5. e,f, The defects were pronounced even more at P0. In the mutants the axons occasionally failed to respect muscle boundaries and grew onto the tendon organ. Note that some axons had abnormal nerve terminals with a round morphology (d, f, arrowheads). Scale bars: a, b, 200 μm; c-f, 300 μm.
AChR prepatterning and maturation of NMJs
Defects in axon growth and arborization could be caused by defects in synapse formation. We therefore asked whether the loss of β1 integrins in muscle could interfere with the formation of AChR clusters in the muscle membrane before innervation by motor neurons or with the subsequent consolidation of this AChR prepattern into stable synaptic contacts.
AChR clusters that were concentrated in the medial part of the diaphragm muscle were evident at E14.5 in wild-type and HSAItgb1Ko mice (Fig. 7a). Many clusters were not opposed by nerve terminals because motor axon terminals had just started to reach muscle. The overall number of AChR clusters was not significantly different in wild-type and HSA-Itgb1Ko mice, but we observed a difference in the size distribution of AChR clusters (Fig. 7b). Although the number of clusters in the size range between 4 and 10 μm was comparable in wild-type and HSA-Itgb1Ko mice, the mutants had approximately twofold more clusters <2 μm and fourfold less clusters >12 μm. These findings are consistent with the interpretation that the AChR prepattern was established normally in HSA-Itgb1Ko mice by E14.5; the clusters remained small in the mutants and started to disperse, whereas clusters in wild-type mice started to be consolidated into larger AChR patches, presumably in response to neural agrin that was released from the first motor neuron terminals that had invaded muscle by E14.5.
Figure 7.
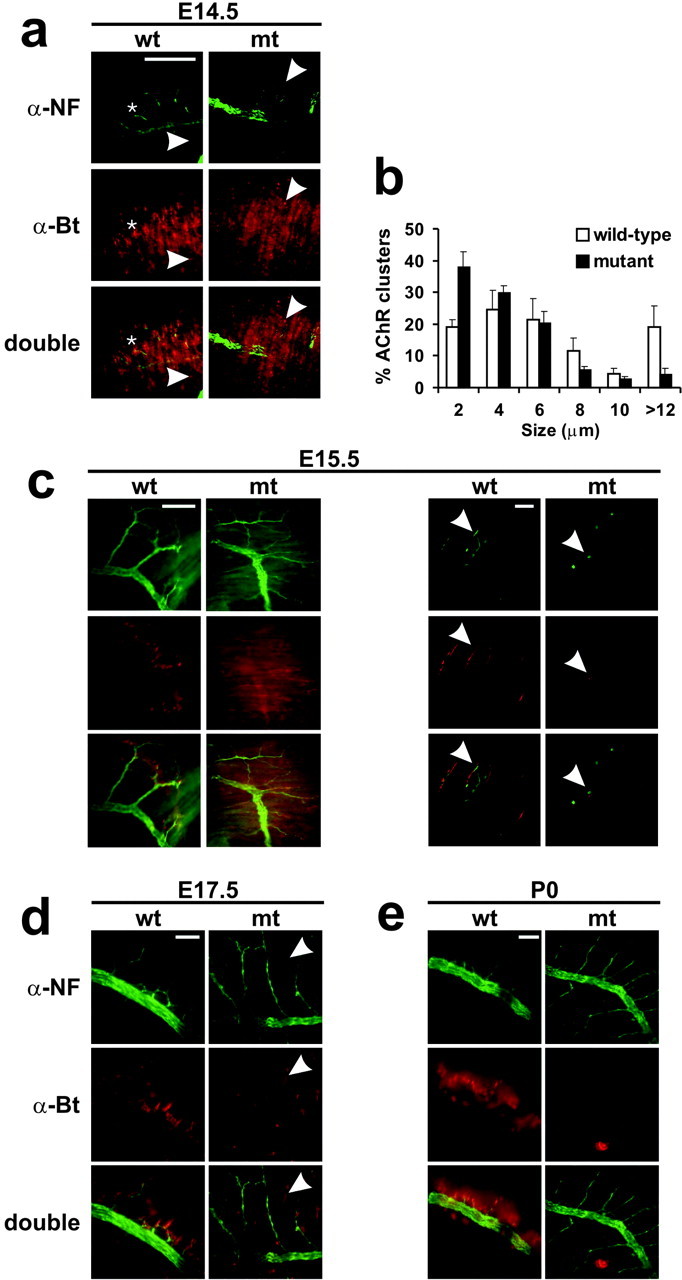
Defects in AChR clustering and presynaptic differentiation. Diaphragm muscles from wild-type and HSA-Itgb1Ko embryos were stained with antibodies to neurofilament (green) and with TRITC-α-bungarotoxin (red) to reveal motor neurons and AChRs, respectively. a, At E14.5 the clustering of AChRs was observed in a broad endplate band in muscle of wild-type and HSA-Itgb1Ko embryos. Small aneural AChR clusters were present (arrowheads) in muscle of wild-type and mutant animals, whereas bigger clusters that were opposed by nerve were visible only in wild-type animals (asterisks). b, The size distribution of AChR clusters was compared in diaphragm muscle from wild-type and HSA-Itgb1Ko embryos. (Figure legend continues.) (Figure legend continued.) Wild-type mice had a higher percentage of clusters >12 μm and fewer clusters <2 μm. Error bars indicate SEM. c, By E15.5 the AChR clusters in muscle in wild-type embryos had grown in size and were concentrated in the endplate zone, opposing flat terminal nerve boutons. In contrast, muscle of HSA-Itgb1Ko embryos displayed only a few and small AChR clusters, which were opposed by nerve terminals with a ball-like shape (arrowheads). d, At E17.5 some dispersed AChR clusters were present in muscle of mutant animals (arrowheads). e, At P0 all AChR clusters in muscle of wild-type embryos were opposed by nerve. Only a few small and weakly stained AChR clusters remained in the muscle of mutant animals. Motor axons grew extensively in P0 mutants. Scale bars: a, 100 μm; c, left, 100 μm; c, right, 5 μm; d, e, 100 μm.
At E15.5 many nerve fibers in wild-type mice had invaded the muscle, and AChR clusters were abundant and opposed by nerve terminals. In contrast, in HSA-Itgb1Ko mice substantially fewer clusters were evident (Fig. 7c). The AChR clusters in the mutants no longer were restricted to a medial band but scattered along the length of muscle fibers. They also were reduced markedly in size and staining intensity (Fig. 7c, arrowheads) and usually not opposed by nerve terminals. Motor nerve terminals were still visible, but they were abnormally shaped and not opposed by AChR clusters. Terminal boutons in the mutants frequently exhibited a ball-like shape or ended without apparent specializations, in contrast to the rather flat nerve endings in wild-type mice (Fig. 7c). At E17.5 many mutant muscle fibers bore no or few AChR clusters, and the clusters that formed were mostly not opposed by nerve (Fig. 7d). At birth the muscle in the mutant mice basically was devoid of synapses, suggesting that the AChR clusters had dispersed (Fig. 7e).
We conclude that in HSA-Itgb1Ko mice the AChR clusters form before innervation by motor neurons. However, the AChR prepattern is not consolidated in mature synaptic contacts. These defects in postsynaptic differentiation are accompanied by defects in presynaptic nerve terminals that show an aberrant morphology and fail to terminate their growth in the prospective endplate band. Similar defects have been reported for mice lacking agrin (Gautam et al., 1996; Lin et al., 2001; Yang et al., 2001), suggesting that agrin signaling may be defective in HSA-Itgb1Ko mice.
Agrin-dependent induction of AChR clusters in myotubes in culture
Previous in vivo and in vitro studies have shown that expression of MuSK and rapsyn in muscle fibers is essential for agrin-induced AChR clustering (Gautam et al., 1995; DeChiara et al., 1996; Glass et al., 1996). Function perturbation studies with β1 integrin-specific antibodies and myotubes in culture have provided evidence that β1 integrins modulate responses to agrin (Martin and Sanes, 1997). Therefore, defects in the maturation of NMJs in HSA-Itgb1Ko mice could be caused by defects in agrin signaling. To test genetically whether β1 integrins in muscle fibers are essential for the formation of AChR clusters in response to agrin, we cultured myoblasts from mutant and control embryos, allowed them to form myotubes, and treated them with recombinant agrin. As previously described, only a few myotubes formed in cultures containing β1-deficient myoblasts (Schwander et al., 2003). Nevertheless, small AChR clusters, so-called AChR “hot spots”(Anderson and Cohen, 1977; Frank and Fischbach, 1979), could be visualized in both wild-type and β1-deficient myotubes before the addition of agrin (Fig. 8a,b). In response to recombinant agrin, large and numerous AChR clusters formed that contained additional postsynaptic proteins such as utrophin (Fig. 8c-h). These data provide strong evidence that essential components of the agrin signaling pathway, such as MuSK/rapsyn (Gautam et al., 1995; DeChiara et al., 1996; Glass et al., 1996), were expressed in β1-deficient myotubes. The data also suggest that β1 integrins are not essential for agrin-induced postsynaptic differentiation. However, β1 integrins still may modulate responses to agrin, as previously demonstrated in function perturbation studies with β1 integrin-specific antibodies (Martin and Sanes, 1997). Unfortunately, we could not address this issue further because only a few β1-deficient myotubes formed in culture, and they showed defects in their cytoskeletal architecture (Schwander et al., 2003). Therefore, potential primary defects in agrin signaling could not be distinguished from secondary effects caused by structural defects in β1-deficient myotubes.
Figure 8.

The β1-deficient primary myotubes are responsive to recombinant agrin. Myotubes were generated in vitro with cells from wild-type (a, c, e, g) or HSA-Itgb1Ko embryos (b, d, f, h). Some myotubes (c-h) were treated for 24 hr with recombinant neural agrin. Cultures were stained with TRITC-α-bungarotoxin (a-d, red) or antibodies to utrophin (e-h, green) and DAPI (blue). Wild-type and β1-deficient myotubes formed few AChR clusters in the absence of recombinant agrin (a, b, arrow). Wild-type and β1-deficient myotubes were similarly responsive to recombinant agrin (c, d, arrows), and utrophin was concentrated at AChR clusters (e-h, arrows). Scale bar, 20 μm.
Together, the data suggest that defects in NMJ formation and nerve branching in HSA-Itgb1Ko mice are probably not a consequence of the inability of β1-deficient myotubes to respond to agrin. The data are more consistent with a model in which a retrograde signal that regulates presynaptic differentiation is not presented appropriately by β1-deficient muscle fibers to motor neurons.
Discussion
We show here that β1 integrins in motor neurons are in vivo not essential for axon outgrowth and the formation of NMJ. The β1 expression in skeletal muscle is also not essential for the formation of the AChR prepattern that develops before innervation by motor neurons. However, motor neurons fail to consolidate the AChR clusters into stable synaptic contacts and instead branch excessively on β1-deficient muscle fibers. These defects are likely a consequence of perturbations in interactions between motor axons and muscle fibers that are required to restrict the outgrowth of motor axons and to induce presynaptic differentiation.
The β1 integrins and motor axon outgrowth
In vitro studies with function-blocking antibodies have shown that integrins regulate neurite outgrowth of motor neurons on purified ECM substrates (Tomaselli and Reichardt, 1988; Varnum-Finney et al., 1995). Using motor neurons genetically modified to lack β1 integrin expression, we could confirm these findings. However, one major novel finding of our study is that β1 integrins in motor neurons are not essential for axon outgrowth and for synaptic differentiation during neuromuscular development in the in vivo environment. In nestin-Itgb1Ko embryos the outgrowth of peripheral nerve fibers was not obviously affected; the phrenic nerve invaded the diaphragm muscle, formed normal branches, and established synaptic contacts with muscle fibers. Importantly, β1-deficient neurons were impaired in their ability to grow neurites on LN substrates, but not on fibroblast monolayers; this shows that failure to grow neurites in vitro was not a consequence of a general inability of the cells to extend neurites but specifically of defects in a LN/integrin-dependent pathway. Function-blocking antibodies against β1 integrins and other cell surface receptors such as L1 and N-cadherin have additive effects on the inhibition of neurite outgrowth on astrocyte surfaces (Tomaselli et al., 1988). Therefore, motor neurons may use several receptors to promote axon outgrowth in the more complex in vivo environment. Recent studies also have provided evidence that receptors and signaling molecules that regulate axonal pathfinding, including netrins and semaphorins, may exert their effects in part in cooperation with β1 integrin-dependent pathways (Pasterkamp et al., 2003; Serini et al., 2003; Yebra et al., 2003). In the future it will be important to analyze interactions between β1 integrins and axonal guidance molecules that could cause more subtle defects, such as perturbation in the formation of topographically organized axonal projections.
The β1 integrin functions at presynaptic sites
Presynaptic differentiation is affected in mice that lack synaptic LNs (Noakes et al., 1995; Patton et al., 2001). Our findings suggest that β1 integrins in motor neurons are mainly not essential to mediate effects of synaptic LNs on presynaptic differentiation. In nestin-Itgb1Ko mice the synaptic marker proteins are localized appropriately at NMJ, including synaptophysin that is expressed presynaptically and aligned with AChRs in the postsynaptic membrane. In mice that lack synaptic LNs, terminal Schwann cell processes invade the synaptic cleft (Noakes et al., 1995). As one possibility, β1 integrins may regulate interactions of Schwann cells with ECM components at synaptic sites; presynaptic defects in motor nerve terminals therefore may be a consequence of Schwann cell defects. Alternatively, receptors other than β1 integrins may mediate interactions of motor nerve terminals with synaptic LNs. Interestingly, calcium channels in the presynaptic membrane and vesicle-associated proteins copurify with synaptic LNs, suggesting that LNs may affect presynaptic differentiation in part by interacting with components of the vesicle release apparatus (Son et al., 2000; Sunderland et al., 2000).
The β1 integrins and postsynaptic differentiation
In wild-type mice AChR clusters form at E14.5 in muscle fibers in the absence of innervation by motor neurons and, therefore, in the absence of neural agrin. Agrin subsequently is required to consolidate this AChR prepattern into a stable postsynaptic structure. Intriguingly, the formation of AChR clusters before innervation, although independent of agrin, is still dependent on MuSK, suggesting that MuSK is activated by an agrin-independent pathway (Lin et al., 2001; Yang et al., 2001). In myotubes LN-1 can induce clustering of AChRs via a pathway that involves tyrosine kinases downstream of MuSK (Marangi et al., 2002). Because LN-1, but not neural agrin, is present at early developmental stages, it has been suggested that LN/integrin-mediated signaling may be involved in the agrin-independent AChR clustering at E14.5 (Marangi et al., 2002; Willmann and Fuhrer, 2002). Our results do not support this model. At E14.5 small AChR clusters form in β1-deficient diaphragm muscle, many of which are not associated with nerve terminals. Defects become obvious subsequently, when AChRs fail to assemble into stable complexes that oppose nerve terminals. We have shown previously that the cytoskeleton of β1-deficient skeletal muscle fibers shows structural defects. F-actin filaments are assembled and aligned parallel to the long axis of muscle fibers, but the formation of sarcomeres is defective (Schwander et al., 2003). Previous studies also have shown that the formation of AChR clusters is regulated by interactions of AChR with the F-actin cytoskeleton and that AChR clusters are disrupted by drugs that interfere with F-actin assembly (Borges and Ferns, 2001). As one possibility, defects in the muscle fiber cytoskeleton in the β1 mutants, although not affecting AChR prepatterning before innervation, may have contributed to the observed defects in synaptic maturation. Alternatively, β1 integrins may modulate agrin signaling during maturation of the AChR prepattern into mature synaptic contacts. This interpretation is consistent with previous experiments that have shown that β1 integrin-specific antibodies modulate agrin signaling in myotubes in culture (Martin and Sanes, 1997). Although we show here that β1-deficient myotubes still respond to agrin signaling, we cannot exclude at present a modulatory role for β1 integrins. The structural defects in β1-deficient myotubes prevented us from analyzing the cultures quantitatively in a meaningful way and distinguishing primary defects in agrin signaling from secondary effects caused by structural defects in β1-deficient myotubes. Additional studies will be necessary to resolve this issue.
The β1 integrins and presynaptic differentiation
One intriguing finding of our study is that the phenotype of HSA-Itgb1Ko mice closely resembles the phenotype of mice null for agrin. In these mice the development of postsynaptic specializations is defective, and motor neurons fail to terminate their growth at the muscle midline and instead form exuberant branches on muscle fibers (Gautam et al., 1996). These findings and the observation that agrin and synaptic LNs inhibit neurite outgrowth in vitro have led to the hypothesis that agrin and/or LNs could provide a stop signal for motor axons (Campagna et al., 1995; Porter et al., 1995). Basement membrane (BM) components such as neural agrin and synaptic LNs interact with β1 integrins (Hunter et al., 1991; Martin and Sanes, 1997; Burgess et al., 2002), and assembly of the muscle fiber BM is defective in the absence of β1 integrins in muscle (Schwander et al., 2003). Therefore, the excessive branching of motor neurons over β1-deficient muscle fibers could be caused because β1 integrins are required for the appropriate presentation of agrin and/or LN to motor nerve terminals. We have attempted to analyze the distribution of putative “stop signals” such as neural agrin and synaptic LNs in β1-deficient muscle. Unfortunately, available antibodies do not allow us to distinguish between agrin isoforms that are expressed by nerve and muscle. In addition, although readily detectable in muscle at postnatal day 0 (P0), we failed to detect synaptic LNs on the surface of E14.5 diaphragm muscle fibers, presumably because of low expression levels at this developmental stage. Finally, it has been shown recently that motor neurons branch excessively on muscle in mice in which expression of the gene encoding choline acetyltransferase is inactivated (Misgeld et al., 2002). In these mice neurotransmission at NMJs is blocked. Some of the defects that we are reporting here could be the result of defects in neurotransmission that arise as a consequence of defects in the assembly of the presynaptic and postsynaptic apparatus. Additional studies will be necessary to distinguish among these possibilities.
In conclusion, we provide here several novel insights into the function of β1 integrins in neuromuscular development. We demonstrate that β1 integrins in spinal motor neurons are not essential for axon outgrowth and for the development of NMJs. The β1 integrins in muscle are also not essential for the establishment of the AChR prepattern. However, in the absence of β1 integrins in muscle, interactions between motor neurons and muscle are defective, preventing normal presynaptic differentiation and the cessation of growth of motor axons in the endplate zone of muscle. The defects likely are caused in part because β1 integrins regulate aspects of cytoskeletal differentiation and BM organization in skeletal muscle fibers that are essential to organize the postsynaptic apparatus and to present stop signals to growing motor axons.
Footnotes
This research was funded by the Novartis Research Foundation(U.M.), the Schweizer Stiftung zur Erforschung der Muskelkrankheiten (U.M. and M.S.), Japan Society for the Promotion of Science Fellowship for Research Abroad (R.S.), and National Institutes of Health Grants 1RO1 NS046456 (U.M.) and 5RO1 NS037116 (S.L.P.). We thank S. C. Froehner, U. Mayer, and M. Ruegg for their kind gift of antibodies; M. Ruegg for recombinant agrin; S. Orkin for Rosa26lacZflox mice; C. Damsky for Itgb1+/- mice; R. Klein for nestin-Cre mice; and D. Graus-Porta and M. Senften for their contribution in the analysis of nestin-Cre mice.
Correspondence should be addressed to U. Müller, The Scripps Research Institute, Department of Cell Biology, 10550 North Torrey Pines Road, La Jolla, CA 92073. E-mail: umueller@scripps.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248181-11$15.00/0
References
- Anderson MJ, Cohen MW (1977) Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol (Lond) 268: 757-773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick GS, Nicholson LV, Young C, O'Donnell E, Slater CR (1992) Different distributions of dystrophin and related proteins at nerve-muscle junctions. NeuroReport 3: 857-860. [DOI] [PubMed] [Google Scholar]
- Borges LS, Ferns M (2001) Agrin-induced phosphorylation of the acetylcholine receptor regulates cytoskeletal anchoring and clustering. J Cell Biol 153: 1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RW, Dickman DK, Nunez L, Glass DJ, Sanes JR (2002) Mapping sites responsible for interactions of agrin with neurons. J Neurochem 83: 271-284. [DOI] [PubMed] [Google Scholar]
- Burkin DJ, Kim JE, Gu M, Kaufman SJ (2000) Laminin and α7β1 integrin regulate agrin-induced clustering of acetylcholine receptors. J Cell Sci 113[Pt 16]: 2877-2886. [DOI] [PubMed] [Google Scholar]
- Campagna JA, Ruegg MA, Bixby JL (1995) Agrin is a differentiation-inducing “stop signal” for motoneurons in vitro Neuron 15: 1365-1374. [DOI] [PubMed] [Google Scholar]
- Cohen MW, Hoffstrom BG, DeSimone DW (2000) Active zones on motor nerve terminals contain α3β1 integrin. J Neurosci 20: 4912-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (1996) The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo Cell 85: 501-512. [DOI] [PubMed] [Google Scholar]
- Drazba J, Lemmon V (1990) The role of cell adhesion molecules in neurite outgrowth on M∫ller cells. Dev Biol 138: 82-93. [DOI] [PubMed] [Google Scholar]
- Farinas I, Yoshida CK, Backus C, Reichardt LF (1996) Lack of neurotrophin-3 results in death of spinal sensory neurons and premature differentiation of their precursors. Neuron 17: 1065-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E, Fischbach GD (1979) Early events in neuromuscular junction formation in vitro: induction of acetylcholine receptor clusters in the postsynaptic membrane and morphology of newly formed synapses. J Cell Biol 83: 143-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froehner SC (1984) Peripheral proteins of postsynaptic membranes from Torpedo electric organ identified with monoclonal antibodies. J Cell Biol 99: 88-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP (1995) Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 377: 232-236. [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (1996) Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85: 525-535. [DOI] [PubMed] [Google Scholar]
- Georgiou J, Robitaille R, Trimble WS, Charlton MP (1994) Synaptic regulation of glial protein expression in vivo Neuron 12: 443-455. [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (1996) Agrin acts via a MuSK receptor complex. Cell 85: 513-523. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, M∫ller U (2001) β1-Class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron 31: 367-379. [DOI] [PubMed] [Google Scholar]
- Greer JJ, Allan DW, Martin-Caraballo M, Lemke RP (1999) An overview of phrenic nerve and diaphragm muscle development in the perinatal rat. J Appl Physiol 86: 779-786. [DOI] [PubMed] [Google Scholar]
- Hunter DD, Cashman N, Morris-Valero R, Bulock JW, Adams SP, Sanes JR (1991) An LRE (leucine-arginine-glutamate)-dependent mechanism for adhesion of neurons to S-laminin. J Neurosci 11: 3960-3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Jurata LW, Funahashi J, Ruiz EC, Pfaff SL (2004) Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131: 3295-3306. [DOI] [PubMed] [Google Scholar]
- Leu M, Bellmunt E, Schwander M, Farinas I, Brenner HR, M∫ller U (2003) Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development 130: 2291-2301. [DOI] [PubMed] [Google Scholar]
- Lin W, Burgess RW, Dominguez B, Pfaff SL, Sanes JR, Lee KF (2001) Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 410: 1057-1064. [DOI] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Orkin SH (1999) Improved reporter strain for monitoring Cre recombinase-mediated DNA excisions in mice. Proc Natl Acad Sci USA 96: 5037-5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi PA, Wieland ST, Fuhrer C (2002) Laminin-1 redistributes postsynaptic proteins and requires rapsyn, tyrosine phosphorylation, and Src and Fyn to stably cluster acetylcholine receptors. J Cell Biol 157: 883-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Sanes JR (1997) Integrins mediate adhesion to agrin and modulate agrin signaling. Development 124: 3909-3917. [DOI] [PubMed] [Google Scholar]
- Martin PT, Kaufman SJ, Kramer RH, Sanes JR (1996) Synaptic integrins in developing, adult, and mutant muscle: selective association of α1, α7A, and α7B integrins with the neuromuscular junction. Dev Biol 174: 125-139. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635-648. [DOI] [PubMed] [Google Scholar]
- Montanaro F, Gee SH, Jacobson C, Lindenbaum MH, Froehner SC, Carbonetto S (1998) Laminin and α-dystroglycan mediate acetylcholine receptor aggregation via a MuSK-independent pathway. J Neurosci 18: 1250-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes PG, Phillips WD, Hanley TA, Sanes JR, Merlie JP (1993) 43K protein and acetylcholine receptors colocalize during the initial stages of neuromuscular synapse formation in vivo Dev Biol 155: 275-280. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP (1995) Aberrant differentiation of neuromuscular junctions in mice lacking S-laminin/laminin β2. Nature 374: 258-262. [DOI] [PubMed] [Google Scholar]
- Pasterkamp RJ, Peschon JJ, Spriggs MK, Kolodkin AL (2003) Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature 424: 398-405. [DOI] [PubMed] [Google Scholar]
- Patton BL, Cunningham JM, Thyboll J, Kortesmaa J, Westerblad H, Edstrom L, Tryggvason K, Sanes JR (2001) Properly formed but improperly localized synaptic specializations in the absence of laminin α4. Nat Neurosci 4: 597-604. [DOI] [PubMed] [Google Scholar]
- Porter BE, Weis J, Sanes JR (1995) A motoneuron-selective stop signal in the synaptic protein S-laminin. Neuron 14: 549-559. [DOI] [PubMed] [Google Scholar]
- Reist NE, Werle MJ, McMahan UJ (1992) Agrin released by motor neurons induces the aggregation of acetylcholine receptors at neuromuscular junctions. Neuron 8: 865-868. [DOI] [PubMed] [Google Scholar]
- Ruegg MA (1996) Agrin, laminin β2 (S-laminin), and ARIA: their role in neuromuscular development. Curr Opin Neurobiol 6: 97-103. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation, and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791-805. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Marshall LM, McMahan UJ (1978) Reinnervation of muscle fiber basal lamina after removal of myofibers. Differentiation of regenerating axons at original synaptic sites. J Cell Biol 78: 176-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander M, Leu M, Stumm M, Dorchies OM, Ruegg UT, Schittny J, M∫ller U (2003) Beta1 integrins regulate myoblast fusion and sarcomere assembly. Dev Cell 4: 673-685. [DOI] [PubMed] [Google Scholar]
- Serini G, Valdembri D, Zanivan S, Morterra G, Burkhardt C, Caccavari F, Zammataro L, Primo L, Tamagnone L, Logan M, Tessier-Lavigne M, Taniguchi M, Puschel AW, Bussolino F (2003) Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424: 391-397. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Katsumata R, Murakami F (1998) Change in chemoattractant responsiveness of developing axons at an intermediate target. Science 279: 105-107. [DOI] [PubMed] [Google Scholar]
- Son YJ, Trachtenberg JT, Thompson WJ (1996) Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci 19: 280-285. [DOI] [PubMed] [Google Scholar]
- Son YJ, Scranton TW, Sunderland WJ, Baek SJ, Miner JH, Sanes JR, Carlson SS (2000) The synaptic vesicle protein SV2 is complexed with an α5-containing laminin on the nerve terminal surface. J Biol Chem 275: 451-460. [DOI] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH (1995) Deletion of β1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev 9: 1883-1895. [DOI] [PubMed] [Google Scholar]
- Sugiyama JE, Glass DJ, Yancopoulos GD, Hall ZW (1997) Laminin-induced acetylcholine receptor clustering: an alternative pathway. J Cell Biol 139: 181-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland WJ, Son YJ, Miner JH, Sanes JR, Carlson SS (2000) The presynaptic calcium channel is part of a transmembrane complex linking a synaptic laminin (α4β2γ1) with non-erythroid spectrin. J Neurosci 20: 1009-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomaselli KJ, Reichardt LF (1988) Peripheral motoneuron interactions with laminin and Schwann cell-derived neurite-promoting molecules: developmental regulation of laminin receptor function. J Neurosci Res 21: 275-285. [DOI] [PubMed] [Google Scholar]
- Tomaselli KJ, Neugebauer KM, Bixby JL, Lilien J, Reichardt LF (1988) N-cadherin and integrins: two receptor systems that mediate neuronal process outgrowth on astrocyte surfaces. Neuron 1: 33-43. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schutz G (1999) Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23: 99-103. [DOI] [PubMed] [Google Scholar]
- Varnum-Finney B, Venstrom K, M∫ller U, Kypta R, Backus C, Chiquet M, Reichardt LF (1995) The integrin receptor α8β1 mediates interactions of embryonic chick motor and sensory neurons with tenascin-C. Neuron 14: 1213-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CD, Yoshida CK, de Curtis I, Reichardt LF (1995) Expression and in vitro function of β1-integrin laminin receptors in the developing avian ciliary ganglion. J Neurosci 15: 5275-5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann R, Fuhrer C (2002) Neuromuscular synaptogenesis: clustering of acetylcholine receptors revisited. Cell Mol Life Sci 59: 1296-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Arber S, William C, Li L, Tanabe Y, Jessell TM, Birchmeier C, Burden SJ (2001) Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 30: 399-410. [DOI] [PubMed] [Google Scholar]
- Yebra M, Montgomery AM, Diaferia GR, Kaido T, Silletti S, Perez B, Just ML, Hildbrand S, Hurford R, Florkiewicz E, Tessier-Lavigne M, Cirulli V (2003) Recognition of the neural chemoattractant Netrin-1 by integrins α6β4 and α3β1 regulates epithelial cell adhesion and migration. Dev Cell 5: 695-707. [DOI] [PubMed] [Google Scholar]