

Abstract
Mild traumatic brain injuries are of major public health significance. Neurons in such injuries often survive the primary mechanical deformation only to succumb to subsequent insults. To study mechanisms of vulnerability of injured neurons to secondary insults, we used an in vitro model of sublethal mechanical stretch. Stretch enhanced the vulnerability of the neurons to excitotoxic insults, causing nuclear irregularities, DNA fragmentation, and death suggestive of apoptosis. However, the DNA degradation was not attributable to classical (caspase mediated) or caspase-independent apoptosis. Rather, it was associated with profound stretch-induced mitochondrial dysfunction and the overproduction of reactive oxygen species (ROS). Sublethally stretched neurons produced surprisingly high levels of ROS, but these in isolation were insufficient to kill the cells. To be lethal, the ROS also needed to combine with nitric oxide (NO) to form the highly reactive species peroxynitrite. Peroxynitrite was not produced after stretch alone and arose only after combining stretch with an insult capable of stimulating NO production, such as NMDA or an NO donor. This explained the exquisite sensitivity of sublethally stretched neurons to a secondary NMDA insult. ROS scavengers and NO synthase (NOS) inhibitors prevented cell death and DNA degradation. Moreover, inhibiting neuronal NOS activation by NMDA using peptides that perturb NMDA receptor-postsynaptic density-95 interactions also reduced protein nitration and cell death, indicating that the reactive nitrogen species produced were neuronal in origin. Our data explain the mechanism of enhanced vulnerability of sublethally injured neurons to secondary excitotoxic insults and highlight the importance of secondary mechanisms to the ultimate outcome of neurons in mild neurotrauma.
Keywords: neurotrauma, excitotoxicity, mitochondria, reactive oxygen species, apoptosis, PSD-95
Introduction
Traumatic brain injury (TBI) afflicts people of all ages and places a significant socioeconomic burden on Western society (Marshall, 2000). It is likely that this burden is underestimated, because milder forms of TBI and its consequences are underre-ported (National Center for Health Statistics, 1997; NIH, 1999; Centers for Disease Control and Prevention, 2001). TBI can result in neurological impairment because of immediate CNS tissue disruption (primary injury), and surviving cells may be secondarily damaged by mechanisms triggered by the primary event, leading to additional damage and disability (Teasdale and Graham, 1998; Amar and Levy, 1999).
Unlike in severe TBI, neurons in mild TBI are less affected by primary mechanical disruption but may be vulnerable to subsequent events. Accordingly, to study secondary injury mechanisms in vitro, we developed a model of sublethal stretch-induced injury in cultured cortical neurons (Arundine et al., 2003). The sublethal stretch approach removes the confounding effects of neuronal loss from primary tissue disruption. We have extensively characterized this model. In brief, sublethally stretched neurons maintained cell membrane integrity, viability, and electrophysiological function. However, stretching evoked in the neurons a heightened vulnerability to subsequent challenges with l-glutamate or NMDA. This heightened vulnerability was specifically mediated by NMDA receptors (NMDARs), because stretched neurons did not become more vulnerable to either AMPA/kainate toxicity or to that induced by a Ca2+ ionophore. Stretch-enhanced vulnerability to NMDA occurred independently of endogenous glutamate release but required Ca2+ influx through NMDARs. Stretch did not affect the electrophysiological properties of NMDARs nor excitatory synaptic activity, indicating that specificity of enhanced vulnerability to NMDA involves postsynaptic mechanisms downstream from NMDARs. The data indicated that sublethal in vitro stretch injury triggers distinct secondary injury signaling pathways rather than causing a generalized increase in vulnerability to secondary insults. These were the focus of this study.
Here, we demonstrate a major, previously unappreciated, mechanism underlying the enhanced vulnerability of sublethally stretched neurons to subsequent insults. We show that sublethal stretch injury caused neurons to exhibit nuclear irregularities and DNA fragmentation suggestive of a switch in the mode of cell death from necrosis to apoptosis. However, the increased vulnerability of stretched neurons to secondary insults was not apoptotic. Rather, it was associated with mitochondrial dysfunction and an unexpectedly high production of reactive oxygen species (ROS). The high ROS levels primed neurons to be vulnerable to additional insults so long as these insults generated the free radical nitric oxide (NO). Consequently, secondary insults with even low concentrations of NMDA induced sufficient NO that combined with mitochondrial ROS (superoxide) to form peroxynitrite, causing DNA degradation and cell death.
Materials and Methods
Cortical neuronal cultures. Mixed cortical cell cultures containing both neurons and glia were prepared from embryonic Swiss mice at 15 d gestation, as described previously (Sattler et al., 1997), with minor modifications from Choi (1987). In brief, cerebral cortices from 10 to 12 embryos were incubated for 10 min in 0.05% trypsin-EDTA, dissociated by trituration, and plated on flexible membranes coated with poly-l-ornithine in 6-well plates (Flexcell International, Hillsborough, NC) at a density of 3.25 × 106 cells per well. Plating medium consisted of DMEM supplemented with 10% heat-inactivated horse serum (Invitrogen, San Diego, CA), 2 mm glutamine, 25 mm glucose, and 26 mm bicarbonate. The cultures were maintained at 37°C in a humidified 5% CO2 atmosphere. After 3 d in vitro, growth of non-neuronal cells was halted by a 48 hr exposure to a 10 μm solution (5 μm uridine and 5 μm (+)-5-fluor-2′-deoxyuridine). This produces cultures in which >85% of the cells are neurons, based on immunohistochemical staining for glial fibrillary-associated protein (exclusive to astrocytes) and for the NMDAR1 subunit (data not shown). The cultures were used for experiments after 12-14 d in vitro.
Drugs and solutions. The control solution contained (in mm) 121 NaCl, 5 KCl, 20 d-glucose, 10 HEPES acid, 7 HEPES-Na salt, 3 NaHCO3, 1 Na-pyruvate, 1.8 CaCl2, and 0.01 glycine, adjusted to pH 7.4 with NaOH. All drug stock solutions were kept at -20°C. Stocks of the following were prepared in DMSO (10-20 mm) and diluted to their indicated final concentrations in control buffer: nimodipine (L-type Ca2+ channel antagonist; 2 μm; Miles, Elkhart, IN), 6-cyano-7-nitroquinoxaline (CNQX; AMPA/kainate antagonist; 10 μm; Research Biochemicals, Natick, MA), tetramethylrhodamine methyl ester (TMRM; mitochondrial potential probe; 10 nm; Molecular Probes, Eugene, OR), dihydrorhodamine-123 (DHR; ROS indicator; 5 μm; Molecular Probes), z-val-ala-aspfluorometylketone (z-VAD-FMK; a pan-caspase inhibitor; 200 μm; Calbiochem, La Jolla, CA), and z-val-phe-CHO (calpain inhibitor III; 10 μm; Calbiochem).
Stocks of the following were prepared in double-distilled H2O (ddH2O; 10-20 mm) and diluted to their final concentrations in control buffer: MK-801 (NMDAR antagonist; 10 μm), sodium nitroprusside (SNP; an NO donor; 300 μm; Sigma, St. Louis, MO), and NG-nitro-l-arginine methyl ester [l-NAME; a neuronal NO synthase (nNOS) inhibitor; 100 μm; Sigma].
Propidium iodide (PI; viability indicator; Molecular Probes) was prepared as a 1 mg/ml stock and dissolved to a 50 μg/ml final concentration. Manganese(III) tetrakis(4-benzoic acid) porphyrin (MnTBAP; O2- scavenger; 10 mm) stock was prepared by first dissolving the crystals in 100 μl of 1 m NaOH and then adding an equal volume of 1 m HCl. This stock was diluted to its final concentration (200 μm) in control solution, and pH of 7.4 was confirmed before each use.
MnTBAP, SNP, and nimodipine were protected from light at all times. Nimodipine, CNQX, and MK-801 were always applied at concentrations of 2, 10, and 10 μm, respectively (Sattler et al., 1998). All solutions were prepared under sterile conditions. All other chemicals were obtained from Sigma.
Stretching of cultured neurons. The cultures, grown on SILASTIC membranes as above, were placed in the Flexercell FX-3000 Strain unit (Flexcell International), a computer-driven instrument that uses a vacuum pulse to deform the culture substrate. In all experiments, the cortical cultures were stretched to 130% of their original length for 1 sec, inducing a sublethal injury as recently characterized by us (Arundine et al., 2003). The stretched neurons maintained cell membrane integrity, viability, and electrophysiological function.
Determination of cell death. Cell death was determined by serial quantitative measurements of PI fluorescence using a multiwell plate fluorescence scanner (Cytofluor II; PerSeptive Biosytems, Framingham, MA), as described previously (Sattler et al., 1997, 1998). In brief, the culture medium in each tissue culture well was replaced with control solution containing 50 μg/ml PI, and a baseline fluorescence reading was taken. Sequential readings were then taken at appropriate intervals over the 20 hr observation period. The fraction of dead cells in each culture at a given time was calculated as follows: fraction dead = (Ft - Fo)/FNMDA, where Ft is the PI fluorescence at time t, Fo is the initial PI fluorescence at time 0, and FNMDA is the background-subtracted PI fluorescence of identical cultures from the same dissection and plating, 20-24 hr after a 60 min exposure to 1 mm NMDA at 37°C. Based on manual observations at the time of validation of this technique, this NMDA exposure routinely produced near complete neuronal death in each culture but had no effect on surrounding glia (Bruno et al., 1994; David et al., 1996; Sattler et al., 1997). Adding Triton X-100 (0.1%) to cultures treated in this manner produced an additional 10-15% increase in PI fluorescence because of permeabilization of non-neuronal cell membranes, consistent with a 10-15% glial component in the cultures.
Experimental protocols. All experiments were performed at 37°C. Unless indicated otherwise, all solutions contained nimodipine (2 μm) and CNQX (10 μm) to restrict the actions of applied NMDA to NMDARs by preventing the secondary activation of other pathways (Sattler et al., 1998). In general, the cultures were washed two times with control solution and immediately stretched to 130% of their original length for 1 sec. After the stretch, they were washed once with control solution alone or challenged with an additional treatment (e.g., NMDA) and placed at 37°C for 1 hr. They were then washed once with control solution and then with control solution containing 50 μg/ml PI to obtain a baseline reading. In pilot studies, baseline readings obtained at this stage were similar to those obtained by staining the cells with PI before stretch. Because the stretch causes splashing of the bath solution, staining with PI after stretch reduced biohazard potential and artifacts associated with PI outside the culture well. Unstretched controls were manipulated identically to stretched cultures.
Assessment of mitochondrial membrane potential. Mitochondrial potential was measured with TMRM because it is believed to reduce mitochondrial respiration to a lesser extent than other dyes such as rhodamine-123 or tetramethylrhodamine ethyl ester (Scaduto and Grotyohann, 1999). Cultures were preincubated at 37°C with 10 nm TMRM for 30 min. This concentration is well below that which is anticipated to quench TMRM fluorescence, and reductions in mitochondrial potential are manifest by reductions in whole-cell fluorescence (see Fig. 5B for effect of carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP), a mitochondrial depolarizing agent). TMRM was maintained in the solution throughout the experiment. TMRM fluorescence was imaged on an Eclipse TE-2300 microscope (Nikon) using 20× objective, a 510-560 nm bandpass excitation, and 590 nm long-pass emission filter. Images were acquired using an ORCA-ER digital camera (Hamamatsu, Middlesex, NJ) and Simple PCI software (Nikon). Fluorescence from 10-30 cells was averaged and background subtracted for each image.
Figure 5.

Stretch causes a reduction in mitochondrial membrane potential. Cultures were pretreated with 10 nm TMRM for 30 min before each experiment (see Materials and Methods). A, Effects of the indicated conditions on TMRM fluorescence at the indicated times after insult. The bars indicate background-subtracted fluorescence of cultures exposed to an insult (Ft) normalized to background-subtracted TMRM fluorescence in unstretched cells (Fo) at the indicated time points. The bars represent mean + SE of three to four cultures from n = 3 separate experiments. The asterisks indicate the difference from unstretched cultures at the indicated time point (Bonferroni t test; p < 0.05). B, Representative TMRM fluorescence images from the indicated condition. TMRM fluorescence in cultures exposed to stretch only recovered, whereas TMRM fluorescence from cultures exposed to stretch plus NMDA did not. Treatment with the mitochondrial uncoupler FCCP abolished TMRM fluorescence.
Assessment of ROS production. The production of ROS was measured with DHR. In brief, DHR is oxidized to a fluorescent rhodamine-123 (Royall and Ischiropoulos, 1993), and the consequent rise in fluorescence may be used as an indicator of ROS production. DHR has been shown to be oxidized by superoxide anions (Rothe et al., 1991; Bueb et al., 1995; Ostrovidov et al., 1998), hydrogen peroxide (Royall and Ischiropoulos, 1993; Gow et al., 1999), peroxide radicals (Royall and Ischiropoulos, 1993; Gow et al., 1999), and peroxynitrite (Gilad et al., 1997).
ROS were measured as described previously (Tymianski et al., 1998; Aarts et al., 2003). In brief, cultures were preloaded for 30 min with 5 μm DHR and subjected to the experimental insult. DHR fluorescence was measured in a multiwell plate fluorescence scanner (Cytofluor II; 485 nm excitation; 530 emission; PerSeptive Biosytems). DHR fluorescence was normalized to baseline using the formula (Ft - Fo)/Fo, where Ft is the DHR fluorescence at time t and Fo is the DHR fluorescence at time 0. DHR fluorescence images were also acquired using a microscope (465-495 nm excitation; 515-555 nm emission) as described above for TMRM.
DNA fragmentation assays. DNA fragmentation was examined using the terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) method (Gavrieli et al., 1992; Didenko and Hornsby, 1996; Didenko et al., 1998) and by DNA agarose gel electrophoresis (Hill et al., 2000).
TUNEL assay. Nuclear DNA strand breaks were detected by enzymatically labeling free 3′-OH termini with modified nucleotides provided in the Apoptag kit (Intergen, Purchase, NY), using the manufacturer's instructions. Visualization of TUNEL-stained nuclei was achieved using an anti-digoxigenin antibody conjugated to a fluorescein or peroxidase reporter molecule (ApopTag Peroxidase kits S7110 and S7100, respectively; Intergen). In fluorescence experiments, nuclei were counterstained using 5 ng/ml Hoechst 33258 (1:5000; Transduction Laboratories, Lexington, KY) and visualized using 465-495 nm excitation and 515-555 nm emission for fluorescein and 340 nm excitation and 510-40 nm emission for Hoechst. The number of TUNEL-positive cells was expressed as a fraction of the total cell number in the field. In each culture, four to eight random fields were quantified. Approximately 100-200 cells were counted per culture, and at least three experiments were performed for each set of counts.
DNA gel electrophoresis. DNA laddering was evaluated as described previously (Hill et al., 2000). In brief, all tissue was collected from each culture well and pelleted by centrifugation (1000 rpm; 1 min). The cells were digested for 2 hr at 56°C in buffer containing (in mm) 10 Tris, pH 8.0, 2 EDTA, pH8, 400 NaCl, 0.5% SDS, and 1 mg/ml proteinase K. Digest solution was subjected to phenol/chloroform extraction, and total DNA was precipitated from the aqueous layer with an equal volume of 100% ethanol. The DNA pellet was dried, dissolved in 50 μl of ddH20, and kept overnight at 4°C. Ten micrograms of DNA were prepared on a 2% agarose gel. Gels were stained with ethidium bromide and photographed.
Protein harvest for immunoblots. All tissue was collected from each culture well and centrifuged to obtain a total cell pellet. The pellet was washed with phosphate buffered saline (PBS) containing phenylmethylsulfonyl fluoride (PMSF) to inhibit intracellular proteases. Subsequent to the final wash, the pellet was resuspended in Triton lysis buffer containing (in mm) 150 NaCl, 20 Tris, pH 7.4, 20 NaF, 0.1 sodium vanadate, 1% Triton X-100, and 1 PMSF). In experiments necessitating cytosolic and nuclear protein fractions, fractioned lysates were prepared according to the method of Borer et al. (1989). In brief, harvested cells were reconstituted with hypotonic buffer (in mm: 10 HEPES, pH 7.9, 10 KCl, 0.1 EDTA, 0.1 EGTA, and 1 PMSF) and incubated on ice for 15 min. NP-40 (0.5% final concentration) was added to cell suspension, vortexed, and incubated for 1 min at room temperature. Suspension was centrifuged (10,000 rpm; 30 sec; room temperature), and supernatant and pellet fractions were separated. The supernatant fraction (cytosolic fraction) was reconstituted in Triton lysis buffer by adding 5× stock buffer directly to the fraction. The pellet fraction (nuclear fraction) was washed two times with hypotonic buffer (containing 0.5% NP-40), and the final pellet was reconstituted with 1× Triton lysis buffer.
The protein content of all fractions was assessed using the DC Protein Assay kit (Bio-Rad, Mississauga, Ontario, Canada), and the samples were stored at -80°C until the time of analysis.
Immunoblotting. Protein samples were immunoblotted as described previously (Jones et al., 1997; Sattler et al., 2000). The blotted proteins were probed using the following primary antibodies: rabbit polyclonal anti-caspase 3 (1:1000; StressGen, Sidney, Canada); mouse monoclonal anti-apoptosis-inducing factor (AIF; 1:800; Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal anti-nNOS (NOS type 1) IgG2a (1:2000; Transduction Laboratories); mouse monoclonal anti-endonuclease g (endo g; 1:1000; ProSci). The secondary antibodies were sheep anti-mouse Ig or donkey anti-rabbit Ig conjugated to horseradish peroxidase (diluted 1:3000 in TBS with 0.05% Tween 20; Amersham Biosciences, Oakville, Ontario, Canada).
Immunostaining. Cultures were fixed with warm 4% paraformaldehyde and 4% sucrose in PBS for 20 min, permeabilized with 0.1% Triton X-100 for 10 min at 4°C, and blocked with 10% goat serum in PBS for 1 hr at room temperature. Immunofluorescence was visualized with an inverted Nikon microscope using a 40× oil immersion lens. FITC, rhodamine, and Hoechst were visualized using 465-495 nm excitation and 515-555 nm emission, 510-560 nm excitation and 590 nm emission, and 340 nm excitation and 510 nm emission, respectively. Active caspase 3 was labeled by incubating the cultures with a purified rabbit monoclonal anti-active caspase 3 antibody (1:300; BD PharMingen San Diego, CA) and a fluorescein-conjugated goat anti-mouse IgG secondary antibody (1:500; Transduction Laboratories). Nitrotyrosine was labeled by incubating the cultures with a rabbit polyclonal α-nitrotyrosine primary antibody (1:300; Upstate Biotechnology, Lake Placid, NY) and a rhodamine-conjugated goat anti-rabbit IgG secondary antibody (1:500; Transduction Laboratories). AIF was labeled by incubating the cultures with an AIF antibody (1:200; Santa Cruz Biotechnology) (Zhang et al., 2002) and a rhodamine-conjugated goat anti-mouse IgG secondary antibody (1:500; Transduction Laboratories). In some experiments, nuclei were counterstained with with Hoechst 33258 (1:5000; Transduction Laboratories).
Synthetic Tat peptides and plasmid construction. Tat-conjugated peptides were synthesized at the Advanced Protein Technology Centre (Hospital for Sick Kids, Toronto, Ontario, Canada). Tat peptides were conjugated to either the terminal nine amino acids of the NMDAR NR2B subunit (Tat-NR2B9c) or to the mutated form (serine and valine replaced with alanine; Tat-NR2B-AA). The Tat peptide sequences are as shown in Figure 10B.
Figure 10.
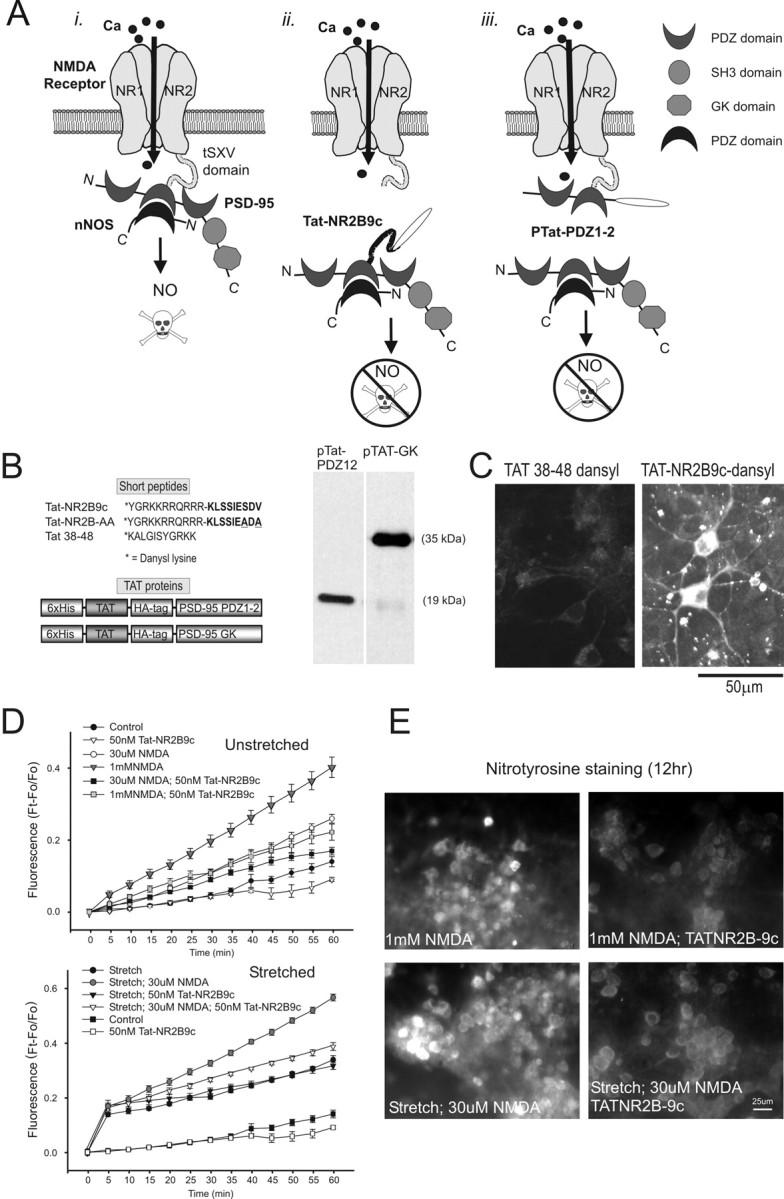
Effect of uncoupling NMDAR NR2B from PSD-95 on ROS production and protein nitration. A, Schematic illustrating the approach: i, NMDARs associate with nNOS via PSD-95; ii, iii, dissociating nNOS from NMDARs using Tat fused either to the C terminus of NR2B (Tat-NR2B9c; ii) or to the first and second PDZ domains of PSD-95 (pTat-PDZ1-2; iii). B, Tat peptides and fusion proteins (pTat) used in these experiments. Inset, Representative immunoblots obtained during purification of pTat-PDZ1-2 and pTat-GK proteins. C, Visualization of intraneuronal accumulation of Tat-NR2B9c-dansyl (10 μm) but not Tat-38-48-dansyl (10 μm) 30 min after application to cortical cultures. D, Effect on DHR fluorescence of pretreating cultures with Tat-NR2B9c 30 min before the indicated insult. Cultures were simultaneously preincubated with 10 μm DHR for 30 min before the insult. Pretreatment with 50 nm TatNR2B9c reduced ROS production in all stretched (bottom) and unstretched (top) cultures treated with NMDA (30 μm or 1 mm; Boneferroni t test; p < 0.05). However, Tat-NR2B9chas no effect on ROS production by stretch alone (t24 = 1.10; p = 0.284). Symbols are the means ± SE of 5-18 cultures from three separate experiments. E, Tat-NR2B9c pretreatment reduces NMDAR-mediated protein nitration. Nitrotyrosine immunostaining was performed 12 hr after the insult under the indicated conditions (representative of 3 experiments).
Transducible proteins were constructed by cloning PCR products in frame into the pTat-HA bacterial expression vector containing an N-terminal 6-histidine leader, followed by the 11 amino acid Tat protein transduction domain, a hemagglutinin (HA) tag, and a polylinker site (a kind gift from S. Dowdy, Washington University, St. Louis, MO). The transducible pTat-PDZ1-2 and pTat-GK fusion proteins were constructed as follows: oligonucleotides PDZ1-2 (sense, 5′-ggtaccgaggagarcacattggaa-3′; antisense, 5-gaattctgggggagcatagctgtc-3′) and GK (sense, 5′-cgggtaccgctcgtcccatcatcatc-3′; antisense, 5′-gaattctcagagtctctctcgggctgg-3′) were used to generate postsynaptic density-95 (PSD-95) fragments with the 5′ KpnI site and 3′ EcoRI site for subcloning into the pTAT-HA vector polylinker site. Both the PCR products and pTAT-HA plasmid were digested with KpnI and EcoRI, ligated, and subsequently transformed into competent DH5α bacterial cells (Invitrogen, Burlington, Ontario, Canada). Plasmids were isolated and retransformed into BL21(DE3)pLysS bacterial cells (Invitrogen). To determine whether the transformations were successful, homogenates of AMP-resistant colonies were run on SDS-PAGE, transferred to nitrocellulose membranes, and probed with mouse HA.11 antibody against the HA tag (Covance/BabCo, Princeton, NJ). HA-tagged proteins were detected using a goat anti-mouse secondary antibody conjugated to horseradish peroxidase and visualized using enhanced chemiluminescence. Clones expressing the fusion protein were harvested by sonication in 8 m urea, and a Ni-NTA column (Amersham Biosciences) was used to extract the fusion proteins. The proteins were desalted on PD-10 columns (Amersham Biosciences), protease inhibitors were added (1 μg/ml aprotinin and 1 μg/ml leupeptin), and the protein concentration was determined using the DC Protein Assay kit (Bio-Rad).
Results
Sublethal stretch injury model
The model used to study mechanisms of neuronal vulnerability after stretch was detailed by Arundine et al. (2003). The stretch severity was titrated downward until no cells died after insult for a 24 hr observation period (sublethal injury). This eliminated the confounding effects of mechanical cell disruption (primary injury) on any damage that might be imposed by a subsequent insult (secondary injury). The cultures were stretched to 130% of their original length for 1 sec. Preliminary studies demonstrated that at static peak deflection, the neurons and dendrites remained attached to the culture membrane (data not shown) (but see Smith et al., 1999). Therefore, the membrane stretch likely correlated with cell stretch. We did not calculate the culture membrane strain, an estimate of the strain imparted to the cells, because the latter may differ from the strain of the underlying substrate. Sublethally stretched cells did not take up the cell viability indicator PI, were morphologically unchanged, and exhibited normal whole-cell ionic currents (Arundine et al., 2003).
Stretch renders neurons more vulnerable to secondary insults with NMDA
Although stretch to 130% for 1 sec (hereafter referred to as “stretch”) did not kill the neurons, it made them more vulnerable to a subsequent application of NMDA at concentrations that were tolerated by most unstretched controls. This occurred both when NMDA was applied just before (data not shown) or within 10 min after the stretch (Fig. 1A,B). We showed previously that this enhanced vulnerability to a second challenge was not ubiquitous to all types of insults. Rather, stretched neurons exhibited an enhanced mortality when challenged with agonists of NMDARs (NMDA and l-glutamate), but not when challenged with selective AMPA/kainate receptor agonists or with a Ca2+ ionophore. Moreover, the enhanced vulnerability of stretched neurons to NMDA was not attributable to synaptic or nonsynaptic release of excitatory amino acids from stretched cells, nor attributable to alterations of NMDAR function (Arundine et al., 2003).
Figure 1.

Sublethal stretch injury renders cortical cultures vulnerable to low concentrations of NMDA. NMDA was applied for 1 hr with in 10 min of stretch. A, Effects of NMDA at the indicated concentration with or without previous stretch (130% for 1 sec). Cell death was measured at 20 hr. The asterisk indicates the difference from unstretched (t61 = 13.23; p < 0.001). The bars show the mean ± SE of 27-34 cultures from three separate dissections. B, Representative phase-contrast and PI fluorescence images of unstretched and stretched cultures 20 hr after challenge, as indicated. C, High magnification of Hoechst-stained neuronal nuclei 20 hr after the indicated challenge. NMDA (1 mm or 30 μm) applied to unstretched cultures did not affect the round nuclear morphology (white arrows). However, stretch caused condensation and irregularity of nuclear morphology (open arrows). Images were obtained using identical excitation, emission, and camera gain settings (representative of 3 separate experiments).
Sublethal stretch injury produces irregular nuclear morphology
To examine the effects of stretch on nuclear morphology, the cells were stained with the cell-permeant nuclear dye Hoechst 33258 (see Materials and Methods). Nuclei of unstretched neurons exposed to lethal (1 mm) or sublethal (30 μm) NMDA concentrations remained round (Fig. 1C, white arrows). However, neurons exposed to sublethal stretch or to the lethal combination of stretch and 30 μm NMDA exhibited irregular and condensed nuclei (Fig. 1C, red arrows). This morphology has been interpreted as representing apoptosis in some studies examining the effects of low NMDA concentrations (Bonfoco et al., 1995, 1996) and in past studies of in vitro traumatic neuronal damage (Shah et al., 1997). Moreover, apoptosis has been implicated as a causative death mechanism in many animal studies of TBI (Rink et al., 1995; Clark et al., 1997; Conti et al., 1998; Newcomb et al., 1999; Raghupathi et al., 2000; Wennersten et al., 2003). Because NMDA alone did not produce irregular nuclei even at lethal (1 mm) concentrations, we questioned whether stretch biased the neurons to an apoptotic death. Thus, we next sought additional markers of apoptosis in these cells.
NMDA challenge to sublethally stretched neurons produces DNA fragmentation
Internucleosomal DNA fragmentation is an important biochemical feature of apoptosis and can be determined using in situ nick-end labeling (TUNEL) or by DNA fragmentation analysis by gel electrophoresis (Gavrieli et al., 1992; Gerschenson and Rotello, 1992; Allen et al., 1997). We used both techniques to detect DNA fragmentation in stretched cells 20 hr after the insult (Fig. 2A-C). As a positive control for apoptosis, the cultures were treated with staurosporine (1 μm for 48 hr) because this is an established method for triggering an apoptotic death in cortical cultures (Yu et al., 1997; Budd et al., 2000). TUNEL staining was observed only in staurosporine-treated cultures and in stretched cultures that were also challenged with NMDA (Fig. 2A,B). Cultures exposed to the nonlethal conditions of sham (unstretched controls), stretch alone, or 30 μm NMDA without stretch did not TUNEL stain (Fig. 2A,B). Interestingly, cultures exposed to 1 mm NMDA, which is lethal (Fig. 1A), also failed to TUNEL stain (Fig. 2A,B), a finding consistent with a lack of effect of 1 mm NMDA on nuclear morphology (Fig. 1C). Complementary results were obtained when DNA fragmentation was evaluated by DNA gel electrophoresis: DNA ladders suggestive of intenucleosomal DNA fragmentation were only observed in staurosporine-treated positive controls and in stretched neurons that were also treated with 30 μm NMDA (Fig. 1C). These results show that DNA fragmentation was uniquely displayed by neurons undergoing the combination of sublethal stretch, followed by NMDA at concentrations that would have been tolerated by most cells in the absence of previous stretch. A lethal excitotoxic challenge without stretch was insufficient to produce DNA fragmentation.
Figure 2.
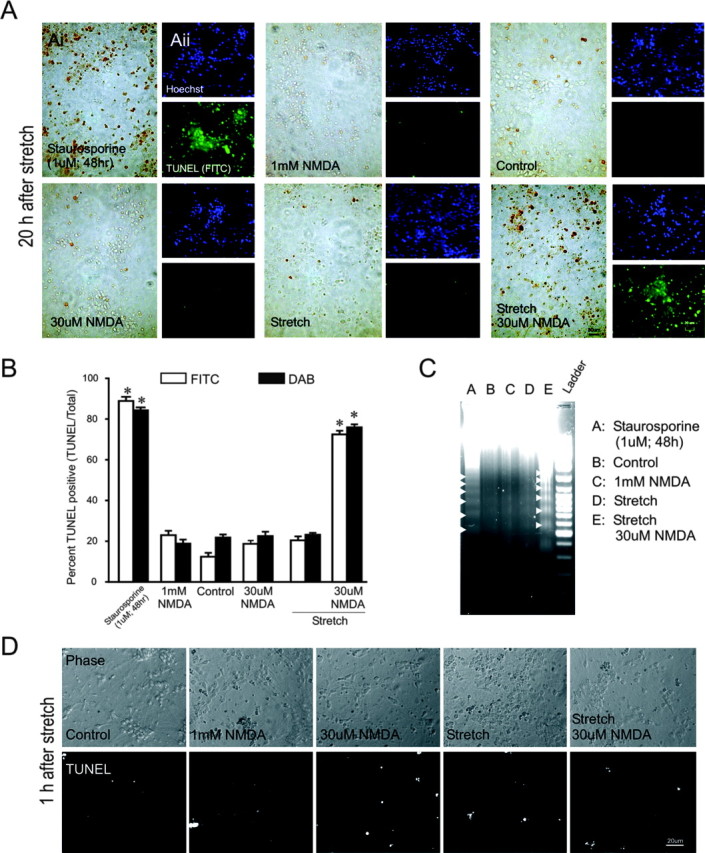
Challenging sublethally stretched neurons with low NMDA concentrations produces DNA damage. NMDA was applied for 1 hr within 10 min of stretch. A, TUNEL staining 20 hr after injury under the indicated conditions using the DAB (Ai) or FITC (Aii) methods. Nuclei in Aii were also counterstained with Hoechst. Each panel is representative of three experiments. B, Quantification of TUNEL staining at 20 hr by each method. TUNEL-positive cells were normalized to total cell number. Approximately 100-200 cells were counted per culture. Compared with unstretched controls, treatment with 1 μm staurosporine for 48 hr and stretch plus NMDA for 1 hr resulted in increased TUNEL staining (staurosporine: FITC-t18 = 7.63, p < 0.0001; DAB-t53 = 30.70, p < 0.0001; stretch plus NMDA: FITC-t29 = 10.298, p < 0.0001; DAB-t83 = 23.923, p < 0.0001). Treatment with NMDA alone (30 μm to 1 mm) or stretch alone did not result in increased TUNEL staining. The asterisks indicate the difference from unstretched control (Bonferroni t test; p < 0.05). The bars represent the mean + SE of four to eight fields in each of three cultures in each of three (FITC) or six (DAB) experiments. C, Staurosporine and stretch plus 30 μm NMDA, but not 1 mm NMDA alone or stretch alone, induced DNA laddering (arrowheads) 20 hr after insult. The data are representative of three separate experiments. D, TUNEL staining does not occur within 1 hr of stretch. Phase-contrast and fluorescent TUNEL stainimages taken 1 hr after sham (nostretch) or stretch are shown. The data are representative of three separate experiments.
Because stretch is a physical insult, we questioned whether the mechanical deformation of nuclei might directly induce chromatin damage at the weakest points (internucleosomal breaks), thus initiating programmed cell death. To test this, we surmised that if the DNA damage is mechanically induced, then it should be evident immediately after stretch. Accordingly, we examined cells at 1 hr after injury by TUNEL staining. However, TUNEL positivity 1 hr after stretch was no different than in unstretched controls (Fig. 2D). Similar results were obtained using DNA gel electrophoresis (data not shown). Thus, it is unlikely that the DNA fragmentation induced after stretch plus NMDA is caused by direct mechanical damage.
Although apoptosis causes DNA fragmentation, the converse is not necessarily true. For example, in traumatic head injury, DNA fragmentation analysis (TUNEL staining or gel electrophoresis) was of no value because these tests were positive for both processes in both necrotic and apoptotic cells (Ishimaru et al., 1999). This is likely because DNA of necrotic cells also undergoes degradation and reacts with the terminal transferase used in the TUNEL assay (Charriaut-Marlangue and Ben Ari, 1995; Grasl-Kraupp et al., 1995). Thus, to seek additional evidence of apoptosis, we next examined other indicators of apoptotic pathways.
Role of classical apoptosis in DNA fragmentation of sublethally stretched neurons exposed to NMDA
Previous studies have suggested that in models of glutamate toxicity, TBI, and ischemia, internucleosomal DNA fragmentation is mediated by pathways involving effector caspases, including caspase 3 (Tenneti et al., 1998; Eldadah and Faden, 2000; Pike et al., 2000). In response to various apoptotic stimuli, the pro-form of caspase 3 (i.e., inactive form) is cleaved into a smaller fragment (active caspase 3) that has proteolytic activity resulting in eventual endonuclease activation and DNA fragmentation (Enari et al., 1998; Sakahira et al., 1998; Yakovlev et al., 2001). Thus, we questioned whether caspase-associated pathways were involved in the DNA fragmentation observed after stretch.
First, we used immunofluorescence to examine the cultures for the presence of active caspase 3 20 hr after stretch, a time point at which we found increased internucleosomal DNA fragmentation in neurons exposed to stretch plus NMDA (Fig. 2A-C). Although cultures exposed to staurosporine (positive control) stained for active caspase 3, we failed to observe a concomitant increase in active caspase 3 under any other conditions (Fig. 3A). To examine this further, we next checked for the presence of active caspase 3 by immunoblotting. However, at 20 hr after stretch, there was no evidence of the cleaved form of caspase 3 (17 kDa) (Fig. 3Bi). All conditions were immunopositive for the pro-form of caspase 3, as represented by the 32 kDa band (Fig. 3Bi). Next, we tested whether active caspase 3 was detectable at earlier time points in stretched neurons treated with 30 μm NMDA. However, this also failed to reveal evidence of active caspase 3 at 1, 4, 8, r or 12 hr after the insult.
Figure 3.
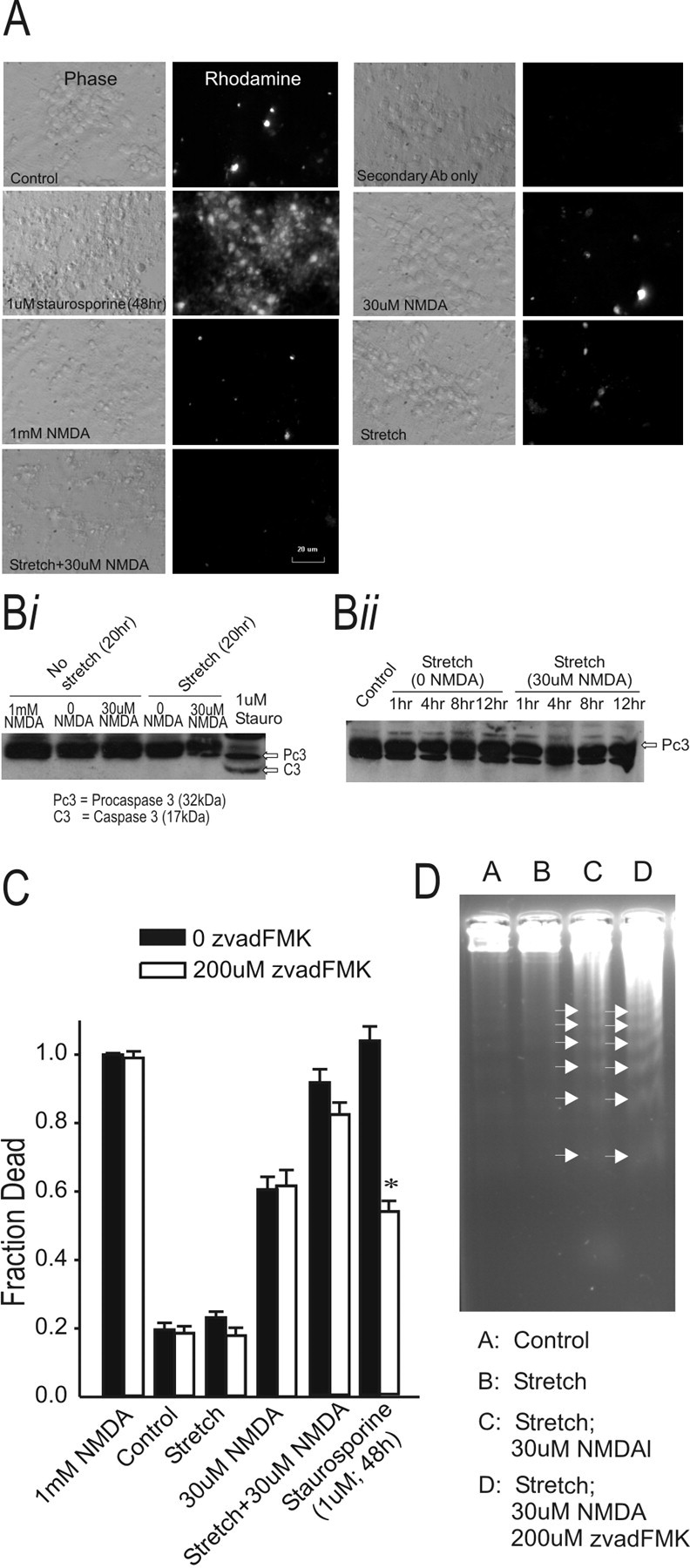
Stretch plus NMDA-dependent cell death is not caspase mediated. A, Active caspase 3 immunoreactivity in fixed cortical neuronal cultures at 20 hr after the insult using the indicated conditions. Only cultures treated with staurosporine exhibited pronounced active caspase 3 immunofluorescence. Bi, Immunoblot of caspase 3 (both pro and active/cleaved forms) 20 hr after the indicated insult. The pro-caspase 3 form (32 kDa) was detectable under all culture conditions, whereas only cultures treated with stauro sporine displayed the active caspase 3 band (17 kDa). The data are representative of three experiments. Bii, Immunoblot of the time course of caspase 3 expression (both pro and active/cleaved forms) after stretch with or without NMDA. Although pro-caspase 3 (32 kDa) was detectable at all time points, neither stretch nor stretch plus 30 μm NMDA induced active caspase 3 (17 kDa) at any time point. The data are representative of two experiments. C, Stretch-induced vulnerability to NMDA toxicity is not attenuated by the pan caspase inhibitor z-VAD-FMK. Treatment with 200 μm z-vad-FMK for 48 hr attenuated staurosporine-induced death by 47% (t17 = 9.561; p < 0.001). The asterisks indicate the difference from paired control (Bonferroni t test; p < 0.05). The bars show the mean + SE of 9-13 cultures obtained from three separate dissections. D, z-vad-FMK treatment did not reduce DNA laddering (arrowheads) 20 hr after NMDA challenge of stretched cultures (representative of 2 experiments).
Although active caspase 3 was not detected, we next questioned whether other caspases might have been involved. Thus, we examined the effects of the pan caspase inhibitor z-VAD-FMK (Fearnhead et al., 1995). Previous studies suggest that z-VAD-FMK treatment of cortical neurons protects them from NMDA-induced apoptosis (Charriaut-Marlangue and Ben Ari, 1995; Grasl-Kraupp et al., 1995; Tenneti et al., 1998). Indeed, we found that cell death in cultures treated with staurosporine (1 μm; 48 hr) can be significantly reduced by pretreatment with 200 μm z-VAD-FMK (Fig. 3C). Two hundred micromoles of z-VAD-FMK were not toxic to unstretched cultures or to those that underwent stretch alone. However, when treating stretched cultures with both 30 μm NMDA and z-VAD-FMK, we found no significant reduction in cytotoxicity (Fig. 3C). Similarly, z-VAD-FMK did not reduce DNA fragmentation of neurons exposed to stretch plus NMDA as measured by DNA gel electrophoresis (Fig. 3D). Taken together, our results suggest that caspase-mediated apoptosis is unlikely to explain the DNA fragmentation observed in sublethally stretched neurons challenged with NMDA.
Role of caspase-independent apoptosis in DNA fragmentation of sublethally stretched neurons exposed to NMDA
In addition to caspase-mediated apoptosis, there also exists caspase-independent mechanisms of DNA degradation. These are initiated by the cytoplasmic release of mitochondrial proteins such as endo g or AIF. The endo g protein is a 30 kDa nuclease involved in mitochondrial DNA replication (Cote and Ruiz-Carrillo, 1993). AIF is a mitochondrial flavoprotein (Susin et al., 1999) that translocates from the mitochondria to the nucleus after insults and activates nucleases that induce large-scale DNA fragmentation (>50 kbp) and cell death (Yu et al., 2003; Lorenzo and Susin, 2004). Released endo g can produce nuclear DNA cleavage directly (Wang, 2001). Recent studies of experimental TBI have implicated AIF translocation from mitochondria to cell nuclei in the resulting damage (Zhang et al., 2002).
To explore this, we first used immunohistochemistry to determine whether AIF translocated to nuclei (Fig. 4A). Cultures that underwent stretch under the indicated conditions (Fig. 4) were then fixed and stained 20 hr after insult. Although AIF immunofluorescence was detectable in the cells (Fig. 4A, red), we could not detect it in the nuclei (Fig. 4A, blue) by this means. Therefore, we next used Western blots to determine whether AIF or endo g translocated to the nucleus after stretch. The cultures underwent a stretch insult using the different conditions indicated in Figure 4B and were harvested at the indicated times (6 or 20 hr). The tissue was then used to prepare cytoplasmic and nuclear fractions (see Materials and Methods) to examine them separately. These fractions were immunoblotted for AIF and endo g, as well as for nNOS. The latter is a cytoplasmic protein not expected in the nuclear fraction and was thus used as a control for contamination of the nuclear fraction by cytoplasmic proteins. Using this approach, we failed to detect any significant quantities of nuclear AIF or endo g immunoreactivity in any of the stretched cells at either 6 or 20 hr after injury (Fig. 4 B). Thus, it is unlikely that these caspase-independent apoptotic mechanisms explain the DNA fragmentation observed in neurons exposed to stretch plus NMDA.
Figure 4.
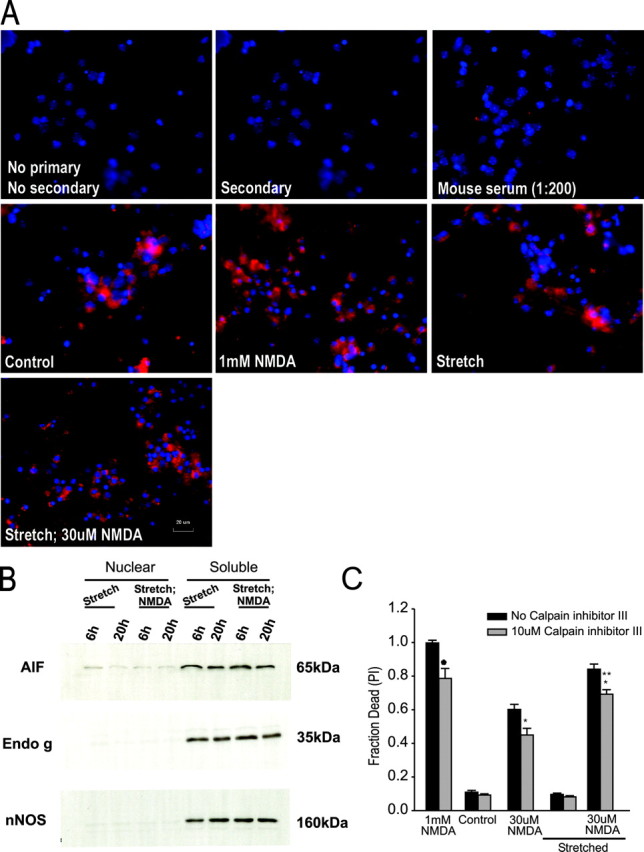
NMDA challenge of stretched neurons does not cause death through caspase-independent apoptotic pathways. A, Lack of nuclear localization of AIF by immunofluorescence. AIF was visualized in fixed cultures at 20 hr after insult. B, Immunoblots of AIF (65 kDa), endo g (35 kDa), and nNOS (160 kDa) in nuclear and cytosolic fractions taken from cells at 6 or 20 hr after insult (stretch ± 30 μm NMDA). All cytoplasmic fractions were positive for immunoreactivity of AIF, endo g, and nNOS. However, nuclear fractions showed only trace immunoreactivity for any of these proteins (representative of 3 separate experiments). C, Inhibiting calpain does not reduce stretch-induced vulnerability to NMDA toxicity. Cultures were prencubated for 30 min with 10 μm calpain inhibitor III and then exposed to the indicated challenge. Calpain inhibitor III remained in the bath until cell death was measured at 20 hr. Calpain inhibition reduced slightly the toxicity of NMDA in all conditions but failed to reduce the vulnerability of the cells to stretch. The asterisks indicate the difference from paired control (Bonferroni t test; p < 0.05). The bars show the mean + SE of 9-12 cultures obtained from three separate experiments.
Role of calpains in sublethally stretched neurons exposed to NMDA
The Ca2+-activated neutral cysteine protease calpain has long been implicated in excitotoxic damage (Siman and Noszek, 1988; Siman et al., 1989). Calpain activity causes cleavage of cytoskeletal proteins, enzymes, and transcription factors. We have already determined that cytoskeletal protein breakdown is unlikely to explain the enhanced vulnerability of stretched neurons to NMDA (Arundine et al., 2003). However, calpains also share some common substrates with the caspases including cleavage of caspases themselves, thus raising the possibility of protease-induced apoptosis (Gil-Parrado et al., 2002; Danial and Korsmeyer, 2004). Accordingly, we examined whether calpain activation might explain the enhanced vulnerability of sublethally stretched neurons to NMDA. We examined the effect of 10 μm z-val-phe-CHO (calpain inhibitor III), which provides neuroprotection against UV-induced neuronal death, reduces spectrin degradation in primary cortical neuronal cultures (McCollum et al., 2002), and reduces hippocampal culture cell mortality from glutamate toxicity (Rami et al., 1997). Calpain inhibitor III was applied 1 hr before stretch and remained in all solutions thereafter. It had no toxic effects in control or in stretched cultures (Fig. 4C). Consistent with other studies, cells challenged either with 30 μm or 1 mm NMDA were slightly protected by this compound, whether or not they underwent stretch. However, the heightened vulnerability of stretched neurons to NMDA was not reduced (Fig. 4C). These data suggest that although calpain activity may partly mediate NMDA toxicity, it does not mediate the increased vulnerability of stretched cultures to NMDA.
Our results to this point show that although sublethally stretched neurons exhibit an enhanced vulnerability to NMDA toxicity, irregular nuclear morphology, and DNA fragmentation, these are unlikely to be caused by classical or caspase-independent apoptotic mechanisms. Thus, we sought alternative explanations for why sublethal stretch causes neurons to be more vulnerable to subsequent insults with NMDA and why DNA fragmentation occurs.
Mitochondrial potential measurements predict survival after sublethal stretch
Mitochondrial dysfunction in cultured neurons has been observed after excitotoxin exposure (Wang and Thayer, 1996; Nicholls and Budd, 1998) and also after in vitro stretch (Ahmed et al., 2000, 2002). Accordingly, we examined whether the lethality of combining sublethal stretch with excitotoxin exposure could be explained by mitochondrial dysfunction as gauged by mitochondrial potential measurements. We used TMRM, a cell-permeant cationic mitochondrial potential indicator. When applied at low concentrations (10 nm), TMRM is extruded from depolarized mitochondria, resulting in a net reduction in whole-cell TMRM fluorescence (Petronilli et al., 2001). As with many potentiometric probes, TMRM is affected by both mitochondrial and membrane potentials (Ehrenberg et al., 1988; Loew et al., 1993). Accordingly, these experiments can only indicate a relative rather than absolute change in mitochondrial depolarization.
In the absence of insults, neurons maintained their mitochondrial potential throughout the 20 hr observation period (Fig. 5). As a positive control, we applied the protonophore FCCP (10 μm; 10 min), which causes a profound mitochondrial depolarization and a corresponding loss of TMRM fluorescence (Fig. 5). Cultures exposed to stretch also exhibited a rapid loss of TMRM fluorescence. However, this recovered within hours (Fig. 5). In contrast, in stretched neurons that were subsequently challenged with NMDA, TMRM fluorescence never recovered (Fig. 5). Thus, although all stretched cells exhibited a drop in mitochondrial potential, only those that died (stretch plus NMDA) exhibited an inability of mitochondrial potential to recover. Moreover, mitochondrial dysfunction in these cells started within minutes and thus preceded cell death.
Sublethal stretch produces high levels of nonlethal ROS
Mitochondria are a significant source of neuronal ROS after anoxic or excitotoxic insults (for review, see Lipton, 1999; Nicholls and Budd, 2000), and ROS mediate cell injury in neurotrauma (Lewen et al., 2000). Because cell death in stretch plus NMDA was associated with irrecoverable loss of mitochondrial function, we questioned whether this could be related to the ROS production in the cell.
ROS levels were gauged over 60 min using the fluorescent indicator DHR (Royall and Ischiropoulos, 1993; Dugan et al., 1995), as described previously (Tymianski et al., 1998). Treatment with the mitochondrial uncoupler FCCP abolished all DHR fluorescence (Fig. 6B, top right), thus excluding any possibility that an increase in DHR signal could be caused by stretch-induced mitochondrial depolarization.
Figure 6.
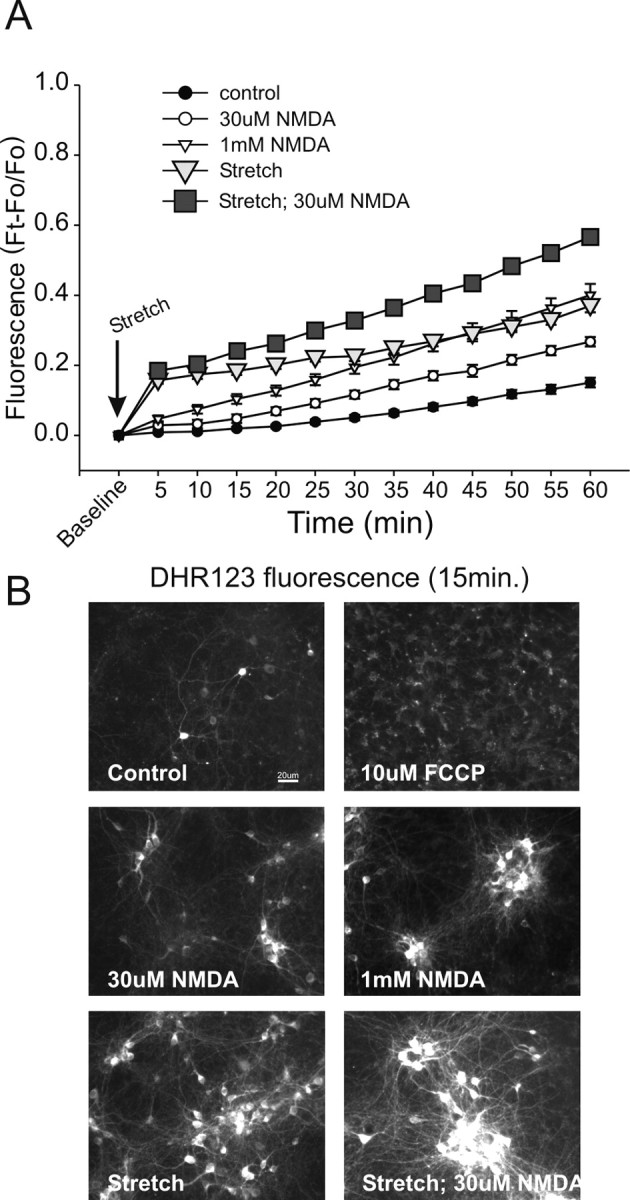
Sublethal stretch injury causes extensive ROS production. Cultures were preincubated with DHR (5 μm) for 30 min before insult. DHR fluorescence was measured at 5 min intervals. Statistical comparisons were made at 60 min. A, Applying either 30 μm or 1 mm NMDA to unstretched cultures caused significant ROS production by 60 min compared with controls (30 μm NMDA-t32 = 5.82, p < 0.0001; 1 mm NMDA-t35 = 7.57, p < 0.0001). Stretch alone also caused extensive ROS production (t34 = 15.13; p < 0.0001), similar in magnitude to that of 1 mm NMDA (t31 = 0.817; p = 0.420). Applying 30 μm NMDA to stretched cultures further increased DHR fluorescence compared with stretch alone (t28 = 7.65; p < 0.0001). Symbols represent the mean ± SE of 14-20 cultures obtained from three separate dissections. The error bars are shown where they exceed symbol size. B, Representative DHR fluorescence images at 15 min after the indicated insult. Treating cultures with the mitochondrial uncoupler FCCP did not increase rhodamine-123 fluorescence at 15 min.
Treatment of unstretched cultures with 30 μm or 1 mm NMDA caused the expected progressive increase in ROS, with a larger rise produced by the 1 mm concentration of NMDA (Fig. 6A). Stretch plus 30 μm NMDA produced the largest amount of DHR fluorescence, a finding commensurate with the lethality of this insult (Fig. 6A,B). Surprisingly, however, lethality was not solely a function of the quantity of ROS produced: sublethal stretch alone produced a rapid increase in ROS production that matched that produced bya1mm NMDA insult (Fig. 6A). However, stretch alone was nonlethal, whereas all neurons challenged with 1 mm NMDA ultimately died (Fig. 1A). Thus, different insults can evoke similar levels of ROS, but similar levels of ROS do not necessarily dictate a similar lethality for different insults. This could be explained if ROS production was not the cause of NMDA toxicity, or if the stretch insult induced the production of different, less toxic, free radical species than the NMDA treatment. Individual species of oxygen-free radicalts are difficult to resolve with fluorescent indicators. For example, DHR is sensitive to superoxide but also to other ROS such as hydrogen peroxide and peroxynitirite (Rothe et al., 1991; Royall and Ischiropoulos, 1993; Bueb et al., 1995; Gilad et al., 1997; Ostrovidov et al., 1998; Gow et al., 1999). Thus, we turned to pharmacological approaches to further examine the contribution of ROS to the enhanced vulnerability of sublethally stretched neurons to NDMAR-mediated toxicity.
Pretreatment with a superoxide dismutase mimic reduces stretch-induced ROS production and NMDAR-mediated toxicity
ROS are produced in large quantities during excitotoxic or anoxic insults (Reynolds and Hastings, 1995; Bindokas et al., 1996; Lipton, 1999). To suppress their effects, we used the superoxide dismutase (SOD) mimetic MnTBAP, which scavenges superoxide, hydrogen peroxide, and peroxynitrite (Patel et al., 1996; Day et al., 1997; Zingarelli et al., 1997; Hill et al., 2000).
Accordingly, we first studied the effect of MnTBAP treatment on the survival of stretched neurons treated with NMDA. Cultures underwent stretch under the conditions indicated in Figure 7A. Cell death was gauged at 20 hr thereafter. Treatment with MnTBAP completely eliminated cell death in stretched cells treated with 30 μm NMDA (Fig. 7A,B). This confirmed that ROS are key in the vulnerability of the neurons to NMDA and thus in the vulnerability of stretched cells to NMDA.
Figure 7.
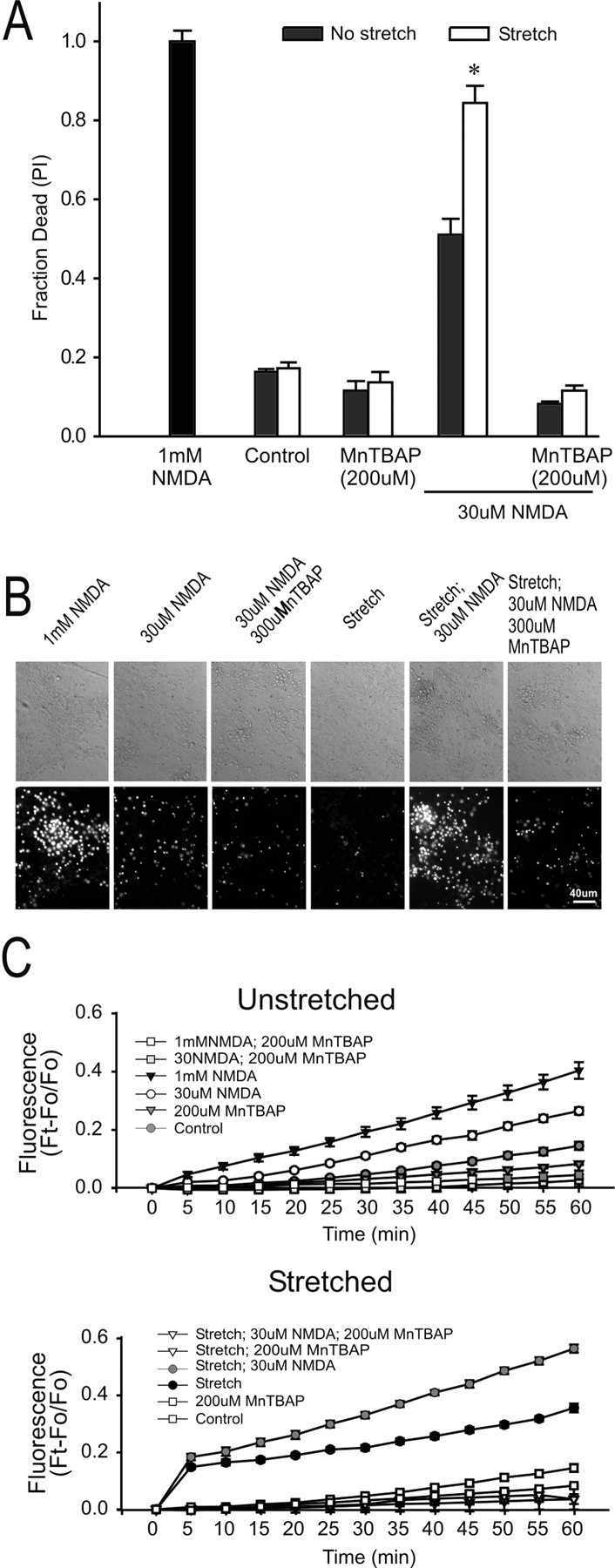
Elimination of stretch-induced vulnerability NMDA toxicity by pretreatment with an ROS scavenger, MnTBAP. Cultures were preincubated for 30 min with 200 μm MnTBAP, which remained in the bath thereafter. Cell death was measured at 20 hr. A, Effects of MnTBAP on cell death under the indicated conditions. The bars represent the mean + SE of 6-12 cultures obtained from three separate dissections. The asterisk indicates the difference from unstretched cultures in same group (t21 = 5.63; p < 0.001). B, Representative phase-contrast and PI fluorescence images of unstretched and stretched cultures 20 hr after the indicated insult. C, Effect of Mn TBAP on ROS levels in unstretched (top) and stretched (bottom) cultures. Pretreatment with MnTBAP reduced the 60 min DHR fluorescence in stretched and unstretched cultures under all insult conditions (Bonferroni t test; p < 0.05). Each symbol represents the mean ± SE of 9-19 cultures obtained from three separate dissections.
To confirm that MnTBAP protected cells by reducing ROS production, we repeated the stretch experiments with MnTBAP and subsequently measured ROS with DHR. MnTBAP pretreatment reduced DHR fluorescence in all culture conditions, indicative of reduced ROS production (Fig. 7C). These findings support the use of DHR as a ROS probe in these experiments and confirm the role of ROS in the toxic effects of NMDA in stretched cells.
NO is required for the enhanced vulnerability of sublethally stretched neurons to NMDA
The most common ROS produced by mithochondria is superoxide (Nicholls and Budd, 2000). However, in some instances, NMDAR-mediated toxicity has been ascribed to NO (Dawson et al., 1991; Sattler et al., 1999), produced by nNOS. To determine the role of NO in the vulnerability of stretched neurons to NMDA, we first used the NOS inhibitor l-NAME, which protects our cultures against NMDAR toxicity (Sattler et al., 1999). Treating the cultures with l-NAME reduced the toxicity of 30 μm NMDA in unstretched neurons (Fig. 8A), although not to the extent achieved by the SOD mimic MnTBAP (compare Figs. 7A, 8). However, treatment with l-NAME completely eliminated the enhanced vulnerability of stretched neurons to NMDA (Fig. 7A). The disproportionate protective effect of l-NAME on the toxicity induced in stretched neurons by NMDA suggests that this enhanced vulnerability might be mediated by NO production.
Figure 8.
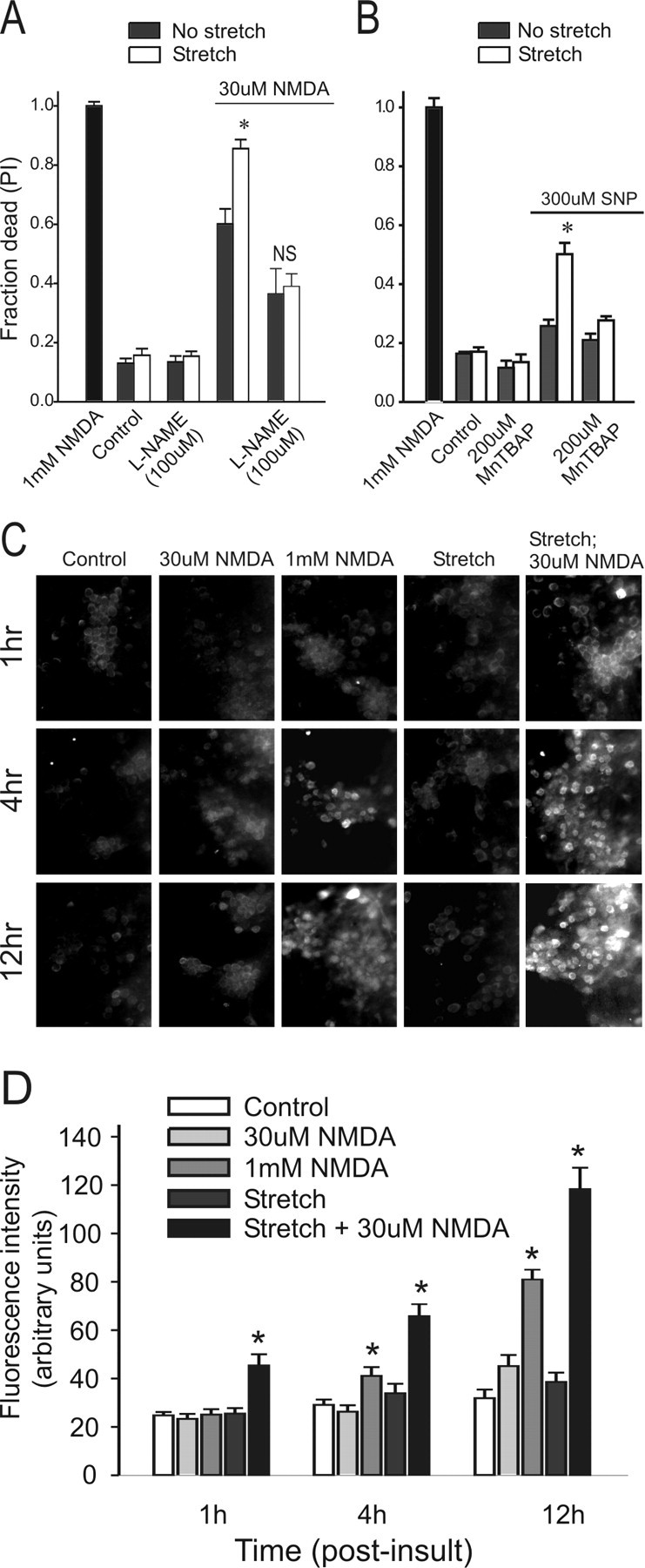
NO production is key instretch-mediated vulner ability to NMDA toxicity. Cell death in A and B was measured at 20 hr. A, Effects of l-NAME, a NOS inhibitor, on cell death under the indicated conditions. Cultures were preincubated with 100 μm l-NAME for 30 min. The asterisk inicates the difference from paired control (t18 = 4.141; p < 0.001). NS, Not different from paired control (t25 = 1.14; p = 0.313). The bars represent the mean + SE of 6-19 cultures obtained from three separate dissections. B, Attenuation of SNP (300 μm; an NO donor) toxicity by the ROS scavenger MnTBAP. The solutions contained MK-801 (10 μm), CNQX (10 μm), and nimodipine (2 μm) to block Ca influx through these pathways. SNP was applied for 1.5 hr. Previous stretch enhanced the vulnerability of neurons to SNP (*t16 = 5.583; p < 0.001), and this was abolished with a 30 min pretreatment with 200 μm MnTBAP. The bars represent the mean + SE of 6-12 cultures obtained from three separate dissections. C, Effect of NMDA treatment on nitrotyrosine staining at the indicated time and conditions. D, Quantification of nitrotyrosine staining intensity at the indicated times. Background-subtracted fluorescence intensity measurements were taken from 5-15 randomly chosen fields from each culture using identical excitation wavelengths, microscope, and camera settings. The bars indicate the mean + SE of two cultures from each of two separate experiments. The asterisks indicate the difference from unstretched controls at the same time point (Bonferroni t test; p < 0.05).
To examine this further, we used the NO donor SNP to generate this radical in stretched neurons (Sattler et al., 1999). Pilot experiments using the NO-sensitive dye DAF2-DA (4,5, diaminofluoresceine) (Kojima et al., 1998) confirmed that SNP treatment enhanced NO levels in the cultures (data not shown). If the enhanced vulnerability of stretched neurons to NMDA is mediated by NO, then stretched neurons should also be more vulnerable to the direct addition of the NO donor independently of NMDA. To test this, stretched and unstretched cultures were exposed to 300 μm SNP. Consistent with our hypothesis, stretched cultures exhibited increased vulnerability to SNP (Fig. 8B). Taken together, the selective effects of l-NAME and SNP on the vulnerability of stretched cultures strengthens the notion that NO production is important for this effect and that NO is a mediator of the enhanced vulnerability of stretched cultures to secondary insults.
NO reacts with mitochondrially derived superoxide to form peroxynitrite (Bonfoco et al., 1995), a potent radical to which have been ascribed the damaging effects of NO signaling (Royall and Ischiropoulos, 1993; Trackey et al., 2001). Indeed, treating the cultures with MnTBAP, which scavenges superoxide and peroxynitrite (Zingarelli et al., 1997), reduced SNP toxicity to baseline (Fig. 8B), suggesting that this toxicity might involve peroxynitrite formation. Accordingly, we next examined the role of peroxynitrite in the vulnerability of stretched neurons to NMDA toxicity.
Peroxynitrite is the lethal ROS responsible for enhanced NMDA toxicity in stretched cultures
Peroxynitrite nitrates tyrosine residues found on many proteins, and this can be detected immunohistochemically by anti-nitrotyrosine antibodies (Trackey et al., 2001). To determine whether peroxynitrite is the ROS mediating the vulnerability of stretched neurons to NMDA, cultures were exposed to stretch, stretch plus 30 μm NMDA, or NMDA at low (30 μm) or high (1 mm) concentrations. Nitrotyrosine staining was performed at 1, 4, and 12 hr after the indicated experimental manipulations (Fig. 8C,D). Unstretched cultures that had been challenged with 1 mm NMDA displayed significantly increased nitrotyrosine immunofluorescence compared with unstretched cultures controls, cultures challenged with 30 μm NMDA, or with cultures challenged only with stretch. This indicates that stretch alone does not produce peroxynitrite, a finding consistent with the lack of cell death with stretch alone. Furthermore, the data indicate that an NMDA insult evokes the production of ROS that are different from those elicited by stretch. The results point to peroxynitrite as the likely culprit necessary for cell death in these paradigms and explain why stretch and 1 mm NMDA insults each evoke similar levels of ROS as measured with DHR (Fig. 6A) but the NMDA insult is lethal while the stretch is not. As suggested in a previous study (Trackey et al., 2001), cortical cells may be better able to cope with an oxidative but not a nitrosative stress.
An application of 30 μm NMDA to unstretched neurons, or exposure only to sublethal stretch, did not evoke significant nitrotyrosine staining. However, the lethal combination of the two caused an even greater amount of nitrotyrosine staining than the 1 mm NMDA insult (Fig. 8C,D). This suggests that the production of superoxide by previous stretch sensitizes the neurons so that the NO produced by low NMDA concentrations (30 μm) is channeled into peroxynitrite production.
Pretreatment with l-NAME or MnTBAP protects neurons from internucleosomal DNA fragmentation
We have shown that DNA fragmentation occurs to a significant extent only in stretched neurons subsequently challenged with NMDA, compared with stretch alone or NMDA alone (Fig. 2A-C). However, this DNA fragmentation was not the result of apoptosis (Figs. 3, 4). Moreover, we showed that death of stretched neurons challenged with NMDA requires ROS that include NO and peroxynitrite and that these radicals explained the enhanced vulnerability of the stretched cultures to NMDA. Previous studies have shown that these radicals, on their own, can also induce DNA fragmentation (Hill et al., 2000). If so, then this fragmentation should be reversible by NO and peroxynitrite inhibition.
To test this, cultures were exposed to stretch, followed by NMDA as before, but in the presence of MnTBAP or the NOS inhibitor l-NAME. TUNEL staining and DNA gel electrophoresis were performed as before at 20 hr after insult. Treatment with either compound virtually abolished TUNEL staining (Fig. 9A,B) and DNA laddering (Fig. 9C), confirming that DNA fragmentation was the result of ROS produced through the NO signaling pathway.
Figure 9.
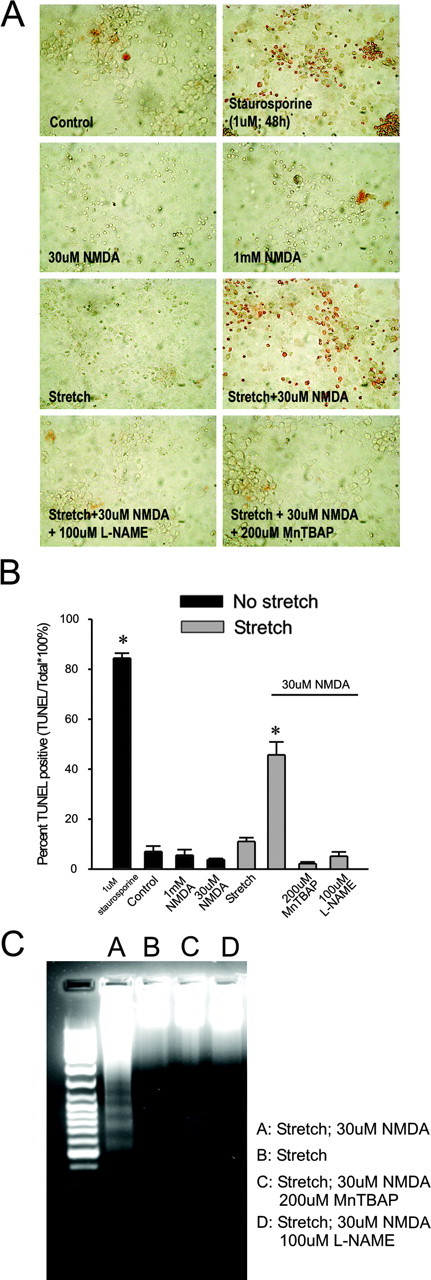
Pretreatment with MnTBAP or l-NAME results reduces TUNEL staining and DNA laddering in sublethally stretched cultures challenged with NMDA. Representative images (A) and quantification (B) of TUNEL staining using the DAB method at 20 hr after the indicated insult. The cultures were princubated for 30 min with either 200 μm MnTBAP or 100 μm l-NAME, as indicated. Staurosporine was applied for 48 hr. The asterisks indicate the difference from unstretched control (Bonferroni t test; p < 0.05). Each plotted data represents TUNEL-positive cells normalized to total cell number. The bars represent the mean + SE of two to four randomly selected fields in each of three cultures from each of three experiments. C, Representative DNA gel of the effect of pretreating stretched cultures with either MnTBAP or l-NAME on DNA laddering. The data are representative of three separate experiments.
Disrupting NMDAR-PSD-95 interactions attenuates stretch-induced enhancement of NMDA toxicity
If NO formation is responsible for enhancing the vulnerability of stretched neurons to NMDA, then blocking NO signaling should reduce this. Although NOS inhibition with l-NAME is one approach, it may not be specific to neurons because inducible NOS in glia may also mediate excitotoxic and anoxic damage to cortical neurons (Hewett et al., 1994, 1996).
To inhibit NO signaling with greater specificity to neurons, we focused on inhibiting neuronal NO formation by NMDA by perturbing the interactions of NMDARs with the submembrane scaffolding protein PSD-95 (Sattler et al., 1999). Through its second PDZ [PSD-95/Discs large/zona occludens-1] domain (PDZ2), PSD-95 binds NMDAR NR2 subunits as well as nNOS, thus keeping this signaling enzyme in a close functional association with NMDARs (Kornau et al., 1995; Brenman et al., 1996; Brenman and Bredt, 1997). The interaction between NMDAR NR2B subunits and the PDZ2 domain of PSD-95 depends on a conserved C terminus tSXV motif of NR2B (Fig. 10Ai) (Kornau et al., 1995). This interaction can be disrupted by the intracellular introduction of exogenous proteins that competitively bind to either the NR2B or the PDZ2 interaction domains (Fig. 10Aii,iii). We used a nine-residue peptide encompassing the PDZ-binding motif of NR2B (KLSSIESDV; NR2B9c), which binds the PSD-95 PDZ2 domain (Fig. 10Aii). This peptide prevents the association of PSD-95 with NR2 subunits (Kornau et al., 1995) and with Kv1.4 channels (Brenman et al., 1998). NR2B9c also increases the binding activity of full-length recombinant PSD-95 protein to MAP1A (Brenman et al., 1998). Although the significance of this is unclear, and microtubules are unlikely to participate in increasing the vulnerability of stretched cultures to NMDA toxicity, we controlled for this additional effect of NR2B9c by developing an alternative means to interfere with the NMDAR-PSD-95 interaction. We constructed a protein comprising residues 65-248 of PSD-95 encompassing the first and second PDZ domains (PDZ1-2), which contains the principal binding domain in PSD-95 for the C terminus of NR2B. This protein should also affect NR2B/PSD-95 binding by interacting with native NR2B C terminus motifs (Fig. 10Aiii).
NR2B9c or PDZ1-2 on their own were not anticipated to enter cells, and therefore we fused each to a peptide corresponding to the cell membrane transduction domain of the HIV-1-Tat protein (YGRKKRRQRRR; Tat) to obtain a 20 amino acid peptide (Tat-NR2B9c) and the fusion protein pTat-PDZ1-2 (Fig. 10B). These transduce cell membranes in a rapid, dose-dependent manner independent of receptors and transporters (Schwarze et al., 1999; Aarts et al., 2002). As a control for cell transduction, we created a peptide comprising HIV-1-Tat residues 38-48 (KALGISYGRKK; Tat38-48) outside the Tat transduction domain (Mann and Frankel, 1991), which does not pass through cell membranes (Aarts et al., 2002). As a control for the Tat-NR2B9c peptide, we synthesized a peptide in which the C-terminal tSXV motif of NR2B contained a double point mutation to alanines (Tat-KLSSIEADA; Tat-NR2BAA), rendering it incapable of binding PSD-95 (Kornau et al., 1995). As a control for pTat-PDZ1-2, we made pTat-GK, a Tat fusion protein containing residues 534 -724 of PSD-95 comprising the C-terminal guanylate kinase homology domain that lacks enzymatic activity (Kistner et al., 1995). A listing of the active and control proteins constructed to target either side of the NR2B-PSD-95 interaction is provided in Figure 10B.
To examine intracellular delivery of Tat peptides, they were conjugated to the fluorophore dansyl chloride (excitation, 360 nm; emission, >510 nm). Intracellular accumulation of dansyl-Tat-NR2B9c (10 μm), but not control peptide (dansyl-Tat-38-48; 10 μm), was observed 30 min after application to cortical neuronal cultures using confocal microscopy (Fig. 10C; representative of five experiments). Fluorescence of cultures treated with dansyl-Tat-38-48 was similar to background (data not shown). In a previous study, dansyl-Tat-NR2B9c was detectable in the neurons within 10 min of the start of the application, reaching a peak level over the next 20 min. This level was maintained until the dansyl-Tat-NR2B9c was washed from the bath, and the peptide remained detectable within the neurons for >5 hr thereafter (Aarts et al., 2002). In previously characterizing the effects of Tat-NR2B9c in vitro, we found that applying Tat-NR2B9c reduced the coimmunoprecipitation of PSD-95 with NR2B. In hippocampal CA1 neurons, Tat-NR2B9c (50 nm) had no effect on synaptic responses, on patch recordings of total EPSC, or on AMPA or NMDA components of the EPSC. In cultured cortical neurons, Tat-NR2B9c or pTat-PDZ1-2 (each at 50 nm) did not alter the uptake of 45Ca2+ produced by applying NMDA (Aarts et al., 2002). However, Tat-NR2B9c (50 nm) treatment dissociated NMDAR activity from NO production by nNOS (Aarts et al., 2002), which is bound to the PDZ2 domain of PSD-95 (Brenman et al., 1996).
Having previously shown that Tat-NR2B9c treatment reduced NO production in cultures challenged with NMDA (Aarts et al., 2002), we next evaluated the effects of Tat-NR2B9c on NMDA-mediated free radical production and nitrotyrosine staining in both unstretched (Fig. 10D, top) and stretched (Fig. 10D, bottom) cultures. Tat-NR2b9c pretreatment reduced NMDA-evoked ROS production in both paradigms, as measured by DHR oxidation (Fig. 10D). Also, Tat-NR2B9c pretreatment resulted in reduced nitrotyrosine immunoreactivity at 12 hr after NMDA challenge, indicating that this approach successfully reduces peroxynitrite formation in the neurons (Fig. 10E).
Next, we examined the effect of pretreating the cortical neuronal cultures with 50 nm Tat-NR2B9c or with the control peptide Tat-NR2BAA. The peptides were applied 30 min before stretch, and NMDA (30 μm) was applied for 1 hr thereafter. Neuronal cell death was quantified 20 hr after stretch (Fig. 11A,B; top). The peptides had no toxic effects in control or in stretched cultures (Fig. 11A). On exposure to low NMDA concentrations, stretched cultures that were untreated with the peptides exhibited the anticipated increased vulnerability to NMDA toxicity (Fig. 11A). However, this heightened vulnerability was completely abolished in cultures pretreated with Tat-NR2B9c (Fig. 11A). The effects of Tat-NR2B9c were not seen with the control peptide Tat-NR2BAA (Fig. 11A).
Figure 11.

Pretreatment and post-treatment with Tat peptides and fusion proteins reduces the stretch-induced increased vulnerability to NMDA toxicity. A, Effect of pretreatment with Tat peptides on survival 20 hr after the indicated insults. Cultures were preincubated with 50 nm Tat peptides for 30 min. The peptides remained in the bath thereafter. Inset, Experimental time course. The asterisks indicate the different from paired control (Bonferroni t test; p < 0.05). N.S., Not significantly different. The bars are the mean + SE of 6-20 cultures obtained from four different experiments. B, Representative phase-contrast and PI fluorescence images of unstretched (left) and stretched (right) cultures 20 hr after challenge with 30 μm NMDA. Pretreatment with Tat-NR2B9c, but not with Tat-NR2B-AA, resulted in decreased PI fluorescence. C, D, Effect of post-treatment with Tat peptides (C) or fusion proteins (D) on cell survival 20 hr after the indicated insult. The peptides or fusion proteins were added 1 hr after insult onset (after termination of the NMDA challenge). Post-treatment with 50 nm Tat-NR2B9c or pTat-PDZ1-2 reduced the vulnerability of neurons to NMDA after stretch. The asterisks indicate the differences from paired controls (Bonferroni t test; p < 0.05). The bars are the mean + SE of 7-22 cultures obtained from four different experiments. Inset, Experimental time course.
Our data indicate that NMDARs, through their specific interactions with PSD-95 protein, are able to trigger distinct downstream signaling cascades responsible for the enhanced vulnerability of neurons to NMDA toxicity. Because these signaling mechanisms, once activated, may persist beyond the duration of the NMDA challenge, we next examined whether post-treatment of cultures with Tat peptides or fusion proteins could also be protective. Experiments in cultured cortical neurons were repeated as in Figure 11A, but the Tat peptides were applied after terminating the 1 hr NMDA challenge (Fig. 11C,D; top). Post-treatment with Tat-NR2B9c, but not Tat-NR2B-AA, reduced the enhanced cell mortality produced in stretched neurons by the NMDA challenge (Fig. 11C). Similar results were obtained by post-treating the cultures with pTat-PDZ1-2, the fusion protein that targets the opposite side of the NR2-PSD-95 interaction from Tat-NR2B9c (Fig. 11D). In contrast, the control fusion protein pTat-GK, which does not target this interaction, was ineffective (Fig. 11D). In conclusion, introducing into the cells an exogenous peptide containing the C-terminal nine amino acids of the NR2B NMDAR subunit has profound effects on excitotoxic signaling pathways downstream of NMDAR activation. The effects of this peptide are lost by mutating amino acids that are essential for mediating PDZ binding to PSD-95. In addition, a protein comprising PDZ1-2 of PSD-95 shares the effects of the NR2B C-terminal peptide. These findings imply that the cytotoxic signals downstream from NMDARs may be interrupted by these cell-permeant peptides.
Discussion
Here, we describe the mechanism of increased vulnerability of sublethally stretched neurons to secondary glutamatergic insults, which are common after CNS trauma (Faden et al., 1989; Brown et al., 1998; Lewen et al., 2000). Also, we described the death mechanisms underlying the DNA fragmentation that was so suggestive of apoptosis in these cells. Sublethal stretch injury was tolerated by the neurons but rendered them vulnerable to low concentrations of NMDA that otherwise would have been tolerated (Fig. 1). Unstretched neurons challenged with lethal NMDA concentrations exhibited the usual morphological features of necrotic death, whereas stretched cells exhibited nuclear irregularities (Fig. 1C) and DNA fragmentation (Fig. 2) suggestive of a switch to apoptosis. However, the DNA degradation was not attributable to classical (caspase mediated) (Fig. 3) or caspase-independent (Fig. 4) apoptosis. Rather, it was related to profound mitochondrial dysfunction (Fig. 5) and ROS production (Fig. 6). Indeed, sublethal stretch resulted in surprisingly high levels of ROS, matching those created by a lethal insult with 1 mm NMDA (Fig. 6). However, to be lethal, the ROS produced also needed to include reactive nitrogen species, especially peroxynitrite (Figs. 7, 8C,D). ROS scavengers and NOS inhibitors prevented cell death and DNA degradation (Figs. 7, 8A,B, 9). Moreover, inhibiting NMDAR-evoked nNOS activation with peptides that perturbed NMDAR-PSD-95 interactions reduced protein nitration (Fig. 10) and cell death (Fig. 11). Collectively, these data indicate that increased vulnerability of stretched neurons to secondary insults is attributable to the surprisingly high ROS produced in the cells even when stretch is sublethal. This primes the neurons to additional insults such as with NMDA, which produces NO. The combination of mitochondrial ROS (superoxide) and NO into peroxynitrite (Reiter et al., 2000) leads to the protein nitration, DNA degradation, and cell death (Fig. 12). Thus, the DNA fragmentation seen in these cells is not apoptotic but is attributable to DNA damage by reactive nitrogen species.
Figure 12.

Proposed mechanism of cell death in sublethally stretched neurons exposed to NMDA. Stretch results in increased superoxide production at a level that is still tolerated by the cells. However, subsequent NMDAR activation causes NO production, which permits the formation of peroxynitrite. This, in turn, causes DNA fragmentation by a process independent of classical caspase-dependent apoptosis, caspase-independent apoptosis (AIF, endo g), or caplains.
Numerous studies have implicated apoptotic mechanisms in spinal cord injury (SCI) and TBI (for review, see Beattie et al., 2000; Eldadah and Faden, 2000; Keane et al., 2001; Faden, 2002; Liou et al., 2003), and the present study does not exclude a role for apoptosis in these conditions. Also, our data do not exclude the possibility that apoptotic mechanisms play key roles in the death of non-neuronal cells in CNS trauma. For example, in SCI, neurological dysfunction is attributable in part to oligodendrocyte apoptosis (Beattie et al., 2002; Dong et al., 2003; Stirling et al., 2004). Rather, our focus here was on mechanisms that can lead to neuronal damage in mild in vitro stretch injury (Arundine et al., 2003). The strain rates experienced by the neurons in our injury model are considerably less than those in previously published reports, because most of these used models that reproduced strains experienced during severe TBI in vivo (Thibault et al., 1992; Ellis et al., 1995; Zhang et al., 1996; Laplaca et al., 1997). Thus, our findings in a milder traumatic insult are significant in several respects. First, we showed that even sublethal injuries produce changes in neurons that render them vulnerable to secondary insults, highlighting the importance of preventing secondary perturbations that might otherwise have been tolerated. Second, we illustrated that in mild neurotrauma, apoptotic-like features are attributable entirely to nonapoptotic mechanisms, highlighting the potential difficulties in attributing the relative contribution of apoptosis to neurons survival in more severe mechanical trauma. Third, we determined the mechanisms of enhanced vulnerability of the neurons to secondary insults and of the DNA damage produced, thus raising the possiblility of treatment based on these mechanisms. Finally, we demonstrated a means of treating stretch-induced injury and preventing the vulnerability of neurons to secondary insults, using peptides that reduce this vulnerability even after the injury onset.
Previous studies have suggested that ROS production and apoptosis may occur concurrently in cultured neurons exposed to excitotoxicity (Bonfoco et al., 1996) or stretch injury (Shah et al., 1997). Similar findings have been published in animal models of neurotrauma (for review, see Raghupathi et al., 2000; Leker and Shohami, 2002). However, when the primary insult can itself be lethal, it is difficult to distinguish whether the ROS detected were the cause of apoptosis or whether apoptotic mechanisms occurred independently of ROS production. If so, then at least part of the DNA fragmentation and nuclear irregularities observed in these studies may have been attributable to ROS rather than to apoptosis. Our approach of sublethally stretching neurons removed the confounding effects of cell death because of the primary (stretch) insult, and the data suggest that caution should be exercised in drawing inferences about the mechanism of cell death in more severe forms of injury.
Excitotoxicity has long been implicated in mediating traumatic damage. For example, neuronal damage induced in mixed cultures by tearing with a plastic stylet is attenuated by NMDA antagonists (Tecoma et al., 1989). In rats, brain injury induced by fluid percussion causes a marked elevation in extracellular glutamate and aspartate adjacent to the trauma site, which correlates to the severity of the injury and cellular bioenergetic failure (Faden et al., 1989). Neurotrauma also induces CNS ischemia in animals and humans (Tator and Koyanagi, 1997; Zwienenberg and Muizelaar, 2001), and excitotoxicity attributable to an excessive release of excitatory amino acids is anticipated. Furthermore, glutamate is present in millimolar quantities in neurons and glial cells, and it is anticipated that primary mechanical disruption of cell membranes would raise extracellular glutamate levels and cause excitotoxicity. However, in the present study, we show that the levels of extracellular excitotoxic substances are not the sole determinant of survival outcome. Whereas high concentrations of NMDA are lethal to unstretched cells, very low amounts are needed to kill neurons that had undergone mild trauma because of their previous priming through mitochondrial ROS production. This may be an additional important mechanism of damage in neurotrauma.
Our previous characterization of the present model (Arundine et al., 2003) demonstrated that the enhanced vulnerability of sublethally stretched cortical neurons is not ubiquitous to all secondary insults. Rather, it exhibited specificity to NMDAR-associated signaling pathways. We have previously demonstrated this phenomenon in excitotoxicity in cultured spinal and cortical neurons (Tymianski et al., 1993; Sattler et al., 1998). Although it is generally accepted that NMDARs play key roles in excitotoxicity because of their high Ca2+ permeability (Choi, 1992; Tymianski, 1996), the mechanism by which Ca2+ influx triggers neuronal cell death is no longer felt to be a simple function of the quantity of Ca2+ entering the cell. Rather, there is now compelling evidence that various calcium-dependent processes are regulated via distinct signal pathways linked to specific routes of Ca2+ influx (Bading et al., 1993; Ghosh et al., 1994). The “source specificity hypothesis” (reviewed in Tymianski and Tator, 1996; Hardingham and Bading, 2003) reasons that Ca2+ toxicity occurs not simply as a function of increased Ca2+ concentration but is linked to the route of Ca2+ entry and the distinct second messenger pathways that are activated as a result. Because this applies also to mechanical neuronal damage (Arundine et al., 2003), this suggests that despite the large number of processes that might be perturbed in neurotrauma, only some might require therapeutic intervention. In the case of the present paradigm, we reason that the preferential association of NMDARs with PSD-95 over other submembrane molecules underlies the specificity of stretch-induced vulnerability of neurons to NMDA toxicity.
PSD-95 binds cytoskeletal organizing proteins (Brenman et al., 1998; Niethammer et al., 1998), other scaffolding molecules (Kim et al., 1997; Naisbitt et al., 1999), and signaling enzymes such as SynGAP (Kim et al., 1998). Through its second PDZ domain (PDZ2), PSD-95 binds both NR2 subunits and nNOS (Kornau et al., 1995; Brenman et al., 1996). This configuration couples NMDAR activity with the production of NO, a signaling molecule that also mediates NMDAR-dependent excitotoxicity (Dawson et al., 1991). Research has shown that NMDAR function is unaffected by genetically disrupting PSD-95 in vivo (Migaud et al., 1998) or by suppressing its expression in vitro (Sattler et al., 1999). Nonetheless, PSD-95 deletion dissociates NMDAR activity from NO production and suppresses NMDAR-dependent excitotoxicity (Sattler et al., 1999). We have previously demonstrated in cultured cortical neurons that suppressing PSD-95 expression selectively attenuated excitotoxicity triggered via NMDARs but not by other glutamate or Ca2+ channels (Sattler et al., 1999). More recently, we disrupted preformed NMDAR-PSD-95 complexes in cultured neurons and in vivo, which also attenuated NMDA toxicity in vitro and ischemic brain damage in rats (Aarts et al., 2002). Given the data in the present study, it is plausible to attribute the affects of perturbing NMDAR-PSD-95 interactions to a reduction in NO signaling. However, PSD-95 also binds a range of additional anchoring and signaling proteins, the functions of which are incompletely understood and which may also participate in enhancing the enhanced vulnerability of the neurons to NMDA after stretch (Sheng, 2001; Sheng and Kim, 2002). Thus, it is possible that targeting other intracellular proteins using the approach described here could be used to study and modulate signaling pathways responsible for other components of traumatic and ischemic CNS damage.
Footnotes
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP 53353 and National Institutes of Health Grant RO1 NS 48956 to M.T. M.Ar. is a student of the CIHR, M.Aa. is a fellow of the Ontario Heart and Stroke Foundation, and M.T. is a CIHR Clinician-Scientist.
Correspondence should be addressed to Dr. Michael Tymianski, Toronto Western Hospital, Laboratory 11-416, McLaughlin Pavilion, 399 Bathurst Street, Toronto, Ontario, M5T 2S8 Canada. E-mail: mike_t@uhnres.utoronto.ca.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248106-18$15.00/0
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298: 846-850. [DOI] [PubMed] [Google Scholar]
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M (2003) A key role for TRPM7 channels in anoxic neuronal death. Cell 115: 863-877. [DOI] [PubMed] [Google Scholar]
- Ahmed SM, Rzigalinski BA, Willoughby KA, Sitterding HA, Ellis EF (2000) Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem 74: 1951-1960. [PubMed] [Google Scholar]
- Ahmed SM, Weber JT, Liang S, Willoughby KA, Sitterding HA, Rzigalinski BA, Ellis EF (2002) NMDA receptor activation contributes to a portion of the decreased mitochondrial membrane potential and elevated intracellular free calcium in strain-injured neurons. J Neurotrauma 19: 1619-1629. [DOI] [PubMed] [Google Scholar]
- Allen RT, Hunter III WJ, Agrawal DK (1997) Morphological and biochemical characterization and analysis of apoptosis. J Pharmacol Toxicol Methods 37: 215-228. [DOI] [PubMed] [Google Scholar]
- Amar AP, Levy ML (1999) Pathogenesis and pharmacological strategies for mitigating secondary damage in acute spinal cord injury. Neurosurgery 44: 1027-1039. [DOI] [PubMed] [Google Scholar]
- Arundine M, Chopra GK, Wrong AW, Lei S, Aarts M, MacDonald JF, Tymianski M (2003) Enhanced vulnerability to NMDA toxicity in sublethal traumatic neuronal injury in-vitro. J Neurotrauma 20: 1377-1395. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260: 181-186. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Farooqui AA, Bresnahan JC (2000) Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma 17: 915-925. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO (2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36: 375-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ (1996) Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci 16: 1324-1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA (1995) Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 92: 7162-7166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Leist M, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P (1996) Cytoskeletal breakdown and apoptosis elicited by NO donors in cerebellar granule cells require NMDA receptor activation. J Neurochem 67: 2484-2493. [DOI] [PubMed] [Google Scholar]
- Borer RA, Lehner CF, Eppenberger HM, Nigg EA (1989) Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56: 379-390. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Bredt DS (1997) Synaptic signaling by nitric oxide. Curr Opin Neurobiol 7: 374-378. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS (1996) Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 84: 757-767. [DOI] [PubMed] [Google Scholar]
- Brenman JE, Topinka JR, Cooper EC, McGee AW, Rosen J, Milroy T, Ralston HJ, Bredt DS (1998) Localization of postsynaptic density-93 to dendritic microtubules and interaction with microtubule-associated protein 1A. J Neurosci 18: 8805-8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JI, Baker AJ, Konasiewicz SJ, Moulton RJ (1998) Clinical significance of CSF glutamate concentrations following severe traumatic brain injury in humans. J Neurotrauma 15: 253-263. [DOI] [PubMed] [Google Scholar]
- Bruno VMG, Goldberg MP, Dugan LL, Giffard RG, Choi DW (1994) Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem 63: 1398-1406. [DOI] [PubMed] [Google Scholar]
- Budd SL, Tenneti L, Lishnak T, Lipton SA (2000) Mitochondrial and extra-mitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA 97: 6161-6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueb JL, Gallois A, Schneider JC, Parini JP, Tschirhart E (1995) A double-labelling fluorescent assay for concomitant measurements of [Ca2+]i and O2 production in human macrophages. Biochim Biophys Acta 1244: 79-84. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2001) Traumatic injury in the United States: an interim report to congress. Atlanta: Centers for Disease Control and Prevention.
- Charriaut-Marlangue C, Ben Ari Y (1995) A cautionary note on the use of the TUNEL stain to determine apoptosis. NeuroReport 7: 61-64. [PubMed] [Google Scholar]
- Choi DW (1987) Ionic dependence of glutamate neurotoxicity. J Neurosci 7: 369-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW (1992) Excitotoxic cell death. J Neurobiol 23: 1261-1276. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Dixon CE, Chen M, Marion DW, Heineman S, DeKosky ST, Graham SH (1997) Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J Neurotrauma 14: 179-189. [DOI] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK (1998) Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci 18: 5663-5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote J, Ruiz-Carrillo A (1993) Primers for mitochondrial DNA replication generated by endonuclease G. Science 261: 765-769. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205-219. [DOI] [PubMed] [Google Scholar]
- David JC, Yamada KA, Bagwe MR, Goldberg MP (1996) AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci 16: 200-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH (1991) Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA 88: 6368-6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day BJ, Fridovich I, Crapo JD (1997) Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys 347: 256-262. [DOI] [PubMed] [Google Scholar]
- Didenko VV, Hornsby PJ (1996) Presence of double-strand breaks with single-base 3′ overhangs in cells undergoing apoptosis but not necrosis. J Cell Biol 135: 1369-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didenko VV, Tunstead JR, Hornsby PJ (1998) Biotin-labeled hairpin oligo-nucleotides: probes to detect double-strand breaks in DNA in apoptotic cells. Am J Pathol 152: 897-902. [PMC free article] [PubMed] [Google Scholar]
- Dong H, Fazzaro A, Xiang C, Korsmeyer SJ, Jacquin MF, McDonald JW (2003) Enhanced oligodendrocyte survival after spinal cord injury in Bax-deficient mice and mice with delayed Wallerian degeneration. J Neurosci 23: 8682-8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW (1995) Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci 15: 6377-6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM (1988) Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J 53: 785-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldadah BA, Faden AI (2000) Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma 17: 811-829. [DOI] [PubMed] [Google Scholar]
- Ellis EF, McKinney JS, Willoughby KA, Liang S, Povlishock JT (1995) A new model for rapid stretch-induced injury of cells in culture: characterization of the model using astrocytes. J Neurotrauma 12: 325-339. [DOI] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S (1998) A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391: 43-50. [DOI] [PubMed] [Google Scholar]
- Faden AI (2002) Neuroprotection and traumatic brain injury: theoretical option or realistic proposition. Curr Opin Neurol 15: 707-712. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink P (1989) The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 244: 798-800. [DOI] [PubMed] [Google Scholar]
- Fearnhead HO, Dinsdale D, Cohen GM (1995) An interleukin-1 beta-converting enzyme-like protease is a common mediator of apoptosis in thymocytes. FEBS Lett 375: 283-288. [DOI] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Indentification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerschenson LE, Rotello RJ (1992) Apoptosis: a different type of cell death. FASEB J 6: 2450-2455. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Ginty DD, Bading H, Greenberg ME (1994) Calcium regulation of gene expression in neuronal cells. J Neurobiol 25: 294-303. [DOI] [PubMed] [Google Scholar]
- Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Best-vater F, Holloschi A, Knoch TA, Auerswald EA, Welsh K, Reed JC, Fritz H, Fuentes-Prior P, Spiess E, Salvesen GS, Machleidt W (2002) Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem 277: 27217-27226. [DOI] [PubMed] [Google Scholar]
- Gilad E, Cuzzocrea S, Zingarelli B, Salzman AL, Szabo C (1997) Melatonin is a scavenger of peroxynitrite. Life Sci 60: L169-L174. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Branco F, Christofidou-Solomidou M, Black-Schultz L, Albelda SM, Muzykantov VR (1999) Immunotargeting of glucose oxidase: intracellular production of H(2)O(2) and endothelial oxidative stress. Am J Physiol 277: L271-L281. [DOI] [PubMed] [Google Scholar]
- Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R (1995) In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21: 1465-1468. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2003) The yin and yang of NMDA receptor signalling. Trends Neurosci 26: 81-89. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Csernansky CA, Choi DW (1994) Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron 13: 487-494. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Muir JK, Lobner D, Symons A, Choi DW (1996) Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of iNOS. Stroke 27: 1586-1591. [DOI] [PubMed] [Google Scholar]
- Hill IE, Murray C, Richard J, Rasquinha I, MacManus JP (2000) Despite the internucleosomal cleavage of DNA, reactive oxygen species do not produce other markers of apoptosis in cultured neurons. Exp Neurol 162: 73-88. [DOI] [PubMed] [Google Scholar]
- Ishimaru MJ, Ikonomidou C, Tenkova TI, Der TC, Dikranian K, Sesma MA, Olney JW (1999) Distinguishing excitotoxic from apoptotic neurode-generation in the developing rat brain. J Comp Neurol 408: 461-476. [PubMed] [Google Scholar]
- Jones OT, Bernstein GM, Jones EJ, Jugloff DG, Law M, Wong W, Mills LR (1997) N-Type calcium channels in the developing rat hippocampus: subunit, complex, and regional expression. J Neurosci 17: 6152-6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane RW, Kraydieh S, Lotocki G, Alonso OF, Aldana P, Dietrich WD (2001) Apoptotic and antiapoptotic mechanisms after traumatic brain injury. J Cereb Blood Flow Metab 21: 1189-1198. [DOI] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M (1997) GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol 136: 669-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Liao D, Lau LF, Huganir RL (1998) SynGAP: a synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 20: 683-691. [DOI] [PubMed] [Google Scholar]
- Kistner U, Garner CC, Linial M (1995) Nucleotide binding by the synapse associated protein SAP90. FEBS Lett 359: 159-163. [DOI] [PubMed] [Google Scholar]
- Kojima H, Nakatsubo N, Kikuchi K, Urano Y, Higuchi T, Tanaka J, Kudo Y, Nagano T (1998) Direct evidence of NO production in rat hippocampus and cortex using a new fluorescent indicator: DAF-2 DA. NeuroReport 9: 3345-3348. [DOI] [PubMed] [Google Scholar]
- Kornau HC, Schenker LT, Kennedy MB, Seeburg PH (1995) Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science 269: 1737-1740. [DOI] [PubMed] [Google Scholar]
- Laplaca MC, Lee VM, Thibault LE (1997) An in vitro model of traumatic neuronal injury: loading rate-dependent changes in acute cytosolic calcium and lactate dehydrogenase release. J Neurotrauma 14: 355-368. [DOI] [PubMed] [Google Scholar]
- Leker RR, Shohami E (2002) Cerebral ischemia and trauma-different etiologies yet similar mechanisms: neuroprotective opportunities. Brain Res Brain Res Rev 39: 55-73. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH (2000) Free radical pathways in CNS injury. J Neurotrauma 17: 871-890. [DOI] [PubMed] [Google Scholar]
- Liou AK, Clark RS, Henshall DC, Yin XM, Chen J (2003) To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol 69: 103-142. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- Loew LM, Tuft RA, Carrington W, Fay FS (1993) Imaging in five dimensions: time-dependent membrane potentials in individual mitochondria. Biophys J 65: 2396-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo HK, Susin SA (2004) Mitochondrial effectors in caspase-independent cell death. FEBS Lett 557: 14-20. [DOI] [PubMed] [Google Scholar]
- Mann DA, Frankel AD (1991) Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J 10: 1733-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall LF (2000) Epidemiology and cost of central nervous system injury. Clin Neurosurg 46: 105-112. [PubMed] [Google Scholar]
- McCollum AT, Nasr P, Estus S (2002) Calpain activates caspase-3 during UV-induced neuronal death but only calpain is necessary for death. J Neurochem 82: 1208-1220. [DOI] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RGM, Morrison JH, O'Dell TJ, Grant SGN (1998) Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396: 433-439. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M (1999) Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23: 569-582. [DOI] [PubMed] [Google Scholar]
- National Center for Health Statistics (1997) Data file documentation, National Hospital Discharge Survey 1980-1995. Atlanta: Centers for Disease Control and Prevention.
- Newcomb JK, Zhao X, Pike BR, Hayes RL (1999) Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp Neurol 158: 76-88. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (1998) Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta 1366: 97-112. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315-360. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Valtschanoff JG, Kapoor TM, Allison DW, Weinberg RJ, Craig AM, Sheng M (1998) CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron 20: 693-707. [DOI] [PubMed] [Google Scholar]
- NIH (1999) Consensus conference. Rehabilitation of persons with traumatic brain injury. NIH Consensus Development Panel on Rehabilitation of Persons With Traumatic Brain Injury. JAMA 282: 974-983. [PubMed] [Google Scholar]
- Ostrovidov S, Franck P, Capiaumont J, Dousset B, Belleville F (1998) Effects of H2O2 on the growth, secretion, and metabolism of hybridoma cells in culture. In Vitro Cell Dev Biol Anim 34: 259-264. [DOI] [PubMed] [Google Scholar]
- Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO (1996) Requirement for superoxide in excitotoxic cell death. Neuron 16: 345-355. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F (2001) The mito-chondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem 276: 12030-12034. [DOI] [PubMed] [Google Scholar]
- Pike BR, Zhao X, Newcomb JK, Glenn CC, Anderson DK, Hayes RL (2000) Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J Neurotrauma 17: 283-298. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after traumatic brain injury. J Neurotrauma 17: 927-938. [DOI] [PubMed] [Google Scholar]
- Rami A, Ferger D, Krieglstein J (1997) Blockade of calpain proteolytic activity rescues neurons from glutamate excitotoxicity. Neurosci Res 27: 93-97. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Teng RJ, Beckman JS (2000) Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem 275: 32460-32466. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG (1995) Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci 15: 3318-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK (1995) Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol 147: 1575-1583. [PMC free article] [PubMed] [Google Scholar]
- Rothe G, Emmendorffer A, Oser A, Roesler J, Valet G (1991) Flow cytometric measurement of the respiratory burst activity of phagocytes using dihydrorhodamine 123. J Immunol Methods 138: 133-135. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H (1993) Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys 302: 348-355. [DOI] [PubMed] [Google Scholar]
- Sakahira H, Enari M, Nagata S (1998) Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391: 96-99. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M (1997) Determination of the time-course and extent of neurotoxicity at defined temperatures in cultured neurons using a modified multi-well plate fluorescence scanner. J Cereb Blood Flow Metab 17: 455-463. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M (1998) Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem 71: 2349-2364. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M (1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284: 1845-1848. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, MacDonald JF, Tymianski M (2000) Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci 20: 22-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto Jr RC, Grotyohann LW (1999) Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 76: 469-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285: 1569-1572. [DOI] [PubMed] [Google Scholar]
- Shah PT, Yoon KW, Xu XM, Broder LD (1997) Apoptosis mediates cell death following traumatic injury in rat hippocampal neurons. Neuroscience 79: 999-1004. [DOI] [PubMed] [Google Scholar]
- Sheng M (2001) Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA 98: 7058-7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek JC (1988) Excitatory amino acids activate calpain I and induce structural protein breakdown in vivo. Neuron 1: 279-287. [DOI] [PubMed] [Google Scholar]
- Siman R, Noszek C, Kegerise C (1989) Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci 9: 1579-1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999) High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci 19: 4263-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W (2004) Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci 24: 2182-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441-446. [DOI] [PubMed] [Google Scholar]
- Tator CH, Koyanagi I (1997) Vascular mechanisms in the pathophysiology of human spinal cord injury. J Neurosurg 86: 483-492. [DOI] [PubMed] [Google Scholar]
- Teasdale GM, Graham DI (1998) Craniocerebral trauma: protection and retrieval of the neuronal population after injury. Neurosurgery 43: 723-737. [DOI] [PubMed] [Google Scholar]
- Tecoma ES, Monyer H, Goldberg MP, Choi DW (1989) Traumatic neuronal injury in vitro is attenuated by NMDA antagonists. Neuron 2: 1541-1545. [DOI] [PubMed] [Google Scholar]
- Tenneti L, D'Emilia DM, Troy CM, Lipton SA (1998) Role of caspases in N-methyl-d-aspartate-induced apoptosis in cerebrocortical neurons. J Neurochem 71: 946-959. [DOI] [PubMed] [Google Scholar]
- Thibault LE, Meaney DF, Anderson BJ, Marmarou A (1992) Biomechanical aspects of a fluid percussion model of brain injury. J Neurotrauma 9: 311-322. [DOI] [PubMed] [Google Scholar]
- Trackey JL, Uliasz TF, Hewett SJ (2001) SIN-1-induced cytotoxicity in mixed cortical cell culture: peroxynitrite-dependent and -independent induction of excitotoxic cell death. J Neurochem 79: 445-455. [DOI] [PubMed] [Google Scholar]
- Tymianski M (1996) Cytosolic calcium concentrations and cell death in vitro. In: Advances in neurology: cellular and molecular mechanisms of ischemic brain damage (Siesjo BK, Wieloch T, eds), pp 85-105. Philadelphia: Lippincott-Raven. [PubMed]
- Tymianski M, Tator CH (1996) Normal and abnormal calcium homeostasis in neurons: a basis for the pathophysiology of traumatic and ischemic central nervous system injury. Neurosurgery 38: 1176-1195. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH (1993) Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci 13: 2085-2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tymianski M, Sattler R, Zabramski JM, Spetzler RF (1998) A characterization of neuroprotection from excitotoxicity by moderate and profound hypothermia in cultured cortical neurons unmasks a temperature-insensitive component of glutamate neurotoxicity. J Cereb Blood Flow Metab 18: 848-867. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA (1996) Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol 76: 1611-1621. [DOI] [PubMed] [Google Scholar]
- Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev 15: 2922-2933. [PubMed] [Google Scholar]
- Wennersten A, Holmin S, Mathiesen T (2003) Characterization of Bax and Bcl-2 in apoptosis after experimental traumatic brain injury in the rat. Acta Neuropathol (Berl) 105: 281-288. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Di X, Movsesyan V, Mullins PG, Wang G, Boulares H, Zhang J, Xu M, Faden AI (2001) Presence of DNA fragmentation and lack of neuroprotective effect in DFF45 knockout mice subjected to traumatic brain injury. Mol Med 7: 205-216. [PMC free article] [PubMed] [Google Scholar]
- Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW (1997) Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 278: 114-117. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Dawson TM, Dawson VL (2003) Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol Dis 14: 303-317. [DOI] [PubMed] [Google Scholar]
- Zhang L, Rzigalinski BA, Ellis EF, Satin LS (1996) Reduction of voltage-dependent Mg2+ blockade of NMDA current in mechanically injured neurons. Science 274: 1921-1923. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS (2002) Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem 82: 181-191. [DOI] [PubMed] [Google Scholar]
- Zingarelli B, Day BJ, Crapo JD, Salzman AL, Szabo C (1997) The potential role of peroxynitrite in the vascular contractile and cellular energetic failure in endotoxic shock. Br J Pharmacol 120: 259-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwienenberg M, Muizelaar JP (2001) Cerebral perfusion and blood flow in neurotrauma. Neurol Res 23: 167-174. [DOI] [PubMed] [Google Scholar]