

Abstract
Disruption of cAMP response element (CRE)-dependent transcription has been hypothesized to contribute to neuronal death and dysfunction in Huntington's disease (HD) and other polyglutamine repeat disorders. Whether dysregulation of CRE-dependent transcription actually occurs in vivo in response to expression of expanded polyglutamine repeats has not been tested. We directly tested whether CRE-dependent transcription is affected in vivo by cross breeding a transgenic mouse model of HD (line R6/2) with a transgenic mouse that expresses a CRE-regulated reporter gene. Instead of compromised CRE-dependent transcription in HD mice, we found a robust upregulation of CRE-dependent transcription in several brain regions (striatum, hippocampus, cortex). CRE-mediated transcription was also evoked by striatal forskolin infusion and by photic stimulation in HD animals. Increased cAMP response element-binding protein (CREB) phosphorylation and elevated levels of the CREB-regulated gene product, CCAAT/enhancer binding protein β, were also found in HD mice. Significant alterations in CREB binding protein expression and localization were not observed in symptomatic R6/2 mice. Thus, rather than repressing CRE-mediated transcription, mutant huntingtin appears to facilitate transcription via a CRE-dependent mechanism in vivo.
Keywords: Huntington's disease, polyglutamine, transcription, CREB, CBP, transgenic
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder that is caused by an increased polyglutamine repeat domain in the huntingtin protein (The Huntington's Disease Collaborative Research Group, 1993). Striatal degeneration and difficulty in controlling motor function are prominent features of this disease. The function of normal huntingtin is unknown, and the mechanism by which the increased glutamine repeat size (>37 repeats) in the huntingtin protein causes neurodegeneration is also not known. It has been proposed that mutant huntingtin-forming intranuclear inclusions interfere with transcription, and that aberrant transcriptional regulation could be an underlying component in HD and other polyglutamine repeat disease pathologies (McCampbell et al., 2000; Shimohata et al., 2000; Steffan et al., 2000, 2001; Holbert et al., 2001; Nucifora et al., 2001; Wyttenbach et al., 2001; Chai et al., 2002; Dunah et al., 2002; Kegel et al., 2002; Li et al., 2002; Luthi-Carter et al., 2002; Jiang et al., 2003). In particular, inhibition of cAMP response element (CRE)-mediated gene transcription has been hypothesized to contribute to HD pathology (Kazantsev et al., 1999; McCampbell et al., 2000, 2001; Shimohata et al., 2000; Steffan et al., 2000, 2001; Nucifora et al., 2001; Jiang et al., 2003; Taylor et al., 2003). Alterations in CRE-mediated gene expression are thought to result from the ability of huntingtin to associate with, sequester, or trigger cAMP response element-binding protein (CREB) binding protein (CBP) degradation, although there have been conflicting reports (Yu et al., 2002). Much of the work implicating altered CRE-dependent transcription has been performed using in vitro binding studies and cell culture transient transfection assays, and, thus, the effect of this interaction on CRE-dependent transcription in vivo remains unclear.
To directly test the hypothesis that mutant huntingtin expression and formation of intranuclear inclusions interferes with CRE-dependent transcription in vivo, we generated a double-transgenic mouse that expresses mutant huntingtin and a CRE-β-galactosidase reporter construct (Impey et al., 1996; Obrietan et al., 2002). The HD line used to generate this double transgenic strain expresses exon 1 and ∼150 glutamine repeats of human huntingtin (line R6/2) (Mangiarini et al., 1996). This line exhibits a characteristic decline in motor function, weight loss, and formation of intranuclear inclusions; death occurs by 14–15 weeks. The CRE transgenic line is a well established model system that has been used to explore the relationship between CRE-dependent transcription and a number of physiological and behavioral states (Impey et al., 1996, 1998; Obrietan et al., 1999; Barth et al., 2000). Instead of the predicted decrease in CRE-dependent transcription in HD transgenic mice exhibiting intranuclear inclusions, we found a robust upregulation of CRE-dependent transcription in neurons of these double-transgenic mice.
Materials and Methods
Animals. HD transgenic mice (line R6/2; Jackson Laboratories, Bar Harbor, ME) (Mangiarini et al., 1996) were bred with CRE-β-galactosidase reporter gene transgenic mice (obtained from Dr. Daniel Storm, University of Washington, Seattle, WA). Genotyping was performed as described previously (Mangiarini et al., 1996; Obrietan et al., 2002). The Ohio State University Animal Care and Use Committee approved all surgical procedures. Brain tissue was collected and processed as described by Butcher et al. (2002).
Immunohistochemistry and immunofluorescence. Free-floating sections were processed using the techniques described by Butcher et al. (2002). The following primary antibodies were used: rabbit anti-β-galactosidase (1:10,000, immunohistochemistry; 1:2000, immunofluorescence; Cortex Biochemicals, San Leandro, CA), rabbit anti-CBP (a-22; 1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-ubiquitin (1: 500; Dako, Carpinteria, CA), mouse anti-human huntingtin, (mEM-48; 1:500; Chemicon, Temecula, CA), and mouse anti-neuronal nuclear antigen neuronal-specific nuclear protein (NeuN; 1:250; Chemicon). X-gal histochemistry was performed as described by Barth et al. (2000).
All images were captured under identical settings using a 16-bit digital camera (Princeton Instruments, Trenton, NJ) connected to a Leica (Trenton, NJ) DMIRB microscope or a Zeiss (Nussloch, Germany) 510 Meta confocal microscope (Z section, 1 μm) and analyzed using Metamorph (Universal Imaging Corporation, West Chester, PA). A macrobased routine was used to count diaminobenzidine-labeled cells in an area of fixed size. Cells were counted in three consecutive coronal sections per mouse, and the cell counts from each region were averaged. These data were then averaged for each genotype to arrive at the values used for genotype comparisons. For immunofluorescence analysis, the fluorescence intensity from each region was averaged for each animal (three sections per animal) and used for comparison between the genotypes. As noted previously (Impey et al., 1996; Barth et al., 2000), β-galactosidase expression was not observed in all animals genotyped as CRE positive. We found that ∼30% of the CRE-positive animals lacked β-galactosidase expression. Importantly, the absence of β-galactosidase expression was not concordant with the human huntingtin exon 1 (HTT)-positive (hd) genotype; both the hd and wild-type (wt, HTT-negative) lines had the same percentage of β-galactosidase nonexpressing animals. Tissues from animals that lacked β-galactosidase expression were not used.
Western blotting. Twenty micrograms of protein from each sample was electrophoresed using the procedures outlined by Butcher et al. (2002). Antibodies against the following proteins were used: CBP (1:10000; Santa Cruz Biotechnology), ERK (extracellular signal-regulated kinase; 1:1000; Santa Cruz Biotechnology), CCAAT/enhancer binding protein β (C/EBPβ) (1: 5000; Santa Cruz Biotechnology), and anti-human huntingtin (1:5000; Chemicon). Triton X-100 soluble fractions were prepared as described previously (Yu et al., 2002).
Cannulation. The technique described by Butcher et al. (2002) was used to implant mice with indwelling guide cannulas. Cannulas were placed in the striatum using the following coordinates: anterior, 1.0 mm from bregma; lateral, 1.45 mm from the midline; dorsoventral, –3.0 mm. Mice were cannulated at 6 weeks of age and infused 5 weeks later with 500 nL of forskolin (2 mm, diluted in 90% artificial CSF, 10% DMSO; Sigma, St. Louis, MO). A subset of animals was infused with bodipy-forskolin (2 mm; Molecular Probes, Eugene, OR). Tissue was collected 8 hr after infusion. Forskolin elicited a much more robust β-galactosidase signal than the basal expression signal examined above. To clearly differentiate these two signals, sections from forskolin-infused brains were underdeveloped so that only the forskolin-induced signal could be clearly observed.
Light-pulse protocol. Animals were dark adapted for 1 d and then exposed to light (500 lux, 60 min) 2 hr after the beginning of the subjective night. Animals were killed 8 hr after light-pulse onset. The diaminobenzidine development conditions described in the cannulation section were used to examine β-galactosidase expression.
Results
We identified double-transgenic mice by PCR for both HTT and the β-galactosidase reporter gene (CRE) (Fig. 1C). All results reported here used mice between 11–12 weeks of age, comparing HTT-positive and HTT-negative (wild type) CRE transgenic mice. As expected, mice expressing both the HTT gene and the CRE transgene mice developed intranuclear inclusions (Fig. 1A) and a decrease in brain size (Fig. 1B), as well as decreased body weight and shortened survival time paralleling the pathological changes observed in the parental R6/2 line.
Figure 1.
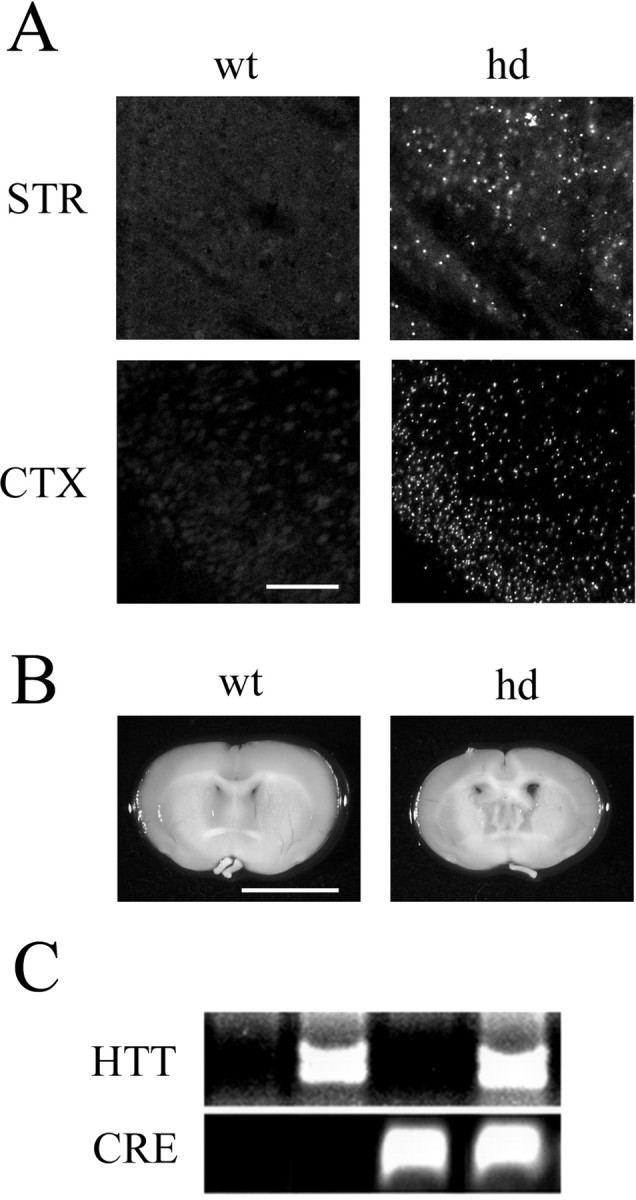
Phenotype of mice expressing both mutant huntingtin and the CRE reporter gene. A, Ubiquitin immunofluorescence reveals characteristic intranuclear inclusions as punctate spots of fluorescent labeling in hd mice. Representative data from the piriform cortex (CTX) and striatum (STR) are shown. Scale bar, 100 μm. B, Comparison of coronal brain sections from hd and wt mice demonstrates that hd mice have reduced brain size. Scale bar, 4 mm. C, Validation of the double-transgenic hd–CRE line. Cross-bred mice were genotyped for the human huntingtin gene and for the CRE-β-galactosidase reporter gene. Agarose gel electrophoresis of the PCR reaction products reveals the four possible genotypes resulting from the cross breeding. Only mice expressing the CRE transgene with (hd) or without HTT (wt) were used in this study.
Although there was widespread intranuclear inclusion formation in hd mice, a comparison of hd and wt mice revealed no differences in CBP subcellular localization (Fig. 2A). Rather, CBP labeling in hd mice was diffuse and exclusively nuclear, overlapping with the nuclear immunolabeling boundary formed by the neuronal nucleus-specific marker NeuN (data not shown). Although a punctate CBP expression pattern was detected in some nuclei, this pattern was similarly present in wt brains (Fig. 2A). Quantification of the fluorescence intensity of CBP labeling revealed no significant difference between hd and wt mice (Fig. 2B). Western analysis of the Triton X-100-soluble protein fraction revealed no difference in CBP expression between hd and wt mice (Fig. 2C). These data reveal that CBP expression is not significantly reduced in hd animals. Confocal microscopy was also used to examine whether intranuclear inclusions depleted CBP levels (Fig. 2D). Quantification of CBP levels within and outside inclusions reveals no difference in CBP localization in hd mice.
Figure 2.

CBP expression in hd and wt brain sections. A, Coronal sections (STR, striatum; CTX, piriform cortex) were labeled by immunofluorescence using an antibody to CBP. Scale bar, 50 μm. B, Corresponding sections from hd and wt mice were analyzed for fluorescence intensity of CBP labeling. No significant difference in the fluorescence intensity of CBP labeling between the two lines was observed (n = 5 mice per genotype; mean ± SEM). C, Western analysis revealed that equivalent levels of detergent-soluble CBP (hd vs wt) were detected. Tissue from individual mice was run, two lanes per genotype. D, CBP (red) was not concentrated within intranuclear inclusions (green). CBP immunofluorescence was quantified within regions containing inclusions and in nuclear regions lateral to inclusions. Analysis was only performed on cells with inclusions (n = 88 cells). E, pCREB levels are increased in hd mice. Each lane represents extracts from individual mice in this Western blot. As a protein-loading control, the blot was stripped and probed for total ERK expression. F, Expression of the CREB-regulated gene, C/EBPβ, was also increased in hd mice, as revealed by Western analysis.
Next, we examined whether mutant huntingtin may affect other events within the CREB–CRE transcriptional pathway. To this end, hd brain tissue was probed for the Ser-133 phosphorylated form of CREB. Phosphorylation of CREB at Ser-133 is a necessary step for CREB to stimulate CRE-mediated transcription (Gonzalez and Montminy, 1989). Surprisingly, the phosphorylated state of CREB (pCREB) was significantly upregulated in hd animals (Fig. 2E) (pCREB/ERK2 ratio was 0.44 ± 0.07 in wt and 0.89 ± 0.06 in hd brain tissue; n = 3 mice per group; p < 0.01; Student's t test). If the elevated level of CREB phosphorylation in hd animals is physiologically relevant, it follows that CREB-regulated genes may also be upregulated in hd animals. To address this issue, expression of the CREB-regulated gene product C/EBPβ (Niehof et al., 1997) was assessed. In accordance with the pCREB data, C/EBPβ expression was significantly increased in hd animals (Fig. 2F) (C/EBPβ/ERK2 ratio was 0.57 ± 0.03 in wt and 1.2 ± 0.18 in hd brain tissue; n = 3 mice per group; p < 0.05; Student's t test).
To directly test whether CRE-dependent transcription was altered in symptomatic hd mice, the expression of the CRE-regulated β-galactosidase reporter construct was examined. Consistent with the increased pCREB and C/EBPβ levels, we found that hd mice had an upregulation in the number of cells that express the reporter gene compared with wt mice. Prominent increases were observed in the cortex as well as in the dentate gyrus and areas CA1 and CA3 of the hippocampus. Notably, CRE-mediated gene expression was also elevated in the striatum, the brain region most severely effected by HD (Fig. 3A,B). We confirmed that the β-galactosidase antibody was specific by comparing its staining pattern with another β-galactosidase detection method, X-gal enzyme histochemistry. These two detection methods gave similar staining patterns (Fig. 3C), thus supporting the specificity of the immunohistochemical staining technique. At the antibody concentration used, background labeling was not detected in nontransgenic C57/Bl6 mice.
Figure 3.
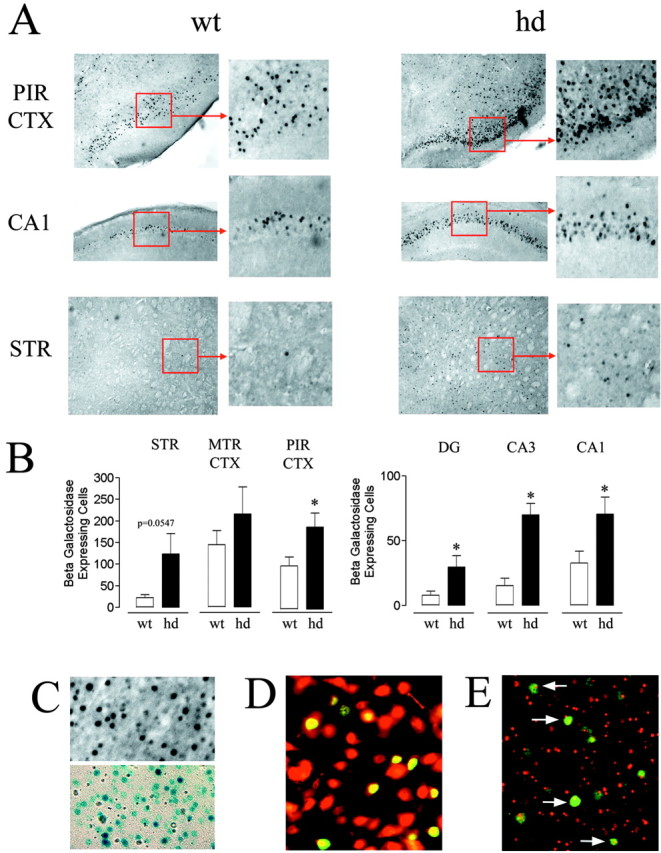
CRE-mediated gene expression is increased in hd mice.A, Coronal brain sections (PIRCTX, piriform cortex; hippocampal area CA1; STR, striatum) from hd and wt mice were labeled forβ-galactosidase expression and visualized using diaminobenzidine immunohistochemistry. There was a marked increase in the number of β-galactosidase-expressing cells (labeled black against gray background) in hd mice compared with wt mice. B, The number of β-galactosidase-expressing cells was counted in seven mice per genotype in the following brain regions: STR, motor cortex (MTR CTX), PIR CTX, and hippocampal regions CA1 and CA3, and dentate gyrus (DG). *, Significantly different from wt (p < 0.05; Student's t test). C, Immunolabeling (top, black cells) and X-gal histochemistry (bottom, blue cells) generated similar β-galactosidase staining patterns in brain tissue of CRE-LacZ mice. D, Confirmation that CRE-mediated gene expression occurs in neurons. Merged image of β-galactosidase labeling (green) and NeuN labeling (red) in a striatal section from an hd mouse. The yellow hue indicates coexpression of the two antigens. E, CRE-mediated gene expression occurs in striatal neurons that contain intranuclear inclusions. The merged image reveals that nuclear huntingtin inclusions (red) are observed in the subset of cells expressing β-galactosidase (green, arrows).
Although these results reveal that CRE-mediated gene expression occurs in hd animals, the phenotype of the β-galactosidase-expressing cells was not known. To determine whether CRE-mediated transcription occurred in neurons, hd brain tissue was double labeled for β-galactosidase and NeuN expression. Figure 3D reveals the colocalized expression of β-galactosidase and NeuN, indicating that CRE-mediated gene expression occurs in neurons during the later stages of HD. To determine whether CRE-dependent transcription occurs in cells with intranuclear huntingtin inclusions, tissue from symptomatic hd animals was colabeled for inclusions and β-galactosidase. Inclusions were labeled using a huntingtin antibody (mEM-48) (Li et al., 2000). Figure 3E shows that robust β-galactosidase expression is readily detected in neurons containing intranuclear inclusions. Collectively, these data indicate that CRE-dependent transcription is not suppressed but rather enhanced in hd mice. Furthermore, these data indicate that the development of intranuclear inclusions does not limit the ability of neurons to stimulate transcription via a CRE-dependent mechanism.
Finally, two approaches were used to examine inducible CRE-mediated gene expression in hd mice. The goal of these experiments was to determine whether HD-dependent alterations in cellular signaling pathways may affect the capacity of a stimulus to couple to CRE-mediated gene expression. In the first approach, mice were implanted with indwelling guide cannulas aimed at the striatum, and the adenylyl cyclase agonist forskolin was infused. Administration of forskolin triggered a robust increase in CRE-mediated gene expression within the region surrounding the tip of the guide cannula. Marked increases in β-galactosidase expression were observed in both hd and wt mice (Fig. 4A,B). A fluorescent forskolin conjugate (bodipyforskolin) was used to approximate the region of forskolin diffusion (Fig. 4C). β-galactosidase expression colocalized with the neuronal marker NeuN (Fig. 4D), indicating that forskolin stimulated CRE-dependent transcription in neurons. In the second approach, mice were exposed to light during the night. Photic stimulation has been shown to stimulate CRE-mediated gene expression in the suprachiasmatic nucleus (SCN) (Obrietan et al., 1999), the locus of the circadian clock. To determine whether this noninvasive exogenous stimulus triggers CRE-mediated gene expression, hd animals were exposed to light during the early part of the night. Relative to control animals, light exposure triggered a marked increase in CRE-dependent transcription in the SCN (Fig. 4E,F). Control experiments verified that intranuclear huntingtin inclusions are located within the SCN (data not shown). These data indicate that hd animals possess the cell surface receptors and intracellular signaling machinery required to couple an extracellular signal to CRE-mediated gene expression.
Figure 4.
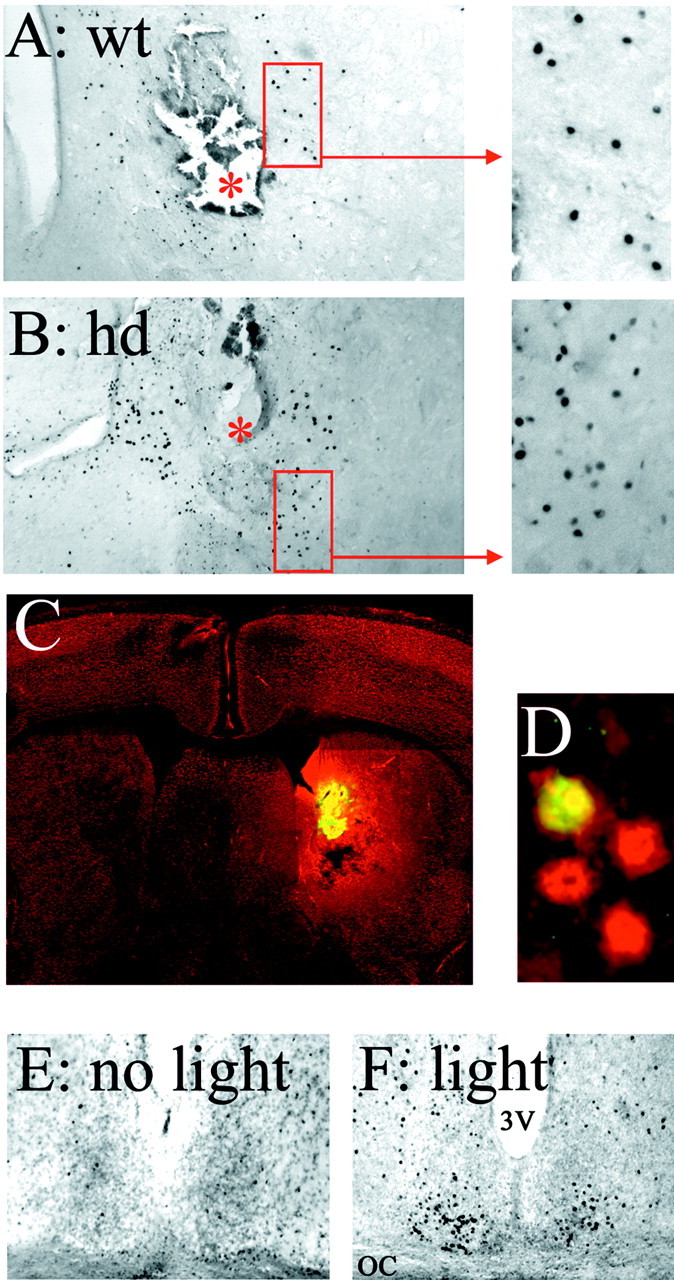
Induction of CRE-mediated gene expression in hd mice. A, B, Intrastriatal infusion of forskolin-stimulated robust β-galactosidase expression in hd (B) and wt (A) mice around the site of the infusion. The red asterisks indicate the infusion sites. C, A fluorescent forskolin conjugate (bodipy-forskolin) was used to approximate the drug diffusion range. In this representative coronal brain section, yellow fluorescence reveals the site of infusion and the diffusion pattern of bodipy-forskolin. NeuN immunostaining (red) is the counter label. D, Colocalization of β-galactosidase (green) with NeuN (red) reveals that forskolin elicited CRE-mediated gene expression in neurons. This photomicrograph was captured ∼100 μm from the infusion site. E, F, Light stimulates CRE-mediated gene expression in the SCN of hd mice. Relative to control animals not exposed to light (E), photic stimulation (F) elicited β-galactosidase expression within the retinoreceptive region of the SCN. OC, Optic chiasm; 3V, third ventricle.
Discussion
Although there are well documented alterations in the expression pattern of a large number of genes in HD transgenic mice (Luthi-Carter et al., 2002), the causative nature of these changes remains elusive. Recent work has suggested that aberrant transcriptional regulation plays a central role in the pathological development of HD. Prominent among these studies was work showing that sequestration of CBP interrupts CRE-mediated transcription (Nucifora et al., 2001) in vitro. Here, we addressed the specific question of whether CRE-dependent transcription is modified by mutant huntingtin expression and aggregation in a symptomatic mouse model of HD. We were surprised to find that, in contrast to the predicted result that CRE-dependent transcription would be decreased, there was an increase in CRE-dependent reporter gene expression in hd mice.
There are a number of possible explanations for the discrepancy between our results and those reported previously. Previous studies used transient transfection techniques to overexpress mutant huntingtin in cultured cells (McCampbell et al., 2000; Shimohata et al., 2000; Steffan et al., 2000; Nucifora et al., 2001; Wyttenbach et al., 2001; Chai et al., 2002; Jiang et al., 2003). An acute accumulation of high mutant huntingtin levels could trigger effects not observed in vivo. For example, in an overexpression system, there may be a sufficient interaction between weakly associating proteins, such as huntingtin and CBP (Steffan et al., 2000), that a pathophysiological state is created. Likewise, rapid aggregate formation may sequester proteins so quickly that the transcription–translation machinery of the cell may not be able to compensate. In this regard, the R6/2 transgenic mouse may be a better tool to model HD. Along these lines, the R6/2 line expresses mutant human huntingtin at physiological levels, ∼75% of the endogenous mouse huntingtin level (Mangiarini et al., 1996). Thus, although in a cell culture system there may be a measurable disruption in CBP function–expression and CRE-dependent transcription, when the system is reconstituted in vivo, this inhibitory effect is lost.
The upregulation of CRE-dependent transcription in hd transgenic mice is intriguing. It has been reported that NMDA receptor-mediated signaling may be enhanced in neurons from HD transgenic mice. This increased excitatory signaling could contribute to the increased pCREB levels, which in turn would facilitate both β-galactosidase and C/EBPβ expression. As a plasticity-associated transcriptional pathway that drives the expression of a number of cell survival and antiapoptotic genes (Impey and Goodman, 2001; West et al., 2001; Lonze and Ginty, 2002), CRE-mediated transcription may also be elevated as a compensatory response to the pathophysiological state created by HD. It should be noted that we have not excluded the possibility that other transcriptional pathways, such as those mediated by Sp1, p53, and REST–NRSF, may be affected by the polyglutamine-expanded form of huntingtin (Steffan et al., 2000; Dunah et al., 2002; Zuccato et al., 2003) and thus contribute to changes in expression of the various genes in HD. Furthermore, the histone acetylase function of CBP was not addressed in our study. Huntingtin has been shown to interact with the acetyltransferase domain of CBP (Steffan et al., 2001) and diminish its inherent acetyltransferase activity. The effects of altering acetyltransferase activity are likely to be multifaceted, influencing a wide array of both cellular signaling and transcriptional events. In this regard, it is interesting to note that changing the relative level of acetylation has been shown to facilitate the expression of some CREB-dependent genes and repress the expression of other genes (Fass et al., 2003).
The data presented here, showing normal CBP distribution and a functional CRE signaling cassette in mice with advanced stage HD, raise doubts about the role of CRE-mediated gene expression in the etiology of the disease. Rather, the work reported here reveals an upregulation of CRE-dependent transcription in HD transgenic animals. The mechanism(s) underlying this increase in CRE-dependent transcription in HD transgenic mice requires additional study.
Footnotes
This work was supported by National Institutes of Health Grants MH62335 (K.O.) and NS41003 (K.R.H.) and the Robert A. Vaughan Family Fund for Huntington's Disease Research. We thank Jessica Buescher, Heather Dziema, and Ben Oatis for their technical assistance.
Correspondence should be addressed to Dr. Kari R. Hoyt, Division of Pharmacology, The Ohio State University, 412 Riffe Building, 496 West Twelfth Avenue, Columbus, OH 43210. E-mail: hoyt.31@osu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240791-06$15.00/0
DOI: 10.1523/JNEUROSCI.3493-03.2004
References
- Barth AL, McKenna M, Glazewski S, Hill P, Impey S, Storm D, Fox K (2000) Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J Neurosci 20: 4206–4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher GQ, Dziema H, Collamore M, Burgoon PW, Obrietan K (2002) The p42/44 mitogen-activated protein kinase pathway couples photic input to circadian clock entrainment. J Biol Chem 277: 29519–29525. [DOI] [PubMed] [Google Scholar]
- Chai Y, Shao J, Miller VM, Williams A, Paulson HL (2002) Live-cell imaging reveals divergent intracellular dynamics of polyglutamine disease proteins and supports a sequestration model of pathogenesis. Proc Natl Acad Sci USA 99: 9310–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296: 2238–2243. [DOI] [PubMed] [Google Scholar]
- Fass DM, Butler JE, Goodman RH (2003) Deacetylase activity is required for cAMP activation of a subset of CREB target genes. J Biol Chem 278: 43014–43019. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR (1989) Cyclic-AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675–680. [DOI] [PubMed] [Google Scholar]
- Holbert S, Denghien I, Kiechle T, Rosenblatt A, Wellington C, Hayden MR, Margolis RL, Ross CA, Dausset J, Ferrante RJ, Neri C (2001) The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: neuropathologic and genetic evidence for a role in Huntington's disease pathogenesis. Proc Natl Acad Sci USA 98: 1811–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Goodman RH (2001) CREB signaling–timing is everything. Sci STKE 82: PE1. [DOI] [PubMed] [Google Scholar]
- Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR (1996) Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron 16: 973–982. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR (1998) Stimulation of cAMP-response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1: 595–601. [DOI] [PubMed] [Google Scholar]
- Jiang H, Nucifora Jr FC, Ross CA, DeFranco DB (2003) Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum Mol Genet 12: 1–12. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404–11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JR, Aronin N, DiFiglia M (2002) Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem 277: 7466–7476. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ (2000) Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet 25: 385–389. [DOI] [PubMed] [Google Scholar]
- Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H, Li XJ (2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22: 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35: 605–623. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM (2002) Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet 11: 1911–1926. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH (2000) CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 9: 2197–2202. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH (2001) Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA 98: 15179–15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehof M, Manns MP, Trautwein C (1997) CREB controls LAP/C/EBP-beta transcription. Mol Cell Biol 17: 3600–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora Jr FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423–2428. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Smith D, Athos J, Storm DR (1999) Circadian regulation of cAMP-response element-mediated gene expression in the suprachiasmatic nuclei. J Biol Chem 274: 17748–17756. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR (2002) cAMP response element-mediated gene expression in transgenic reporter gene mouse strain. Methods Enzymol 345: 570–584. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, et al. (2000) Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet 26: 29–36. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97: 6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila Nature 413: 739–743. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT (2003) Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev 17: 1463–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024–11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC (2001) Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet 10: 1829–1845. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Nguyen HP, Li XJ (2002) Huntingtin inclusions do not deplete polyglutamine-containing transcription factors in HD mice. Hum Mol Genet 11: 905–914. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35: 76–83. [DOI] [PubMed] [Google Scholar]