

Abstract
Patch-clamp recordings from small-diameter rat dorsal root ganglion (DRG) neurons maintained in culture demonstrated preferential inhibition by ATP of high-voltage-activated, but not low-voltage-activated, Ca2+ currents (ICa). The rank order of agonist potency was UTP > ADP > ATP. ATP depressed the ω-conotoxin GVIA-sensitive N-type current only. Pyridoxal-5-phosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) and 2′-deoxy-N6-methyladenosine 3′,5′-bisphosphate tetraammonium, two P2Y1 receptor antagonists, almost abolished the ATP-induced inhibition. Both patch-clamp recordings and immunocytochemistry coupled with confocal laser microscopy indicated a colocalization of functional P2X3 and P2Y1 receptors on the same DRG neurons. Because the effect of ATP was inhibited by intracellular guanosine 5′-O-(2-thiodiphosphate) or by applying a strongly depolarizing prepulse, P2Y1 receptors appear to block ICa by a pathway involving the βγ subunit of a Gq/11 protein. Less efficient buffering of the intracellular Ca2+ concentration ([Ca2+]i) by reducing the intrapipette EGTA failed to interfere with the ATP effect. Fura-2 microfluorimetry suggested that ATP raised [Ca2+]i by a Gα-mediated release from intracellular pools and simultaneously depressed the high external potassium concentration-induced increase of [Ca2+]i by inhibiting ICa via Gβγ. Adenosine 5′-O-(2-thiodiphosphate) inhibited dorsal root-evoked polysynaptic population EPSPs in the hemisected rat spinal cord and prolonged the nociceptive threshold on intrathecal application in the tail-flick assay. These effects were not antagonized by PPADS. Hence, P2Y receptor activation by ADP, which is generated by enzymatic degradation of ATP, may decrease the release of glutamate from DRG terminals in the spinal cord and thereby partly counterbalance the algogenic effect of ATP.
Keywords: ATP, P2Y receptor, calcium channel, dorsal root ganglion, patch clamp, analgesia
Introduction
Nociceptive information is carried from the peripheral processes of small-diameter dorsal root ganglion (DRG) neurons to their central terminals in the dorsal horn of the spinal cord. This information is then relayed to the secondary sensory neurons projecting into the upper CNS. Recent studies classified voltage-activated calcium channels (VACCs) in DRG neurons into three groups: (1) high-voltage-activated calcium channels (HVACCs), which included L-, N-, P-, and Q-types; (2) intermediate (R-type) calcium channel; and (3) low-voltage-activated calcium channel (LVACC; T-type) (Varadi et al., 1999). On depolarization, VACCs allow the entry of calcium into the cell interior, shaping excitability and initiating neurotransmitter release. Inhibition of calcium channel activity in DRG cells was shown to play an important role in pain transmission (Malmberg and Yaksh, 1994; Diaz and Dickenson, 1997).
Extracellular nucleotides (ATP, ADP, UTP, and UDP) have been shown to be signaling molecules in the CNS and PNS by acting via two types of P2 receptors termed P2X (ligand-gated cationic channels) and P2Y (G-protein-coupled receptors) (Abbracchio and Burnstock, 1994; Ralevic and Burnstock, 1998). Currently, eight mammalian P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) have been cloned (von Kugelgen and Wetter, 2000; Communi et al., 2001; Nicholas, 2001; Abbracchio et al., 2003). A number of studies have demonstrated that P2Y receptors inhibit VACCs in neurons or in neuronally differentiated endocrine cell lines (Boehm, 2003). Activation of Gi-coupled endogenous P2Y12 receptors has been shown to inhibit N-type VACCs in PC12 cells (Kulick and von Kugelgen, 2002; Kubista et al., 2003). Moreover, agonists at heterologously expressed P2Y1, P2Y2, P2Y4, and P2Y6 receptors inhibited N-type VACCs in rat superior cervical ganglion neurons (Fillippov et al., 1998, 1999, 2000, 2003).
P2X3 receptor mRNA is selectively expressed by a subpopulation of small-diameter DRG cells binding isolectin B4 and possessing also capsaicin-sensitive VR1 vanilloid receptors (Burgard et al., 1999; Ueno et al., 1999; Chizh and Illes, 2001). This neuronal population is thought to be involved in pain transmission; it displays rapidly desensitizing agonist-evoked currents most closely matching those of recombinant homomeric P2X3 receptors (Burgard et al., 1999). These small-fiber nociceptive neurons are believed to coexpress P2Y1 (Tominaga et al., 2001) or P2Y2 (Moriyama et al., 2003) and VR1 receptor mRNA; ATP may cause thermal hypersensitivity via a protein kinase C-mediated positive interaction between P2Y1/P2Y2 and VR1 receptors.
The possible inhibition of VACCs by P2Y receptors could be a key element in the regulation of neurotransmitter release from the central terminals of the sensory neurons and, thereby, in the modulation of pain transmission. Here, we report for the first time that endogenous Gq-coupled P2Y (P2Y1) receptors selectively inhibit the N-type VACC by a voltage-dependent, membrane-delimited pathway. This effect, being observed in DRG neurons, leads to antinociception rather than pronociception under in vivo conditions. Part of these results have been published previously as a short communication (Borvendeg et al., 2003).
Materials and Methods
Animal experimentation. The present investigation was conducted in accordance with the principles outlined in the Declaration of Helsinki and the German law governing the care and use of laboratory animals and was approved by the representatives for animal care and use both in Leipzig (Rudolf-Boehm-Institute of Pharmacology, University of Leipzig) and Aachen (Department of Pharmacology, Grünenthal GmbH).
Preparation of DRG neuronal cultures. One-day-old and 7-week-old Wistar rats (own breed; WIST/Lei) were killed under CO2 and decapitated to obtain cell cultures of DRG neurons for electrophysiology and Ca2+ imaging, respectively. Previous experiments indicated that rats of the above age groups are optimal sources of neurons for each type of investigation. The isolation and culturing procedures of thoracic and lumbar DRG cells have been described in detail previously (Himmel et al., 2002). These cells were plated at a density of 2 × 10 4 cells/ml onto poly-l-lysine-coated (25 μg/ml) glass coverslips. Cultures were maintained for 2–4 d at 37°C in a humidified atmosphere with 5% CO2 before experimentation.
Whole-cell patch-clamp recordings in DRG neurons. Calcium currents (ICa) were recorded using the whole-cell configuration of the patch-clamp technique by means of an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Patch electrodes (1.5–3 MΩ) were filled with intracellular solution of the following composition (mm): 135 CsCl, 2 MgCl2, 20 HEPES, 11 EGTA, 1 CaCl2, 1.5 Mg-ATP, and 0.3 Li-GTP, pH 7.3 adjusted with CsOH. When indicated, GTP was substituted with guanosine 5′-O-(2-thiodiphosphate) (GDP-β-S) (0.3 mm). In some experiments, 1.1 mm EGTA and 9.9 mm CsCl were used to substitute 11 mm EGTA. The seal resistance was routinely 1 GΩ. Residual pipette capacitance was compensated in the cell-attached configuration. The series resistance (Rs) was reduced by 60–80% using the inbuilt series resistance compensation circuit of the amplifier. The liquid junction potential (VLJ) between the bath and pipette solutions at 22°C was calculated according to Barry (1994) and was found to be –9.3 mV. Holding potential values given in this study were corrected for VLJ. The external recording solution consisted of (in mm) 155 TEA, 5 CaCl2, 0.8 MgCl2, 10 HEPES, and 11 glucose, pH 7.3 adjusted with TEA-OH. Under these conditions, sodium and potassium currents were suppressed. In some experiments, a modified TEA solution was used containing (mm) 120 NaCl, 20 TEA, 5 CaCl2, 0.8 MgCl2, 10 HEPES, 11 glucose, and 0.5 μm TTX, pH 7.3 adjusted with TEA-OH.
ICa was elicited repetitively from a holding potential of –90 mV by rectangular voltage steps, 80 msec in duration, to –10 mV, one voltage step every 20 sec. In some experiments, strongly depolarizing prepulses to +120 mV from the holding potential of –90 mV were applied for 20 msec. The interval between the prepulse and the test pulse was 5 msec. Current amplitudes were measured isochronically 15 msec from the onset of the test pulse near to the peak of the currents. Current–voltage relationships for ICa were obtained by applying fast (320 msec) ramp-like voltage pulses from –90 to +70 mV every 30 sec.
Current recordings were low-pass filtered at 2 kHz using the built-in four-pole low-pass Bessel filter of the amplifier and sampled at 10 kHz via a Digidata 1200 interface (Axon Instruments). Recordings were corrected for linear capacitance and leakage currents by the –P/4 function of the pClamp 8.0 software (Axon Instruments) used otherwise for data acquisition and analysis.
All experiments were performed at room temperature (20–24°C). A pressure-operated, computer-controlled rapid drug application device (DAD-12; Adams and List, Westbury, NY) was used for drug administration. After a control period (1–2 min), P2Y receptor agonists were applied for 2 min and then washed out. P2Y receptor antagonists were added 7 min before agonist application and were maintained until the end of the experiment. VACC blockers were added as described in the Results.
Concentration–response curves were fitted using the following logistic function (Sigma Plot; SPSS, Erkrath, Germany):
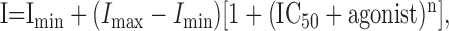 |
(1) |
where I is the steady-state inhibition produced by the agonist; Imax and Imin are the maximal and minimal inhibition, respectively; n is the Hill coefficient; and IC50 is the concentration of agonist producing 50% of Imax.
In a few experiments, DRG neurons of Sprague Dawley rats were prepared and cultured by using identical procedures to those described for Wistar rats. Both the inhibition of ICa by ATP and the interaction of this agonist with PPADS were indistinguishable in DRGs obtained from the two different strains of rats.
Intracellular Ca2+ measurements in DRG neurons. DRG cell cultures were loaded for 50–60 min at 37°C in the dark with the cell permeant acetoxymethyl ester of the fluorescent Ca2+ indicator fura-2 (1 μm). Simultaneously, cells were exposed to FITC-labeled Bandeireia simplicifolia isolectin IB4 (0.25 μg/ml; Sigma). To remove excess extracellular fura-2 and IB4, glass coverslips were washed several times with extracellular medium (in mm: 140 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH adjusted to 7.4 with NaOH) and were allowed to rest for 30 min at room temperature protected from light. Ca2+ imaging experiments and the quantification of FITC-IB4 labeling of DRG cells were performed as described previously (Himmel et al., 2002). In short, intracellular fura-2 was alternately excited at 340 nm and at 380 nm, and the emitted light was measured at a wavelength of 510 nm. The fluorescence ratio (340/380) provides a relative measure of the cytosolic-free Ca2+ concentration ([Ca2+]i). Drugs and the washout solution were applied via the flush valves of DAD-12. The TILL vision software (release 3.3; Till Photonic CS, Graefelfing, Germany) was used for data acquisition, system control, and off-line analysis.
DRG neurons were depolarized with a high K+-containing extracellular medium (in mm: 94.5 NaCl, 50 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH adjusted to 7.4 with NaOH) for 2 sec every 5 min. P2 receptor agonists were applied 60 sec before, during, and after the third K+ superfusion for a total of 90 sec. The last response to K+ before superfusion with the P2 receptor agonist was considered to be 100% (control); the response to a P2 receptor agonist, or the response to K+ in the presence of a P2 receptor agonist, was expressed as a percentage of the control response. P2 receptor antagonists were applied 10 min before the first stimulation with a high K+ medium and were maintained until the end of the experiment.
In the experimental protocols aimed at examining the effect of a Ca2+-free extracellular medium, K+ or the P2 receptor agonists were administered five times for 10 sec every 10 min. A Ca2+-free external medium supplemented with EGTA (1 mm) was applied 3 min before, during, and after the third superfusion with K+ or the P2 receptor agonists, for a total of 5 min. The last response to K+ or the P2 receptor agonists before the application of a Ca2+-free external medium was considered to be 100% (control); responses to these stimuli in a Ca2+-free medium were expressed as percentages of the control response.
Electrophysiology in hemisected spinal cord preparations. Immature Sprague Dawley rats (Janvier, Le Genest Saint Isle, France) of either sex (2–10 d old; 8–20 gm) were anesthetized with urethane (1.5 gm/kg, i.p.), and after decapitation, spinal cords were removed and hemisected along the midline. Sprague Dawley, rather than Wistar, rats were used to have a better comparability with the behavioral data (see below). The preparation was then placed into a recording chamber and perfused at room temperature at 2 ml/min with external medium containing (mm): 118 NaCl, 3 KCl, 1.5 CaCl2, 1.25 MgSO4, 24 NaHCO3, and 12 glucose, gassed with 95% O2/5% CO2. The stimulating electrode was in contact with the L4 or L5 dorsal root and the recording electrode with the corresponding ipsilateral ventral root (Faber et al., 1997).
Two different protocols of electrical stimulation were used: low-intensity stimuli (single pulses of 0.5 msec duration at three times the threshold intensity) and high-intensity stimuli (single pulses at 16 times the threshold intensity). In the hemisected spinal cord preparation, several components of population dorsal root-evoked ventral root potentials were observed. The low-intensity stimulation evoked an initial monosynaptic compound action potential [monosynaptic reflex (MSR); duration up to 200 msec] superimposed on a polysynaptic population EPSP that lasted for up to 4 sec. These two responses were mediated by A-fibers. Single high-intensity stimuli evoked prolonged, presumably both A- and C-fiber-mediated, polysynaptic EPSPs lasting ∼40 sec. The averages of five consecutive low-intensity sweeps (interpulse interval, 6 sec), followed by the averages of five consecutive high-intensity sweeps (interpulse interval, 18 sec) were obtained. Signals were digitized, stored, and analyzed using commercially available software (DASYLAB; Synotech, Linnich, Germany). The following parameters were analyzed: the peak amplitude of the MSR as well as the areas under the curve of low- and high-intensity EPSPs. Drugs were dissolved in external medium and applied via superfusion. Increasing concentrations were tested on the same preparation in a cumulative manner with equilibration periods sufficient to reach a plateau of the effect (typically 20 min for each drug concentration or compound). The long equilibration period is attributable to the slow diffusion of the ligands into the tissue.
Immunofluorescence. After fixation with ice-cold methanol for 10 min at 4°C, washing with HBSS for 5 min, and blocking with 5% FCS (Seromed, Berlin, Germany) and 0.1% Triton X-100 in TBS (0.05 m; pH 7.6), the cell cultures were incubated in a first step with an antibody mixture of rabbit anti-P2Y1 (1:400; Alomone, Jerusalem, Israel) and guinea pig anti-P2X3 receptor antibodies (1:1000; Neuromics, Minneapolis, MN) in TBS containing 0.1% Triton X-100 and 5% FCS overnight at 4°C. The secondary antibodies used for the simultaneous localization of the two primary antisera were Cy5-conjugated goat anti-rabbit IgG (1: 400; Jackson ImmunoResearch, Baltimore, MD) and Cy2-conjugated goat anti-guinea pig IgG (1:400; Jackson ImmunoResearch), respectively, and the cultures were incubated for 2 hr at room temperature. Then, after washing with TBS, the cultures were incubated with goat anti-VR1 IgG (R-18; 1:1500; Santa Cruz Biotechnology, Santa Cruz, CA) in TBS containing 0.1% Triton X-100 and 5% FCS for 24 hr at 4°C. As a secondary antibody, Cy3-conjugated donkey anti-goat IgG (1:1000; Jackson ImmunoResearch) was used for 2 hr at room temperature.
After washing, all stained cultures were dehydrated in a series of graded ethanol, processed through N-butylacetate and covered with entellan (Merck, Darmstadt, Germany). Control experiments were performed without primary antibody or by preadsorption of the antibody with the immunizing peptides. The suppliers of all commercially available antibodies confirmed the selectivity of their preparations by Western blotting.
The triple immunofluorescence was investigated by means of a laser scanning confocal microscope (LSM 510; Zeiss, Oberkochen, Germany) at an excitation wavelength of 633 nm (helium/neon2, blue Cy5 immunofluorescence), 543 nm (helium/neon1, red Cy3 immunofluorescence), and 488 nm (argon, yellow–green Cy2 immunofluorescence). The three reaction products were distinguished by their different fluorescence: the P2X3 receptor by the green Cy2 immunofluorescence, the VR1 receptor by the red Cy3 immunofluorescence, and the P2Y1 receptor by the blue Cy5 immunofluorescence. Quantification of immunofluorescence was performed by randomly selecting neurons of 20–35 μm diameter At least 100 neurons were counted in four DRG cultures, each obtained from a different animal.
Tail-flick test. Male Sprague Dawley rats (Janvier) weighing 204–270 gm were used. The animals were housed under standardized humidity, temperature, and lighting conditions. The animals had access to water and food ad libitum.
The tail-flick test was performed according to D'Amour and Smith (1941) with a light beam analgesia meter (Rhema Labortechnik, Hofheim, Germany). Briefly, various doses of adenosine 5′-O-(2-thiodiphosphate) (ADP-β-S) were injected intrathecally between L1 and L3 under light diethylether (Merck) anesthesia. A radiant heat beam was focused onto the dorsal surface of the restrained rat's tail ∼2–4 cm apart from the root. The intensity of the radiant heat beam was adjusted to 40% of the maximum value, resulting in a mean control latency for tail withdrawal of 7.5 ± 1.5 sec (n = 100). To avoid tissue damage, the cutoff time was adjusted to 30 sec. The antinociceptive effect was calculated according to the following equation:
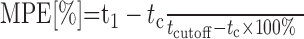 |
(2) |
Where MPE is the maximum possible effect, tl is the latency time of tail withdrawal, tc is the control latency, and tcutoff is the cutoff time. The ED50 value is the agonist dose producing 50% of the MPE. Both ADP-β-S and PPADS were dissolved in 0.9% NaCl.
The latency of the tail withdrawal response was measured before (tc) and at 10, 20, 30, 60, 90, and 120 min after drug application. Control animals received an intrathecal injection of 0.9% NaCl. Various doses of ADP-β-S were applied intrathecally under light ether anesthesia; the antagonist PPADS was also injected intrathecally, 10 min before the application of ADP-β-S. Accordingly, PPADS controls were injected with 0.9% NaCl intrathecally instead of ADP-β-S. The application volumes were 5 μl/animal. Ten animals were used per group.
In these series of experiments, Sprague Dawley rather than Wistar rats were used, because long-lasting experience indicates that this strain is more appropriate for all kinds of behavioral investigations. Preliminary experiments showed that ADP-β-S applied intrathecally prolonged the tail-flick latency in Wistar and Sprague Dawley rats to a comparable extent.
Data analysis. Data were presented as means ± SEM. Student's t test (unpaired) or ANOVA, followed by Dunnett's post hoc test, as appropriate, was applied to the data to determine statistical significance. A probability level of ≤0.05 was considered to be statistically significant.
Drugs. Chemicals used were: ATP, ADP, ADP-β-S, α,β-methylenadenosine 5′-triphosphate (α,β-meATP), 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP), GTP, GDP-β-S, BAPTA, suramin hexasodium salt, TEA, TTX, ω-conotoxin GVIA (ω-CTX), ω-agatoxin IVA (ω-Aga), isolectin B4 (Sigma, Deisenhofen, Germany), 2′-deoxy-N 6-methyladenosine 3′,5′-bisphosphate tetraammonium (MRS 2179), PPADS (Tocris Cookson, Bristol, UK), fura-2 (Molecular Probes, Leiden, The Netherlands), and AR-C69931MX (AstraZeneca, Wilmington, DE; a gift from Dr. M. Wayne).
Samples of ATP (99% ATP, 1% ADP) and UTP (82% UTP, 18% UDP) were checked for purity by HPLC before use. According to information from the supplier, the only significant impurity in ADP-β-S was identified as AMP. Therefore, an impurity of ATP was certainly not responsible for any effect of the UTP or ADP-β-S samples.
Results
Inhibition of the N-type Ca2+ current of DRG neurons by P2Y1 receptors
Figure 1Aa shows representative tracings of whole-cell ICa flowing through VACCs in cultured small-diameter (20–35 μm) DRG neurons. The currents were evoked by a depolarizing ramp from a holding potential of –90 to +70 mV. Currents through LVACCs were activated at –60 mV and reached their maximum at –40 mV. The HVACCs had an activation threshold at –40 mV (as estimated by extrapolation) and peaked at –10 mV. The mixed P2X/P2Y receptor agonist ATP (100 μm) markedly inhibited HVACCs and only slightly affected LVACCs (Fig. 1Aa,Ab). The effect of ATP was reversible on washout. ATP by itself did not alter the holding current, indicating that the opening of the cationic P2X3 receptor channels failed to cause inward current when TEA+ was used to completely replace Na+ in the external medium. HVACC currents were also activated by a single depolarizing step from –90 to –10 mV for 80 msec (Fig. 1Ba). The cationic species entering the cells appeared to be predominantly Ca2+, because the inorganic Ca2+ antagonist Co2+ (1 mm) almost abolished the inward current (data not shown). Voltage steps delivered every 20 sec caused reproducible inward HVACC current amplitudes during the whole measurement period (Fig. 1Bb). ATP (30 μm) inhibited the current amplitude in a manner completely reversible on washout with ATP-free medium (Fig. 1Ba,Bb). In the presence of ATP (30 μm), the current activation showed a tendency to become slower, which, however, did not reach the level of statistical significance. The rise time from 10 to 90% of the peak current amplitude (τ(10–90)) was 6.3 ± 1.5 msec before, 8.9 ± 2.1 msec during, and 6.6 ± 2.0 msec after the application of ATP (n = 6; p > 0.05) (Fig. 1Ba). This tendency did not become statistically significant despite increasing the concentration of ATP from 30 to 100 μm (n = 6; data not shown).
Figure 1.

Inhibition of VACCs by ATP. Calcium currents were recorded in rat DRGs using the whole-cell configuration of the patch-clamp technique. A, ATP (100 μm) inhibited HVACCs but not LVACCs. Depolarizing voltage ramps of 320 msec duration from the holding potential of –90 to + 70 mV were applied every 30 sec to evoke Ca2+ currents. Currents through LVACCs were activated at –60 mV and reached their maximum at –40 mV. HVACCs had an activation threshold at –40 mV (as estimated by extrapolation) and peaked at –10 mV. Aa, Representative tracing. Current responses before (control), during, and 2 min after (washout) the application of ATP (100 μm) for 2 min. Ab, Peak HVA and LVA Ca2+ current amplitudes normalized with respect to the mean of the first five control responses evoked by repetitive ramp-like voltage pulses. Data are mean ± SEM of five experiments. B, HVACC currents were evoked by depolarization from –90 to –10 mV for 80 msec every 20 sec. Ba, Representative tracings. Current responses before (control), during, and 2 min after (washout) the application of ATP (100 μm) for 2 min. Bb, Ca2+ current amplitudes normalized with respect to the mean of the first seven control responses evoked by repetitive voltage steps. Data are mean ± SEM of six experiments.
A second series of experiments were designed to determine which HVACC current types were inhibited by ATP. ω-CTX (0.5 μm), a selective blocker of N-type HVACCs, caused a rapid inhibition of Ca2+ current amplitudes that did not recover within 2 min after washout of the toxin (Fig. 2Aa). The ω-CTX-sensitive N-type fraction contributed ∼65% of the total ICa. The ω-CTX-induced maximum inhibition was not increased further by the application of ATP (30 μm) (Fig. 2Aa,Ab), suggesting that ATP only acts at the N-type Ca2+ current (ICa(N)). In fact, the P/Q-type HVACC blocker ω-Aga (0.4 μm) did not influence Ca2+ current amplitudes when applied alone (Fig. 2Ab), arguing against a major contribution of this current type to total ICa under the present experimental conditions (but see Mintz et al., 1992).
Figure 2.

Inhibition of HVACCs by the P2Y receptor agonists ATP, ADP, and UTP, and interaction of ATP with the P2Y receptor antagonists PPADS and MRS 2179. Calcium currents were recorded in rat DRGs using the whole-cell configuration of the patch-clamp technique. Voltage steps were made from a holding potential of –90 to –10 mV every 20 sec to induce inward currents. Aa, ATP (30 μm) did not affect the ω-CTX-insensitive HVACC currents. ω-CTX alone caused a rapidly developing inhibition of ICa which did not recover on washout of the toxin for 2 min. Ca2+ current amplitudes in Aa and Bb were normalized with respect to the mean of the first seven control responses evoked by repetitive voltage steps. Data are mean ± SEM of six (⋄) and seven (♦) experiments. Ab, Summary data showing the effect of ATP (30 μm; n = 6) after a 2 min application. Respective data forω-CTX (0.5 μm; n = 6), ω-CTX (0.5 μm) plus ATP (30 μm; n = 7), and ω-Aga (0.4 μm; n = 6) after a 4 min application. The bars are means ± SEM expressed as a percentage of the mean control currents (n = 7) before drug application. *p < 0.05, statistically significant difference from 100% (predrug control value). Ba, Concentration–response curves for the cumulative application of ATP (○; n=9), ADP (•; n=8), and UTP (▿; n = 6). After recording of seven stable control currents, cumulatively increasing concentrations of ATP, ADP, or UTP were applied for 2 min each. The agonist-induced depression of the HVACC current was normalized to the mean of the seven control currents. Bb, Antagonism by PPADS and MRS 2179 of inhibition by ATP (30 μm) of ICa. PPADS (30 μm) and MRS 2179 (30 μm) were applied 7 min before the application of ATP.
To investigate the possible participation of different P2Y receptors in the inhibition of HVACCs in DRG neurons, various P2Y receptor agonists and antagonists were tested. Single concentrations of ATP (0.1–1000 μm; n = 5–6) caused a steady-state inhibition of ICa within the 2 min application time (for 30 μm ATP, see Fig. 2Bb). ICa was depressed maximally by ∼70% of the original amplitude; the concentration producing IC50 was 28.1 μm. Cumulative application of increasing concentrations of ATP (0.1–1000 μm) every 2 min resulted in a concentration–response curve similar to that obtained with the single-dose application protocol (Fig. 2Ba); the IC50 value was 19.1 μm. Cumulative application of the P2Y1,12,13 receptor agonist ADP (0.1–1000 μm), concentration-dependently inhibited the calcium current with an IC50 value of 664 nm (Fig. 2Ba), indicating that the potency of ADP was higher than that of ATP. It is noteworthy that UTP (0.001–100 μm) also inhibited ICa with an IC50 value of 103 nm (Fig. 2Ba). In the following experiments, a 7 min superfusion with P2Y receptor antagonists was followed by a 2 min application of ATP (30 μm) (Fig. 2Bb). The P2Y1 receptor-selective antagonists PPADS (30 μm) and MRS 2179 greatly reduced the ATP-induced inhibition of HVACCs (Fig. 2Bb).
Colocalization of P2X3 and P2Y1 receptor subtypes on small-diameter DRG neurons
To demonstrate the functional coexistence of P2X and P2Y receptors on the same DRG neuron, a NaCl-containing extracellular solution was used instead of the usual medium in which all Na+ was completely replaced by TEA+. Under these conditions, voltage-dependent K+ and Na+ channels were inhibited by TEA (20 mm) and TTX (0.5 μm), respectively. A single depolarizing step from –90 to –10 mV evoked a current that included two components attributable to sodium and calcium ions flowing through the voltage-dependent TTX-insensitive Na+ channels (Roy and Narahashi, 1992) and HVACCs, respectively (Fig. 3Aa). Omission of Ca2+ from the extracellular solution (no added Ca2+ plus 1 mm EGTA) inhibited the late component of the current response by 57.6 ± 6.8% (n = 7; p < 0.05) (Fig. 3Aa). Application of ATP (30 μm) caused a rapidly desensitizing inward current that was produced conceivably via activation of P2X3 receptor channels. ATP, given for 2 min, failed to affect the early component of the current response to depolarizing stimuli in a statistically significant manner (0.1 ± 5.6%; n = 8; p > 0.05) but inhibited its late component (19.2 ± 3.3%; n = 8; p < 0.05). Both the effects of a Ca2+-free medium and of ATP (30 μm) were reversed on washout with normal external medium. Subtraction of the currents recorded in the nominal absence of Ca2+ from the control currents as well as from those recorded in the presence of ATP (30 μm) demonstrated the inhibition of ICa by ATP (Fig. 3Ab).
Figure 3.
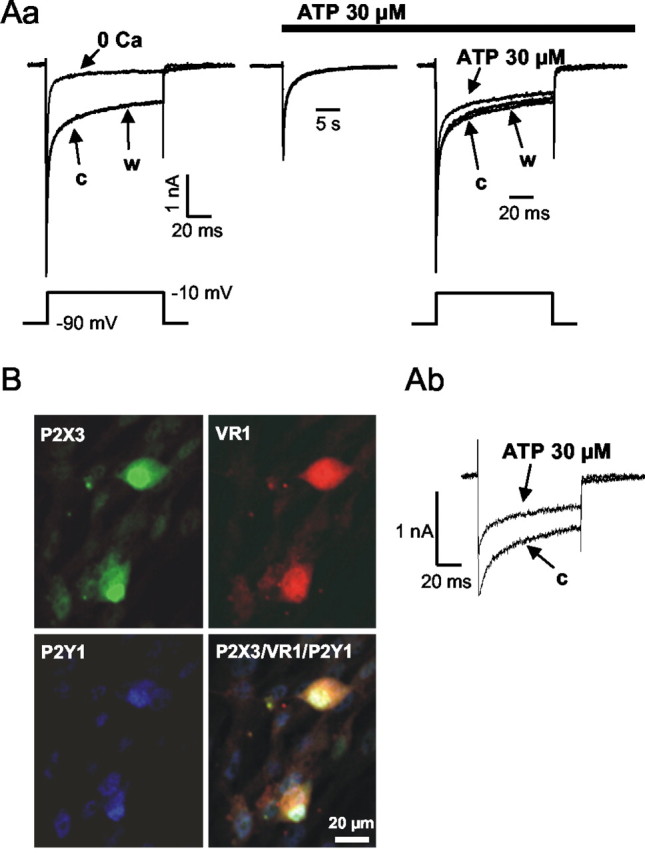
Functional and immunhistochemical evidence for the coexistence of P2X3, VR1, and P2Y1 receptors on the same DRG cell population. Aa, Calcium currents and TTX-insensitive sodium currents were recorded in rat DRGs using the whole-cell configuration of the patch-clamp technique. A NaCl- and TTX-containing extracellular solution (see Materials and Methods) was used. Voltage steps were made from a holding potential of –90 to –10 mV every 20 sec to induce inward currents. After three control currents (c), Ca2+-free extracellular solution (no added Ca2+ plus 1 mm EGTA) was superfused for 2 min (0 Ca) and then was washed out (w). Subsequently, ATP (30 μm) induced an inward current that recovered within seconds to baseline but continued to inhibit the depolarization-evoked currents. This effect was washed out within 1 min (w). Ab, When ICa was calculated by subtraction of the currents recorded in the absence of Ca2+ from the control currents as well as from those recorded in the presence of ATP (30 μm), ATP was found to cause inhibition. Note the different current calibrations in Aa and Ab. B, Triple immunofluorescence for P2X3, VR1, and P2Y1 receptors of rat DRG neurons investigated by means of a laser scanning confocal microscope. Images are of fluorescence for P2X3 (green Cy2 immunofluorescence), VR1 (red CY3-immunofluorescence), and P2Y1 (blue Cy5 immunofluorescence) receptor subtypes. Colocalization of P2X3, VR1, and P2Y1 receptors is shown.
Immunofluorescence in combination with confocal laser scanning microscopy confirmed the colocalization of the P2X3 and P2Y1 receptor subtypes on the cell bodies of VR1-immunoreactive cells (Fig. 3B). Almost every small-diameter neuron was immunoreactive for VR1 receptors (97.8 ± 1.3%; n = 4). A costaining for P2Y1, P2X3, and VR1 receptors was observed in 82.5 ± 3.1% (n = 4) of this neuronal population (P2X3 and P2Y1 receptor colocalization) (Ruan and Burnstock, 2003).
Inhibition of the N-type Ca2+ current by Gβγ
The above experiments demonstrate that, in an extracellular solution containing both TEA and TTX to block voltage-dependent K+ and Na+ channels, respectively, the P2X3 receptor-mediated inward current had already been desensitized completely at the time point when ATP still caused a clear depression of ICa. Hence, the direct participation of an endogenous P2X receptor in the inhibition of the N-type Ca2+ current is most unlikely. The indirect inhibition of the Ca2+ current [e.g., by Ca2+ entering into the cell via P2X3 receptor channels and, thereby, evoking Ca2+-dependent inactivation of ICa (Budde et al., 2002)] could be excluded; ATP depressed ICa despite failing to alter the holding current when the external medium contained Cs+ in the place of Na+.
To assess the possible involvement of different transduction pathways in the inhibition of ICa by G-protein-coupled P2Y receptors, a strongly depolarizing prepulse to +120 mV (20 msec duration), known to convert channels inhibited through the membrane-delimited pathway from the “willing” to the “reluctant” mode (Bean, 1989), was applied. This prepulse markedly counteracted the effect of ATP (Fig. 4Ab,Ba,Bb). In additional experiments, instead of GTP, GDP-β-S (0.3 mm), an inhibitor of G-protein-dependent processes (Sternweis and Pang, 1990), was added to the standard pipette solution. Ten minutes after establishing whole-cell configuration with a GDP-β-S-containing micropipette, ATP (100 μm) failed to inhibit ICa (Fig. 4Ac,Ba,Bb). Finally, the reduction of the internal EGTA from 11 to 1.1 mm, which results in less efficient buffering of the intracellular-free Ca2+ concentration, did not change the ATP-induced inhibition of ICa (Fig. 4Ad,Bb). These findings indicate that after the activation of the G-protein-coupled P2Y receptors by ATP, the βγ subunit of the G-protein, directly inhibits the N-type VACCs through a membrane-delimited pathway.
Figure 4.
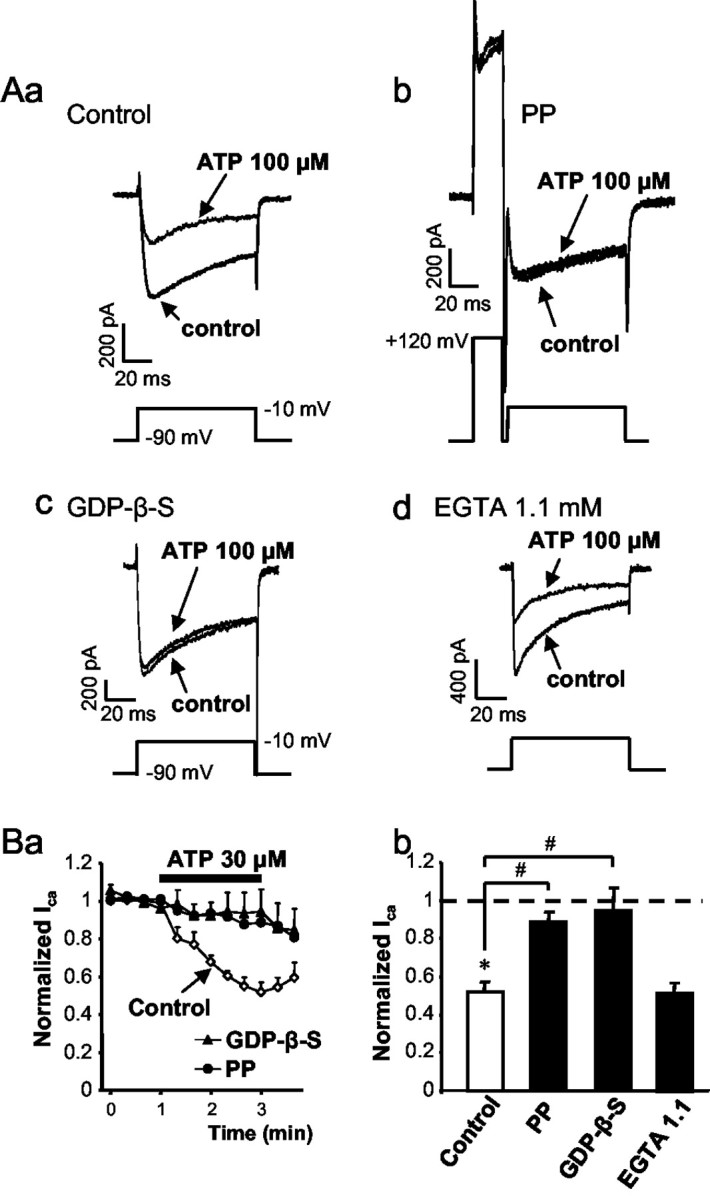
The possible transduction pathway of the ATP-induced inhibition of HVACCs. Calcium currents were recorded in rat DRGs using the whole-cell configuration of the patch-clamp technique. Voltage steps were made from a holding potential of –90 to –10 mV every 20 sec to induce inward currents. A, Representative tracings. Aa, HVACC currents before (control) and during the application of ATP (100 μm) for 2 min. A standard pipette solution containing GTP (0.3 mm; n = 6) was used. Ab, The regular pulse preceded by a strongly depolarizing prepulse (PP) to +120 mV abolished the inhibitory effect of ATP (100 μm; n = 5). Ac, HVACC currents recorded with a pipette solution containing GDP-β-S (0.3 mm; n = 7) instead of GTP. Ad, HVACC currents recorded with an intracellular solution containing 1.1 mm EGTA instead of the usual 11 mm (n = 10). Ba, Data were normalized to the first four points of each series of experiments and expressed as means ± SEM. ICa before, during, and after superfusion of ATP (30 μm), recorded with a pipette solution containing GTP (0.3 mm; control) or GDP-β-S (0.3 mm) is shown; the effect of a strongly depolarizing prepulse (PP) on the ATP-induced inhibition is shown. Bb, Summary data. Bars are means ± SEM expressed as a percentage of the four control currents before drug application. The number of experiments is indicated in A. *p < 0.05, statistically significant difference from the average of the first four currents measured before the application of ATP; #p < 0.05, statistically significant difference between the indicated bars.
Inhibition of depolarization-induced increase of intracellular Ca2+ by P2Y receptors
All DRG neurons used for Ca2+ imaging were labeled with IB4 and had diameters (20–35 μm) similar to those used for patch-clamp experiments. The application of a 50 mm K+-containing medium for 2 sec, as well as the application of ATP (30 μm), α,β-meATP (30 μm), or ADP-β-S (30 μm) for 10 sec each, every 10 min, caused reproducible increases of intracellular Ca2+ ([Ca2+]i). Apparently, a high external K+ concentration depolarizes the neuronal membrane, leads to the opening of VACCs, and allows Ca2+ to enter the cell. In support of this assumption, omission of Ca2+ from the bath solution for 3 min abolished the high K+-induced increase in [Ca2+]i (99.4 ± 0.3% inhibition; n = 16; p < 0.05) identifying the extracellular solution as the sole source of [Ca2+]i. In contrast, superfusion with the Ca2+-free medium for 3 min only approximately halved the response to ATP (30 μm; 54.3 ± 8.7% inhibition; n = 15; p < 0.05), suggesting that ATP leads to the entry of Ca2+ via P2X receptor channels and, in addition, releases Ca2+ from intracellular pools via P2Y receptor activation (Ralevic and Burnstock, 1998). ADP-β-S (30 μm), despite its assumed selectivity for P2Y1,12,13 receptors, exhibited, at the rather high concentration used, a considerable residual activity on P2X receptors, because the ADP-β-S-induced [Ca2+]i transients were partially dependent on external Ca2+ (52.5 ± 14.0% inhibition in a Ca2+-free medium; n = 9; p < 0.05). In contrast, α,β-meATP (30 μm) induced [Ca2+]i transients that were absolutely dependent on external Ca2+ (97.6 ± 0.6% inhibition in a Ca2+-free medium; n = 9; p < 0.05), identifying the site of agonist action as the P2X3 receptor channel.
ATP (30 μm) was applied 60 sec before, during, and after high K+ superfusion for 90 sec in total (Fig. 5Aa). The last response to K+ before the application of ATP was considered to be 100%. ATP (30 μm) raised [Ca2+]i exemplified by the corresponding change of the fluorescence ratio and, at the same time, depressed the effect of the high K+ stimulus (Fig. 5Aa,B; Table 1). Both effects depended on the concentration of ATP used (0.3, 3, 30, or 300 μm), with a maximum at 30 μm ATP (Table 1). PPADS (30 μm) applied for 10 min before the first K+ stimulation and throughout reduced the increase of [Ca2+]i by ATP and, in addition, reversed the ATP-induced inhibition of the high K+ effect (Table 1). ADP-β-S (30 μm) increased the fluorescence ratio and simultaneously inhibited the response to K+, thereby acting in a manner indistinguishable from ATP itself (Fig. 5Ab,B; Table 1). The effect of ADP-β-S was antagonized fully by the selective P2Y1 receptor antagonist MRS 2179 (30 μm) (Table 1). Finally, α,β-meATP (30 μm), an agonist at P2X1,3 receptors, increased [Ca2+]i and potentiated the response to high K+ (Fig. 5Ac,B; Table 1). Both effects were concentration dependent and seemed to be caused by the activation of P2X3 receptors, because the P2X1,3 receptor-selective TNP-ATP (30 μm) was clearly antagonistic (Table 1).
Figure 5.
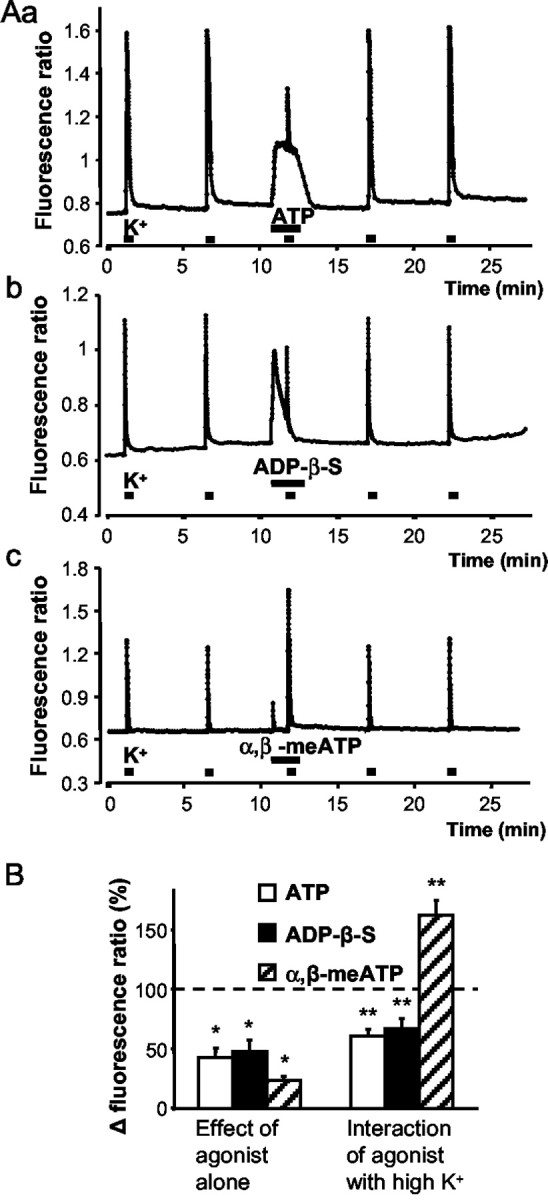
Dual effect of P2Y receptor agonists on the intracellular calcium concentration of rat DRG neurons by P2Y receptor agonists. Cells were loaded with the fluorescent indicator fura-2 acetoxymethyl ester (1 μm). The fluorescence ratio (340/380 nm) was taken as a measure of the cytosolic free Ca2+ concentration ([Ca2+]i). DRG neurons were depolarized by a 50 mm K+-containing extracellular medium for 2 sec. ATP, ADP-β-S, or α,β-meATP (30 μm each) was applied 60 sec before, during, and after high K+ superfusion for 90 sec in total. A, Representative experiments. Constant increase by ATP (Aa) and transient increases by ADP-β-S (Ab) and especially by α,β-meATP (Ac) of basal [Ca2+]i is shown. Inhibition by ATP (Aa) and ADP-β-S (Ab) but potentiation by α,β-meATP (Ac) of the high K+-induced [Ca2+]i transients is shown. B, Percentage changes of the Δ fluorescence ratio by ATP, ADP-β-S, and α,β-meATP when the second response to high K+ before superfusion with drugs was considered to be 100%. Data are means ± SEM of 15–38 experiments. *p < 0.05, statistically significant difference from 0% (first set of columns); **p < 0.05, statistically significant difference from 100% (second set of columns).
Table 1.
Increase of the intracellular free Ca2+ concentration by P2 receptor agonists when given alone and inhibition by the same agonists of the high K+-induced entry of Ca2+ into the cell, and interaction with P2 receptor antagonists.
|
Agonist |
μm |
Antagonist |
μm |
Percentage (%) increase of [Ca2+]i |
Percentage (%) inhibition of the high K+-induced [Ca2+]i elevation |
Number |
|---|---|---|---|---|---|---|
| ATP | 0.3 | 7.8 ± 1.1a | -14.9 ± 6.2 | 25 | ||
| 3 | 16.6 ± 3.1a | 20.5 ± 5.1 | 12 | |||
| 30 | 42.5 ± 8.2a | 38.9 ± 5.4b | 38 | |||
| 300 | 55.1 ± 10.1a | 30.1 ± 4.3b | 39 | |||
| 30 | PPADS | 18.1 ± 4.9a,c | -1.4 ± 6.0c | 27 | ||
| ADP-β-S | 30 | 47.9 ± 9.4a | 32.8 ± 8.6a | 32 | ||
| 30 | MRS 2179 | 30 | 14.9 ± 6.3a,c | -15.6 ± 5.0c | 11 | |
| α,β-meATP | 3 | 19.7 ± 3.8a | -4.4 ± 5.2 | 18 | ||
| 30 | 23.6 ± 3.6a | -62.6 ± 12.7b | 15 | |||
| 300 | 37.8 ± 8.6a | -125.4 ± 18.2b | 16 | |||
|
|
30 |
TNP-ATP |
0.01 |
1.3 ± 0.2c
|
37.9 ± 5.5a,c
|
16 |
p<0.05; significant difference from 0%.
p<0.05; significant difference from 100%.
p<0.05; significant difference from the effect of the respective agonist in the absence of the respective antagonist.
Inhibition of spinal nociceptive transmission by P2Y receptors
Low- and high-intensity electrical stimuli were applied to the dorsal root of the hemisected spinal cord preparation. The low-intensity stimulation evoked an initial monosynaptic compound action potential (MSR), superimposed on a polysynaptic population EPSP. These two potentials were mediated by A-fibers. The high-intensity stimuli evoked prolonged, presumably both A- and C-fiber-mediated, polysynaptic EPSPs. Low-intensity EPSPs were facilitated by ADP-β-S at 1 μm, unaltered at 10 μm, and inhibited at 100 μm (Fig. 6Ab). In contrast, high-intensity EPSPs were markedly depressed by ADP-β-S at 100 μm, with a threshold of inhibition at 10 μm of the agonist (Fig. 6Aa,Ab). The inhibitory effect of ADP-β-S (100 μm) of both low- and high-intensity EPSPs was completely reversed after its washout. The MSR was not significantly affected by ADP-β-S at any of the concentrations tested (Fig. 6Ab).
Figure 6.
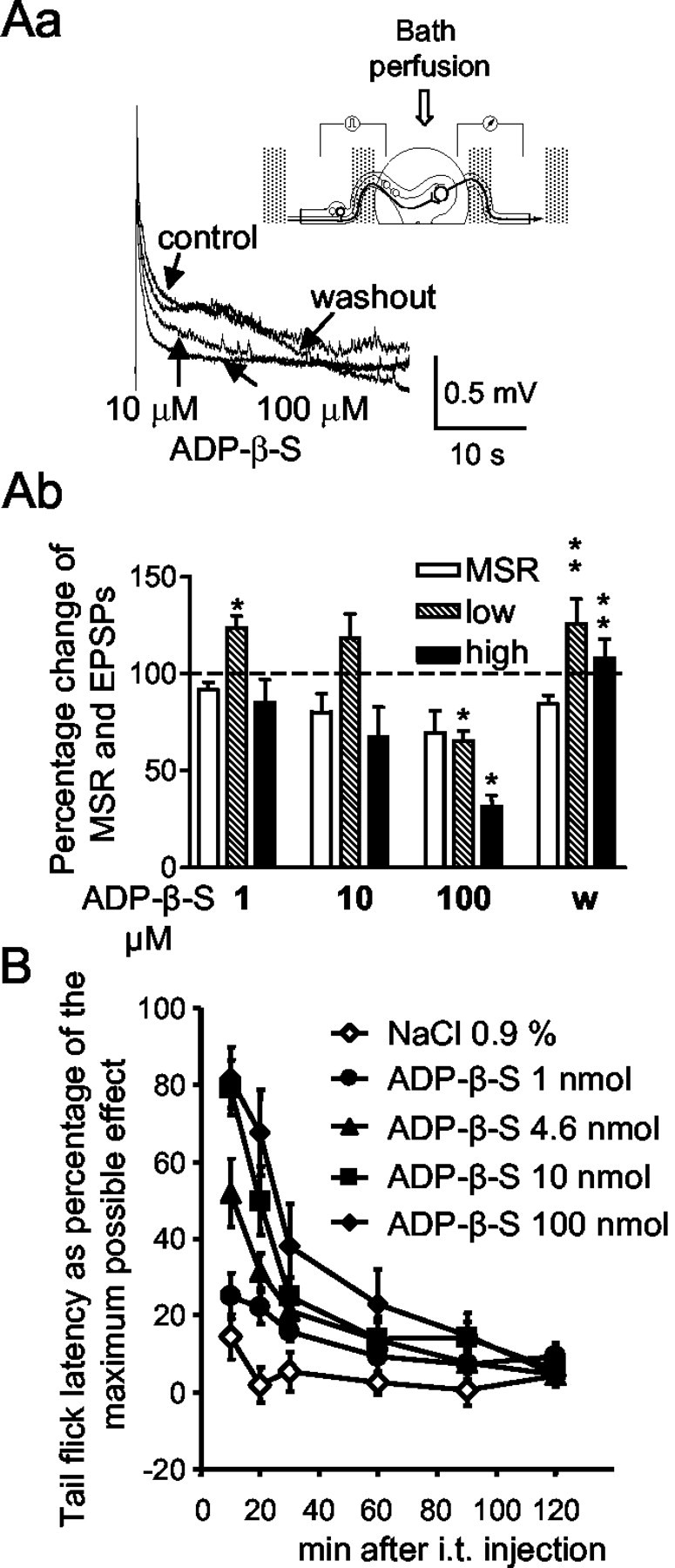
Modulation of segmental nociceptive spinal transmission by ADP-β-S in vitro and acute antinociceptive effect of ADP-β-S in vivo. Aa, Representative recordings showing high-intensity-evoked A- and C-fiber-mediated polysynaptic EPSPs in the hemisected spinal cord preparation of a juvenile rat before (control) and after the cumulative addition of 1, 10, and 100 μm ADP-β-S to the perfusion medium, followed by washout. Each concentration of ADP-β-S as well as the control medium (washout) was superfused for 20–25 min each. A scheme of the recording set-up is presented in the inset. The neuronal pathways involved in the monosynaptic and polysynaptic transmission are also shown. Ab, Summary data showing the effects of ADP-β-S on dorsal root-evoked MSR and polysynaptic (low, low-intensity EPSP; high, high-intensity EPSP) dorsal root responses. Data are means ± SEM of six experiments expressed as percentages of predrug control values. *p < 0.05, statistically significant difference from control responses (100%); **p < 0.05, statistically significant difference from the effect of the respective agonist concentration before washout. B, Measurement of tail-flick latency as a percentage of the maximum possible effect; dose-dependent antinociception caused by ADP-β-S. ADP-β-S was dissolved in 0.9% NaCl and was injected intrathecally (i.t.) in the indicated doses per animal. The effect of an equivalent amount of solvent was also tested. Data are means ± SEM of 10 experiments each.
The following experiments were designed to identify the individual P2Y receptor(s) involved in the ADP-β-S-induced inhibition of high-intensity EPSPs. Neither the P2Y1 receptor-preferential antagonist PPADS (30 μm) (ADP-β-S, 51.8 ± 6.4%; ADP-β-S plus PPADS, 70.7 ± 14.1; n = 10 each; p > 0.05) nor the P2Y12,13 receptor-selective antagonist AR-C69931MX (1 μm; a concentration about three orders of magnitude higher than its IC50 value against ADP-induced platelet aggregation) (Ingall et al., 1999) antagonized the effect of ADP-β-S (100 μm) (ADP-β-S, 56.2 ± 18.5%; ADP-β-S plus AR-C69931MX, 56.0 ± 14.1%; n = 7 each; p > 0.05). The P2Y1-selective antagonist MRS 2179 (1, 10, and 30 μm, at the highest concentration ∼100 times its Ki value at the P2Y1 receptor) (Boyer et al., 1998) produced a substantial inhibition of EPSPs on its own (data not shown). Therefore, this antagonist could not be used to investigate the involvement of P2Y1 receptors. Finally, the nonselective P2 receptor antagonist suramin (30 μm) also failed to antagonize the inhibitory effect of ADP-β-S (100 μm) (ADP-β-S, 57.0 ± 6.6%; ADP-β-S plus suramin, 48.1 ± 11.0%; n = 5 each; p > 0.05). It is interesting to note that the P2Y2,4,6 receptor agonist UTP did not change the high-intensity EPSPs at lower concentrations (0.01–0.1 μm; n = 5; data not shown) but facilitated them at the higher concentration of 1 μm (137.3 ± 8.6%; n = 5; p < 0.05). Finally, PPADS (30 μm) slightly increased the high-intensity EPSP when given alone (137.3 ± 8.6%; n = 5; p < 0.05), whereas both AR-C69931MX (1 μm) and suramin (30 μm) had no effect (data not shown).
ADP-β-S is antinociceptive in the tail-flick test after intrathecal application
To find out whether ADP-β-S exerts antinociceptive effects in vivo, the compound was tested in the tail-flick assay after intrathecal administration. ADP-β-S (1, 4.6, 10, and 100 nmol/animal) increased the nociceptive threshold in response to an acute thermal stimulus, because tail-withdrawal latencies were prolonged in a dose-dependent manner (Fig. 6B). Because 10 min after drug application ether anesthesia still slightly interfered with withdrawal latencies (Fig. 6B, 0.9% NaCl), the ED50 value was calculated at 20 min after application. Vehicle (0.9% NaCl) injection exerted no effect on nociceptive thresholds in control animals. The ED50 value of the acute antinociceptive effect of ADP-β-S amounted to 16.1 nmol/animal. Intrathecal ADP-β-S (100 nmol/animal; 65.2 ± 4.9% of the MPE; n = 10) was not antagonized by pretreatment with intrathecal PPADS (30 nmol/animal; 74.1 ± 10.0% MPE; n = 10; p > 0.05). In PPADS (30 nmol/animal, intrathecal) control experiments, only a negligible effect on withdrawal latencies was observed (19.1 ± 5.5% MPE; n = 10; p < 0.05). PPADS at 10 nmol/animal also did not antagonize the antinociceptive effect of ADP-β-S (100 nmol/animal), although this concentration of intrathecal PPADS has been shown to antagonize the decrease by α,β-meATP of the thermal nociceptive threshold (Tsuda et al., 1999).
Discussion
The main finding here is that P2Y1 receptor activation inhibits VACCs and the subsequent increase of the intracellular Ca2+ concentration in small nociceptive DRG neurons of rats. It has been suggested repeatedly that the properties of the DRG cell bodies and their terminals are, in many respects, similar (Belmonte and Gallego, 1983). Hence, it is assumed that P2Y receptors interfere with excitatory synaptic transmission from the central terminals of sensory neurons onto spinal cord neurons. In accordance with this idea, ADP-β-S, a P2Y1,12,13 receptor-preferential agonist inhibited C-fiber-mediated polysynaptic EPSPs in the rat spinal cord and thereby increased the nociceptive threshold when pain-induced behavior was measured with the tail-flick test.
A subtype of ionotropic ATP receptors, the P2X3 receptor (Chen et al., 1995; Lewis et al., 1995) is expressed with considerable selectivity by small-diameter, nonpeptidergic DRG neurons specifically binding the isolectin B4 (Chizh and Illes, 2001). Immunocytochemistry studies indicate that these neurons project to lamina II of the dorsal horn (Vulchanova et al., 1998). There is relatively high level of colocalization of P2X3 receptor immunoreactivity with the vanilloid VR1 receptor (Guo et al., 1999), which confers sensitivity to noxious heat (Caterina et al., 1997).
In the present patch-clamp study, when calcium was the sole charge carrier, ramp-like depolarizations evoked both LVACC and HVACC currents of rat DRG neurons. The HVACC current was preferentially depressed by the endogenous P2 receptor agonist ATP. In addition to the characteristic voltage at which they are activated and their differing single-channel properties, selective antagonists are known to block each channel type (N, ω-CTX; P/Q, ω-Aga). We found that HVACC current responses to depolarizing voltage steps were blocked by ω-CTX but not by ω-Aga, thereby identifying the current as belonging to the N type but not to the P/Q type (but see Mintz et al., 1992). Moreover, the application of ATP in the presence of ω-CTX did not cause any additional depression, supposedly because only the N-type current was sensitive to the inhibitory action of ATP.
The next series of experiments were designed to find out which of the eight known mammalian P2Y receptors is involved in the VACC inhibition. Interaction between ATP and the P2Y1 receptor-selective antagonists MRS 2179 and PPADS (von Kugelgen and Wetter, 2000) identified the blockade of VACCs as a P2Y1 receptor-function. In situ hybridization studies indicated a limited colocalization of mRNA for P2Y1 and VR1 receptors on small-diameter DRG neurons, whereas PY2 and VR1 receptor mRNA colocalized to a much larger extent (Moriyama et al., 2003). In contrast, we demonstrated the coexpression of P2X3, P2Y1, and VR1 receptor immunopositivities in ∼80% of small-diameter DRG cells. A mismatch between the nucleic acid message and its expression at the protein level is a possible explanation for this discrepancy. In addition, P2X3 (induction of a rapidly desensitizing inward current) and P2Y1 receptor (inhibition of VACCs) functions were also found to be confined to identical DRG neurons. The blockade of ICa by both ADP and UTP conformed with the presence of P2Y1 and P2Y2 receptors on the same DRG neuron; both receptors were reported to lead to the phosphorylation of VR1 receptor channels and thereby increase their conductance (Tominaga et al., 2001; Moriyama et al., 2003). This may cause thermal hypersensitivity and partly counteract the antinociception induced by the blockade of ICa(N), as proved for P2Y2, but not for P2Y1, receptors (Moriyama et al., 2003).
Previous studies in neurons have identified different G-protein-dependent pathways involved in the inhibition of ICa(N) (Hille, 1994). One mechanism may involve diffusible second messengers and the subsequent activation of protein kinases (Boehm et al., 1996). A second mechanism is membrane delimited and involves neurotransmitter receptors, G-proteins, and voltage-dependent calcium channels. A key role of the Gβγ rather than Gα subunits had been proposed (Herlitze et al., 1996). In the present experiments, ATP time- and concentration-dependently inhibited Ca2+ currents and shifted the current–voltage curve to the right, which are characteristic reactions after stimulation of G-protein-coupled receptors (Bean, 1989). Application of a strong depolarizing prepulse prevented ATP-induced inhibition of the Ca2+ current, indicating a G-protein-mediated mechanism of Ca2+ channel modulation (Bean, 1989). Intracellular dialysis with GDP-β-S, an inhibitor of G-protein-dependent reactions (Sternweis and Pang, 1990), also prevented the action of ATP. A Ca2+-dependent inactivation of the N-type Ca2+ channel (Budde et al., 2002) as a consequence of the P2Y receptor-induced increase of intracellular Ca2+ (Ralevic and Burnstock, 1998) could be excluded by the use of a lower concentration of EGTA in the pipette solution. Under these conditions, the calcium-buffering capacity of EGTA decreases and a Gα-mediated triggering of the phospholipase C–IP3–Ca2+ pathway could lead to a larger inhibition of ICa(N). However, this was apparently not the case, indicating the involvement of Gβγ rather than Gα in this process.
In experiments directly measuring [Ca2+]i by means of fura-2 and a Ca2+ imaging setup, the nonselective P2 receptor agonist ATP raised [Ca2+]i when given alone and, at the same time, depressed the high K+-induced increase of [Ca2+]i. Whereas the former effect is attributable both to a P2Y1 (Gα)-mediated release of Ca2+ from its intracellular storage sites and the entry of Ca2+ via the P2X3 receptor channel, the second effect seems to be a consequence of the membrane-delimited inhibition of ICa(N) by P2Y1 (Gβγ) (Fig. 7). The use of preferential agonists (ADP-β-S, P2Y1,12,13; α,β-meATP, P2X1,2,3) and antagonists (MRS 2179, P2Y1; TNP-ATP, P2X1,3) supported these conclusions.
Figure 7.

Schematic drawing demonstrating the multiple modulatory effects of ATP on intracellular Ca2+ in DRG neurons. ATP may activate P2X3 receptor channels, thereby facilitating the entry of Ca2+ via these channels into the cell. Moreover, the activation of P2Y1 receptors may lead via the Gα subunit to the generation of IP3 and the subsequent release of Ca2+ from the endoplasmatic reticulum. Finally, P2Y1 receptors may also via Gβγ close voltage-operated Ca2+ channels and thereby inhibit the passage of Ca2+ through the VACCs. Hence, the intracellular Ca2+ concentration may be modified by three simultaneous and partly opposing effects of ATP, two of which are algogenic (P2X3, P2Y1/Gα) and one antinociceptive (P2Y1/Gβγ). PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacylglycerol; ER, endoplasmatic reticulum.
The preferential inhibition of C-fiber-induced population polysynaptic EPSPs in the hemisected spinal cord by ADP-β-S localized the site of inhibition of synaptic responses to the dorsal horn of the spinal cord. P2X3 receptors have been suggested to be expressed on the central terminals of C-fibers innervating dorsal horn lamina II (Gu and MacDermott, 1997). The activation of these receptors leads to an enhanced release of glutamate. Based on patch-clamp and Ca2+ imaging experiments in cultured DRG cells, we hypothesized that P2Y1 receptors situated on the central axon terminals of DRG neurons may cause analgesia by decreasing the release of glutamate. However, the P2Y1 receptor-preferential antagonist PPADS, the P2Y12,13 receptor-selective antagonist AR-C69931MX (Barnard and Simon, 2001), and the nonselective P2 receptor antagonist suramin all failed to interact with the inhibitory effect of ADP-β-S in this system.
ATP has been reported to depress slow depolarization induced by repetitive dorsal root stimulation via P2Y receptors in substantia gelatinosa neurons of the rat spinal cord (Yoshida et al., 2002). In agreement with our results, in the hemisected spinal cord, the broad-spectrum P2 receptor antagonist suramin did not interact with ATP in the experiments of Yoshida et al. (2002). An additional argument for the absence of P2Y1 receptors on the central terminals of C-fibers is supplied by the failure of immunoreactivities for the vesicular glutamate transporter VGLUT2 (Bai et al., 2001) and P2Y1 receptors to colocalize in layer I–II of the spinal cord dorsal horn (W. Schröder, H. Franke, and P. Illes, unpublished observations). Hence, it is concluded that P2Y receptors, with a higher sensitivity for ADP than for ATP, are expressed on the central C-fiber terminals. However, this P2Y receptor does not conform in its ligand-binding characteristics with any of the presently known subtypes and still awaits structural determination.
As a final argument for the pharmacological significance of the supposedly presynaptically localized, and presently unclassified, inhibitory P2Y receptor, intrathecally applied ADP-β-S dose-dependently increased the tail-flick latency of rats. In perfect accordance with data obtained in the hemisected spinal cord, PPADS did not interfere with ADP-β-S, once again confirming the involvement of a P2Y receptor distinct from P2Y1. ADP-β-S rather than ADP itself was used in these experiments in view of the higher enzymatic stability of the ADP structural analog. Because the intrathecal injection of UTP and UDP has been shown to prolong thermal nociceptive latency in the tail-flick test (Okada et al., 2002), the ADP(ATP)-sensitive P2Y receptor described by us may be colocalized with a UDP(UTP)-sensitive P2Y receptor mediating spinal analgesia.
Hence, in conclusion, P2X3 receptor activation by ATP may lead to increased firing of DRG cells and subsequently to increased release of the sensory transmitter glutamate from their central processes. An opposite effect on DRG neurons is also conceivable. P2Y receptor activation by ADP, which is generated by the enzymatic degradation of ATP, may decrease the release of glutamate onto secondary sensory neurons of the spinal cord and may thereby partly counterbalance the algogenic effect of ATP. Finally, we hypothesize that the P2Y1 receptors found on immature cultured DRG neurons change their phenotype with development to a presently unclassified P2Y receptor (P2X receptors) (Inokuchi and McLachlan, 1995).
Footnotes
This work was supported by the Bundesministerium für Bildung, Forschung und Technologie (Leitprojekt “Molekulare Schmerzforschung,” 01GG981/0; Interdisziplinäres Zentrum für Klinische Forschung, 01KS9504, Projects C14 and Z10) and by the Deutsche Forschungsgemeinschaft (IL 20/11-1). We are grateful to H. Sobottka, S. Rosenow, H. Fischer, and B. Petter for technical assistance and Dr. J. Grosche for help with confocal microscopy. The expert linguistic correction of this manuscript by Dr. E. Wade is gratefully acknowledged.
Correspondence should be addressed to Dr. Peter Illes at the above address. E-mail: illp@medizin.uni-leipzig.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240797-11$15.00/0
DOI: 10.1523/JNEUROSCI.4019-03.2004
References
- Abbracchio MP, Burnstock G (1994) Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol Ther 64: 445–475. [DOI] [PubMed] [Google Scholar]
- Abbracchio MP, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G (2003) Characterization of the UDP-glucose receptor (re-named here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends Pharmacol Sci 24: 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai I, Xu H, Collins JF, Ghishan FK (2001) Molecular and functional analysis of a novel neuronal vesicular glutamate transporter. J Biol Chem 276: 36764–36769. [DOI] [PubMed] [Google Scholar]
- Barnard E, Simon J (2001) An elusive receptor is finally caught: P2Y12, an important drug target in platelets. Trends Pharmacol Sci 22: 388–391. [DOI] [PubMed] [Google Scholar]
- Barry PH (1994) JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods 51: 107–116. [DOI] [PubMed] [Google Scholar]
- Bean BP (1989) Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340: 153–156. [DOI] [PubMed] [Google Scholar]
- Belmonte C, Gallego R (1983) Membrane properties of cat sensory neurones with chemoreceptor and baroreceptor endings. J Physiol (Lond) 342: 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S (2003) P2Ys go neuronal: modulation of Ca2+ and K+ channels by recombinant receptors. Br J Pharmacol 138: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Huck S, Freissmuth M (1996) Involvement of a phorbol esterinsensitive protein kinase C in the α2 adrenergic inhibition of voltagegated Ca2+ current in chick sympathetic neurons. J Neurosci 16: 4596–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borvendeg SJ, Gerevich Z, Gillen C, Illes P (2003) P2Y receptor-mediated inhibition of voltage-dependent Ca2+ channels in rat dorsal root ganglion neurons. Synapse 47: 159–161. [DOI] [PubMed] [Google Scholar]
- Boyer JL, Mohanram A, Camaioni E, Jacobson KA, Harden TK (1998) Competitive and selective antagonism of P2Y1 receptors by N6-methyl 2′-deoxyadenosine 3′,5′-bisphosphate. Br J Pharmacol 124: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Meuth S, Pape H-C (2002) Calcium-dependent inactivation of neuronal calcium channels. Nat Rev 3: 873–883. [DOI] [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Touma E, Metzger RE, Kowaluk EA, Jarvis MF (1999) P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J Neurophysiol 82: 1590–1598. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Chen C-C, Akopian AN, Sivilotti L, Colquhoun D, Burnstock G, Wood JN (1995) A P2X purinoceptor expressed by a subset of sensory neurons. Nature 377: 428–431. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Illes P (2001) P2X receptors and nociception. Pharmacol Rev 53: 553–568. [PubMed] [Google Scholar]
- Communi D, Gonzalez NS, Detheux M, Brezillon S, Lannoy V, Parmentier M, Boeynaems JM (2001) Identification of a novel human ADP receptor coupled to Gi J Biol Chem 276: 41479–41485. [DOI] [PubMed] [Google Scholar]
- D'Amour FE, Smith DL (1941) A method for determining loss of pain sensation. J Pharmacol Exp Ther 72: 74–79. [Google Scholar]
- Diaz A, Dickenson AH (1997) Blockade of spinal N- and P-type, but not L-type, calcium channels inhibits the excitability of rat dorsal horn neurones produced by subcutaneous formalin inflammation. Pain 69: 93–100. [DOI] [PubMed] [Google Scholar]
- Faber ES, Chambers JP, Brugger F, Evans RH (1997) Depression of A and C fibre-evoked segmental reflexes by morphine and clonidine in the in vitro spinal cord of the neonatal rat. Br J Pharmacol 120: 1390–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA (1998) P2Y2 nucleotide receptors expressed heterologously in sympathetic neurons inhibit both N-type Ca2+ and M-type K+ currents. J Neurosci 18: 5170–5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Webb TE, Barnard EA, Brown DA (1999) Dual coupling of heterologously-expressed rat P2Y nucleotide receptors to N-type Ca2+ and M-type K+ currents in rat sympathetic neurones. Br J Pharmacol 126: 1009–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Brown DA, Barnard EA (2000) The P2Y1 receptor closes the N-type Ca2+ channel in neurones, with both adenosine triphosphates and diphosphates as potent agonists. Br J Pharmacol 129: 1063–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippov AK, Simon J, Barnard EA, Brown DA (2003) Coupling of the nucleotide P2Y4 receptor to neuronal ion channels. Br J Pharmacol 138: 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu JG, MacDermott AB (1997) Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature 389: 749–753. [DOI] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R (1999) Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci 11: 946–958. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Gardia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G protein βγ subunits. Nature 380: 258–262. [DOI] [PubMed] [Google Scholar]
- Hille B (1994) Modulation of ion channel function by G protein-coupled receptors. Trends Neurosci 17: 531–536. [DOI] [PubMed] [Google Scholar]
- Himmel HH, Kiss T, Borvendeg SJ, Gillen C, Illes P (2002) The argininerich hexapeptide R4W2 is a stereoselective antagonist at the vanilloid receptor 1: a Ca2+ imaging study in adult rat dorsal root ganglion neurons. J Pharmacol Exp Ther 301: 981–986. [DOI] [PubMed] [Google Scholar]
- Ingall AH, Dixon J, Bailey A, Coombs ME, Cox D, McInally JI, Hunt SF, Kindon ND, Teobald BJ, Willis PA, Humphries RG, Leff P, Clegg JA, Smith JA, Tomlinson W (1999) Antagonists of the platelet P2T receptor: a novel approach to antithrombotic therapy. J Med Chem 42: 213–220. [DOI] [PubMed] [Google Scholar]
- Inokuchi H, McLachlan EM (1995) Lack of evidence for P2X-purinoceptor involvement in fast synaptic responses in intact sympathetic ganglia isolated from guinea-pigs. Neuroscience 69: 651–659. [DOI] [PubMed] [Google Scholar]
- Kubista H, Lechner SG, Wolf AM, Boehm S (2003) Attenuation of the P2Y receptor-mediated control of neuronal Ca2+ channels in PC12 cells by antithrombotic drugs. Br J Pharmacol 138: 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulick MB, von Kugelgen I (2002) P2Y-receptors mediating an inhibition of the evoked entry of calcium through N-type calcium channels at neuronal processes. J Pharmacol Exp Ther 303: 520–526. [DOI] [PubMed] [Google Scholar]
- Lewis C, Neidhart S, Holy C, North RA, Buell G, Surprenant A (1995) Coexpression of P2X2 and P2X3 receptor subunits can account for ATP-gated currents in sensory neurons. Nature 377: 432–435. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL (1994) Voltage-sensitive calcium channels in spinal nociceptive processing: blockade of N- and P-type channels inhibits formalin-induced nociception. J Neurosci 14: 4882–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP (1992) P-type calcium channels in rat central and peripheral neurons. Neuron 9: 85–95. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, Leon C, Suzuki N, Inoue K, Gachet C, Noguchi K, Tominaga M (2003) Possible involvement of P2Y2 metabotropic receptor in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci 23: 6058–6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RA (2001) Identification of the P2Y12 receptor: a novel member of the P2Y family of receptors activated by extracellular nucleotides. Mol Pharmacol 60: 416–420. [PubMed] [Google Scholar]
- Okada M, Nakagawa T, Minami M, Satoh M (2002) Analgesic effects of intrathecal administration of P2Y nucleotide receptor agonists UTP and UDP in normal and neuropathic pain model rats. J Pharmacol Exp Ther 303: 66–73. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413–492. [PubMed] [Google Scholar]
- Roy ML, Narahashi T (1992) Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 12: 2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan HZ, Burnstock G (2003) Localisation of P2Y1 and P2Y4 receptors in dorsal root, nodose and trigeminal ganglia of the rat. Histochem Cell Biol 120: 415–426. [DOI] [PubMed] [Google Scholar]
- Sternweis PC, Pang IH (1990) The G protein-channel connection. Trends Neurosci 13: 122–126. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98: 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno S, Tsuda M, Iwanaga T, Inoue K (1999) Cell type-specific ATP-activated responses in rat dorsal root ganglion neurons. Br J Pharmacol 126: 429–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi G, Strobeck M, Koch S, Caglioti L, Zucchi C, Palyi G (1999) Molecular elements of ion permeation and selectivity within calcium channels. Crit Rev Biochem Mol Biol 34: 181–214. [DOI] [PubMed] [Google Scholar]
- von Kugelgen I, Wetter A (2000) Molecular pharmacology of P2Y receptors. Naunyn-Schmiedeberg's Arch Pharmacol 362: 310–323. [DOI] [PubMed] [Google Scholar]
- Vulchanova L, Riedl MS, Shuster SJ, Stone LS, Hargreaves KM, Buell G, Surprenant A, North RA, Elde R (1998) P2X3 is expressed by DRG neurons that terminate in inner lamina II. Eur J Neurosci 10: 3470–3478. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Nakagawa T, Kaneko S, Akaike A, Satoh M (2002) Adenosine 5′-triphosphate inhibits slow depolarization induced by repetitive dorsal root stimulation via P2Y purinoceptors in substantia gelatinosa neurons of the adult rat spinal cord slices with the dorsal root attached. Neurosci Lett 320: 121–124. [DOI] [PubMed] [Google Scholar]