

Abstract
In an effort to understand the complexity of genomic responses within selectively vulnerable regions after experimental brain injury, we examined whether single apoptotic neurons from both the CA3 and dentate differed from those in an uninjured brain. The mRNA from individual active caspase 3(+)/terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling [TUNEL(–)] and active caspase 3(+)/TUNEL(+) pyramidal and granule neurons in brain-injured mice were amplified and compared with those from nonlabeled neurons in uninjured brains. Gene analysis revealed that overall expression of mRNAs increased with activation of caspase 3 and decreased to below uninjured levels with TUNEL reactivity. Cell type specificity of the apoptotic response was observed with both regionally distinct expression of mRNAs and differences in those mRNAs that were maximally regulated. Immunohistochemical analysis for two of the most highly differentially expressed genes (prion and Sos2) demonstrated a correlation between the observed differential gene expression after traumatic brain injury and corresponding protein translation.
Keywords: apoptosis, traumatic brain injury, hippocampus, CA3, dentate, single-cell amplification, microarray, cell death, differential expression, real-time quantitative PCR
Introduction
Traumatic brain injury (TBI) remains the leading cause of disability and mortality in young adults in the United States and Europe (Sosin et al., 1989; Sauaia et al., 1995). Cognitive function is particularly affected after brain injury attributable, in part, to selective cell death in the hippocampus (Smith et al., 1995; Bramlett et al., 1997), the neuroanatomical substrate of memory and learning (Olton and Papas, 1979). An experimental paradigm of brain injury in animals, the controlled cortical impact (CCI) model, has been shown to consistently reproduce the posttraumatic memory deficits and both the apoptotic and necrotic cell death (Smith et al., 1995; Colicos and Dash, 1996; Clark et al., 1997; Fox et al., 1998) presumed occurring in brain-injured humans (Williams et al., 2001).
Apoptosis is actively regulated through the induction of selective members of the caspase gene family of cysteine proteases (Alnemri, 1997). Activation of caspases and increased expression of caspases 3 and 8 have been observed in the cortex and hippocampus after CCI in the rat (Beer et al., 2000b, 2001; Clark et al., 2000). Although caspases seem to play a central role in TBI-induced apoptosis (Eldadah and Faden, 2000; Raghupathi et al., 2000), the expression of other genes has, likewise, been demonstrated to influence both the initiation or progression of apoptosis (Ashkenazi and Dixit, 1998). For example, increased expression of adaptor proteins such as the Fas or tumor necrosis factor (TNF) receptor, which link the caspases to upstream regulators of apoptosis, have been demonstrated in several experimental models of TBI (Bruce et al., 1996; Beer et al., 2000a). Similarly, transcription of selective members of the Bcl-2 family has been repeatedly shown to either induce or inhibit apoptosis depending on the member differentially expressed (Clark et al., 1997; Raghupathi et al., 2003).
Together, these data indicate that differential expression of certain genes may be essential to either the initiation or progression of apoptotic cell death. The identification of mRNA abundance differences after TBI could elucidate specific molecular events that may mediate programmed cell death. Methods historically used to evaluate mRNA abundances in the injured brain (e.g., RNase protection assays, Northern blots, or reverse transcription-PCR) have been limited in their ability to simultaneously detect multiple mRNA species. These techniques are restricted to the use of tissue homogenates that prevent the localization of the cellular source of the differential expression. The use of amplified antisense mRNA (aRNA) bypasses these restrictions by allowing the detection of multiple mRNA species within individual cells in culture or fixed tissues (Eberwine et al., 1992b; Crino et al., 1996; Morrison et al., 2000; O'Dell et al., 2000). Recently, this methodology was used to generate an expression profile of apoptotic cortical cells at two time points after experimental brain injury in the rat (O'Dell et al., 2000). After analyzing the expression of a few candidate genes, the authors reported that phenotypically identical cells have different underlying molecular profiles dependent on the time point after injury. By elucidating the posttraumatic differential mRNA expression in apoptotic hippocampal neurons through the use of high-density microarrays, we hypothesize that these single-cell mRNA expression profiles may reveal cell type-specific molecular cascades in the posttraumatic brain. In this study, the CA3 subfield and dentate gyrus were selected exclusively because of the high portion of apoptotic neuronal cell death reported previously (Clark et al., 1997; Fox et al., 1998). In contrast, the lack of apoptotic neurons in injured the CA1 subfield rendered this region less optimal for our focus of uncovering apoptotic-specific expression changes ultimately contributing to the design of novel and targeted therapies for human brain injury.
Materials and Methods
Surgical procedure and injury. The model of CCI brain injury used in these studies has been described for the rat (Dixon et al., 1991) and subsequently modified for use with mice (Smith et al., 1995). All procedures described here were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pennsylvania Animal Care and Use Committee. Male C57BL/6 mice, 8–10 weeks of age (n = 30), weighing ∼25–32 gm, were housed in a 12 hr light/dark cycle with ad libitum access to food and water. All mice were anesthetized with intraperitoneal injections of sodium pentobarbital (65 mg/kg), surgically prepared, and subjected to either CCI brain injury (n = 18) using a velocity of 5.0 m/sec and at a depth of 1.0 mm as characterized previously (Smith et al., 1995; Raghupathi et al., 1998) or sham injury (n = 12). Throughout the entire surgery and during recovery, all mice were kept on a heating pad to maintain physiological body temperature.
Tissue processing, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, and Immunohistochemistry. Twenty-four hours after injury (time point chosen because of the robustness of immunohistochemical staining for apoptotic markers while showing little cell loss), animals were anesthetized (65 mg/kg sodium pentobarbital, i.p.) and perfused transcardially with heparinized saline, followed by 4% paraformaldehyde. Mice were decapitated, and heads were fixed in paraformaldehyde at 4°C for 4 hr, after which brains were removed and postfixed for an additional 24 hr at 4°C. Paraffin-embedded blocks were then sectioned serially at 6 μm between 1.5 and 1.6 mm posterior to bregma (the site of maximal cortical injury) and mounted on poly-l-lysine-coated slides. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed using previously described methods (Conti et al., 1998). In brief, after proteinase K digestion, sections were incubated in the labeling solution containing terminal deoxynucleotidyltransferase (0.3 U/μl; Boehringer Mannheim, Indianapolis, IN), biotinylated-16-dUTP (20 μm; Boehringer Mannheim), and 1.5 μm cobalt chloride. The product was visualized using streptavidin-conjugated alkaline phosphatase (BioGenex, San Ramon, CA), followed by Fast Red (Fast Red kit; Sigma, St. Louis, MO).
After the TUNEL reaction, sections were immersed in 10% H2O2, rinsed, blocked in 2% normal horse serum (NHS), and then incubated overnight at 4°C in primary antibody for activated caspase 3 (MF397; Merck-Frosst Quebec, Quebec, Canada) (Gervais et al., 1999) diluted at 1:1000 in 2% NHS/0.1 m Tris buffer. Goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), was applied at 1:200 for 2 hr, followed by ABC (Vector Laboratories, Burlingame, CA). The antibody complex was subsequently visualized enzymatically with DAB (Sigma). The omission of primary antibody on select sections of the mouse brain provided a negative control. Bouins-fixed sections from prenatal bcl-xl knock-out mice (generously provided by Dr. K. A. Roth, Washington University, St. Louis, MO) were used as a positive control for activated caspase 3. For analysis of selected protein expression corresponding to highly differentially expressed genes, additional sections were immunolabeled as described above for the proteins prion (PrPc) and son of sevenless 2 (Sos2). Primary antibodies for PrPc (Serotec, Oxford, UK) and Sos2 (Santa Cruz Biotechnology, Santa Cruz, CA) were applied at dilutions of 1:100, and 1:1000 dilutions of donkey anti-goat and goat anti-rabbit IgG secondary antibodies were used, respectively (Jackson ImmunoResearch).
Single-cell microdissection. Transcription of the first strand of cDNA was performed in situ to increase the yield of cDNA from the endogenous mRNA pool before microdissection (Crino et al., 1996; Morrison et al., 2000). After TUNEL and active caspase 3 immunohistochemistry, sections were incubated overnight with the oligo-dT primer coupled with a T7 RNA polymerase promoter sequence (oligo-dT-T7), as described previously (Eberwine et al., 1992b; Phillips and Eberwine, 1996). After first-strand cDNA synthesis, the sections were washed in 0.5× SSC, and individual labeled cells were viewed on an inverted microscope (IX-70; Olympus America, Lake Success, NY) (Crino et al., 1996; O'Dell et al., 2000). With the use of a RNase-free glass micropipette attached to a micromanipulator (Narishige International, Greenvale, NY), individual neurons were carefully microdissected from the surrounding neuropil, taking care not to disrupt any adjacent cells, and subsequently aspirated into the micropipette for amplification. The microdissected neurons were either unstained, active caspase 3(+)/TUNEL(–), or active caspase 3(+)/TUNEL(+) from both the pyramidal layer of the CA3 region and granule layer of the dentate gyrus (Fig. 1 A). Digital photomicrographs in both fluorescence and bright fields were taken before and after aspiration to assure that >80% of the cell was dissected and that only a single cell was detached (Fig. 1 B). If any part of an adjoining cell was perturbed or detached in the process, the dissected cell was not included in additional analyses.
Figure 1.
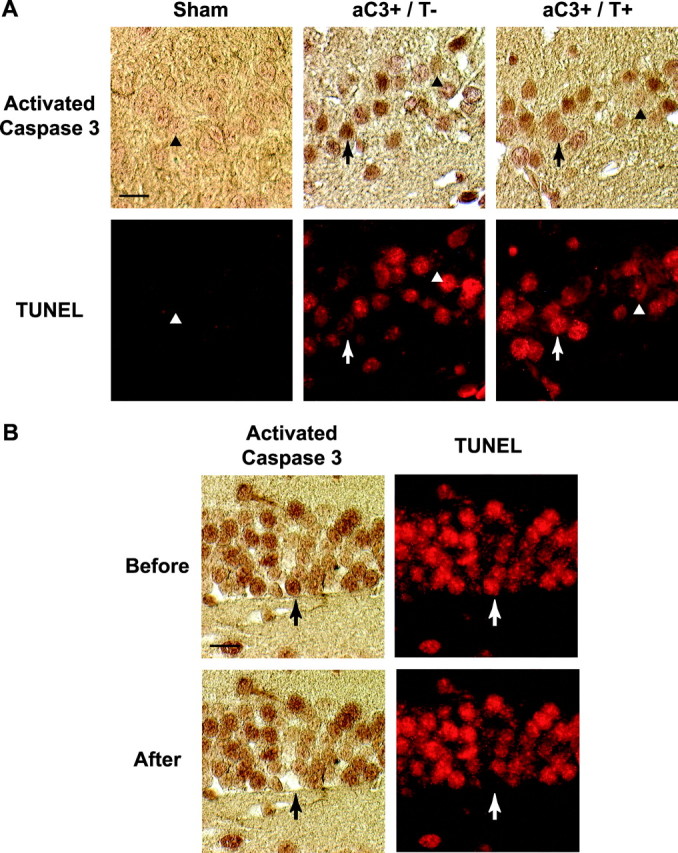
Histopathological analysis of the injured and uninjured brain at 24 hr after injury/surgery with both activated caspase 3 and TUNEL stain. A, In the hippocampal CA3 subfield, some neurons displayed colocalization of activated caspase 3 and TUNEL (aC3+/T+, arrow), whereas others were only immunoreactive with activated caspase 3 (aC3+/T–, arrow). Neurons not stained with either activated caspase 3 antibody or TUNEL were also found (arrowheads). B, Microdissection of a single active caspase 3 (+)/TUNEL(+) granule cell from the dentate gyrus 24 hr after injury. Before and after images were used to demonstrate that >80% of the cell was dissected and that none of the surrounding cells were disrupted. Scale bars, 25 μm.
We chose to examine the hippocampus because we wanted to examine the downstream influence of injury on neuronal functioning. Examination of the cortex would have required making subjective choices with regard to the area examined (e.g., distance from the injury) that would make microarray data analysis more difficult.
Single-cell amplification. The mRNA from individual neurons was amplified according to previously described methods (Eberwine et al., 1992a,b; Phillips and Eberwine, 1996). Approximately 75% of the double-stranded DNA material was used for first-round aRNA amplification with T7 RNA polymerase (2000 U/μl; Epicenter Technologies, Madison, WI), and the remaining 25% was kept for the real-time PCR validation. Radiolabeled CTP was used to visualize the probe on an RNA denaturing gel. After the estimated 1000- to 2000-fold amplification, the newly synthesized aRNA was primed with random hexanucleotide primers and served as a template for cDNA synthesis.
cDNA microarrays. The double-stranded DNA products from phenotypically identical cells were pooled to decrease variability attained from non-littermates and from cell-to-cell variation. Each pooled probe was composed of a total of six cells from three different mice: one cell from each of two nonconsecutive sections per animal. Labeled probes synthesized from all three groups [active caspase 3(+)/TUNEL(+), active caspase 3(+)/TUNEL(–), and active caspase 3(–)/TUNEL(–) from sham animals] within each of the two regions (for a total of nine cells) were analyzed and hybridized to the microarrays. Each comparison was replicated with labeled probes composed from a separate group of three different animals, and the dyes linked to each probe were swapped. Consequently, a total of 18 cells per group were used to generate the microarray data. Dye swapping was used to normalize any variation in the fluorescence of the Cy3 or Cy5.
The labeled probe was synthesized by converting the amplified aRNA to either a Cy3- or Cy5-labeled cDNA probe using a custom labeling kit as described by Yue et al. (2001). Briefly, the fluorescently labeled cDNA was synthesized with Cy3- or Cy5-labeled random 9mer (Trilink) and Moloney murine leukemia virus RNase H-free reverse transcriptase (Promega, Madison, WI). Hybridization of labeled cDNA probes was performed in 20 μlof5× SSC, 0.1% SDS, and 1 mm DTT at 60°C for 6 hr to Incyte Mouse GEMs version 1. The arrays were populated with 9024 total unique elements, of which 98 were blank (H2O), 192 were composed of yeast control fragments, and the remaining 8734 were PCR-amplified mouse expression sequence tags (ESTs) selected randomly from The Institute for Genomic Research Mus.ET database and the GenBank mouse database. After hybridization, the microarrays were washed, dried by centrifugation, and scanned with a GenePix 4000A fluorescent reader and GenePix image acquisition software (Axon Instruments, Foster City, CA) at 535 nm for Cy3 and 625 nm for Cy5. The complete analysis used a total of 24 Incyte microarrays.
cDNA microarray data analysis. Once the microarrays were hybridized with the appropriate samples and subsequently scanned, the resulting data consisted of tagged image file format (TIFF) images representing both the Cy3 and Cy5 channels (see Fig. 3 A). A tabular representation of the TIFF images was generated by the software package Spot (Buckley, 2000), which is based on a new addressing, segmentation, and background correction method described previously by Yang et al. (2002a). Briefly, spots are addressed semiautomatically by an algorithm based the “seeded region growing” method of Adams and Bischof (1994), in which no a priori restrictions on the size or the shape of the spots are made. The resulting data were then analyzed to determine both the clones expressed above background and those differentially expressed between injured and uninjured.
Figure 3.
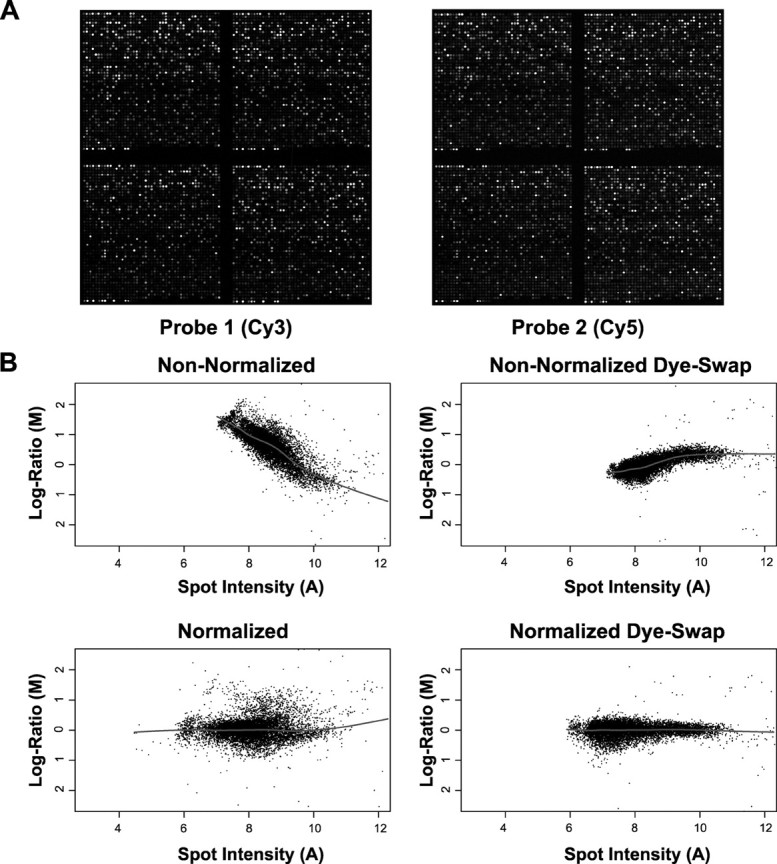
Differential expression in the dentate. A, TIFF images from both active caspase 3(+)/TUNEL(+) granule neurons (Cy3) from an injured brain and nonlabeled granule neurons from an uninjured brain (Cy5). Each channel is segmented and analyzed to extract the intensity and background values for every spot on the array. B, The resulting data are then plotted on log ratio (M = log2 Cy3/Cy5) versus overall spot intensity [A = log2√(Cy3 × Cy5)] to assess effectiveness of normalization of the dye biases and to calculated the differential expression.
To determine the genes expressed, no normalization across arrays was applied, given that the thresholds and lists of spots were calculated for each array (and channel) individually. The foreground measurement of every spot was calculated as the sum of pixel intensities for that spot. The background adjustment is based on “morphological opening,” a technique derived from mathematical image analysis (Soille, 1999), and corrected by the area. The intensities of the blank or buffer only spots dispersed throughout each microarray were used to determine individual levels of noise for each array and channel of interest. Thresholds were set to the maximum of the intensities of the blanks over the array in that channel. For each array considered, a list was compiled of those spots with intensities in the channel of interest (corresponding to a given cell type and condition) that were above such threshold. Intersections of these lists for each cell type [for the biggest four of five in the sham and active caspase 3(+)/TUNEL(+)and for the biggest two of three in active caspase 3(–)/TUNEL(+)] were compiled, and the percentage of clones in common across regions was calculated.
The analysis of the differential expression, however, required both normalization to remove technological sources of variation and ranking to extract maximally expressed genes. The background measurement was determined as stated previously, whereas the foreground measurement was calculated as the median of pixel intensities of that spot. The data were normalized using local normalization methods introduced by Yang et al. (2002b,c), implemented in the publicly available R-package “Statistics for Microarray Analysis.” Normalization amounts to a transformation of the data set by a function estimated by robust locally weighted regression (lowess). The intensities were represented by M versus A plots, by graphing their log ratio (M = log2 sample A/sample B) versus the mean of their logs [A = (log2 sample A + log2 sample B)/2] (see Fig. 4 B). Variation across the slide was further accounted for by performing normalization separately for each print-tip group. The genes were ranked based on the magnitude of differential expression calculated across dye swap experiments, and priority was given to the clones most consistently differentially expressed among the replicates (Dudoit et al., 2002). According to the Minimum Information About A Microarray Experiment protocol, the raw microarray data will be deposited in ArrayExpress.
Figure 4.

Real-time PCR validation of the differential expression determined from the microarrays. Threshold value (dotted line) was set for each EST, and the CT (cycle at which threshold was passed) was determined for both the injured and uninjured samples. The calculated difference in CT represents the relative ratio of the clones in the two templates.
Real-time PCR validation. Real-time PCR was used to quantify the abundances of selected individual clones within the cDNA template pool to validate the differential expression demonstrated by the microarrays. Twenty-four clones were chosen randomly from the differentially expressed genes between the injured activated caspase 3(+)/TUNEL(+) cells compared to uninjured nonlabeled neurons from either region and were processed on an ABI Prism 7000 sequence detection system (Applied Biosystems, Foster City, CA). Primers were constructed using Primer Express version 2.0 program (Applied Biosystems), and the full sequence of the clones was spotted on the array. The primer pairs chosen were staggered to the 3′ end of the sequence and generated an amplicon of 100–150 bp. The concentrations of each primer pair were optimized according to assay performance, and the final primer concentrations were challenged with no template controls to assure for minimal primer dimer product formation. AmpliTaq Gold DNA polymerase was used for the PCR, and SYBR Green was used as the reporter dye (Applied Biosystems). Universal thermal cycling parameters as recommended by the manufacturer were used to allow for the use of multiple primer pairs on each 96-well plate for a total of 60 cycles.
Real-Time PCR experiments for each of the 24 primer pairs were performed with six replicates for each of the four templates. The threshold used to determine the CT (defined as the fractional cycle number at which the fluorescence passes a fixed threshold) of each run was based on the variability of the baseline data between cycles 3 and 15. The resulting ΔRns (defined by the equation ΔRn = Rn + – Rn –, where Rn + is the fluorescence emission of the product at each time point and Rn – is the fluorescence emission of the baseline) were averaged, and a ΔCT was calculated for each clone. The difference in cycle times between the injured and uninjured samples determined both the direction and amount of regulation. A dissociation curve analysis (slowly ramping the temperature of the reaction solutions from 60 to 95°C while continuously collecting fluorescence data) was performed after each completed PCR run to assess the amount of nonspecific amplification occurring. Gel electrophoresis was also performed to assure specific amplification at the predicted amplicon size.
Results
Immunohistochemical analysis
Neurons from uninjured animals exhibited no TUNEL reactivity. Only occasional active caspase 3 immunolabeled cells were observed ipsilaterally to the craniotomy, in both the dentate gyrus and CA3 subfield. These neurons retained normal nuclear morphology with distinct nucleoli, and the activated caspase 3 immunoreactivity was predominantly cytoplasmic. In injured brains, however, the pattern of the activated caspase 3 and TUNEL staining exhibited more heterogeneity. In both regions analyzed, a subset of neurons exhibited both activated caspase 3 immunoreactivity and TUNEL staining that was almost exclusively in the nucleus. In several neurons, TUNEL positivity localized to well-demarcated punctate foci within the nucleus, possibly corresponding to chromatin marginalization (Fig. 1A, aC3+/T+, arrows). In close proximity to double-labeled neurons were both active caspase 3(+)/TUNEL(–) neurons (Fig. 1A, aC3+/T–, arrows) and unlabeled neurons (Fig. 1A, arrowheads). All regions analyzed contained one cell representative of each of these three types of staining in each high-powered field.
cDNA microarrays
Overall mRNA expression calculations demonstrate the number of mRNAs expressed varied between cell types and specific hippocampal regions. Of the 8734 randomly selected ESTs that were spotted in the array, the uninjured pooled pyramidal neurons expressed a total of 3514 mRNAs, whereas uninjured pooled granule neurons expressed 4800 mRNAs (Table 1). Comparison analysis demonstrated that although 3119 mRNAs were expressed in both regions, the remaining 35% (or 1681) of the genes expressed were unique to the dentate, and 11% (or 395) were unique to the CA3 (Fig. 2A). The brain injury and subsequent activation of caspase 3 in the active caspase 3(+)/TUNEL(–) cell increased the total expression in pyramidal and granule neurons to 5985 and 7078, respectively. The intersection of these two sets of expressed genes resulted in an increased overlap of 5377 clones with an associated decreased percentage of unique genes to 10 and 24% in the CA3 and dentate, respectively (Fig. 2A). Colocalization of TUNEL reactivity in the double-labeled active caspase 3(+)/TUNEL(+) resulted in a decreased total expression of mRNAs in both regions to 3659 in pyramidal neurons and 1953 in granule neurons. However, the percentage of mRNAs expressed exclusively in the CA3 increased to 1843 or 50% of those expressed and was decreased to 137 or 7% of those expressed in the dentate (Fig. 3A).
Table 1.
Expression derived from the microarrays
|
Cell type |
Total in CA3 |
Total in dentate |
Unique to CA3 |
Unique to dentate |
Common |
Total |
|---|---|---|---|---|---|---|
| Sham | 3514 | 4800 | 395 | 1681 | 3119 | 5195 |
| aC3+/T- | 5985 | 7078 | 608 | 1701 | 5377 | 7686 |
| aC3+/T+ |
3659 |
1953 |
1843 |
137 |
1816 |
3796 |
Values represent the total number of clones expressed above background of the 8734 spotted on the array.
Figure 2.

Comparison of gene expression across region and cell phenotype. A, Percentage of clones that are expressed exclusively in either pyramidal or granule neurons when to compared with total expression in that cell. B, Overall total expression across the array of both CA3 and dentate increased in active caspase 3(+)/TUNEL(–) neurons and then decreased to below uninjured levels in activated caspase 3(+)/TUNEL(+) neurons.
Total expression across both regions also varied with activation of caspase 3 or TUNEL positivity after brain injury. Uninjured neurons from both the CA3 and dentate expressed a total of 59% of the mRNAs on the array represented (Fig. 2B). After injury and activation of caspase 3, the total mRNA expression across both regions increased to 88% of the array. Conversely, in cells that were double labeled with TUNEL, the total mRNA expression decreased to 43% of the array.
Differential mRNA expression analysis that required comparisons of intensities across arrays necessitated normalization to eliminate biases in the overall fluorescence of the dyes encountered among dye-swap replicates (Fig. 3A). The biases are best visualized when plotting the differential expression (log ratio, M) for each clone on the array by the overall fluorescence (spot intensity, A). Lowess (robust locally linear fits) analysis demonstrates a trend line (Fig. 3B, gray line) that is skewed in one direction for one non-normalized array and in the opposite direction in the dye-swap replicate. This bias appeared to be dependent on the overall fluorescence of the spot, in which the lower intensities had a greater amount of skewing than the higher intensities. Subsequent to the normalization, the resulting log ratio versus the overall spot intensity graphs produced a trend line that was not predominantly skewed in the direction of one dye (Fig. 3B, normalized).
The lists of genes differentially expressed in all three cell types from both regions were ranked based on the absolute magnitude of differential expression. To focus the analysis on those clones shown to be maximally upregulated or downregulated in the injured brain, only the clones differentially expressed by >2 SDs above the mean intensity ratios across all microarrays, or a differential expression of 1.5, are reported in Table 2 (a ratio of 1.5 was used instead of 1.56 to include also those genes close to the cutoff point). The sequence of each spotted EST differentially expressed >1.5-fold was searched for similarity among all the available nucleotide sequence databases, and the resulting genes are listed in Table 2 alongside their respective quantity of differential expression. mRNAs differentially expressed in unlabeled neurons from injured brains when compared with those in sham brains were assumed not to be involved in the apoptotic process and were removed from additional analyses. The data set generated when comparing injured nonlabeled versus sham nonlabeled (data not shown) was subtracted from the apoptotic versus sham nonlabeled data set, therefore removing all injury-related changes in gene expression and leaving those specific to the apoptotic cascade. In the CA3 region, the number of mRNAs upregulated >50% in an active caspase 3(+)/TUNEL(+) neuron was 17, and this number was 13 in active caspase 3(+)/TUNEL(–) neurons when compared to an unlabeled cell from sham mice. The downregulated mRNAs numbered 15 in caspase-3(+)/TUNEL(+) neurons and 7 in caspase 3(+)/TUNEL(–) neurons. In the dentate gyrus, the number of mRNAs upregulated and downregulated >50% in an active caspase 3(+)/TUNEL(+) neuron was 11 and 15, respectively, and 5 and 24, respectively, in an active caspase 3(+)/TUNEL(–) neuron when compared to an unlabeled cell from sham mice. None of the differentially expressed mRNAs were common to either cell type or region analyzed (Table 2).
Table 2.
Differential expression derived from the microarrays
|
|
BLAST |
|
CA3 subfield |
|
Dentate gyrus |
|
|---|---|---|---|---|---|---|
| Clone accession number |
Gene name |
Accession number |
aC3+/T+ |
aC3+/T- |
aC3+/T+ |
aC3+/T- |
| AA086732 | Nuclear inhibitor of protein phosphatase-1 | AL020997 | 2.70 | |||
| W91158 | p53-regulated PA26 nuclear protein | NM_014454 | 2.45 | |||
| AA161816 | Apoptosis inhibitory protein 5 | XM_123850 | 2.21 | |||
| W08616 | EST | AF024647 | 2.17 | |||
| W76728 | EST | XM_124397 | 1.76 | |||
| AI893666 | Shroom | XM_124564 | 1.75 | |||
| AA217294 | EST | NM_058962 | 1.67 | |||
| AI385776 | Calpain 6 | XM_136093 | 1.66 | |||
| AA270270 | Estrogen-related receptor a pseudogene | AL359457 | 1.62 | |||
| AI591874 | EST | AF029875 | 1.61 | |||
| W08432 | Brain protein 44-like protein | AF181116 | 1.59 | |||
| AA397275 | F-box and leucine-rich repeat protein 10 | AF176524 | 1.57 | |||
| AA396001 | EST | AF456412 | 1.56 | |||
| AA543916 | Unc-51 serine/threonin kinase | AF072370 | 1.55 | |||
| AI450438 | EST | AL591074 | 1.54 | |||
| AI595497 | Adult male hippocampus cDNA | AK013461 | 1.54 | |||
| AA276492 | S-adenosylmethionine decarboxylase | AB025024 | 1.50 | |||
| AA241132 | Coatomer protein complex, subunit g1 | NM_017477 | -1.91 | |||
| W13905 | Fibrinogen/angiopoietin-related protein | BC006611 | -1.80 | |||
| AA177260 | Progestin-induced protein | NM_015902 | -1.75 | |||
| AA009187 | 60S acidic ribosomal protein | AF173378 | -1.72 | |||
| AA200282 | Tristetraproline | M57422 | -1.71 | |||
| AI386396 | Vacuolar protein sorting protein 4a | NM_126165 | -1.67 | |||
| AA240082 | Neuronal development-associated protein 7 | AF361435 | -1.64 | |||
| AA207759 | Cleavage stimulation factor | NM 001324 | -1.63 | |||
| W53913 | Transcription factor GATA-4 | AB075549 | -1.60 | |||
| AA097901 | Asparaginase like | AF329099 | -1.60 | |||
| AA272747 | Nuclear factor, erythroid-derived 2 | XM_128255 | -1.55 | |||
| AA499756 | EST | XM_003245 | -1.54 | |||
| W59380 | EST | U80078 | -1.53 | |||
| AA024100 | RaIBP1-associated Eps domain-containing protein | NM_009048 | -1.52 | |||
| AA003646 | Signal recognition particle 68 kDa protein | XM_126768 | -1.51 | |||
| AI386256 | Protein kinase, DNA activated, catalytic polypeptide | XM_148184 | 4.76 | |||
| AA065728 | EST | AF179588 | 2.43 | |||
| AI466677 | Lymphocyte antigen 6 complex | XM_133803 | 2.30 | |||
| AA144535 | EST | M81448 | 2.28 | |||
| AA237920 | EST | NM_015981 | 2.17 | |||
| W40590 | EST | NM_021684 | 2.10 | |||
| W16247 | Putative homology to T-cell g receptor | XM_125725 | 1.79 | |||
| W36541 | Connective tissue growth factor precursor | XM_122007 | 1.71 | |||
| W83271 | ATP-binding cassette, subfamily F | XM_131932 | 1.68 | |||
| AA061359 | Kruppel-like factor 15 | NM_023184 | 1.65 | |||
| AA387537 | ATP-dependent helicase (RNA helicase) | XM_126585 | 1.54 | |||
| AA108457 | Cytochrome P450 monooxygenase | XM_028050 | 1.51 | |||
| AA404092 | Archain 1 | XM_134811 | 1.50 | |||
| AA237828 | Protein S a | XM_148185 | -2.94 | |||
| AA250659 | EST | XM_145607 | -2.86 | |||
| AA271147 | Downregulated by CTNNB1 | XM_124474 | -2.60 | |||
| AA058020 | ENOS interacting protein | NM_015953 | -2.11 | |||
| AA031055 | HLA-B-associated transcript 8 (G9a) | XM_128493 | -1.83 | |||
| AA270201 | Hypothetical protein LOC55565 | BC023180 | -1.73 | |||
| AA403866 | Minor histocompatibility antigen H13 isoform 1 | AF483214 | -1.71 | |||
| W99102 | Prion protein | NM_011170 | 3.26 | |||
| AA184845 | Sos homolog 2 | XM_127051 | 2.84 | |||
| AA049482 | EST | AF336797 | 1.87 | |||
| AA166543 | DUTPase | XM_140593 | 1.83 | |||
| AA259502 | EST | N/A | 1.75 | |||
| AA466235 | Inositol 1,3,4-triphosphate 5/6 kinase | XM_127102 | 1.74 | |||
| AA265234 | EST | BC028723 | 1.73 | |||
| AA266813 | Phosphorylase kinase, g2 | XM_133769 | 1.72 | |||
| AA146090 | EST | NM_133362 | 1.71 | |||
| AI508175 | Partitioning defective 3 homolog | XM_134373 | 1.68 | |||
| AA030153 | EST | BC027770 | 1.67 | |||
| W11965 | Enolase 3, b | BC013460 | -2.74 | |||
| AA017876 | EST | N/A | -2.49 | |||
| AA387095 | EST | XM_033377 | -2.45 | |||
| AA242517 | EST | NM_133491 | -2.17 | |||
| AA050726 | Paired mesoderm homeobox 1 | U03873 | -1.96 | |||
| AA003528 | EST | XP_114002 | -1.81 | |||
| AA066202 | Adducin 3g | NM_013758 | -1.71 | |||
| AA176045 | Forkhead box C2 | XM_134407 | -1.70 | |||
| AA547029 | Inhibitor of growth family, member 3 | AY007791 | -1.69 | |||
| W87183 | Small nuclear RNA activating complex, polypeptide 2 | BC011147 | -1.69 | |||
| AA000819 | Sphingosine kinase 1 | XM_126478 | -1.69 | |||
| AA212339 | Ski interacting protein | AK009218 | -1.68 | |||
| AA241295 | Dimethylarginine dimethylaminohydrolase 1 | XM_131205 | -1.67 | |||
| AA208297 | FXYD domain-containing ion transport regulator 2 | NM_052823 | -1.65 | |||
| AA215144 | Neighbor of TID protein 2 | XM_148657 | -1.63 | |||
| W14371 | EST | AL049866 | 2.96 | |||
| AA170654 | EST | AF162137 | 1.69 | |||
| AA541946 | Homolog to DJ453C12.4 (novel protein) | AK005249 | 1.68 | |||
| W14332 | 6-Pyruvoyl-tetrahydropterin synthase | NM_025273 | 1.64 | |||
| AA415819 | Hypothetical protein FLJ13386 | XM_135158 | 1.57 | |||
| AA030609 | Similar to TLM oncogene | AK009671 | -4.47 | |||
| AA060240 | Serine/threonine kinase 39 | NM_016866 | -4.47 | |||
| AA048619 | PES1 protein | AF289539 | -4.01 | |||
| AA274881 | Similar to KIAA1458 protein | XM_132055 | -3.04 | |||
| AA386657 | EST | AF294729 | -2.53 | |||
| AA162920 | Fasting-inducible integral membrane protein | NM_139107 | -2.49 | |||
| AA268977 | Similar to SEC14-like 2 | XM_126025 | -2.42 | |||
| AA058112 | EST | U16028 | -2.36 | |||
| AI552475 | Phospholipase A2-1b gene | AF094611 | -2.33 | |||
| AA059521 | EST | D26607 | -2.31 | |||
| AA125385 | Suppressor of cytokine signaling 3 | AF314501 | -2.25 | |||
| AA266234 | Claudin 7 | BC008104 | -2.14 | |||
| AA015086 | Similar to RNA binding protein | AK008848 | -2.11 | |||
| AA467585 | Homolog to PBK1 protein | AK012937 | -2.07 | |||
| AA016422 | Cerebellin 1 precursor protein | NM_019626 | -2.05 | |||
| AA423248 | EST | BC008069 | -2.01 | |||
| W91197 | EST | XM_085167 | -1.96 | |||
| AA117414 | Hypothetical protein KIAA0759 | XM_132231 | -1.89 | |||
| AA404083 | EST | NM_014915 | -1.65 | |||
| AA437983 | Translocated promoter region protein | AF490392 | -1.64 | |||
| AA098361 | EST | AF143315 | -1.61 | |||
| AA164038 | Axotrophin | NM_020575 | -1.56 | |||
| AA277646 | EST | N/A | -1.54 | |||
| AA118626 |
Hypothetical protein HSPC195 |
XM_087785
|
|
|
|
-1.54 |
Listed are the differential expression ratios of ESTs upregulated and downregulated > 1.5-fold in active caspase 3(+)/TUNEL(-) and active caspase 3(+)/TUNEL(+) neurons from both the pyramidal and granule cell layers when compared with uninjured nonlabeled cells from the identical region. The GenBank accession numbers are listed for the ESTs spotted on the array, in addition to the resulting gene names and accession numbers from the corresponding Basic Local Alignment Search Tool (BLAST) search (as of December 2002).
To assure precision when assessing levels of differential expression across experiments, the array duplicates were normalized to balance the fluorescent intensities of the dyes. A print-tip lowess normalization procedure in which the normalizing quotient is not fixed, but rather varies with the intensity of the spots surrounding it, was used to account for the intensity-dependent dye biases seen on the log ratio (M) versus overall spot intensity (A) plots (Fig. 3B). To focus only on genes involved in the apoptotic process, all the ESTs differentially regulated in a nonlabeled cell from the injured brain (when compared with a nonlabeled cell from the uninjured brain) were excluded.
Real-time PCR validation
To validate the expression changes, 24 mRNAs were selected randomly from the ESTs differentially regulated >1.5-fold (Table 2) in either direction between injured activated caspase 3(+)/TUNEL(+) cells and nonlabeled cells from uninjured brains across both regions (Fig. 4). Of the 24 mRNAs selected randomly for real-time PCR validation, 3 did not amplify sufficiently to yield results. However, 15 or 71% were differentially expressed in the same direction as determined using the array (Table 3). This percentage of PCR confirmation of microarray data are similar to that seen in other studies (Johnston et al., 2004).
Table 3.
Real-time PCR validation of the microarray changes
|
Region |
Microarray |
Real-time PCR |
Accession number |
Gene name (BLAST) |
|---|---|---|---|---|
| ▴ | ▴ | AA161816 | Apoptosis inhibitory protein 5 | |
| ▴ | ▴ | AI893666 | Shroom | |
| ▴ | ▾ | W08432 | Brain protein 44-like protein | |
| ▴ | ▾ | AA543916 | Unc-51 serine/threonin kinase | |
| ▾ | ▾ | AA241132 | Coatomer protein complex, subunit g1 | |
| ▾ | ▴ | W13905 | Fibrinogen/angiopoietin-related protein | |
| ▾ | ▴ | AA200282 | Tristetraproline | |
| ▾ | ▾ | AA207759 | Cleavage stimulation factor | |
| ▾ | ▾ | AA097901 | Asparaginase like | |
| ▾ | ▾ | AA272747 | Nuclear factor, erythroid-derived 2 | |
| CA3 | ▾ | ▾ | W59380 | EST |
| ▴ | ▴ | W99102 | Prion protein | |
| ▴ | ▴ | AA184845 | Sos homolog 2 | |
| ▴ | ▾ | AA146090 | EST | |
| ▾ | ▾ | W11965 | Enolase 3, b | |
| ▾ | ▴ | AA050726 | Paired mesoderm homeobox 1 | |
| ▾ | ▾ | AA066202 | Adducin 3g | |
| ▾ | ▾ | AA176045 | Forkhead box C2 | |
| ▾ | ▾ | AA000819 | Sphingosine kinase 1 | |
| ▾ | ▾ | AA208297 | FXYD domain-containing ion transport regulator 2 | |
| Dentate |
▾ |
▾ |
AA215144 |
Neighbor of TID protein 2 |
Clones were selected randomly from those maximally differentially expressed between active caspase 3(+)/TUNEL(+) and a nonlabeled neuron in an uninjured brain from both the CA3 subfield and dentate. The columns represent the direction of differential expression in either microarrays or real-time PCR, resulting in an agreement of 71%. The GenBank accession numbers are listed for the ESTs spotted on the array, in addition to the resulting gene names from the corresponding Basic Local Alignment Search Tool (BLAST) search (as of December 2002).
Prion and Sos immunohistochemistry
To analyze the protein correlates of a subset of the differential gene expression we observed in this study, we performed immunohistochemistry for two of the most highly differentially expressed genes in the dentate gyrus, the cellular isoform of prion protein and the Sos2 protein (Fig. 5). Immunohistochemical labeling for Sos2 identified intense protein expression in the subgranular layer of the dentate gyrus of the hippocampus, which consistently appeared greater at 24 hr after injury in the ipsilateral (injured) hemisphere. Whereas many cells in the cortex also stained positive for Sos2, there did not seem to be any overt difference between the ipsilateral (injured) and contralateral (uninjured) hemispheres. Immunohistochemistry for PrPc identified a substantial increase in PrPc immunopositivity in the ipsilateral hemisphere when compared with a near absence of PrPc-positive cells surrounding the subgranular layer of the dentate gyrus in the contralateral hemisphere.
Figure 5.
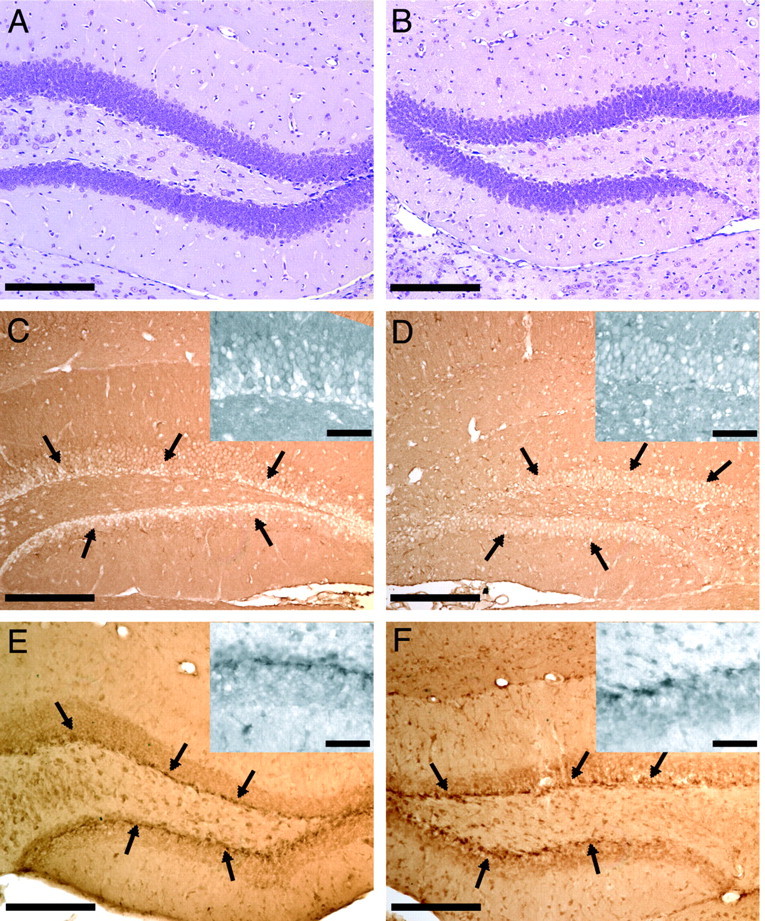
Immunohistochemical analysis of the two most highly differentially expressed genes in the dentate gyrus, the cellular isoform of prion protein (PrPc) and the Sos2 protein. A, B, Nissl-stained sections of the dentate gyrus from an injured brain to illustrate cellular architecture, both contralateral and ipsilateral to the site of fluid percussion injury, respectively. C, A clear absence of PrPc-immunopositive cells is observed surrounding the subgranular layer of the dentate gyrus of the hippocampus contralateral to the site of injury, whereas such laminar organization (best illustrated by higher magnification insets) is not seen in the ipsilateral dentate gyrus, and many more cells immunolabel positively for PrPc (D). E, F, Sos2-immunopositive cells were found bilaterally throughout the subgranular layer of the dentate gyrus after TBI, although immunolabeling is consistently more intense in the subgranular layer ipsilateral to the site of injury (F). Scale bars: larger images, 200 μm; inset photomicrographs, 50 μm.
Discussion
The CCI model of TBI was developed to control for the biomechanical events contributing to posttraumatic cell death and has repeatedly been shown to produce neuronal loss in the CA3 hippocampal subfield and dentate gyrus with relative sparing of the CA1 hippocampal subfield located directly below the impact site (Smith et al., 1995; Hicks et al., 1996; Baldwin et al., 1997; Fox et al., 1998). Our double-labeling experiments corroborate recent investigations (Yakovlev et al., 1997; Beer et al., 2000b) by providing a direct correlation between neuronal loss in both regions and the initiation of apoptosis.
Histopathological analysis revealed three different populations of hippocampal cells at 24 hr after injury: active caspase 3(+)/TUNEL(+), active caspase 3(+)/TUNEL(–), and nonlabeled cells. The minimal activation of caspase 3 observed in the uninjured brain and lack of double staining demonstrates that the initiation of this apoptotic cascade is specific to TBI. The nonlabeled pyramidal and granule neurons from the injured brain were morphologically indistinguishable from those found in the uninjured brain. The remaining neurons all exhibited immunoreactivity to the activated caspase 3 antibody, signifying that they were undergoing apoptosis. In a subset of these neurons, the activation of caspase 3 colocalized with the breakdown of nuclear DNA. This finding is consistent with previous reports that caspase 3 is responsible, in part, for the proteolysis of a large number of substrates, such as lamins, actin, and α-fodrin, that result in disassembly of both the nuclear envelope and cortical cytoskeleton (Lazebnik et al., 1995; Cryns et al., 1996; S. B. Brown et al., 1997; Song et al., 1997). The localization of the active caspase 3 in the nucleus of the neurons from the injured brain further supports the conclusion that caspase 3-mediated cleavage leads to activation of specific DNAses such as DFF40 (Inohara et al., 1999). A significant portion of the neurons from both the CA3 and dentate that were immunoreactive for activated caspase 3 did not concurrently label with TUNEL. These neurons have entered the apoptotic cascade but have yet to break down their nuclear membrane and begin the condensation of their chromatin into the classic apoptotic bodies. This indicates that, at any one time after TBI, there exists a population of nonapoptotic neurons, a population of neurons entering the apoptotic continuum [activated caspase 3(+)/TUNEL(–)], and a third population of neurons well past the commitment step in apoptosis [activated caspase 3(+)/TUNEL(+)].
Previous array expression studies in TBI models have demonstrated that the brain mounts a specific and concerted genomic response to injury, specifically upregulating certain genes and suppressing others (Morrison et al., 2000; O'Dell et al., 2000; Matzilevich et al., 2002; Raghavendra et al., 2003). Because the brain is a complex multicellular organ composed of a variety of cell types that exhibit potentially divergent genomic responses to injury, analyses of whole regions or subregions of the brain will confound the specific contributions from individual neurons. Therefore, the ability to study the mRNA expression profiles of isolated neurons can provide potentially extraordinarily powerful insight in to the pathophysiology of TBI. Further highlighting the need for genomic analysis at the single-cell level is recent evidence by O'Dell et al. (2000), who demonstrate that two phenotypically identical TUNEL(+) cortical neurons have different molecular profiles.
Expression analysis of apoptotic and nonapoptotic single cells creates a comprehensive picture of gene regulation in the hippocampus after TBI. Because it was not possible to analyze every gene in the mouse genome, microarrays selected in this analysis were populated with ESTs selected randomly from the entire mouse genome. This random selection of ESTs allows for the assumption that the expression analysis is indicative of the total genomic expression a neuron without the need to analyze specific genes. Uninjured neurons from both the CA3 subfield and dentate transcribed approximately two-thirds (59%) of the mRNAs represented on the array, indicating that homeostatic or routine metabolic processes as well as cell-specific activities require the transcription of over half the mouse genome. Moreover, the amount of overlap in expression between the two regions was greater than two-thirds, indicating that these processes are genotypically conserved between pyramidal and granule neurons. The activation of caspase 3 in individual injured neurons across both regions increased overall expression to 88%, potentially representing the activation of genes involved in initiating or mediating myriad cascades activated after brain injury. These injury-activated molecular cascades represent the activation of immediate early genes such as c-fos and c-jun (Raghupathi et al., 1995), cytokines such as interleukin-1β and TNFα (Fan et al., 1996; Raghupathi et al., 1998), and neurotropins such as NGF and BDNF (DeKosky et al., 1994; Oyesiku et al., 1999). As these neurons progressed to TUNEL positivity, the overall mRNA abundances declined dramatically. The active caspase 3(+)/TUNEL(+) neurons across both regions expressed the lowest percentage of mRNAs corresponding to ESTs on the arrays when compared with the other two phenotypic states. This decrease in overall expression may represent the attempt of the cell to normalize gene expression after a dramatic insult or may be attributable to DNA damage resulting from the entrance of the cell into the apoptotic cascade. These decreases in mRNA abundances, similarly, are consistent with the histological appearance of the TUNEL positivity and degradation of genomic DNA. Moreover, of the mRNAs expressed in each region, the percentage unique to pyramidal neurons increased to 50%, whereas those unique to granule neurons decreased to 7%, indicating that at this stage of cell death, there are predominantly divergent molecular cascades occurring in each region.
The majority of the changes observed in gene expression likely represent either the molecular pathways involved in initiating apoptosis or those activated by injury but not involved in programmed cell death. Although the resulting differential expression included a significant number of currently unknown ESTs, it is interesting to note that none of the putatively proapoptotic genes, such as the caspases (caspases 1, 2, 8, and 11), were upregulated in either region. Conversely, putative neuroprotective genes (or homologous ESTs) were not observed to be downregulated in these same individual dying cells. For instance, PDGF, TGF, IGF, FGF, epidermal growth factor, ciliary neurotrophic factor, and neurotrophin 3 (NT-3/4) were not differentially regulated in apoptotic cells after brain injury.
After injury to the CNS, genes commonly associated with development are often upregulated as part of response of the cells to the impact stimulus and corresponding secondary neurodegenerative pathology. Members of the son of sevenless (SOS) family of guanine nucleotide exchange factors are widely expressed during development and in adult tissues and are known to activate Ras in response to growth factor stimulation (Nimnual and Bar-Sagi, 2002). Despite ∼65% homology between Sos1 and Sos2 and the embryonic lethality of Sos1 knock-out mice, one recent study showed Sos2 to be dispensable for normal mouse development (Esteban et al., 2000). However, little else is known about Sos2. The intense immunopositivity we observed in the subgranular layer of the dentate gyrus of the hippocampus at 24 hr after injury (Fig. 5E,F) corresponds well with the injury-induced increase in Sos2 gene expression and warrants additional investigation into the function of Sos2 in this region and after brain injury.
Prion protein is most well-recognized for its infectious scrapie isoform (PrPsc). However, recent data has begun to elucidate a neuroprotective role of the ubiquitously expressed and highly conserved cellular isoform of the prion protein (PrPc). Current evidence suggests that the mechanism underlying the neuroprotective properties of PrPc involves a copper-binding activity that protects cells from oxidative damage (Brown et al., 1997a, 2002). In support of this hypothesis, cerebellar cell cultures from PrPc null mice are more susceptible to oxidative stress than wild-type mice (Brown et al., 1997b), and PrPc null mice are more susceptible to acute seizures and kindling induced by different protocols than their wild-type counterparts (Walz et al., 1999). Such a neuroprotective function may indicate a possible reason why we observed PrPc to be highly upregulated in the dentate gyrus after TBI in this study. However, another study recently demonstrated that the accumulation of even small amounts of PrPc in the cytoplasm is strongly neurotoxic in both cultured cells and transgenic mice (Ma et al., 2002; Smith et al., 2002). This indicates that if the normally neuroprotective membrane protein is mistransported or if PrPc-degrading proteases are either disrupted or unable to sufficiently digest overexpressed PrPc, cell death may ensue. Therefore, the increase in PrPc-immunopositive cells we observed in the dentate gyrus of the hippocampus (Fig. 5C,D), one of two selectively vulnerable hippocampal regions after TBI, may have important consequences for posttraumatic cellular vulnerability.
Our data also demonstrates the heterogeneity of the posttraumatic apoptotic cascade at the molecular level, because immunohistochemically identical apoptotic neurons differentially expressed dissimilar genes in response to injury. Although some overlap was observed in genes expressed between apoptotic pyramidal and granule neurons, those genes that were maximally differentially regulated were dramatically different. Regardless of the region or of the direction of expression, no single EST was differentially expressed in more than one physiological state comparison. We conclude that the cell type-specific expression of specific genes correlated with the activation and progression of cell death with that neuron type. Furthermore, the regulation of these maximally differentially regulated genes seems to be dependent on the physiological state of the cell, because the differential expression profiles in the putative early apoptotic active caspase 3(+)/TUNEL(–) neurons from either region did not overlap with profiles from apoptotic cells further along the cell death pathway [active caspase 3(+)/TUNEL(+)].
This study demonstrates immunohistochemical evidence that apoptotic cell death is occurring in the hippocampal formation after TBI in the mouse. Subsequent microarray analysis of neurons progressing through the activation of caspase 3 to TUNEL positivity reveals not only a decrease in overall expression but also an increase in the specificity of the genomic response culminating in regionally specific maximally differentially expressed mRNAs. Apoptotic neurons from the CA3 hippocampal subfield maximally regulated different molecular cascades in response to the initiation of cell death than neurons selected from dentate gyrus. Although proteins are usually the effectors of cellular processes, an expression profile highlights the biochemical potential of the examined cells. Our data demonstrate the diversity of the apoptotic process at the molecular level and suggests that mRNA expression in individual neurons underlies the physiological consequences of injury.
Footnotes
This work was supported in part by Biomedical Research partnership Grant R01-41699 (T.K.M. and J.H.E.) from the National Institute of Child Health and Human Development, National Institutes of Health (NIH)–National Institute of Neurological Disorders and Stroke Grants P50-NS08803 (T.K.M.) and RO1-NS40978 (T.K.M.), National Institute on Aging Grant AG 9900 (J.H.E.), National Institute of General Medical Sciences Grant RO1-GM34790 (T.K.M.), a Merit Review grant from the Veterans Administration (T.K.M.), and NIH Grant HG02296 (E.M.). We thank K. A. Roth for the positive control sections for the activated caspase 3 staining. We thank the Incyte Microarray facilities for array services and Merck-Frosst for the active caspase 3 antibody (MF397).
Correspondence should be addressed to Dr. James H. Eberwine, Department of Pharmacology, 37B John Morgan, 3620 Hamilton Walk, Philadelphia, PA 19104-6084. E-mail: eberwine@mail.med.upenn.edu.
DOI:10.1523/JNEUROSCI.5051-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242866-11$15.00/0
J.H.E. and T.K.M. are senior authors who contributed equally to this work.
References
- Adams R, Bischof L (1994) Seeded region growing. IEEE Trans Pattern Anal Machine Intelligence 16: 641–647. [Google Scholar]
- Alnemri ES (1997) Mammalian cell death proteases: a family of highly conserved aspartate specific cysteine proteases. J Cell Biochem 64: 33–42. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM (1998) Death receptors: signaling and modulation. Science 281: 1305–1308. [DOI] [PubMed] [Google Scholar]
- Baldwin SA, Gibson T, Callihan CT, Sullivan PG, Palmer E, Scheff SW (1997) Neuronal cell loss in the CA3 subfield of the hippocampus following cortical contusion utilizing the optical disector method for cell counting. J Neurotrauma 14: 385–398. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Schopf M, Reindl M, Zelger B, Schmutzhard E, Poewe W, Kampfl A (2000a) Expression of Fas and Fas ligand after experimental traumatic brain injury in the rat. J Cereb Blood Flow Metab 20: 669–677. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Srinivasan A, Hayes RL, Pike BR, Newcomb JK, Zhao X, Schmutzhard E, Poewe W, Kampfl A (2000b) Temporal profile and cell subtype distribution of activated caspase-3 following experimental traumatic brain injury. J Neurochem 75: 1264–1273. [DOI] [PubMed] [Google Scholar]
- Beer R, Franz G, Krajewski S, Pike BR, Hayes RL, Reed JC, Wang KK, Klimmer C, Schmutzhard E, Poewe W, Kampfl A (2001) Temporal and spatial profile of caspase 8 expression and proteolysis after experimental traumatic brain injury. J Neurochem 78: 862–873. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD, Green EJ, Busto R (1997) Chronic histopathological consequences of fluid-percussion brain injury in rats: effects of post-traumatic hypothermia. Acta Neuropathol 93: 190–199. [DOI] [PubMed] [Google Scholar]
- Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H (1997a) The cellular prion protein binds copper in vivo. Nature 390: 684–687. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA (1997b) Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Exp Neurol 146: 104–112. [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L (2002) Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J Neurosci Res 67: 211–224. [DOI] [PubMed] [Google Scholar]
- Brown SB, Bailey K, Savill J (1997) Actin is cleaved during constitutive apoptosis. Biochem J 323: 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP (1996) Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 2: 788–794. [DOI] [PubMed] [Google Scholar]
- Buckley MJ (2000) The Spot user's guide. CSIRO Mathematical and Information Sciences.
- Clark RS, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH (1997) Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci 17: 9172–9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Watkins SC, Chen M, Dixon CE, Seidberg NA, Melick J, Loeffert JE, Nathaniel PD, Jin KL, Graham SH (2000) Caspase-3 mediated neuronal death after traumatic brain injury in rats. J Neurochem 74: 740–753. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dash PK (1996) Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res 739: 120–131. [DOI] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK (1998) Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci 18: 5663–5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino PB, Trojanowski JQ, Dichter MA, Eberwine J (1996) Embryonic neuronal markers in tuberous sclerosis: single-cell molecular pathology. Proc Natl Acad Sci USA 93: 14152–14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryns VL, Bergeron L, Zhu H, Li H, Yuan J (1996) Specific cleavage of alpha-fodrin during Fas- and tumor necrosis factor-induced apoptosis is mediated by an interleukin-1beta-converting enzyme/Ced-3 protease distinct from the poly(ADP-ribose) polymerase protease. J Biol Chem 271: 31277–31282. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Goss JR, Miller PD, Styren SD, Kochanek PM, Marion D (1994) Upregulation of nerve growth factor following cortical trauma. Exp Neurol 130: 173–177. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Clifton GL, Lighthall JW, Yaghmai AA, Hayes RL (1991) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39: 253–262. [DOI] [PubMed] [Google Scholar]
- Dudoit S, Yang YH, Callow MJ, Speed TP (2002) Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Stat Sinica 12: 111–139. [Google Scholar]
- Eberwine J, Spencer C, Miyashiro K, Mackler S, Finnell R (1992a) Complementary DNA synthesis in situ: methods and applications. Methods Enzymol 216: 80–100. [DOI] [PubMed] [Google Scholar]
- Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P (1992b) Analysis of gene expression in single live neurons. Proc Natl Acad Sci USA 89: 3010–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldadah BA, Faden AI (2000) Caspase pathways, neuronal apoptosis, and CNS injury. J Neurotrauma 17: 811–829. [DOI] [PubMed] [Google Scholar]
- Esteban LM, Fernandez-Medarde A, Lopez E, Yienger K, Guerrero C, Ward JM, Tessarollo L, Santos E (2000) Ras-guanine nucleotide exchange factor sos2 is dispensable for mouse growth and development. Mol Cell Biol 20: 6410–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Young PR, Barone FC, Feuerstein GZ, Smith DH, McIntosh TK (1996) Experimental brain injury induces differential expression of tumor necrosis factor-alpha mRNA in the CNS. Brain Res Mol Brain Res 36: 287–291. [DOI] [PubMed] [Google Scholar]
- Fox GB, Fan L, LeVasseur RA, Faden AI (1998) Sustained sensory/motor and cognitive deficits with neuronal apoptosis following controlled cortical impact brain injury in the mouse. J Neurotrauma 15: 599–614. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW (1999) Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 97: 395–406. [DOI] [PubMed] [Google Scholar]
- Hicks R, Soares H, Smith D, McIntosh T (1996) Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol 91: 236–246. [DOI] [PubMed] [Google Scholar]
- Inohara N, Koseki T, Chen S, Benedict MA, Nunez G (1999) Identification of regulatory and catalytic domains in the apoptosis nuclease DFF40/CAD. J Biol Chem 274: 270–274. [DOI] [PubMed] [Google Scholar]
- Johnston R, Nutall R, Doctolero M, Edwards P, Lü J, Wang B, Vainer M, Yue H, Wang X, Minor J, Lash A, Chan C, Goralski T, Parisi M, Oliver B, Eastman S (2004) FlyGEM, a full transcriptome array platform for the Drosophila community. Genome Res, in press. [DOI] [PMC free article] [PubMed]
- Lazebnik YA, Takahashi A, Moir RD, Goldman RD, Poirier GG, Kaufmann SH, Earnshaw WC (1995) Studies of the lamin proteinase reveal multiple parallel biochemical pathways during apoptotic execution. Proc Natl Acad Sci USA 92: 9042–9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S (2002) Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science 298: 1781–1785. [DOI] [PubMed] [Google Scholar]
- Matzilevich DA, Rall JM, Moore AN, Grill RJ, Dash PK (2002) High-density microarray analysis of hippocampal gene expression following experimental brain injury. J Neurosci Res 67: 646–663. [DOI] [PubMed] [Google Scholar]
- Morrison III B, Eberwine JH, Meaney DF, McIntosh TK (2000) Traumatic injury induces differential expression of cell death genes in organotypic brain slice cultures determined by complementary DNA array hybridization. Neuroscience 96: 131–139. [DOI] [PubMed] [Google Scholar]
- Nimnual A, Bar-Sagi D (2002) The two hats of SOS. Science STKE 145: PE36. [DOI] [PubMed] [Google Scholar]
- O'Dell DM, Raghupathi R, Crino PB, Eberwine JH, McIntosh TK (2000) Traumatic brain injury alters the molecular fingerprint of TUNEL-positive cortical neurons in vivo: a single-cell analysis. J Neurosci 20: 4821–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olton DS, Papas BC (1979) Spatial memory and hippocampal function. Neuropsychologia 17: 669–682. [DOI] [PubMed] [Google Scholar]
- Oyesiku NM, Evans CO, Houston S, Darrell RS, Smith JS, Fulop ZL, Dixon CE, Stein DG (1999) Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res 833: 161–172. [DOI] [PubMed] [Google Scholar]
- Phillips J, Eberwine JH (1996) Antisense RNA amplification: a linear amplification method for analyzing the mRNA population from single living cells. Methods 10: 283–288. [DOI] [PubMed] [Google Scholar]
- Raghavendra RV, Dhodda VK, Song G, Bowen KK, Dempsey RJ (2003) Traumatic brain injury-induced acute gene expression changes in rat cerebral cortex identified by GeneChip analysis. J Neurosci Res 71: 208–219. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, McIntosh TK, Smith DH (1995) Cellular responses to experimental brain injury. Brain Pathol 5: 437–442. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Fernandez SC, Murai H, Trusko SP, Scott RW, Nishioka WK, McIntosh TK (1998) BCL-2 overexpression attenuates cortical cell loss after traumatic brain injury in transgenic mice. J Cereb Blood Flow Metab 18: 1259–1269. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Graham DI, McIntosh TK (2000) Apoptosis after traumatic brain injury. J Neurotrauma 17: 927–938. [DOI] [PubMed] [Google Scholar]
- Raghupathi R, Strauss KI, Krajewski S, Reed JC, McIntosh TK (2003) Temporal alteration in cellular Bax: Bcl-2 ratio following traumatic brain injury in the rat. J Neurotrauma 20: 421–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, Pons PT (1995) Epidemiology of trauma deaths: a reassessment. J Trauma 38: 185–193. [DOI] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK (1995) A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12: 169–178. [DOI] [PubMed] [Google Scholar]
- Smith DH, Iwata A, Meaney DF, Chen X (2002) Long-term prion protein accumulation in damaged axons following inertial brain injury in the pig. Paper presented at 20th Annual Meeting of the Society for Neurotrauma, Tampa, FL, October 27 to November 1.
- Soille P (1999) Morphological image analysis: principles and applications. New York: Springer.
- Song Q, Wei T, Lees-Miller S, Alnemri E, Watters D, Lavin MF (1997) Resistance of actin to cleavage during apoptosis. Proc Natl Acad Sci USA 94: 157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosin DM, Sacks JJ, Smith SM (1989) Head injury-associated deaths in the United States from 1979 to 1986. JAMA 262: 2251–2255. [PubMed] [Google Scholar]
- Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR (1999) Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 40: 1679–1682. [DOI] [PubMed] [Google Scholar]
- Williams S, Raghupathi R, MacKinnon MA, McIntosh TK, Saatman KE, Graham DI (2001) In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol 102: 581–590. [DOI] [PubMed] [Google Scholar]
- Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI (1997) Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci 17: 7415–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Buckley MJ, Dudoit S, Speed TP (2002a) Comparison of methods for image analysis on cDNA microarray data. J Comp Graphic Stat 11: 1–29. [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002b) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30: E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Speed TP (2002c) Normalization for cDNA microarray data. In: Microarrays: optical technologies and informatics (Bittner M, Chen Y, Dorsel A, eds), pp 141–152. Bellingham, WA: International Society for Optical Engineering.
- Yue H, Eastman PS, Wang BB, Minor J, Doctolero MH, Nuttall RL, Stack R, Becker JW, Montgomery JR, Vainer M, Johnston R (2001) An evaluation of the performance of cDNA microarrays for detecting changes in global mRNA expression. Nucleic Acids Res 29: E41. [DOI] [PMC free article] [PubMed] [Google Scholar]