

Abstract
The nature of presynaptic calcium (Ca2+) signals that initiate neurotransmitter release makes these signals difficult to study, in part because of the small size of specialized active zones within most nerve terminals. Using the frog motor nerve terminal, which contains especially large active zones, we show that increases in intracellular Ca2+ concentration within 1 msec of action potential invasion are attributable to Ca2+ entry through N-type Ca2+ channels and are not uniformly distributed throughout active zone regions. Furthermore, changes in the location and magnitude of Ca2+ signals recorded before and after experimental manipulations (ω-conotoxin GVIA, diaminopyridine, and lowered extracellular Ca2+) support the hypothesis that there is a remarkably low probability of a single Ca2+ channel opening within an active zone after an action potential. The trial-to-trial variability observed in the spatial distribution of presynaptic Ca2+ entry also supports this conclusion, which differs from the conclusions of previous work in other synapses.
Keywords: neuromuscular junction, calcium, imaging, active zone, nerve terminal, presynaptic
Introduction
Fast exocytosis of chemical neurotransmitters from small synaptic vesicles is the primary basis for communication between neurons. Action potential depolarization is known to activate voltage-gated Ca2+ channels (VGCCs) in specialized “active zone” (AZ) regions of the nerve terminal, with the ensuing influx of Ca2+ ions providing a local trigger for fusion of synaptic vesicles with the plasma membrane (Llinas et al., 1995). At most synapses, and at individual active zones within the frog neuromuscular junction, transmitter release is a low-probability stochastic event (Katz, 1969). This characteristic feature of the release process is believed to be essential to normal brain function (Goda and Sudhof, 1997).
At most nerve terminals, active zones are organized into small disk-like structures <0.5 μm in diameter (Edwards, 1995). This small size, together with Ca2+ influx occurring within 1 msec after an action potential invades the nerve terminal, makes it difficult to study in detail the spatial distribution of Ca2+ entry. For these reasons, many experiments addressing Ca2+ influx during single action potentials have measured time- and volume-averaged elevations in Ca2+ integrated over entire presynaptic terminals. A relatively constant increase in intracellular Ca2+, in response to single action potentials, has been reported at synaptic sites in rat cortical neuron cultures (MacKenzie et al., 1996) and in presynaptic terminals in rat cerebellar slices (Forti et al., 2000). In rat superior collicular neurons, transmitter release was found to fluctuate widely even when trials were matched with regard to the magnitude of total Ca2+ influx, suggesting that a significant source of variability in transmitter release exists downstream of Ca2+ entry (Kirischuk et al., 1999). The spatial extent of single action potential-evoked Ca2+ transients has been a focus for study at the squid giant synapse (Llinas et al., 1992; Smith et al., 1993), lizard motor nerve terminals (David et al., 1997), and embryonic frog neuromuscular junctions in vitro (DiGregorio and Vergara, 1997; DiGregorio et al., 1999). These studies identified spatial gradients in Ca2+ influx but did so in preparations in which it was difficult to detect heterogeneity in Ca2+ influx with subactive zone resolution. Despite some evidence to the contrary (Llinas et al., 1994; Freguelli and Malinow, 1996), many believe that an action potential normally leads to a uniform flooding of active zones with Ca2+ (Sudhof and Scheller, 2001), such that release sites are triggered by overlapping domains of Ca2+ as 70–90% of available Ca2+ channels open (Borst and Sakmann, 1998; Bischofberger et al., 2002).
Unlike the synapses used for the studies described above, the motor nerve terminal of the adult frog features a long series of large active zones arranged as linear arrays (Heuser et al., 1974; Pumplin et al., 1981; Pawson et al., 1998). Each active zone is ∼1 μm in length, with the active zones spaced like railroad ties at regular 1 μm intervals along the length of the nerve terminal. Within each linear active zone there are ∼30 synaptic vesicles associated closely with a double parallel row of intramembraneous particles presumed to include voltage-gated Ca2+ channels (Pumplin et al., 1981; Robitaille et al., 1993). In this study, we took advantage of the size and structural order of active zones at the adult frog neuromuscular junction, in combination with high-speed fluorescence imaging, to examine the spatial distribution of Ca2+ influx sites within an active zone during single action potentials.
Materials and Methods
Tissue preparation. Adult frogs (Rana pipiens) were decapitated and pithed after anesthesia in 0.1% tricaine methane sulfonate solution. The cutaneous pectoris nerve-muscle preparation was removed bilaterally and bathed in normal frog Ringer's solution (NFR) (in mm: 116 NaCl, 1 NaHCO3, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.3). The Ca2+-sensitive dye Calcium Green-1 (3000 MW, dextran conjugate; Molecular Probes, Eugene, OR) was loaded through the cut end of the cutaneous pectoris nerve. The muscle nerve was drawn inside a well, made of petroleum jelly that was filled with dye solution (30 mm in dH20), and the nerve cut (Peng and Zucker, 1993; Wu and Betz, 1996; Narita et al., 1998). After 7 hr of loading at room temperature, the preparation was rinsed in NFR and stored at 4°C for 3 hr. After this loading procedure, the preparation was pinned over an elevated Sylgard (Dow Corning, Midland, MI) platform in a 35 mm dish mounted on a microscope stage. The cutaneous pectoris nerve was drawn into a suction electrode for stimulation at 5× threshold as determined by observation of muscle twitch after a single nerve stimulus. This stimulus-evoked muscle twitch was always present and confirmed functional health of the preparation after the loading procedure. The preparation was exposed to 2 μg/ml tetramethylrhodamine isothiocyanate (TRITC) α-bungarotoxin (α-BTX) for 10 min to label postsynaptic acetylcholine receptors. This labeling was used to aid in locating and focusing on postsynaptic receptor bands adjacent to the presynaptic active zones. Nerve terminals were chosen for study if they were superficially positioned on the muscle and if the majority of the terminal was in a single focal plane as judged by α-BTX staining. All Ca2+ imaging was performed in NFR with 10 μm curare added to prevent nerve-evoked muscle contractions that were not completely blocked by the TRITC α-BTX labeling procedure.
Calcium imaging and analysis. We used an acousto-optic tunable filter (AOTF) approach to control the excitation of the fluorescent dye with submillisecond time resolution (Wachman et al., 1997). This approach also allows for rapid switching of excitation and emission wavelengths. The AOTF filter system (ChromoDynamics, Lakewood, NJ) is based on a custom TeO2 crystal (NEOS Technologies, Melbourne, FL) and was controlled by an arbitrary waveform generator (LW420; LeCroy, Chesnut Ridge, NY) that was used to select wavelengths and gate the output of a krypton-argon laser (Innova 70 spectrum; Coherent, Santa Clara, CA). This light was fiber-coupled into the epi-illumination port of an upright fluorescence microscope equipped with appropriate rejection filters and a 100× water-immersion long-working distance objective with 1.0 numerical aperture (Lumplan–FL-IR; Olympus, Tokyo, Japan). Calcium Green-1 was excited with the 488 nm line of the laser, and TRITC α-BTX was excited with the 567 nm line. Timing and duration of illumination were controlled by activation of the arbitrary waveform generator with a gate pulse obtained from a pulse generator (8013A; Hewlett-Packard, Palo Alto, CA) triggered simultaneously with nerve stimulation. The laser delivered ∼400 μJ of 488 nm light to the area of interest during our brief 1 msec illumination pulses. To provide the high-sensitivity, low-noise detection necessary to measure fluorescence changes during single 1 msec exposures, images were recorded on a liquid nitrogen-cooled, back-thinned CCD camera (LN 1300B; Roper Scientific, Trenton, NJ). Using this camera, we sampled photons with an estimated spatial resolution of 0.275 μm. Using our approach, we did not detect bleaching of our dye, and raw resting photoelectron counts ranged between 3000 and 9000 per pixel within dye-loaded nerve terminals. Shot noise was the major contributor to resting fluctuations in fluorescence intensity. Fluorescence changes during nerve stimulation were clearly above resting fluctuations as shown by plots of the distribution of pixel intensities in the presence and absence of nerve stimulation (see Figs. 1 E, 3B, 4 B, 5B). The histograms in Figures 1 E, 3B, 4 B, and 5B were taken from well focused regions of the nerve terminal (white box; as determined by α-BTX staining of postsynaptic receptor clusters) and restricted to pixels that sampled nerve terminal regions of the image (determined by “masking” the image on the basis of the resting fluorescence intensity) (see Fig. 1C).
Figure 1.

Nerve-evoked Ca2+ entry during a single action potential at the frog neuromuscular junction. A, Gray-scale (top) and pseudocolor representation (bottom) of a difference image generated by subtraction of the mean resting fluorescence from the fluorescence observed in a single trial in the absence of nerve stimulation. B, Gray-scale (top) and pseudocolor representation (bottom) of a difference image generated by subtraction of the mean resting fluorescence from the fluorescence observed immediately after nerve stimulation in a single trial. Images in A and B were collected using 1 msec illumination (in B, this began 1.5 msec after nerve stimulation). The white boxes in the bottom panels of B and C indicate the area of this terminal that is shown in D and used to generate the histogram shown in E. C, The top panel shows a sample single resting fluorescence image of the nerve terminal (same illumination conditions as in A; gray-scale bar in photoelectron counts). The bottom panel shows an image mask (red) indicating pixels in the resting fluorescence image that have intensities between 50 and 100% of maximum. This mask identifies those pixels that sample light from the nerve terminal portion of the image. D, Enlarged region of the stimulus-evoked difference image (from the white box in the bottom panel of B) representing the spatial distribution of Ca2+ entry (left). The right panel shows the same region of the neuromuscular junction labeled with rhodamine-α-BTX to show the location of postsynaptic acetylcholine receptors. E, Histogram showing the distribution of pixel intensities taken from those pixels in the masked nerve terminal portion (red pixels in the bottom panel of C) in the presence and absence of nerve stimulation. A pseudocolor difference image from this same region is shown in D, but the histogram does not include data from pixels outside the nerve terminal (masked region), because these off-nerve data would dominate the distribution. The distribution shown in black represents pixel intensities measured in 10 images recorded in the absence of nerve stimulation (defined as “resting fluctuations”), the distribution shown in red represents pixel intensities measured in 10 images recorded after nerve stimulation, and the dotted blue line represents the limit of two SDs above the resting fluctuation distribution after fit to a single Gaussian. The pseudocolor scale bar is the same for the bottom panels in A and B and the left panel in D, and is expressed as ΔF/F (%). Scale bars, 2 μm.
Figure 3.

Effects of a 7 min exposure to 200 nm ω-CgTX GVIA on Ca2+ entry after nerve stimulation. A, Sample resting fluorescence image of the nerve terminal (gray-scale bar in photoelectron counts) with the well focused region of interest defined by the white box. B, Histogram showing the distribution of pixel intensities in the presence and absence of nerve stimulation as described in Figure 1 E. C, Representative difference images before (top panel) and after (bottom panel) partial blockade of N-type Ca2+ channels. Pseudocolor scale bar applies to both images and is expressed as ΔF/F (%). D, Cumulative frequency distribution of pixel intensity values measured before (solid line) and after (dashed line) partial blockade of N-type Ca2+ channels. In six terminals examined in this manner, there were no significant differences in the distribution of pixel intensities. Scale bars, 2 μm.
Figure 4.
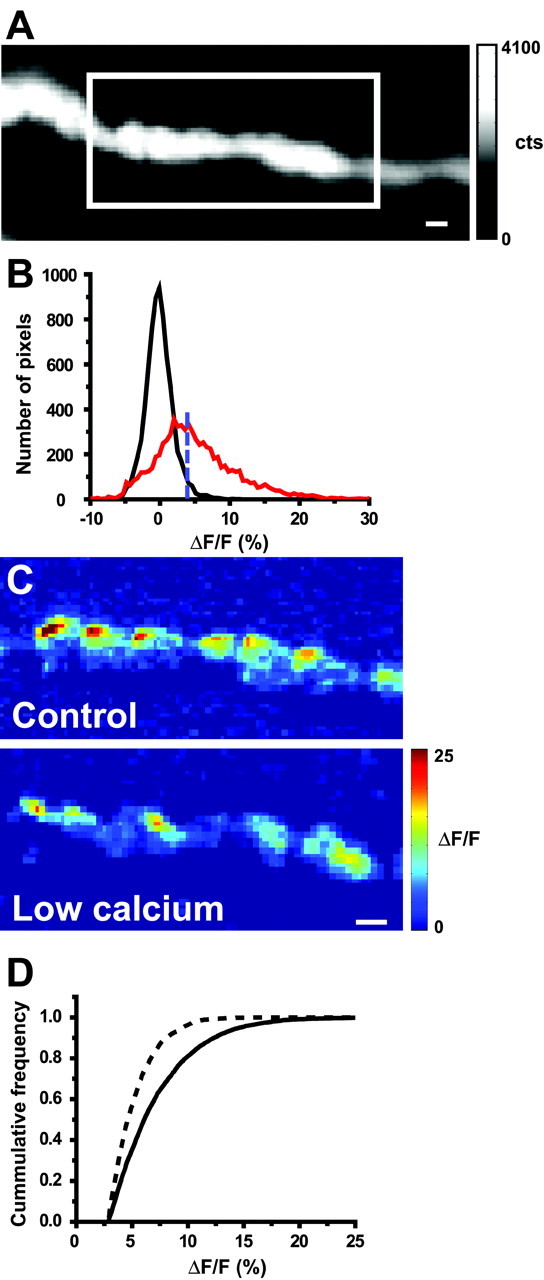
Effects of 5 min exposure to low Ca2+ saline on Ca2+ entry after nerve stimulation. A, Sample resting fluorescence image of the nerve terminal (gray-scale bar in photoelectron counts) with the well focused region of interest defined by the white box. B, Histogram showing the distribution of pixel intensities in the presence and absence of nerve stimulation as described in Figure 1 E. C, Representative difference images in normal Ca2+ (1.8 mm; top panel) and after exposure to 0.5 mm Ca2+ (bottom panel). Pseudocolor scale bar applies to both images and is expressed as ΔF/F (%). D, Cumulative frequency distribution of pixel intensity values measured before (solid line) and after (dashed line) exposure to 0.5 mm Ca2+. In three terminals examined in this manner, there was consistently a significant leftward shift (toward lower intensities) in the distribution of pixel intensities. Scale bars, 2 μm.
Figure 5.
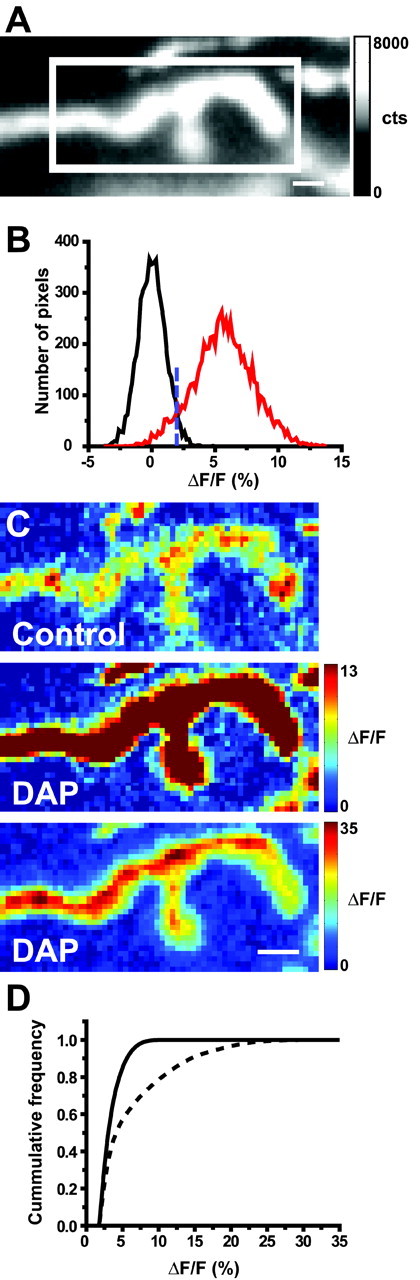
Effects of 15 min exposure to a potassium channel blocker (5 μm DAP) on Ca2+ entry after nerve stimulation. A, Sample resting fluorescence image of the nerve terminal (gray-scale bar in photoelectron counts) with the well focused region of interest defined by the white box. B, Histogram showing the distribution of pixel intensities in the presence and absence of nerve stimulation as described in Figure 1 E. C, Representative difference images before (top panel) and after (middle panel) partial blockade of potassium channels. Pseudocolor scale bar in the middle panel applies to both the top and middle panel images and is expressed as ΔF/F(%). Bottom panel is the same image as shown in the middle panel, except that the pseudocolor scale has been expanded to avoid saturation of the color scale and demonstrate that after DAP treatment, there is a more uniform increase in nerve terminal Ca2+ than was observed before treatment (top panel). D, Cumulative frequency distribution of pixel intensity values measured before (solid line) and after (dashed line) partial blockade of potassium channels. In three terminals examined in this manner, there was consistently a significant rightward shift (toward higher intensities) in the distribution of pixel intensities. Scale bars, 2 μm.
Images were collected at 0.5 Hz in sets of 20. The first 10 images were collected with the stimulator off (background), whereas the next 10 images were collected with the stimulator on (nerve-evoked signals). Images were processed using MATLab. Before analysis, images in each set of 20 were coregistered to correct for slight fluctuations in the lateral position of the preparation during the course of acquisition. Differences in fluorescence above rest were determined for individual images by subtracting mean resting fluorescence (generated by averaging the 10 background images). The resulting “difference images” were displayed using a pseudocolored representation of difference pixel intensity divided by resting fluorescence (ΔF/F; expressed as a percentage above rest).
For quantitative analysis of experimental manipulations (see Figs. 3D, 4 D, 5D), we included only intensities that were, with 95% confidence, brighter than resting fluorescence fluctuations (two SDs above the mean). The range of resting fluorescence fluctuations within the nerve terminal was determined empirically from the intensity histograms for difference images generated by subtracting the average resting signal from each of the 10 resting images (nerve stimulation off). The range over which these resting fluctuations extend is critical for quantifying the effects of the experimental manipulations, because combining data from nerve-evoked signals with contributions from resting fluctuations will skew cumulative frequency plots to the left after a fraction of the nerve-evoked signal is blocked. For the analysis of total signal intensity changes after experimental manipulations, we included only pixel intensities that were brighter than resting fluorescence fluctuations (as defined above). This technique was applied to each terminal individually. This “signal-above-rest” was also used in analysis and characterization of individual “Ca2+ entry sites.” In our images of nerve-evoked Ca2+ signals, Ca2+ entry sites were defined as pixels that detected a signal that was greater than resting fluctuations (as defined above). Because Ca2+ signals within single active zone regions were detected by more than one pixel, and it was difficult to distinguish discrete calcium entry sites when there were several in close proximity to one another, we restricted our analysis of Ca2+ entry sites to a characterization of single pixels that detected a signal above rest. To quantify these sites, we counted the number of pixels that measured a signal above rest and calculated the mean intensity of these pixels. This method allows us to compare quantitative changes after pharmacological manipulations and is an analysis approach that is expected to be proportional to true Ca2+ entry sites.
The use of signal-above-rest in our analysis, as described above, is necessary for plots of the distribution of stimulus-evoked pixel intensities after experimental manipulation and to characterize the number of Ca2+ entry sites. However, use of these thresholded images to quantify graphically spatial variability in the nerve-evoked Ca2+ signal creates some potential problems of interpretation. Although a graphic representation of variability in the spatial location of Ca2+ entry sites with repeated stimulation is easy to demonstrate after thresholding the images (data not shown), the exclusion of pixels using a thresholding approach to create a graphic image may create the appearance of greater variability than actually exists in the raw signal. Therefore, to avoid the potential of overestimating variability, we demonstrated variability in the spatial distribution of Ca2+ entry using difference images that include all data, including pixel intensities that fall within the resting fluctuation range (see Fig. 6).
Figure 6.

Trial-to-trial variability in nerve-evoked Ca2+ entry during single action potentials. A, Representative difference image showing the spatial distribution of nerve-evoked Ca2+ influx in a single stimulus trial. The box indicates the region of the nerve terminal that is enlarged in B. Pseudocolor scale is the same for all images and is expressed as ΔF/F (%). Scale bar, 2 μm. B, Images from four stimulus trials of the enlarged region of the nerve terminal shown in A, represented with pixel intensity on the z-axis. The left-most panel is taken from the trial shown in A. The trial-to-trial variability in the spatial location of Ca2+ entry sites is illustrated by the changes in location of the intensity peaks.
Results
Dextran-conjugated Calcium Green-1 was loaded into presynaptic terminals of the adult frog neuromuscular junction through the cut end of the motor nerve (see Materials and Methods). To detect action potential-evoked changes in intracellular Ca+2, images were collected at low frequency (0.5 Hz) with an illumination window of 1 msec. Ten images were collected at rest, followed by 10 images taken during stimulation of the motor nerve terminal. The first 10 images of the set were averaged, and the resulting mean background image was subtracted from each of the raw images to produce difference images representing changes above average resting fluorescence in the presence and absence of a presynaptic action potential. Figure 1 shows difference images generated by subtracting the mean background image from a single resting image (Fig. 1A) or single nerve-evoked image (Fig. 1B). Stimulus-induced Ca2+ signals were detected in active zone regions of the nerve terminal (as predicted by α-bungarotoxin staining of postsynaptic receptor bands) (Fig. 1D) and exhibited fluorescence intensities well beyond resting fluctuations in fluorescence intensity (Fig. 1E). To minimize the contributions of resting fluctuations in our analyses of the experimental manipulations described below, we defined signal-above-rest as stimulus-evoked pixel intensities greater than two times the SD of the resting fluctuations in that terminal (i.e., those to the right of the dashed blue line in Fig. 1E) (see Materials and Methods).
To identify the source of Ca2+ underlying these stimulus-dependent signals, we applied 8-(N,N-diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride (TMB-8), an inhibitor of Ca2+-induced Ca2+ release (Hunt et al., 1990; Narita et al., 1998) or ω-CgTx GVIA, an N-type Ca2+ channel blocker that completely blocks transmitter release from this synapse (Kerr and Yoshikami, 1984). Action potential-evoked Ca2+ signals were unaffected by a 30 min treatment with 10 μm TMB-8 (data not shown). In contrast, after 30 min of exposure to 500 nm ω-CgTx GVIA, evoked Ca2+ signals were indistinguishable from unstimulated controls (data not shown). From these results, we conclude that the observed stimulus-evoked signals arise from Ca2+ entry through N-type voltage-gated Ca2+ channels, with little or no contribution of Ca2+-induced Ca2+ release from intracellular stores.
We chose the duration and delay of our illumination window to reflect the timing of action potential invasion of the nerve terminal. From our experience with this preparation, we know that the delay between stimulation of the motor nerve and initial rise of the postsynaptic response is ∼1–2 msec (J. Pattillo, R. Poage, and S. Meriney, unpublished observations). We therefore used a 1 msec illumination window delayed by 1.5 msec relative to nerve stimulation. The sites of Ca2+ signals observed under these conditions were highly localized (Fig. 2A). As the illumination time was increased (Fig. 2B) or delayed with respect to a single nerve stimulus (Fig. 2C), the Ca2+ signals that we detected were broadened spatially as the stimulus-evoked Ca2+ elevations diffused through the nerve terminal. We conclude that images of action potential-induced Ca2+ signals acquired with a 1.5 msec delay and 1 msec duration represent an accurate depiction of the spatial distribution of Ca2+ entry in the motor nerve terminal shortly after action potential invasion.
Figure 2.

The observed spatial profile of Ca2+ influx is dependent on the timing of the laser illumination window. A, Representative difference image of Ca2+ entry with an illumination window of 1 msec duration beginning 1.5 msec after nerve trunk stimulation. B, Representative difference image of Ca2+ entry with an illumination window of 2 msec duration beginning 1.5 msec after nerve stimulation. The Ca2+ entry signal is more intense (resulting from a doubling in the dye illumination time) but also more diffusely distributed throughout the nerve terminal. C, Representative raw difference image of Ca2+ entry with an illumination window of 1 msec duration beginning 12 msec after nerve stimulation. The Ca2+ entry signal is slightly reduced in magnitude as compared with A and much more diffusely distributed. The timing of the illumination window is shown schematically in each image. The pseudocolor scale bar is the same for all panels and is expressed as ΔF/F (%). Scale bar, 2 μm.
Experimental manipulation of calcium signals
We evaluated the effects on Ca2+ entry of a partial blockade of N-type Ca2+ channels. Over the time course of our experiments, ω-CgTX GVIA produced an essentially irreversible blockade of N-type Ca2+ channels (Stocker et al., 1997). We used a short exposure (5–10 min) to submaximal concentrations of toxin (200 nm) to evaluate the effects of blocking a fraction of presynaptic Ca2+ channels on Ca2+ entry. These data address the basic question: is each discrete Ca2+ entry site that we detect a result of the Ca2+ flux through very few open Ca2+ channels or the combined flux from clusters of open Ca2+ channels? These two alternatives make clear predictions as to how Ca2+ signals should be changed as a result of the blockade of a subset of channels after submaximal exposure to ω-CgTX GVIA. If each Ca2+ entry site is generated by a single, or very few Ca2+ channel openings, one would predict a toxin-induced decrease in the number of Ca2+ entry sites, with no change in the intensity of signal at sites that remain unblocked. In contrast, if each Ca2+ entry site is generated by Ca2+ flux through a cluster of simultaneously activated Ca2+ channels, one would predict a decrease in the number of Ca2+ entry sites, as some drop below detection threshold, and a decrease in the intensity of signal at those sites that can be detected.
Figure 3A shows the resting fluorescence signal from a sample nerve terminal with the selected region of interest, and Figure 3B shows the distribution of pixel intensities in this region of interest in the presence and absence of nerve stimulation. Figure 3C shows representative pseudocolored difference images before and after partial N-type Ca2+ channel blockade (200 nm ω-CgTX GVIA; 7 min). This treatment significantly decreased the total Ca2+ signal-above-rest by 64.4 ± 8.1% (mean ± SEM; one-sample t test; p < 0.001), but of those Ca2+ entry sites that persisted after blockade, there was no significant change in the average intensity of the fluorescence at each entry site (mean pixel intensity decreased by 1.6 ± 1.9%; p > 0.2; n = 6 nerve terminals). To further quantify these effects, we analyzed the distribution of pixel intensities in the signal-above-rest from these image sets (see Materials and Methods). Cumulative frequency plots from the six nerve terminals examined show no significant change in the distribution of pixel intensities after exposure to ω-CgTX GVIA (Fig. 3D) (Kolmogorov–Smirnov test; p > 0.5). We observed a significant decrease (by 64.6 ± 8.0%; one-sample t test; p < 0.05) in the number of Ca2+ entry sites (see Materials and Methods), consistent with a decrease in total Ca2+ entry but with no change in the intensity distribution of the signal. These data lead us to conclude that the pharmacological elimination of a fraction of N-type Ca2+ channels reduces the number of Ca2+ entry sites without changing the characteristics of the Ca2+ signal at sites that are spared. As outlined above, these data favor the hypothesis that each Ca2+ entry site is generated by the opening of very few Ca2+ channels.
In control experiments, extracellular Ca2+ was decreased from 1.8 to 0.5 mm to reduce Ca2+ flux without altering Ca2+ channel gating during the action potential (Fig. 4). A representative resting fluorescence image is shown with the region of interest used for analysis (Fig. 4A), along with the distribution of control pixel intensities in the presence and absence of nerve stimulation (Fig. 4B). Figure 4C displays representative difference images obtained in both 1.8 and 0.5 mm extracellular Ca2+. Qualitatively, the effect is very different from that after submaximal ω-CgTX GVIA exposure; the remaining Ca2+ signals after low Ca2+ exposure are weaker than the signals that remain after exposure to ω-CgTX GVIA (Fig. 3C). Quantitatively, exposure to low Ca2+ saline resulted in a significant decrease in total Ca2+ signal-above-rest (71.4 ± 11.0% decrease; n = 3; one-sample t test; p < 0.01), coupled with a significant decrease in the average intensity of pixels that led to that signal (31.8 ± 6.0% decrease; one-sample t test; p < 0.05). In this case, there was also a significant decrease in the number of pixels that detected Ca2+ entry above rest (78.1 ± 14.0%) as weaker Ca2+ entry sites slipped into the noise after exposure to the lowered extracellular Ca2+.
To demonstrate that we could detect the effects of increasing Ca2+ influx at entry sites, we exposed the preparation to a potassium channel blocker [3,4-diaminopyridine (DAP)], which increases Ca2+ influx and transmitter release by broadening the presynaptic action potential (Kirsch and Narahashi, 1978; Durant and Marshall, 1980) (Fig. 5). A broadened presynaptic action potential increases Ca2+ entry by providing a longer depolarizing stimulus that activates a greater proportion of available Ca2+ channels and allows activated channels the opportunity to move into the open state a greater number of times during each action potential. A representative resting fluorescence image is shown with the region of interest used for analysis (Fig. 5A), along with the corresponding distribution of control pixel intensities in the presence and absence of nerve stimulation (Fig. 5B). Figure 5C displays representative difference images obtained in control and DAP-treated conditions. After a 10–20 min exposure to 5 μm DAP, the total Ca2+ entry was increased by 410.4 ± 224.0%, and the mean intensity of each pixel that detected a signal-above-rest increased by 170.0 ± 33.2% (n = 3). The number of pixels that detected a signal above rest also increased (by 225.1 ± 92.0%).
Cumulative frequency plots generated from these controls (Figs. 4D, 5D) were very different from those generated after exposure to ω-CgTX GVIA (Fig. 3D). Reduction of extracellular Ca2+ led to a significant shift toward lower intensities (Fig. 4D) (Kolmogorov–Smirnov test; p < 0.001), whereas exposure to DAP led to a significant shift toward higher intensities (Fig. 5D) (Kolmogorov–Smirnov test; p < 0.001). The contrasting effects of these three manipulations demonstrate our ability to detect both increases and decreases in Ca2+ influx at these discrete sites and strengthens our interpretation of the effects of partial blockade of N-type Ca2+ channels using ω-CgTX GVIA. Taken as a whole, these data support the hypothesis that each Ca2+ entry site is generated by the opening of very few, perhaps only one Ca2+ channel (see Discussion).
Variability in the spatial distribution of calcium entry
Based on freeze fracture data from the adult frog neuromuscular junction, it has been suggested that there are many N-type Ca2+ channels in each active zone (Heuser et al., 1974; Pumplin et al., 1981). If only a small number of those channels are activated during an action potential, we expect Ca2+ entry during repeated low frequency stimulation to exhibit prominent spatial variability. In Figure 6A, we show a pseudocolored difference image depicting the spatial distribution of Ca2+ entry sites in a single stimulus trial from a representative nerve terminal. In Figure 6B, we chose a well focused region of interest to display variability in the spatial distribution of Ca2+ entry sites over four stimulus trials. These pseudocolored representations show that the spatial distribution of Ca2+ entry sites is not consistent from one stimulus to the next. In fact, there are many pixels over active zone regions of the nerve terminal where Ca2+ signals are detected in only a subset of stimulus trials (Fig. 6B). Our imaging data show spatial variability during multiple action potential trials that is greater than resting fluctuations, suggesting that Ca2+ entry during an action potential does not uniformly “flood” the active zone region of the nerve terminal (Fig. 6B), at least not until well after transmitter release occurs (Fig. 2). These data are consistent with the hypothesis that Ca2+ entry into the frog motor nerve terminal during action potential depolarization is variable, with respect to spatial location from one stimulus to the next, primarily because very few Ca2+ channels open during any given action potential.
Discussion
Calcium entry sites in nerve terminals of the adult frog neuromuscular junction
We used a fast “snapshot” approach to imaging Ca2+ entry into adult frog motor nerve terminals and studied the spatial distribution of Ca2+ entry during single action potential stimuli at low frequency. Using this approach, we observed spatially isolated sites of Ca2+ entry in active zone regions of the nerve terminal that disperse with time after action potential invasion and vary in their location with repeated trials. Furthermore, a partial blockade of the N-type Ca2+ channels that mediate this Ca2+ entry results in a decrease in the number of Ca2+ entry sites with no change in the intensity of signal at remaining sites.
In interpreting these data, we considered the probable number of calcium channels in the active zone that are sampled by each pixel and the activation of these channels by action potential stimuli. Each linear active zone in the adult frog neuromuscular junction is ∼1 μm long and separated from other active zones by ∼1 μm. Freeze-fracture data demonstrate that there are ∼200 active zone particles distributed in two parallel double rows along the length of the active zone (Heuser et al., 1974; Pumplin et al., 1981; Pawson et al., 1998). Based on fluorescent staining of these linear zones with labeled toxins selective for both calcium and calcium-activated potassium channels (Robitaille et al., 1993), and the expectation that other active zone proteins might also be represented, these active zone particles likely represent several classes of membrane proteins, only some of which may be calcium channels. For the purposes of discussion, we will consider a range of potential calcium channel numbers in a single active zone. On one extreme, on the basis of the number of particles observed in freeze-fracture replicas, 200 calcium channels per active zone must be considered the top limit. On the other extreme, it is possible that each vesicle-docking site might only be associated with a single calcium channel. Because there are ∼30 vesicles associated with the release site membrane along a single active zone, 30 calcium channels might be considered the bottom limit. Because the resolution of our imaging system is 0.275 μm, we can detect differences in the spatial distribution of Ca2+ entry within single active zones. Because, as mentioned, each active zone is ∼1 μm in length, we sampled each linear active zone with a linear array of approximately four pixels. Therefore, in estimating how many Ca2+ channels might be sampled by a single pixel in our experiments, we must divide the range of probable calcium channels sampled in an active zone by four. As such, somewhere between 6 and 50 calcium channels might be under each pixel that samples a portion of a single active zone.
Conceptually, if we consider the range of estimates for the number of calcium channels that might be sampled by a single pixel and the range of possibilities for the likelihood that an N-type calcium channel will open during a single action potential stimulus, we can determine whether we would expect to observe a graded calcium signal or one that includes a significant number of failures during low-frequency action potential stimuli. If there are many calcium channels (∼50) sampled by each of our pixels, and a relatively high probability (greater than ∼0.2) for calcium channel opening during an action potential stimulus, our calculations (see Appendix; available at www.jneurosci.org) predict a calcium signal that is little changed with repeated trials and shows a graded change in intensity after partial blockade of calcium channels using ω-CgTX GVIA. This is clearly not what we observed. In contrast, if there are few calcium channels sampled by each pixel (approximately six) and a relatively low probability (less than ∼0.2) for calcium channel opening during an action potential stimulus, our calculations predict a calcium signal that shows large variability with repeated trials (including the failure to detect signal in some trials), some increase in the number of trials in which there is a failure to detect calcium signal after partial blockade by ω-CgTX GVIA, and no change in the intensity of signal detected at entry sites that remain unblocked. Our data are consistent with this prediction.
The mathematical simulation presented in the Appendix (available at www.jneurosci.org) provides quantitative details of predicted changes in calcium signals for various numbers of calcium channels per active zone and various probabilities for calcium channel opening. We can draw three conclusions from this analysis. First, using a range of reasonable estimates for the number of calcium channels that might be sampled by each pixel, it appears possible that single action potential stimuli could generate a local (subactive zone) calcium signal from a single calcium channel opening, and that these signals would show considerable variability with repeated trials. Second, after a blockade of ∼65% of available calcium channels (using low doses of ω-CgTX GVIA), it is reasonable to expect an increase in the number of trials in which there is a failure of any available calcium channel under a single pixel to open, and that this would be coupled with little or no change in the intensity of signal at sites that remain unblocked. Finally, on the basis of our imaging data, this analysis suggests that very few of the Ca2+ channels that populate a frog motor nerve terminal active zone open with each action potential stimulus, and that this would not occur unless there are relatively few calcium channels in an active zone (∼30), each with a low probability for opening during an action potential (less than ∼0.2).
Relationship between calcium entry and transmitter release at active zones
Our work addresses a point of debate regarding the manner in which Ca2+ triggers neurotransmitter release from the active zone (Dunlap et al., 1995; Neher, 1998). Is each synaptic vesicle fusion event triggered by the Ca2+ flux through one Ca2+ channel, or by the Ca2+ that accumulates when many local Ca2+ channels open near a release-ready synaptic vesicle? It seems likely that either situation can occur, depending mostly on which synapse is being studied. In particular, the organization of the active zone, the fraction of Ca2+ channels that open with each action potential, and the conditions of the experiment may alter the stoichiometry between Ca2+ entry sites and vesicle fusion events.
In the calyx of Held synapse from the auditory brainstem of the rat, a large fraction of the available presynaptic Ca2+ channels is believed to open after a single action potential and transmitter release appears to be triggered by the combined action of multiple Ca2+ channels (Borst and Sakmann, 1996, 1998, 1999). In the calyx of Held, there may be large clusters of Ca2+ channels around which synaptic vesicles are arranged at varying distances from these clusters (Meinrenken et al., 2002). Similarly, at mossy fiber boutons in the rat hippocampus, ∼90% of available Ca2+ channels are thought to be activated by a single action potential (Bischofberger et al., 2002). In contrast, a small percentage of available Ca2+ channels is thought to be activated by an action potential at the squid giant synapse (Llinas et al., 1981), and transmitter release may be triggered by the local flux of Ca2+ through single channel openings (Augustine et al., 1991). At the adult frog neuromuscular junction, where there is a long linear arrangement of Ca2+ channels and release-ready synaptic vesicles, our data support the possibility that an exocytotic event might be triggered by the opening of a single Ca2+ channel (Yoshikami et al., 1989), similar to the stoichiometry proposed for the chick ciliary ganglion calyx (Stanley, 1993; Bertram et al., 1996). Therefore, we hypothesize that the low probability of transmitter release from each active zone at the adult frog neuromuscular junction may be primarily a function of a low probability for Ca2+ channel opening.
Robitaille et al. (1993) showed that EGTA (a relatively slow calcium buffer) introduction into the adult frog motor nerve terminal could reduce transmitter release by a small degree. These results could be interpreted to suggest that transmitter release may be triggered by the summed Ca2+ influx from clusters of Ca2+ channels distributed over a relatively large distance. In the interpretation of these data, it is useful to consider the probability of transmitter release at each active zone. Although the frog neuromuscular junction on the whole is a strong synapse containing hundreds of active zones, within each active zone there are ∼30 potential vesicle-docking–release sites, each with a very low probability of release. After a single action potential, the probability of a single vesicle being released from each of these active zones is less than one, making the probability of release at each release site within an active zone very low. With such a low probability of release at each active zone, it may not be surprising that any Ca2+ buffer will have some effect on release. The stochastic nature of calcium influx and vesicle fusion results in a distribution of latencies to fusion (Katz and Miledi, 1965), and a slow calcium buffer could exert a small effect on long-latency fusion events, even if the calcium signal is localized in space. The effects of EGTA reported by Robitaille et al. (1993) are relatively small (∼20%) such that in some limited number of instances, EGTA will have a chance to compete with the Ca2+ sensor for release. Therefore, even in light of the results of Robitaille et al. (1993), it remains possible that single Ca2+ channel openings provide the Ca2+ trigger for release at this synapse (Yoshikami et al., 1989).
Pharmacologic modulation of nerve terminal function
Potassium channel blockers can increase transmitter release from nerve terminals. These agents broaden the presynaptic action potential and increase the number of Ca2+ channels that open, as well as the time during which individual Ca2+ channels will have the opportunity to move into the open state. These agents have been used to treat neuromuscular diseases, because they enhance acetylcholine release from weak synapses. In particular, DAP has been the target of several clinical trials directed at treating Lambert-Eaton Myasthenic syndrome (Molgo and Guglielmi, 1996; Sanders et al., 2000). If our results from the frog neuromuscular junction can be extrapolated to the mammalian neuromuscular junction, clinical use of DAP may change the stoichiometry between Ca2+ channel openings and vesicle fusion at the neuromuscular synapse. If under normal conditions a small proportion of available Ca2+ channels opens and each single Ca2+ channel opening controls the secretion of one vesicle, we would then predict after DAP treatment that the increased Ca2+ influx through each channel, together with the recruitment of additional Ca2+ channel openings, would lead to more overlap in Ca2+ entry domains. Therefore, after DAP treatment, the combined flux through many local Ca2+ channels would trigger each vesicle fusion event. Under these conditions, Ca2+ would flood the presynaptic active zone (Fig. 5) causing multiple transmitter-containing vesicles to fuse with the plasma membrane at each active zone after each action potential stimulus. In this sense, DAP treatment could change the manner in which Ca2+ triggers vesicle fusion at the active zone.
G-protein modulation of transmitter release
Our interpretations of these data are also relevant to the hypothesized effects of neuromodulatory agents on the regulation of Ca2+ entry and the triggering of transmitter secretion at individual release sites in an active zone. Calcium channels are known to be common targets for G-protein-mediated modulation (Hille, 1994). In terms of physiological importance, it appears that negatively modulated Ca2+ channels open so slowly that they do not contribute to action potential-evoked Ca2+ current (Artim and Meriney, 2000). Therefore, inhibitory G-protein modulation essentially prevents a fraction of the available Ca2+ channels from responding to action potential stimuli (an effect similar to partial blockade by ω-CgTX GVIA) (Fig. 3). The specific effects of G-protein modulation on transmitter release will vary depending on the stoichiometry between open Ca2+ channels and the sensor for transmitter release. If the active zone is flooded with Ca2+ after each action potential stimulus and vesicle fusion is triggered by the combined action of Ca2+ ions from many channel openings, G-protein modulation, which decreased the number of Ca2+ channel openings, would decrease the overall magnitude of this active zone flood. Under this scenario, the probability for vesicle fusion might decrease in a manner consistent with the known nonlinear relationship between Ca2+ and transmitter release (Dodge and Rahamimoff, 1967; but see Takahashi et al., 1996, 1998). In contrast, our data lead us to hypothesize that at the frog neuromuscular junction, G-protein modulation, which decreases the number of Ca2+ channel openings, would eliminate a corresponding fraction of the Ca2+ entry domains entirely and lead to a linear reduction in the number of vesicle fusion events as entire release sites drop out. By this mechanism, we predict that even subtle changes in the number of active zone Ca2+ channels that open during action potential stimulation would significantly vary the number of active transmitter release sites and, thus, the efficacy of synaptic transmission.
Footnotes
This work was supported by grants from the National Institutes of Health (NS043396), Muscular Dystrophy Association, National Science Foundation (BESCO 79483), and The University of Pittsburgh Central Research Development Fund. We thank B. V. Kaminsky, E. Lindsley, and J. James for assistance with image analysis and J. M. Pattillo and A. Hoerder for providing assistance with some of the experiments.
J.R.S. is the principal author of the mathematical model presented in the on-line Appendix.
Correspondence should be addressed to Stephen D. Meriney, Department of Neuroscience, University of Pittsburgh, 446 Crawford Hall, Pittsburgh, PA 15260. E-mail: meriney@bns.pitt.edu.
E. S. Wachman's present address: Chromodynamics, 1195 Airport Road, Number 1, Lakewood, NJ 08701.
R. E. Poage's present address: Department of Biology, University of North Carolina at Pembroke, Pembroke, NC 28372.
D. L. Farkas's present address: Department of Surgery, Cedars-Sinai Medical Center, 8700 Beverly Boulevard, Los Angeles, CA 90048.
DOI:10.1523/JNEUROSCI.1660-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242877-09$15.00/0
E.S.W. and R.E.P. contributed equally to this work.
References
- Artim DE, Meriney SD (2000) G protein modulated Ca2+ current with slowed activation does not alter the kinetics of action potential-evoked Ca2+ current. J Neurophysiol 84: 2417–2425. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Adler EM, Charlton MP (1991) The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann NY Acad Sci 635: 365–381. [DOI] [PubMed] [Google Scholar]
- Bertram R, Sherman A, Stanley EF (1996) Single-domain/bound calcium hypothesis of transmitter release and facilitation. J Neurophys 75: 1919–1931. [DOI] [PubMed] [Google Scholar]
- Bischofberger J, Geiger JR, Jonas P (2002) Timing and efficacy of Ca2+ channel activation in hippocampal mossy fiber boutons. J Neurosci 22: 10593–10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1996) Calcium influx and transmitter release in a fast CNS synapse. Nature 383: 431–434. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1998) Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J Physiol (Lond.) 506: 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1999) Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci 354: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF (1997) Stimulation-induced changes in [Ca2+] in lizard motor nerve terminals. J Physiol (Lond) 504: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Vergara JL (1997) Localized detection of action-potential-induced presynaptic calcium transients at a Xenopus neuromuscular junction. J Physiol (Lond) 505: 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Peskoff A, Vergara JL (1999) Measurement of action potential-induced presynaptic calcium domains at a cultured neuromuscular junction. J Neurosci 19: 7846–7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge Jr FA, Rahamimoff R (1967) Co-operative action of calcium ions in transmitter release at the neuromuscular junction. J Physiol (Lond) 193: 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ (1995) Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 18: 89–98. [PubMed] [Google Scholar]
- Durant NN, Marshall IG (1980) The effects of 3,4-diaminopyridine on acetylcholine release at the frog neuromuscular junction. Eur J Pharmacol 67: 201–208. [DOI] [PubMed] [Google Scholar]
- Edwards FA (1995) Anatomy and electrophysiology of fast central synapses lead to a structural model for long-term potentiation. Physiol Rev 75: 759–787. [DOI] [PubMed] [Google Scholar]
- Forti L, Pouzat C, Llano I (2000) Action potential-evoked Ca2+ signals and calcium channels in axons of developing rat cerebellar interneurons. J Physiol (Lond) 527: 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freguelli BG, Malinow R (1996) Fluctuations in intracellular calcium responses to action potentials in single en passage presynaptic boutons of layer V neurons in neocortical slices. Learn Mem 3: 150–159. [DOI] [PubMed] [Google Scholar]
- Goda Y, Sudhof TC (1997) Calcium regulation of transmitter release: reliability unreliable? Curr Opin Cell Biol 9: 513–518. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS, Landis DM (1974) Functional changes in frog neuromuscular junctions studied with freeze-fracture. J Neurocytol 3: 109–131. [DOI] [PubMed] [Google Scholar]
- Hille B (1994) Modulation of ion-channel function by G-protein coupled receptors. Trends Neurosci 17: 531–536. [DOI] [PubMed] [Google Scholar]
- Hunt JM, Silinsky EM, Hirsh KH, Ahn D, Solsona C (1990) The effect of TMB-8 on acetylcholine release from frog motor nerve: interactions with adenosine. Euro J Pharmacol 178: 259–266. [DOI] [PubMed] [Google Scholar]
- Katz B (1969) The release of neural transmitter substances: the Sherrington lectures. X. Liverpool, UK: Liverpool UP.
- Katz B, Miledi R (1965) The effect of calcium on acetylcholine release from motor nerve terminals. Proc R Soc Lond B Biol Sci 161: 496–503. [DOI] [PubMed] [Google Scholar]
- Kerr LM, Yoshikami D (1984) A venom peptide with a novel presynaptic blocking action. Nature 308: 282–284. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Veselovsky N, Grantyn R (1999) Relationship between presynaptic calcium transients and postsynaptic currents at single γ-aminobutyric acid (GABA)ergic boutons. Proc Natl Acad Sci USA 96: 7520–7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch GE, Narahashi T (1978) 3,4-diaminopyridine. A potent new potassium channel blocker. Biophys J 22: 507–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Steinberg IZ, Walton K (1981) Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J 33: 323–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Silver RB (1992) Microdomains of high calcium concentration in a presynaptic terminal. Science 256: 677–697. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Sugimori M, Silver RB (1994) Localization of calcium concentration microdomains at the active zone in the squid giant synapse. Adv Second Messenger Phosphoprotein Res 29: 133–137. [DOI] [PubMed] [Google Scholar]
- Llinas RR, Sugimori M, Silver RB (1995) The concept of calcium concentration microdomains in synaptic transmission. Neuropharm 34: 1443–1451. [DOI] [PubMed] [Google Scholar]
- MacKenzie PJ, Umemiya M, Murphy TH (1996) Ca2+ imaging of CNS axons in culture indicated reliable coupling between single action potentials and distal functional release sites. Neuron 16: 783–795. [DOI] [PubMed] [Google Scholar]
- Meinrenken CJ, Borst JG, Sakmann B (2002) Calcium secretion coupling at calyx of held governed by nonuniform channel-vesicle topography. J Neurosci 22: 1648–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molgo J, Guglielmi JM (1996) 3,4-Diaminopyridine, an orphan drug, in the symptomatic treatment of Lambert-Eaton myasthenic syndrome. Pflügers Arch 431: R295–R296. [DOI] [PubMed] [Google Scholar]
- Narita K, Akita T, Osanai M, Shirasaki T, Kijima H, Kuba K (1998) Ca2+-induced Ca2+ release mechanism involved in asynchronous exocytosis at frog motor nerve terminals. J Gen Physiol 112: 593–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E (1998) Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron 20: 389–399. [DOI] [PubMed] [Google Scholar]
- Pawson PA, Grinnell AD, Wolowske B (1998) Quantitative freeze-fracture analysis of the frog neuromuscular junction synapse. I. Naturally occurring variability in active zone structure. J Neurocytol 27: 361–377. [DOI] [PubMed] [Google Scholar]
- Peng Y-Y, Zucker RS (1993) Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron 10: 465–473. [DOI] [PubMed] [Google Scholar]
- Pumplin DW, Reese TS, Llinas R (1981) Are the presynaptic membrane particles the Ca2+ channels? Proc Natl Acad Sci USA 78: 7210–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP (1993) Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron 11: 645–655. [DOI] [PubMed] [Google Scholar]
- Sanders DB, Massey JM, Sanders LL, Edwards LJ (2000) A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology 54: 603–607. [DOI] [PubMed] [Google Scholar]
- Smith SJ, Buchanan J, Osses LR, Charlton MP, Augustine GJ (1993) The spatial distribution of calcium signals in squid presynaptic terminals. J Physiol (Lond) 472: 573–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley EF (1993) Single calcium channels and acetylcholine release at a presynaptic nerve terminal. Neuron 11: 1007–1011. [DOI] [PubMed] [Google Scholar]
- Stocker JW, Nadasdi L, Aldrich RW, Tsien RW (1997) Preferential interaction of omega-conotoxins with inactivated N-type Ca2+ channels. J Neurosci 17: 3002–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Scheller RH (2001) Mechanisms and regulation of neurotransmitter release. In: Synapses (Cowan MW, Sudhof TC, Stevens CF, eds), pp 177–215. Baltimore, MD: Johns Hopkins UP.
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K (1996) Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science 274: 594–597. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T (1998) G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. J Neurosci 18: 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachman ES, Niu W, Farkas DL (1997) AOTF microscope for imaging with increased speed and spectral versatility. Biophys J 73: 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Betz WJ (1996) Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron 17: 769–779. [DOI] [PubMed] [Google Scholar]
- Yoshikami D, Bagabaldo Z, Olivera BM (1989) The inhibitory effects of omega-conotoxins on Ca channels and synapses. Ann NY Acad Sci 560: 230–248. [DOI] [PubMed] [Google Scholar]