

Abstract
Pyramidal neurons in the lateral amygdala discharge trains of action potentials that show marked spike frequency adaptation, which is primarily mediated by activation of a slow calcium-activated potassium current. We show here that these neurons also express an α-dendrotoxin- and tityustoxin-Kα-sensitive voltage-dependent potassium current that plays a key role in the control of spike discharge frequency. This current is selectively targeted to the primary apical dendrite of these neurons. Activation of μ-opioid receptors by application of morphine or d-Ala2-N-Me-Phe4-Glycol5-enkephalin (DAMGO) potentiates spike frequency adaptation by enhancing the α-dendrotoxin-sensitive potassium current. The effects of μ-opioid agonists on spike frequency adaptation were blocked by inhibiting G-proteins with N-ethylmaleimide (NEM) and by blocking phospholipase A2. Application of arachidonic acid mimicked the actions of DAMGO or morphine. These results show that μ-opioid receptor activation enhances spike frequency adaptation in lateral amygdala neurons by modulating a voltage-dependent potassium channel containing Kv1.2 subunits, through activation of the phospholipase A2–arachidonic acid–lipoxygenases cascade.
Keywords: anxiolytic, arachidonic, channel, nociception, pain, lipoxygenase, Kv1.2
Introduction
Opioid receptors are involved in a multitude of functions in the CNS, including the regulation of stress, anxiety, and antinociception (Vaccarino et al., 1999). There are three subtypes of opioid receptors that can be distinguished by selective agonists and antagonists: μ, ∂, and κ. These receptors are found throughout the CNS and PNS in distinct but overlapping distributions. The amygdala, a structure that plays a critical role in emotional evaluation, in particular the response to fear and anxiety (LeDoux, 2000; Davis and Whalen, 2001), has one of the highest densities of opioid binding sites in the CNS (Atweh and Kuhar, 1977). The lateral nucleus of the amygdala is particularly enriched in μ-opioid receptors (Paden et al., 1987). The opioid system in the amygdala has been implicated in the modulation of fear (Good and Westbrook, 1995), as well as consolidation of memories and the antinociceptive actions of conditioned stimuli (McGaugh, 1989). In agreement with these modulatory effects, functional imaging studies have shown that activation of μ receptors is associated with inhibition of amygdala function (Liberzon et al., 2002). However, the mechanisms underlying this inhibition are not understood.
Opioid receptors are coupled to pertussis toxin-sensitive G-proteins, and their cellular actions are generally mediated by inhibition of adenyl cyclase, activation of inwardly rectifying potassium conductances, or inhibition of voltage-dependent calcium conductances (Law et al., 2000). Postsynaptically, opioid receptor activation leads to membrane hyperpolarization, whereas presynaptically opioids can inhibit transmitter release at both excitatory and inhibitory synapses (Williams et al., 1982, 2001; Madison and Nicoll, 1988). In the lateral amygdala (LA), activation of μ receptors hyperpolarizes and reduces local GABAergic inhibition in some pyramidal neurons (Sugita and North, 1993). However, these effects would be expected to increase excitation of lateral amygdala neurons.
In response to depolarizing stimuli, central neurons fire trains of action potentials at frequencies that are controlled by activation of two slow calcium-dependent potassium currents, IAHP and sIAHP (Sah, 1996). Of these, the IAHP current controls the interspike interval during repetitive activity, whereas activation of the sIAHP is responsible for spike frequency adaptation (Sah, 1996). The sIAHP current is the target for modulation by a wide variety of neurotransmitters that downregulate the current and thus decrease spike frequency adaptation (Madison and Nicoll, 1984; Nicoll, 1988). Because spike frequency adaptation plays a key role in the transfer of integrated synaptic input to neuronal output, modulation of sIAHP has important implications for the operation of neuronal networks.
Projection neurons in the lateral amygdala are a heterogenous population of cells that exhibit a range of firing properties with varying degrees of spike frequency adaptation. These differences are thought to result from varying levels of expression of the sIAHP (Paré and Gaudreau, 1996; Faber et al., 2001; Faber and Sah, 2002). We show here that these neurons express an α-dendrotoxin (α-DTX)-sensitive, voltage-dependent potassium current that also plays a major role in spike frequency adaptation. This current is selectively expressed in the apical dendrite rather than the soma. Furthermore, it is upregulated by activation of μ-opioid receptors, viaaGi/o-protein-mediated activation of the arachidonic acid pathway, resulting in an enhancement in spike frequency adaptation. Thus, μ receptor activation greatly decreases the number of spikes evoked by depolarizing stimuli, effectively inhibiting the output from lateral amygdala pyramidal neurons. This effect of μ receptor activation may explain the inhibitory actions of opioids on amygdala function.
Materials and Methods
Coronal rat brain slices (400 μm) were obtained from Wistar rats (unsexed, 17- to 28-d-old) and maintained in artificial CSF (aCSF) containing the following (in mm): 118 NaCl, 2.5 KCl, 2.5 NaHCO3, 10 glucose, 2.5 MgCl2, 2.5 CaCl2, and 1.2 NaH2PO4. Procedures used to prepare the slices were in accordance with the guidelines of the Australian National University Animal Experimentation Ethics Committee. Whole-cell recordings were made from neurons in the lateral amygdala in slices continuously perfused with oxygenated aCSF maintained at 28–30°C using infrared differential interference contrast techniques. Electrodes (3–6 MΩ) were filled with pipette solution containing the following (in mm): 135 KMeSO4, 8 NaCl, 10 HEPES, 2 Mg2ATP, and 0.3 Na3GTP, pH 7.3 with KOH (osmolarity, 290 mOsm/kg). Signals were recorded using a patch-clamp amplifier (Axopatch 1-D or MultiClamp 700A; Axon Instruments, Foster City, CA). Responses were filtered at 5 kHz and digitized at 10 kHz (ITC-16; InstruTech, Greatneck, NY). All data were acquired, stored, and analyzed on a Power Macintosh G4 (Apple Computers, Cupertino, CA) using Axograph (Axon Instruments).
Only cells with a membrane potential more hyperpolarized than –55 mV with overshooting action potentials were included in this study. Access resistance was 5–30 MΩ and was monitored throughout the experiment. To investigate the firing properties of neurons, six to eight current injection steps (600 msec) were applied from –100 to +600 pA in 100 pA increments. Afterhyperpolarizations (AHPs) were evoked in current clamp by a 100 msec, 400 pA current injection from a holding potential of –70 or –50 mV. The ability of a cell to adapt was judged by counting the number of spikes fired at threshold current injections, twice threshold current injections (in which threshold was the current injection that first elicited an action potential), or in response to a 400 pA current injection. Spike initiation threshold was measured at the beginning of the upstroke of the action potential. To examine the effect of opioids on transient responses, glutamate (200 μm) was puffed onto the cell soma using a Picospritzer II (General Valve, Fairfield, NJ) for 50–100 msec in the presence of picrotoxin (100 μm) and CGP55845A (1 μm).
For whole-cell voltage-clamp experiments, recordings were restricted to cells in which the series resistance was <15 MΩ and was compensated by 60–80%. Potassium currents were isolated by inclusion of 1 μm tetrodotoxin (TTX) and 5 mm nickel (Ni+) or 0.25–0.4 mm cadmium (Cd2+) to block Na+ and Ca2+ channels, respectively. To reduce the amplitude of outward currents, the internal potassium concentration was reduced to 90 mm by replacing KMeSO4 with NaCl and by the inclusion of either 10 mm EGTA or 10 mm BAPTA to block activation of calcium-dependent currents. Whole-cell current–voltage (I–V) relationships were investigated by giving 400 msec, 10 mV voltage steps from –20 to +90 mV from a holding potential of –60 or –50 mV. The I–V plots derived from these were obtained by measuring the currents at steady state at each potential. Capacitance and leak transients were subtracted using a P/4 protocol. Nucleated somatic patches were obtained once the whole-cell configuration had been established. To obtain these patches, negative pressure was applied to the pipette, which was then slowly withdrawn from the cell. As with the whole-cell recordings, somatic K+ currents were isolated by the inclusion of 10 mm EGTA or 10 mm BAPTA in the internal solution and by the external application of 0.25–0.4 mm Cd2+ or 5 mm Ni+. Sodium currents were not blocked. Drugs were either bath applied or puffed onto nucleated somatic patches as described above, using a micropipette filled with the drug solution, and responses to a 400 msec, 60 mV step were measured every 10 sec. Electrodes (10–12 MΩ) were used for dendritic recordings from rats aged between 13 and 20 d. Having achieved a seal of at least 2 GΩ, a whole-cell configuration was obtained before slowly pulling the electrode away from the cell and out of the slice to record from outside-out patches. To generate activation plots, currents were converted to conductance using a K+ reversal potential calculated using the Nernst equation. Curves were then fitted with the following Boltzmann equation: G = Gmax/(1 + exp((V1/2 – V)/k)), where Gmax is the maximum conductance, V1/2 is the half-maximal voltage, and k is the slope factor. The V1/2 and slope values were obtained by averaging the values obtained from Boltzmann fits of individual experiments. Results are expressed as mean ± SEM. Student's t tests were used for statistical comparisons between groups.
TTX, α-DTX, tityustoxin-Kα, and CGP55845 dendrotoxin-K (DTX-K) were obtained from Alomone Labs (Jerusalem, Israel). EGTA, BAPTA, cadmium, 4-aminopyridine (4-AP), nickel sulfate, noradrenaline, N-ethylmaleimide (NEM), picrotoxin, and tetraethylammonium (TEA) were obtained from Sigma (St. Louis, MO). d-Ala2-N-Me-Phe4-Glycol5-enkephalin (DAMGO) and naloxone were obtained from Tocris Cookson (Ballwin, MO). Eicosatriyonic acid (ETI), arachidonyl trifluoromethyl ketone (AACOCF3), and arachidonic acid were obtained from Cayman Chemical (Ann Arbor, MI).
Results
Whole-cell recordings were made from pyramidal neurons in the lateral amygdala. These cells comprise the projection neurons and form the principal cell type in this nucleus. In the majority of these neurons, depolarizing current injection evokes a train of action potentials that adapt fully after two to five spikes (Mahanty and Sah, 1998; Faber et al., 2001) (Fig. 1a). Action potential trains are followed by a slow AHP that lasts several seconds (Fig. 1g). Application of noradrenaline markedly reduced spike frequency adaptation (n = 8) (Fig. 1a,b) by blocking the slow AHP (Fig. 1g). Similar effects were also seen with acetylcholine, serotonin, and glutamate (Faber and Sah, 2002). In contrast, activation of μ-opioid receptors with morphine (1 μm) or the specific agonist DAMGO (0.5 μm) resulted in an enhancement of spike frequency adaptation (Fig. 1c–f), which was reversible on washout of the agonists. Morphine reduced the number of spikes evoked by a prolonged current injection (600 msec, 400 pA) from 3.2 ± 0.4 to 1.7 ± 0.3 (p < 0.001; n = 10). Similarly, application of DAMGO reduced the number of spikes evoked from 4.6 ± 0.7 to 2.4 ± 0.6 (p < 0.05; n = 11). These actions of morphine and DAMGO were fully blocked by the opioid antagonist naloxone (n = 11; data not shown). The effects of morphine and DAMGO were not associated with a change in input resistance, resting membrane potential, action potential initiation threshold, or enhancement of the slow AHP (n = 24) (Fig. 1h,i, Table 1). In fact, the slow AHP was slightly attenuated in the presence of these agonists (Fig. 1h,i) because fewer spikes were initiated during the depolarizing step used to evoke the AHP (data not shown). These results indicate that two independent mechanisms contribute to spike frequency adaptation in amygdala pyramidal neurons. One of these is attributable to activation of the slow AHP, whereas the other is attributable to activation of another current that is modulated by μ-opioid receptors. In agreement with this finding, application of DAMGO reversed the reduction in spike frequency adaptation observed after blockade of the slow AHP by noradrenaline (n = 4) (Fig. 1j).
Figure 1.

μ-Opioid receptor activation increases spike frequency adaptation. a, Action potentials evoked by a suprathreshold (200 pA) current injection rapidly adapt after three spikes. Application of noradrenaline (10 μm) reduces spike frequency adaptation. b, Mean increase in the number of action potentials for each current injection in control and in the presence of noradrenaline (n = 5). c, The number of action potentials evoked by a suprathreshold current injection (400 pA) is markedly reduced by application of 1 μm morphine. d, Mean number of spikes evoked in control and in morphine for each current injection (n = 10). e, DAMGO at 0.5 μm also enhances spike frequency adaptation. f, Average data for 17 cells showing the reduction in spikes evoked by depolarizing current injections from 100 to 400 pA. g, The effect of noradrenaline on spike frequency adaptation is mediated by blockade of the slow AHP. Morphine (h) and DAMGO (i) do not enhance the slow AHP and, in fact, cause a small but insignificant depression of the slow AHP. j, Application of DAMGO reverses the reduction in spike frequency adaptation produced by noradrenaline. The inset shows that the slow calcium-dependent AHP is blocked by noradrenaline and does not recover in the presence of DAMGO.
Table 1.
Effects of potassium channel blockers and opioids on cell firing properties
|
|
Control |
α-DTX |
Tityustoxin |
DTX-K |
Morphine |
DAMGO |
TEA |
|---|---|---|---|---|---|---|---|
| Membrane potential (mV) | -63 ± 1 | -59 ± 1.3 | -58 ± 1.5 | -61 ± 1.1 | -62 ± 1.8 | -63 ± 1.6 | -66 ± 1.8 |
| Input resistance (MΩ) | 141 ± 7 | 119 ± 18 | 163 ± 28 | 128 ± 11 | 152 ± 14 | 145 ± 11 | 137 ± 16 |
| Spike half-width (msec) | 1.1 ± 0.04 | 1.1 ± 0.05 | 1.0 ± 0.03 | 1.1 ± 0.03 | 0.9 ± 0.1 | 1.1 ± 0.03 | 2.1 ± 0.1** |
| Spike initiation threshold (mV) | -33 ± 0.7 | -37 ± 2.5 | -30 ± 1.5 | -36 ± 2.0 | -37 ± 1.2 | -34 ± 2.5 | -41 ± 2** |
| Number of spikes fired | 5.0 ± 0.6 | 12.5 ± 2* | 9.7 ± 3* | 3 ± 1.2 | 1.7 ± 0.3** | 2.4 ± 0.6* | 2.6 ± 0.7** |
| AHP area (mV/sec) | 5.3 ± 0.8 | 3.8 ± 1.1 | 5.6 ± 3 | 3.8 ± 1 | 4.8 ± 3.1 | 3.9 ± 1 | 6.6 ± 2.2 |
|
n
|
49 |
8 |
3 |
3 |
10 |
11 |
14 |
p < 0.05; **p < 0.005 versus control. Control values are the average of all of the cells.
Voltage-dependent potassium currents have been shown recently to contribute to the discharge properties of central (Bekkers and Delaney, 2001; Dodson et al., 2002) and peripheral (Glazebrook et al., 2002) neurons. We therefore tested whether such currents might also contribute to spike frequency adaptation in lateral amygdala neurons. We initially tested the effects of the nonspecific potassium channel blocker 4-AP (Coetzee et al., 1999). Application of 4-AP (30–100 μm) broadened the action potential (Faber and Sah, 2002) and markedly reduced spike frequency adaptation (Fig. 2a,d). With a 400 pA current injection, 4-AP increased the number of evoked action potentials from 5.0 ± 0.6 to 10.6 ± 1.0 (p < 0.001; n = 36). At these concentrations, 4-AP blocks potassium channels containing Kv1.1, Kv1.2, Kv1.3, Kv1.5, Kv1.6, Kv3.1, and Kv3.2 subunits (Pongs, 1992; Coetzee et al., 1999). To determine which of these subunits were mediating the effects of 4-AP, we examined the actions of the potassium channel blockers α-DTX, DTX-K, tityustoxin-Kα, and TEA. α-DTX is a selective blocker of Kv1.1, Kv1.2, and Kv1.6 subunits (Pongs, 1992; Coetzee et al., 1999). As with 4-AP, application of α-DTX (100–500 nm) significantly increased the number of spikes evoked by a 400 pA current injection from a mean of 5.7 ± 0.9 to 12.5 ± 2.0 (p < 0.05; n = 8) (Fig. 2b). The reduction in spike frequency adaptation can be seen as both an increase in the number of action potentials evoked by a given current injection (Fig. 2d) and an increase in the frequency of action potential discharge (Fig. 2e). In contrast, DTX-K (100 nm), a selective blocker of channels containing Kv1.1 subunits (Wang et al., 1999), had no effect on spike frequency adaptation (3.0 ± 1.0 to 3.0 ± 1.2; n = 3; p > 0.05) (Table 1). However, tityustoxin-Kα (100 nm), a selective blocker of Kv1.2-containing potassium channels (Werkman et al., 1993; Rogowski et al., 1994), blocked spike frequency adaptation, increasing the number of spikes fired from 3.0 ± 0 to 9.7 ± 2.9 (n = 3; p < 0.05) (Table 1), without enhancing the slow AHP (Fig. 2c).
Figure 2.
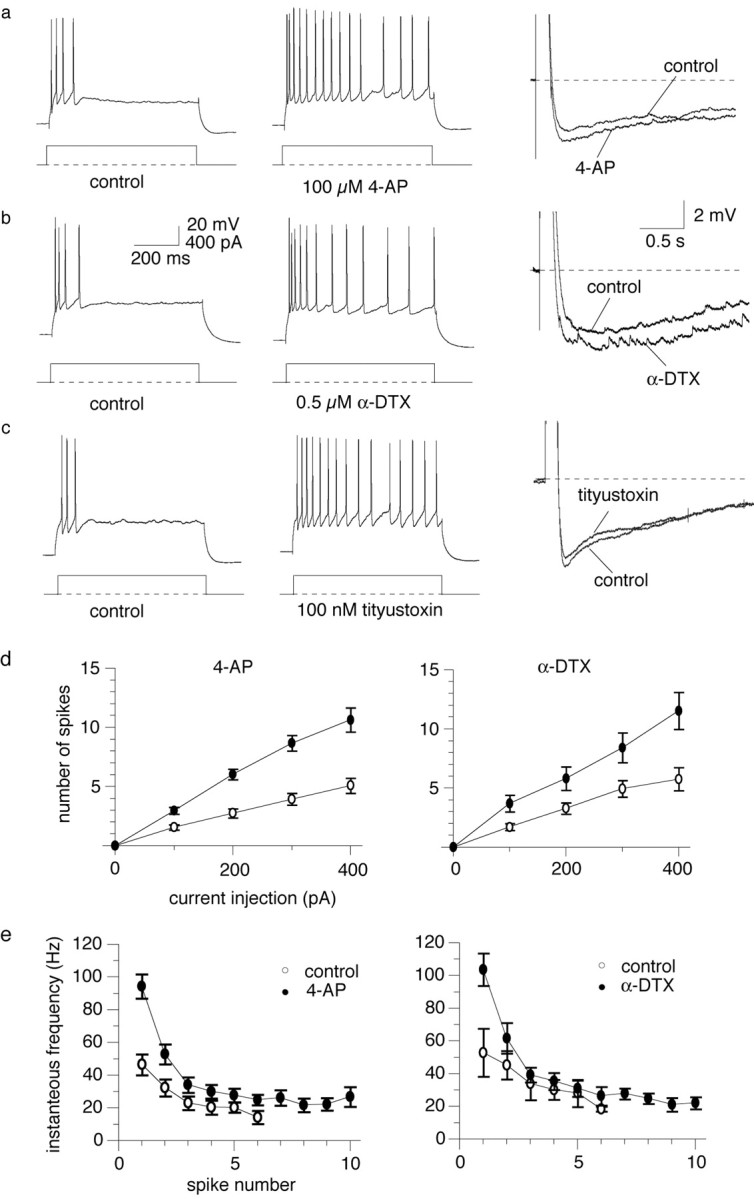
An α-dendrotoxin-sensitive potassium current contributes to spike frequency adaptation in addition to the slow AHP. a, Action potential train evoked by a 600 msec, 400 pA current injection (left) shows marked spike frequency adaptation. Application of 100 μm 4-AP greatly increases the number of evoked action potentials (middle trace) without depressing the slow AHP (right). b, Effects of 4-AP on spike frequency are mimicked by α-DTX. Right, As with 4-AP (a), α-DTX causes a small but insignificant enhancement of the slow AHP. c, The selective Kv1.2 subunit blocker tityustoxin-Kα (100 nm) causes a similar depression of spike frequency adaptation to α-DTX (left) without significantly effecting the slow AHP (right). d, Average data showing the increase in number of action potential-evoked depolarizing current injections in the presence of 4-AP (n = 19) or α-DTX (n = 24). e, Average data for the change in instantaneous frequency with spike number showing the reduction in spike frequency adaptation by 4-AP (n = 19) and α-DTX (n = 7). In control conditions, action potentials stop after six spikes, whereas in the presence of 4-AP or α-DTX, the cells continue to spike throughout the current injection.
TEA (1 mm) also had no effect on the firing patterns of principal lateral amygdala neurons (n = 14; data not shown), with a mean number of spikes fired at threshold of 1.14 ± 0.1 in both control and in the presence of TEA (n = 14). With larger current injections, fewer spikes were evoked in the presence of TEA as a result of the marked spike broadening (Faber and Sah, 2002) and a consequent increase in the AHP (data not shown). At this concentration, TEA has significant effects on K+ channels containing Kv1.1 and Kv1.6, whereas the IC50 for Kv1.2 subunits is >100 mm (Gomez-Hernandez et al., 1997). The pooled data summarizing the effects of morphine, DAMGO, α-DTX, tityustoxin-Kα, DTX-K, and TEA on lateral amygdala neurons is shown in Table 1. These results indicate that, in addition to the calcium-activated sIAHP, activation of voltage-dependent potassium currents also contributes to spike frequency adaptation in lateral amygdala neurons. Our pharmacological results suggest that the channels underlying this current likely contain Kv1.2 subunits (Pongs, 1992; Coetzee et al., 1999).
Potassium channels containing subunits sensitive to α-DTX and low concentrations of 4-AP are present in a number of cell types and form currents that activate rapidly at depolarized potentials and have slow inactivation kinetics (McCormick, 1991; Foehring and Surmeier, 1993; Locke and Nerbonne, 1997; Bekkers and Delaney, 2001). We next tested for the presence of such currents in lateral amygdala neurons. Under whole-cell voltage clamp, depolarizing voltage steps from a holding potential of –50 mV evoked large outward currents that were substantially reduced by application of α-DTX (100 nm; p < 0.001; n = 11) (Fig. 3a,b). The α-DTX-sensitive current activated rapidly and showed little inactivation over 450 msec. Activation plots of the α-DTX-sensitive current generated after converting to conductance (see Materials and Methods) were well fit by a Boltzmann function, with a V1/2 of 5.2 ± 4 mV and a slope of 12.8 ± 1.3 mV (n = 11) (Fig. 3c). The subthreshold activation (action potential threshold being –33 ± 1 mV; n = 49) and slow inactivation kinetics of the α-DTX-sensitive current make it ideally suited to modulate spike frequency adaptation.
Figure 3.

An α-DTX-sensitive current is present in LA pyramidal neurons. a, Whole-cell voltage-clamp recording showing the outward current activated by voltage steps from –70 to +40 mV in 10 mV increments from a holding potential of –50 mV. Application of 100 nm α-DTX markedly reduced the outward current. The α-DTX-sensitive current obtained by digital subtraction is shown on the right. b, Average current–voltage relationship of the α-DTX-sensitive potassium current for 11 neurons. Currents have been normalized to the current measured at +40 mV. c, The data from b has been converted to a conductance–voltage plot, and the data points have been fit with a Boltzmann function (see Materials and Methods). A V1/2 of 5.2 ± 4 mV and a slope of 12.8 ± 1 mV (n=11) for the α-DTX-sensitive current were obtained by averaging the Boltzmann fits of individual experiments.
In cortical pyramidal neurons, an α-DTX-sensitive potassium current is concentrated on the soma and proximal apical dendrites (Bekkers and Delaney, 2001). To examine the location of the α-DTX-sensitive current in lateral amygdala neurons, we tested for the presence of this current in nucleated somatic patches and outside-out patches excised from the apical dendrite. In somatic patches, depolarization activated a fast inactivating inward current, followed by an outward current (Fig. 4a–c). Coapplication of tetrodotoxin (1 μm) and α-DTX (100 nm) led to a reversible blockade of the inward current, demonstrating that it was a sodium current, but had no effect on the outward current in five of five patches (Fig. 4a–c). The mean outward current recorded in response to a 60 mV voltage step from a holding potential of –60 mV was 176 ± 40 pA in control and 177 ± 40 pA in the presence of α-DTX (p > 0.05; n = 5). In an additional two somatic patches in which α-DTX was applied alone, it also had no effect (data not shown).
Figure 4.

The α-DTX-sensitive current is located on the apical dendrite but not on the soma. a–c, Nucleated patches excised from a pyramidal neuron. Currents were evoked by a 400 msec, 60 mV voltage step from a holding potential of –60 mV every 10 sec. This voltage step evoked a fast inward current, followed by a sustained outward current. After obtaining a stable baseline, a combination of α-DTX (100 nm) and TTX (1 μm) were applied by a puffer pipette (arrows). a, The peak amplitudes of the inward current (open circles) and the sustained outward current (filled circles) are plotted for two successive applications of the drugs. b, The mean ± SEM data for the effect of α-DTX on the outward current in seven patches. c, The patch currents evoked on a slow time base are not blocked by α-DTX. The arrows indicate the inward currents (which have been truncated). The inset shows the inward currents evoked on a fast time base. d–f, Voltage-dependent currents evoked by depolarizing voltage steps in an outside-out patch excised from the apical dendrite. Traces on the left show currents under control conditions and after application of α-DTX. The α-DTX-sensitive current obtained by digital subtraction is shown on the right. The inset shows a time course of depression of the outward current evoked by a 60 mV step by puffer application of α-DTX to a dendritic patch. e, Current–voltage relationship of the mean α-DTX-sensitive current. Conductance–voltage plot averaged from six patches is shown in f. The data points have been fit with a Boltzmann function (see Materials and Methods). A V1/2 of –5.1 ± 6 mV and a slope of 11.1 ± 3 mV (n = 6) were obtained by averaging the Boltzmann fits of individual experiments.
In contrast, application of α-DTX (100 nm) to patches excised from the apical dendrite (10–100 μm from the soma) reversibly blocked the outward current in 11 of 19 patches to 40 ± 10% of control (p < 0.001; n = 11) (Fig. 4d). The dendritic α-DTX-sensitive current activated at subthreshold membrane potentials, similar to the whole-cell current, and showed little inactivation (Fig. 4d). Activation plots generated after conversion to conductance gave a V1/2 of –5.1 ± 6 mV and a slope of 11.1 ± 3 mV (Fig. 4c). These values are in good agreement with those measured for whole-cell currents and are within the reported ranges for similar currents in other cell types (McCormick, 1991; Foehring and Surmeier, 1993; Locke and Nerbonne, 1997; Bekkers and Delaney, 2001). These results demonstrate that the α-DTX-sensitive current in amygdala pyramidal neurons contributes to spike frequency adaptation and is located in the proximal apical dendrites but not on the soma.
Was the change in spike frequency adaptation by μ receptor activation attributable to modulation of the α-DTX-sensitive current? In whole-cell recordings, application of DAMGO consistently increased the amplitude of the outward current. The steady-state K+ current at 0 mV was enhanced by 32 ± 11% (p < 0.05; n = 9) (Fig. 5a) in the presence of DAMGO, an effect that was fully blocked by the antagonist naloxone (n = 4) (Fig. 5b). The DAMGO-sensitive current activated at membrane potentials positive to –40 mV and had a V1/2 of –8.6 ± 3 mV and slope of 5.8 ± 1 mV (n = 6) (Fig. 5d). Furthermore, when applied in the presence of α-DTX, DAMGO had no effect on the outward current (n = 4) (Fig. 5c). Consistent with its actions on the α-DTX-sensitive current, application of DAMGO in the presence of α-DTX (n = 3) or 4-AP (n = 5) had no effect on spike frequency adaptation (Fig. 6). The mean number of spikes fired at threshold in the presence of 4-AP or α-DTX was 3.3 ± 0.5 compared with 3.8 ± 0.6 after application of DAMGO (p > 0.05; n = 8). Together, these results show that the actions of DAMGO on the firing properties of these neurons result from enhancement of the α-DTX-sensitive outward current.
Figure 5.

DAMGO enhances the α-DTX-sensitive current through activation of μ-opioid receptors. a, Whole-cell outward currents evoked by 10 mV depolarizing steps from a holding potential of –60 mV, before and after application of DAMGO (0.5 μm). Note the increase in amplitude of the sustained outward current. b, Application of DAMGO has no effect in the presence of naloxone (1 μm; traces on right). c, Application of DAMGO after blockade of the α-DTX-sensitive current with α-DTX (100 nm) has no effect on the outward currents. d, A Boltzmann fit of the DAMGO-sensitive current. A V1/2 of –8.6 ± 3 mV and a slope of 5.8 ± 1 mV (n = 6) were obtained by averaging the Boltzmann fits of individual experiments. e, Average data showing the potentiating effect of DAMGO on outward currents. DAMGO, n = 9; DAMGO plus naloxone, n = 4, DAMGO plus α-DTX, n = 5. All values have been normalized to the peak current recorded before application of DAMGO. *p < 0.05.
Figure 6.

The effects of DAMGO on spike frequency adaptation are blocked by α-DTX and 4-AP. Trains of action potentials evoked by 600 msec depolarizing current injections before and after the application of either 100 nm α-DTX (a) or 100 μm 4-AP (b). Application of DAMGO (0.5 μm) in the presence of either α-DTX or 4-AP has no effect on spike frequency adaptation (traces on right).
We next examined the mechanism through which μ-opioid receptor activation enhances the α-DTX-sensitive current to increase spike frequency adaptation. Opioid receptors are coupled to pertussis-sensitive G-proteins, and their actions have most commonly been reported to result from either inhibition of adenyl cyclase or activation of inwardly rectifying potassium channels. To confirm that the actions of opioids in LA neurons resulted from activation of Gi/o receptors, we first tested the actions of NEM, an alkylating agent that selectively uncouples pertussis-sensitive G-proteins from receptors (Shapiro et al., 1994). Application of NEM (50 μm) completely blocked the actions of morphine on the firing properties. The number of spikes evoked by a 400 pA current injection was 4.6 ± 0.9 in control and 4.2 ± 0.5 in the presence of morphine and NEM (n = 4; p > 0.05) (Fig. 7a). Consistent with β-adrenergic receptor coupling to Gs (Stiles et al., 1984), NEM (50 μm) did not alter the effect of noradrenaline (10 μm) on the slow AHP or spike frequency adaptation (n = 3; data not shown).
Figure 7.

The enhancement of the α-DTX-sensitive current by μ-opioids is mediated by a pertussis toxin-sensitive G-protein and an arachidonic acid-mediated pathway. The enhancement of spike frequency adaptation by morphine is blocked by coapplication with NEM (a), AACOCF3 (b), and ETI (c). d, Arachidonic acid (300 μm; AA) produces a marked enhancement of spike frequency adaptation that is reversible on washout. e, Application of α-DTX blocks the effect of arachidonic acid on spike frequency adaptation (bottom traces). f, Arachidonic acid (300 μm) enhances the whole-cell outward current evoked by a 100 mV voltage step in a reversible manner. g, Summary of the effect of agents acting on the arachidonic acid pathway on the number of spikes evoked by a 400 pA, 600 msec current injection.
μ-Opioid receptor activation has been shown to block transmitter release via an α-DTX-sensitive mechanism by activation of an arachidonic acid-mediated pathway (Vaughan et al., 1997). Therefore, we investigated whether the phospholipase A2 (PLA2)–arachidonic acid pathway is involved in the actions of μ-opioids in the amygdala. We first tested the PLA2 inhibitor AACOCF3 (28 μm) (Balsinde et al., 1999). Application of AACOCF3 blocked the actions of morphine, with 3.0 ± 0.6 spikes evoked in control and 3.3 ± 0.9 in the presence of morphine and AACOCF3 (n = 3; p > 0.05) (Fig. 7b). We then tested whether application of arachidonic acid could mimic the actions of opioids. Arachidonic acid (300 μm) produced a marked enhancement of spike frequency adaptation, reducing the number of spikes evoked by a prolonged current injection (400 pA) from 2.5 ± 0.3 spikes to 0.75 ± 0.25 (p < 0.001; n = 4) (Fig. 7d). This effect was reversible on washout (n = 3) (Fig. 7d). The effect of arachidonic acid was precluded by previous perfusion with α-DTX, with the number of spikes evoked in the presence of α-DTX alone being 5.8 ± 1.8 compared with 6.0 ± 1.8 in the presence of α-DTX and arachidonic acid (n = 4; p > 0.05) (Fig. 7e). Furthermore, application of arachidonic acid enhanced the whole-cell outward current evoked by a 100 mV step to +40 mV in a reversible manner by 14.7 ± 2% (p < 0.001; n = 4) (Fig. 7d), similar to previous reports in CA1 pyramidal neurons (Colbert and Pan, 1999). These findings show that the effect of μ-opioid-mediated enhancement of spike frequency adaptation is mediated by arachidonic acid. Finally, coapplication of morphine with ETI (1 μm), a nonselective blocker of lipoxygenases, also blocked the action of morphine on firing properties. In the presence of these two agents, the number of spikes fired in response to a 400 pA current injection was 3.5 ± 1.0 compared with 3.0 ± 0.6 in control (n = 4; p > 0.05) (Fig. 7f).
These results show that μ-opioids reduce the number of action potentials evoked by prolonged current injections. To test opioid receptor modulation during more physiological stimulation, glutamate (200 μm) was pressure applied to the soma of lateral amygdala neurons in the presence of GABAergic blockers (100 μm picrotoxin and 1 μm CGP55845A) to evoke suprathreshold potentials. Bath application of morphine (1 μm) (Fig. 8) reduced the number of spikes evoked by puffed glutamate application from 3.2 ± 0.5 to 1.4 ± 0.4 (n = 5; p < 0.05) (Fig. 8a). This effect was blocked by coapplication of naloxone (1 μm) (control, 3.8 ± 1.1 spikes; morphine and naloxone, 3.5 ± 1.1 spikes; n = 4; p > 0.05) (Fig. 8b). In contrast, bath application of 4-AP (100 μm) increased the number of spikes evoked from 2.3 ± 0.3 to 4.3 ± 0.3 (n = 3; p < 0.01) (Fig. 8c). As expected, subsequent application of morphine (1 μm) in addition to 4-AP failed to reduce the number of spikes fired (4.3 ± 0.3 spikes evoked in the presence of 4-AP and morphine; n = 3; p > 0.05) (Fig. 8c).
Figure 8.

μ-Opioid activation enhances spike frequency adaptation of spikes evoked on transient depolarizing potentials. Supramaximal responses were evoked by puffer application of glutamate (200 μm; 50–100 msec) onto the soma of lateral amygdala neurons in the presence of CGP55845A (1 μm) and picrotoxin (100 μm). a, Bath application of morphine (1 μm) reduces the number of spikes evoked on the depolarizing potentials. b, Coapplication of naloxone (1 μm) with morphine blocks the reduction in spike initiation. c, 4-AP (100 μm) increases the number of spikes evoked and precludes the action of morphine on spike frequency adaptation. d, Summary of the effects of morphine, naloxone, and 4-AP on the number of spikes evoked by puffer glutamate application. *p < 0.05. e, Schematic diagram showing the pathway through which μ-opioid receptor activation enhances spike frequency adaptation. AA, Arachidonic acid.
Discussion
We have shown that pyramidal neurons in the lateral amygdala express an α-dendrotoxin-sensitive, voltage-dependent potassium current that plays a major role in controlling spike frequency adaptation. This current is not present on the soma but is located on the apical dendritic tree. Activation of μ-opioid receptors enhances the amplitude of the current by activation of an arachidonic acid–lipoxygenase second-messenger pathway and thus increases spike frequency adaptation. The effect of opioids in activating inwardly rectifying potassium channels and inhibiting voltage-dependent calcium channels are well known (Madison and Nicoll, 1988; Williams et al., 1988, 2001; Law et al., 2000). Our results demonstrate a new mechanism of opioid action in reducing the repetitive discharge properties of projection neurons in the lateral amygdala.
Spike frequency adaptation is thought to be primarily mediated by activation of a slow calcium-activated potassium conductance, sIAHP (Sah, 1996). This current is a target of modulation for a variety of neurotransmitters, virtually all of which reduce adaptation by blocking the sIAHP (Nicoll, 1988). We showed that a voltage-dependent potassium current also plays a major role in spike frequency adaptation and is upregulated by at least one class of neurotransmitter. This potassium current is not present on the soma but is selectively targeted to the apical dendrite and blocked by low concentrations of 4-AP, α-DTX, and tityustoxin-Kα, indicating that the underlying channels contain Kv1.2 subunits. Interestingly, the sIAHP current in hippocampal CA1 pyramidal neurons, which is the main determinant of spike frequency adaptation in these cells, has also been suggested to be located at sites distant to the soma (Sah and Bekkers, 1996; Bekkers, 2000). Thus, both currents that are involved in spike frequency adaptation appear to be targeted to the dendritic tree. It would be interesting to determine the location of the sIAHP current in amygdala neurons.
The cellular actions of opioid receptor activation are mediated by the Gi/o class of proteins that, in other cells, either inhibit adenyl cyclase or activate inwardly rectifying potassium channels. This leads to either hyperpolarization of neurons or inhibition of transmitter release at a number of synapses (Law et al., 2000; Williams et al., 2001). However, μ-opioid receptors are also coupled to phospholipase A2 and lead to generation of arachidonic acid (Fukuda et al., 1996). Three pathways have been described for the metabolism of arachidonic acid: the lipooxygenase pathway, the cyclooxgenase pathway, and the epoxygenase pathway, the metabolites of which are known to modulate potassium channels (Piomelli and Greengard, 1990). We showed that μ-opioid receptor activation in the lateral amygdala requires activation of PLA2 via a pertussis toxin-sensitive G-protein. The subsequent generation of arachidonic acid and activation of the lipoxygenase system enhances the α-DTX-sensitive potassium current (Fig. 8e). The V1/2 of the DAMGO-sensitive current is –8.6 mV compared with 5.2 mV for the α-DTX-sensitive current. This is significantly (p < 0.05) shifted to a more negative membrane potentials, indicating that the increase in amplitude of the α-DTX-sensitive current results from a change in the voltage dependence of this current.
In dissociated hippocampal neurons, μ-opioid receptor activation has been reported to potentiate a voltage-dependent potassium current that activated near –60 mV and showed little inactivation (Wimpey and Chavkin, 1991). However, the identity of this current and its physiological role were not determined. A slowly inactivating, voltage-dependent potassium current that is blocked by low concentrations of 4-AP was initially identified in hippocampal pyramidal neurons (Storm, 1988). This current was named ID, because of its action in delaying action potential initiation during long depolarizing current injections (Storm, 1988). Subsequently, potassium currents with similar kinetics and sensitivity to 4-AP but with variable biophysical properties have been shown to also be blocked by α-DTX in a number of different neurons (Wu and Barish, 1992; Foehring and Surmeier, 1993; Golding et al., 1999; Bekkers and Delaney, 2001). Our finding that μ-opioid receptor activation potentiates the α-DTX-sensitive current indicates that the current shown to be upregulated by μ receptors in hippocampal neurons (Wimpey and Chavkin, 1991) was likely to be an ID-like potassium current. Inhibition of transmitter release by opioids has been shown to be blocked by α-DTX at a number of synapses (Zoltay and Cooper, 1993; Simmons and Chavkin, 1996; Vaughan et al., 1997; Lambe and Aghajanian, 2001). Furthermore, the involvement of the arachidonic cascade has been shown to be required in some of these systems (Piomelli, 1994; Vaughan et al., 1997), suggesting that opioid modulation of this current is likely to be widespread.
Unlike inwardly rectifying potassium currents, the α-DTX-sensitive potassium current is not active at resting membrane potentials but requires depolarization for activation. Thus, the physiological actions of upregulation of this current are evident during brief depolarizations, such as during synaptic activation or after somatic puffer application of glutamate. Activation of this current may be the mechanism underlying the reduction in amygdala activity that has been observed during aversive emotional stimuli when μ receptors in the amygdala are occupied (Liberzon et al., 2002). The amygdala has long been implicated in “emotional” processing and particularly in mediating responses associated with fear and anxiety (LeDoux, 2000; Davis and Whalen, 2001). An accumulating body of evidence indicates that it also plays an important role in supraspinal mechanisms of antinociception (Helmstetter, 1992; Tershner and Helmstetter, 2000). Given the involvement of the arachidonic acid–lipoxygenase system in mediating these opioid actions in the amygdala, it is tempting to suggest that nociceptive pathways may share a common mechanism in different brain regions because this mechanism is also used in the periaqueductal gray matter (Christie et al., 2000). The amygdala has one of the highest concentrations of opioid receptors in the CNS, and activation of opioid receptors in the basolateral complex has been implicated in the role of the amygdala in the processing of fear (Good and Westbrook, 1995), defensive behavior (Shaikh et al., 1991), and nociception (Helmstetter et al., 1998; Kang et al., 1999; Manning et al., 2001). Our results show that activation of μ receptors in the lateral amygdala will greatly attenuate the output of these cells in response to depolarizing stimuli. This mechanism may underlie the antinociceptive and anxiolytic actions of opioid receptor activation in the basolateral amygdala.
Footnotes
This work was supported by recurrent funding from the John Curtin School of Medical Research. We thank Jeff Isaacson and Mac Christie for comments on this manuscript.
Correspondence should be addressed to Pankaj Sah, Queensland Brain Institute, University of Queensland, Brisbane QLD 4072, Australia. E-mail: pankaj.sah@uq.edu.au.
DOI:10.1523/JNEUROSCI.4496-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243031-09$15.00/0
References
- Atweh SF, Kuhar MJ (1977) Autoradiographic localization of opiate receptors in rat brain. III. The telencephalon. Brain Res 134: 393–405. [DOI] [PubMed] [Google Scholar]
- Balsinde J, Balboa MA, Insel PA, Dennis EA (1999) Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol 39: 175–189. [DOI] [PubMed] [Google Scholar]
- Bekkers JM (2000) Distribution of slow AHP channels on hippocampal CA1 pyramidal neurons. J Neurophysiol 83: 1756–1759. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Delaney AJ (2001) Modulation of excitability by α-dendrotoxin-sensitive potassium channels in neocortical pyramidal neurons. J Neurosci 21: 6553–6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Connor M, Vaughan CW, Ingram SL, Bagley EE (2000) Cellular actions of opioids and other analgesics: implications for synergism in pain relief. Clin Exp Pharmacol Physiol 27: 520–523. [DOI] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B (1999) Molecular diversity of K+ channels. Ann NY Acad Sci 868: 233–285. [DOI] [PubMed] [Google Scholar]
- Colbert CM, Pan E (1999) Arachidonic acid reciprocally alters the availability of transient and sustained dendritic K+ channels in hippocampal CA1 pyramidal neurons. J Neurosci 19: 8163–8171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Whalen PJ (2001) The amygdala: vigilance and emotion. Mol Psychiatry 6: 13–34. [DOI] [PubMed] [Google Scholar]
- Dodson PD, Barker MC, Forsythe ID (2002) Two heteromeric Kv1 potassium channels differentially regulate action potential firing. J Neurosci 22: 6953–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ESL, Sah P (2002) Physiological role of calcium-activated potassium currents in the rat lateral amygdala. J Neurosci 22: 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ESL, Callister RJ, Sah P (2001) Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro. J Neurophysiol 85: 714–723. [DOI] [PubMed] [Google Scholar]
- Foehring RC, Surmeier DJ (1993) Voltage-gated potassium currents in acutely dissociated rat cortical neurons. J Neurophysiol 70: 51–63. [DOI] [PubMed] [Google Scholar]
- Fukuda K, Kato S, Morikawa H, Shoda T, Mori K (1996) Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J Neurochem 67: 1309–1316. [DOI] [PubMed] [Google Scholar]
- Glazebrook PA, Ramirez AN, Schild JH, Shieh C-C, Doan T, Wible BA, Kunze DL (2002) Potassium channels Kv1.1, Kv1.2 and Kv1.6 influence excitability of rat visceral sensory neurons. J Physiol (Lond) 541: 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Jung HY, Mickus T, Spruston N (1999) Dendritic calcium spike initiation and repolarization are controlled by distinct potassium channel subtypes in CA1 pyramidal neurons. J Neurosci 19: 8789–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Hernandez JM, Lorra C, Pardo LA, Stuhmer W, Pongs O, Heinemann SH, Elliott AA (1997) Molecular basis for different pore properties of potassium channels from the rat brain Kv1 gene family. Pflügers Arch 434: 661–668. [DOI] [PubMed] [Google Scholar]
- Good AJ, Westbrook RF (1995) Effects of a microinjection of morphine into the amygdala on the acquisition and expression of conditioned fear and hypoalgesia in rats. Behav Neurosci 109: 631–641. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ (1992) The amygdala is essential for the expression of conditional hypoalgesia. Behav Neurosci 106: 518–528. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Tershner SA, Poore LH, Bellgowan PS (1998) Antinociception following opioid stimulation of the basolateral amygdala is expressed through the periaqueductal gray and rostral ventromedial medulla. Brain Res 779: 104–118. [DOI] [PubMed] [Google Scholar]
- Kang W, Wilson SP, Wilson MA (1999) Changes in nociceptive and anxiolytic responses following herpes virus-mediated preproenkephalin overexpression in rat amygdala are naloxone-reversible and transient. Ann NY Acad Sci 877: 751–755. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Aghajanian GK (2001) The role of Kv1.2-containing potassium channels in serotonin-induced glutamate release from thalamocortical terminals in rat frontal cortex. J Neurosci 21: 9955–9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH (2000) Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol 40: 389–430. [DOI] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Zubieta JK, Fig LM, Phan KL, Koeppe RA, Taylor SF (2002) μ-Opioid receptors and limbic responses to aversive emotional stimuli. Proc Natl Acad Sci USA 99: 7084–7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke RE, Nerbonne JM (1997) Three kinetically distinct Ca2+-independent depolarization-activated K+ currents in callosal-projecting rat visual cortical neurons. J Neurophysiol 78: 2309–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1984) Control of repetitive discharge of rat CA1 pyramidal neurones in vitro. J Physiol (Lond) 354: 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA (1988) Enkephalin hyperpolarizes interneurones in the rat hippocampus. J Physiol (Lond) 398: 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty NK, Sah P (1998) Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 394: 683–687. [DOI] [PubMed] [Google Scholar]
- Manning BH, Merin NM, Meng ID, Amaral DG (2001) Reduction in opioidand cannabinoid-induced antinociception in rhesus monkeys after bilateral lesions of the amygdaloid complex. J Neurosci 21: 8238–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA (1991) Functional properties of a slowly inactivating potassium current in guinea pig dorsal lateral geniculate relay neurons. J Neurophysiol 66: 1176–1189. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (1989) Involvement of hormonal and neuromodulatory systems in the regulation of memory storage. Annu Rev Neurosci 12: 255–287. [DOI] [PubMed] [Google Scholar]
- Nicoll RA (1988) The coupling of neurotransmitter receptors to ion channels in the brain. Science 241: 545–551. [DOI] [PubMed] [Google Scholar]
- Paden CM, Krall S, Lynch WC (1987) Heterogeneous distribution and upregulation of mu, delta and kappa opioid receptors in the amygdala. Brain Res 418: 349–355. [DOI] [PubMed] [Google Scholar]
- Paré D, Gaudreau H (1996) Projection cells and interneurons of the lateral and basolateral amygdala: distinct firing patterns and differential relation to theta and delta rhythms in conscious cats. J Neurosci 16: 3334–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli D (1994) Eicosanoids in synaptic transmission. Crit Rev Neurobiol 8: 65–83. [PubMed] [Google Scholar]
- Piomelli D, Greengard P (1990) Lipoxygenase metabolites of arachidonic acid in neuronal transmembrane signalling. Trends Pharmacol Sci 11: 367–373. [DOI] [PubMed] [Google Scholar]
- Pongs O (1992) Molecular biology of voltage-dependent potassium channels. Physiol Rev 72: S69–S88. [DOI] [PubMed] [Google Scholar]
- Rogowski RS, Krueger BK, Collins JH, Blaustein MP (1994) Tityustoxin K alpha blocks voltage-gated noninactivating K+ channels and unblocks inactivating K+ channels blocked by alpha-dendrotoxin in synaptosomes. Proc Natl Acad Sci USA 91: 1475–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P (1996) Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci 19: 150–154. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM (1996) Apical dendritic location of slow-afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of LTP. J Neurosci 16: 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh MB, Lu CL, Siegel A (1991) An enkephalinergic mechanism involved in amygdaloid suppression of affective defence behavior elicited from the midbrain periaqueductal gray in the cat. Brain Res 559: 109–117. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B (1994) Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. J Neurosci 14: 7109–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons ML, Chavkin C (1996) κ-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol 50: 80–85. [PubMed] [Google Scholar]
- Stiles GL, Caron MG, Lefkowitz RJ (1984) Beta-adrenergic receptors: biochemical mechanisms of physiological regulation. Physiol Rev 64: 661–743. [DOI] [PubMed] [Google Scholar]
- Storm JF (1988) Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature 336: 379–381. [DOI] [PubMed] [Google Scholar]
- Sugita S, North RA (1993) Opioid actions on neurons of rat lateral amygdala in vitro. Brain Res 612: 151–155. [DOI] [PubMed] [Google Scholar]
- Tershner SA, Helmstetter FJ (2000) Antinociception produced by mu opioid receptor activation in the amygdala is partly dependent on activation of mu opioid and neurotensin receptors in the ventral periaqueductal gray. Brain Res 865: 17–26. [DOI] [PubMed] [Google Scholar]
- Vaccarino AL, Olson GA, Olson RD, Kastin AJ (1999) Endogenous opiates: 1998. Peptides 20: 1527–1574. [DOI] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ (1997) How opioids inhibit GABA-mediated neurotransmission. Nature 390: 611–614. [DOI] [PubMed] [Google Scholar]
- Wang FC, Bell N, Reid P, Smith LA, McIntosh P, Robertson B, Dolly JO (1999) Identification of residues in dendrotoxin K responsible for its discrimination between neuronal K+ channels containing Kv1.1 and 1.2 alpha subunits. Eur J Biochem 263: 222–229. [DOI] [PubMed] [Google Scholar]
- Werkman TR, Gustafson TA, Rogowski RS, Blaustein MP, Rogawski MA (1993) Tityustoxin-K alpha, a structurally novel and highly potent K+ channel peptide toxin, interacts with the alpha-dendrotoxin binding site on the cloned Kv1.2 K+ channel. Mol Pharmacol 44: 430–436. [PubMed] [Google Scholar]
- Williams JT, Egan TM, North RA (1982) Enkephalin opens potassium channels on mammalian central neurones. Nature 299: 74–77. [DOI] [PubMed] [Google Scholar]
- Williams JT, North RA, Tokimasa T (1988) Inward rectification of resting and opiate-activated potassium currents in rat locus coeruleus neurons. J Neurosci 8: 4299–4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O (2001) Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 81: 299–343. [DOI] [PubMed] [Google Scholar]
- Wimpey TL, Chavkin C (1991) Opioids activate both an inward rectifier and a novel voltage-gated potassium conductance in the hippocampal formation. Neuron 6: 281–289. [DOI] [PubMed] [Google Scholar]
- Wu RL, Barish ME (1992) Two pharmacologically and kinetically distinct transient potassium currents in cultured embryonic mouse hippocampal neurons. J Neurosci 12: 2235–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoltay G, Cooper JR (1993) Dendrotoxin blocks a class of potassium channels that are opened by inhibitory presynaptic modulators in rat cortical synaptosomes and slices. Cell Mol Neurobiol 13: 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]