

Abstract
Protein kinase A (PKA) exerts a profound influence on axon extension during development and regeneration; however, the molecular mechanisms underlying these effects of PKA are not understood. Here, we show that DCC (deleted in colorectal cancer), a receptor for the axon guidance cue netrin-1, is distributed both at the plasma membrane and in a pre-existing intracellular vesicular pool in embryonic rat spinal commissural neurons. We hypothesized that the intracellular pool of DCC could be mobilized to the plasma membrane and enhance the response to netrin-1. Consistent with this, we show that application of netrin-1 causes a modest increase in cell surface DCC, without increasing the intracellular concentration of cAMP or activating PKA. Intriguingly, activation of PKA enhances the effect of netrin-1 on DCC mobilization and increases axon extension in response to netrin-1. PKA-dependent mobilization of DCC to the plasma membrane is selective, because the distributions of transient axonal glycoprotein-1, neural cell adhesion molecule, and trkB are not altered by PKA in these cells. Inhibiting adenylate cyclase, PKA, or exocytosis blocks DCC translocation on PKA activation. These findings indicate that netrin-1 increases the amount of cell surface DCC, that PKA potentiates the mobilization of DCC to the neuronal plasma membrane from an intracellular vesicular store, and that translocation of DCC to the cell surface increases axon outgrowth in response to netrin-1.
Keywords: exocytosis, growth cone, regeneration, cAMP, endocytosis, outgrowth
Introduction
The direction taken by an extending axon depends on extracellular cues, the repertoire of receptors for these cues on the axonal growth cone, and the state of signal transduction mechanisms within the growth cone. DCC (deleted in colorectal cancer) is a type I transmembrane protein and a receptor for netrins (Keino-Masu et al., 1996; Stein et al., 2001). Netrins are a family of secreted axon guidance cues that attract some axons and repel others (for review, see Yu and Bargmann, 2001). DCC is required for the attractant response to netrin-1 (de la Torre et al., 1997; Fazeli et al., 1997), whereas both DCC and the UNC5 homolog family of netrin receptors mediate chemorepellent responses to netrin-1 (Ackerman et al., 1997; Leonardo et al., 1997; Hong et al., 1999; Keleman and Dickson, 2001; Merz et al., 2001).
Protein kinase A (PKA) plays a key role regulating the response of axonal growth cones to netrin-1 (for review, see Song and Poo, 1999). Inhibition of PKA in neurons dissociated from either the embryonic Xenopus spinal cord (Ming et al., 1997) or retina (Hopker et al., 1999) switches their response to netrin-1 from attraction to repulsion. Further investigation of growth cone turning using Xenopus CNS neurons suggests that manipulation of phosphatidylinositol 3-kinase, phospholipase Cγ (Ming et al., 1999), MAP kinases (Forcet et al., 2002; Ming et al., 2002), protein synthesis (Campbell and Holt, 2001), and activity (Ming et al., 2001) can all influence the response of a growth cone to netrin-1.
Activating PKA also promotes axon growth in the presence of myelin-associated inhibitors of axon extension (Cai et al., 2001), including promoting axonal regeneration in the adult mammalian CNS after injury (Neumann et al., 2002; Qiu et al., 2002). Although PKA exerts profound effects on axon growth, the mechanisms underlying these actions during either neural development or axon regeneration are not known.
Here, we examined the role of PKA in the response of embryonic rat spinal commissural neurons to netrin-1. We report that DCC is normally present on both the neuronal surface and within an intracellular pool in these cells. We describe two mechanisms that regulate the amount of DCC on the neuronal plasma membrane. First, application of netrin-1 produced a modest increase in the amount of cell surface DCC. Netrin-1 did not increase the intracellular concentration of cAMP or activate PKA. Inhibiting PKA did not affect the netrin-1-induced increase in cell surface DCC or netrin-1-evoked axon outgrowth, providing evidence that PKA is not required for signaling downstream of netrin-1 in these cells. Activating PKA enhanced netrin-1-dependent insertion of DCC into the plasma membrane and increased axon outgrowth evoked by netrin-1. Inhibition of adenylate cyclase, PKA, or exocytosis, but not protein synthesis, blocked the PKA-induced increase in cell surface DCC, consistent with DCC being mobilized from a pre-existing intracellular vesicular pool. Activated PKA did not alter the distribution of other membrane proteins, such as transient axonal glycoprotein-1 (TAG-1), neural cell adhesion molecule (NCAM), or trkB, revealing a selective effect on the mobilization of DCC. These findings demonstrate that PKA activation potentiates the response to netrin-1 by translocating DCC to the plasma membrane.
Materials and Methods
Reagents. Polyclonal anti-trkB was provided by Dr. Louis Reichardt (University of California, San Francisco, CA). Polyclonal anti-TAG-1 (TG3) for Western blot analysis was provided by Dr. Thomas Jessell (Columbia University, New York, NY). Monoclonal anti-TAG-1 (4D7) for immunocytochemistry was obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Polyclonal anti-NCAM (AB5032) and anti-cAMP (AB306) were from Chemicon (Temecula, CA). Monoclonal DCC anti-DCCEX G92-13 and anti-DCCIN G97-449 were obtained from PharMingen (Mississauga, Canada), and anti-DCCFB AF5, tetanus toxin (TeTx) KT5720, and SQ22536 were from Calbiochem (La Jolla, CA). Anti-phospho cAMP response element-binding protein (CREB) (Ser133, 1B6, P-CREB) and anti-CREB were obtained from Cell Signaling Tech (Beverly, MA). Cyclohexamide (CHX), forskolin (FSK), poly-d-lysine (PDL), and 5′-N-ethylcarboxamidoadenosine (NECA) were obtained from Sigma-Aldrich (Oakville, Canada), Minimum Essential Medium was from BioWhittaker (Walkersville, MD), Neurobasal media and B27 supplement were from Invitrogen (Burlington, Canada), and Glutamax, inactivated fetal bovine serum (IFBS), and Penstrep were from Bio Media (Boussens, France). Recombinant netrin-1 protein was purified from a HEK293T cell line secreting netrin-1, as described (Serafini et al., 1994; Shirasaki et al., 1996).
Commissural neuron culture. Staged pregnant Sprague Dawley rats were obtained from Charles River (St. Constant, Canada). The dorsal half of embryonic day (E) 13 rat spinal cords were isolated by microdissection (Serafini et al., 1994) and dissociated to produce a suspension of single cells. In brief, dorsal spinal cords were incubated at 37°C for 30 min in 0.0002% DNase (Sigma-Aldrich) in Ca2+/Mg2+-free Hanks'solution (Invitrogen). The tissue was then triturated to yield a suspension of single cells.
Dissociated cells were plated and cultured for either 2 d (∼25,000 cells/well; growth cone analysis) or 6 d (∼50,000 cells/well; neurite analysis) in 24-well plates (Sarstedt, Quebec, Canada). Cells were grown in the wells on PDL-coated (70–150 kDa, 20 μg/ml) 12 mm round glass coverslips (number 0 Deckgläser; Carolina Biological, Burlington, NC). The first 24 hr, cells were cultured in Neurobasal media containing 10% IFBS, 2 mm glutamine, 1 U/ml penicillin, and 1 μg/ml streptomycin. The second day, the medium was changed to serum-free Neurobasal supplemented with 1% B27, 0.4 mm glutamine, 1 U/ml penicillin, and 1 μg/ml streptomycin. Inhibitors (1 mm SQ22536, 200 nm KT5720, 1.6 nm TeTx, and 100 μm CHX) or their respective vehicles were added to medium 15 min before the addition of netrin-1. Fifteen minutes later, the medium was supplemented with either 10 μm FSK or vehicle.
For Western blot analysis (Harlow and Lane, 1988), cells were plated and cultured for 6 d at a density of ∼250,000 cells per 35 mm PDL-coated tissue culture dish. After treatments, cells were washed once with ice-cold PBS, pH 7.4, and lysed with Laemmli sample buffer or radioimmunoprecipitation assay (RIPA) buffer (Barker and Shooter, 1994). Protein content was quantified using BCA (Pierce, Rockford, IL). Results were visualized using chemiluminescence (NEN Life Science Products, Boston, MA), and quantification was performed on scanned images of immunoblots (ScanJet 5300C; Hewlett Packard, Mississauga, Canada) using NIH Image software (National Institutes of Health, Bethesda, MD).
Immunocytochemistry. Cultures were washed with ice-cold PBS, pH 7.4, fixed with ice-cold 4% paraformaldehyde in PBS, pH 7.4, and blocked with 2% goat serum and 2% BSA in PBS, pH 7.4, for 2 hr at room temperature. Cells were permeabilized by using 0.1% Tween 20 in PBS (PBST) instead of PBS alone. Antibodies were used in blocking solution overnight at 4°C at the following dilutions: anti-DCCIN, 1:500; anti-DCCEX, 1:500; anti-TAG-1, 1:500; anti-Tau, 1:500; anti-trkBECD, 1:500; anti-cAMP, 1:1000. The binding specificities of anti-DCCIN and anti-DCCEX have been characterized (Reale et al., 1994; Shibata et al., 1996; Meyerhardt et al., 1999). Cultures were subsequently washed with PBS (nonpermeabilized cells) or PBST (permeabilized cells) and labeled with Cy2, Cy3, Alexa 488, or Alexa 546 secondary antibodies (Molecular Probes, Eugene, OR). Nuclei were stained with Hoechst 33258 (Sigma-Aldrich).
Quantification of surface receptor density or cAMP immunoreactivity. All micrographs used for quantification were taken using the same Axiovert microscope (Zeiss, Oberkochen, Germany), 100× objective lens, and exposure time to allow comparison of measurements. Fluorescence was quantified using Northern Eclipse image analysis software (Empix Imaging, Mississauga, Canada) by an observer blind to the experimental conditions. For image analysis of neurites or growth cones, both differential interference contrast and fluorescent images were taken. Fluorescence intensity per micrometer squared of the process was quantified and expressed as the mean ± SEM. Statistical significance was evaluated by a one-way ANOVA with a Sheffe's post hoc test (Systat, Chicago, IL).
Surface biotinylation. E13 dorsal spinal cords were dissociated, and commissural neurons were plated and cultured for 6 d at a density of ∼2,000,000 cells per 100 mm PDL-coated tissue culture dish. On day 6, cells were treated with 1 mm SQ22536, 200 nm KT5720, 1.6 nm TeTx, or vehicle for 15 min, followed by the addition of 50 ng/ml netrin-1 or vehicle to the culture media for 15 min. Neurons were exposed for 15 min to 10 μm FSK. Cells were then washed with ice-cold PBS containing 0.1 mm calcium chloride and 1 mm magnesium chloride, pH 7.4, to halt protein trafficking (Meyer-Franke et al., 1998). Surface biotinylation was performed by adding EZ-Link Sulfo-NHS-LC-biotin (Pierce), 5 ml per plate at 0.5 mg/ml in PBS at 4°C for 30 min (Lisanti et al., 1989), removed, and the reaction was quenched by the addition of 5 ml of 10 mm ice-cold glycine in PBS at 4°C for two 10 min periods. Subsequently, cells were washed twice with 5 ml of ice-cold PBS and lysed with RIPA buffer. Biotinylated proteins were precipitated with streptavidin–agarose (Pierce) and analyzed by Western blot.
Embryonic spinal cord explant culture. Dorsal spinal cord explants were dissected from E13 rat embryos (Serafini et al., 1994) and cultured for 16 hr in three-dimensional collagen gels (Tessier-Lavigne et al., 1988) at 37°C in Neurobasal, 10% IFBS, 2 mm glutamine, 1 U/ml penicillin, and 1 μg/ml streptomycin. Inhibitors (1 mm SQ22536, 200 nm KT5720, 1.6 nm TeTx, or 1–100 μm NECA), anti-DCCFB, or vehicle were added to the medium 15 min before the addition of netrin-1. After 15 min of treatment, the medium was supplemented with 10 μm FSK. All drugs were present throughout the experiment.
Segments of E11 rat spinal cord were dissected as described (Placzek et al., 1990), embedded in collagen, and cultured in Neurobasal containing 2% B27, 2 mm Glutamax I, 100 U/ml penicillin, and 100 μg/ml streptomycin for 40 hr. TAG-1 immunoreactivity was visualized as described (Kennedy et al., 1994).
Photomicrographs were taken using an Axiovert microscope (Zeiss), phase-contrast optics, a 20× objective lens, and a Magnafire CCD camera (Optronics, Goleta, CA) and analyzed using Northern Eclipse image analysis software (Empix Imaging). The total length of axon bundles or the length of TAG-1-immunopositive axons was measured and expressed as the mean ± SEM. Statistical significance of differences between means was evaluated by one-way ANOVA with Sheffe's post hoc test (Systat).
Results
Cell surface and intracellular pools of DCC
Little is known about the subcellular distribution of the DCC protein. To investigate this, we developed a dissociated cell culture enriched in embryonic spinal commissural neurons. Greater than 90% of the cultured cells derived from E13 rat dorsal spinal cord were TAG-1 positive (Fig. 1A), a marker for embryonic spinal commissural neurons (Dodd et al., 1988). Immunolabeling with anti-DCCIN, a monoclonal antibody raised against an intracellular epitope of DCC, showed that these neurons also express dcc (Fig. 1B). Immunocytochemical and Western blot analyses demonstrated that these cells do not express ChAT, a marker for motoneurons (data not shown). These findings indicate that these cultures are enriched with embryonic spinal commissural neurons. Confocal analysis of the distribution of DCC immunoreactivity revealed a punctate distribution of the DCC protein in the cytoplasm of the cell bodies and proximal neurites (Fig. 1B), consistent with a subset of the DCC protein being associated with an intracellular vesicular pool.
Figure 1.
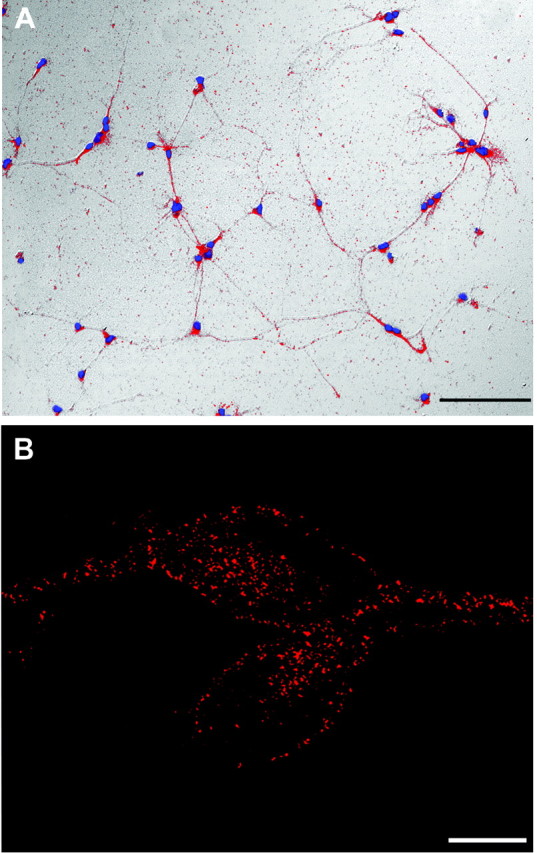
Embryonic spinal commissural neurons in vitro. A, Neurons derived from dissociated dorsal E13 rat spinal cord and cultured for 6 d in vitro. Cells were fixed, permeabilized, and immunostained for TAG-1 (red). Nuclei were stained with Hoechst 33258 dye (blue). Scale bar, 100 μm. B, Confocal microscopy revealed a punctate distribution of DCC immunoreactivity in the cytoplasm and outlining the surface of two commissural neurons cultured as in A (anti-DCCIN, Cy3 secondary). Scale bar, 5 μm.
To specifically visualize cell surface DCC protein, dissociated commissural neurons were cultured for 6 d, fixed, and then processed with or without detergent (0.1% Tween 20). To verify that the cells were not permeabilized in the absence of detergent, they were labeled with either anti-DCCIN or anti-DCCEX, a monoclonal antibody raised against the extracellular domain of DCC, and with polyclonal anti-Tau, a MAP and intracellular marker (Fig. 2). Anti-DCCIN and anti-Tau produced a signal only in permeabilized cells (Fig. 2A). Whereas, anti-DCCEX produced a signal in permeabilized and nonpermeabilized cells (Fig. 2C,D), the intensity of the signal in nonpermeabilized cells was much weaker than in permeabilized cells, consistent with the presence of an intracellular pool of the DCC protein. These findings suggested that much of the DCC protein expressed by commissural neurons is present intracellularly.
Figure 2.

Plasma membrane and intracellular DCC in commissural neurons. Dissociated commissural neurons grown 6 d in vitro (A–D). After fixation, cells in A and C were permeabilized with 0.1% Tween. Cells in B and D were not permeabilized. Cells were labeled with either anti-DCCIN (A, B), anti-DCCEX (C, D), or anti-Tau (A–D). A–D, Middle, Differential interference contrast (DIC) optics. Magnification, 100×. Scale bar, 25 μm.
Netrin-1 increases the amount of DCC on the surface of commissural neurons
We then determined whether netrin-1 alters the subcellular distribution of DCC, using cell surface biotinylation as an assay. Commissural neurons isolated from the E13 rat dorsal spinal cord were cultured for 6 d and then treated for 15 min with netrin-1 (50 ng/ml) or vehicle control. Cell surface proteins were then biotinylated, isolated using streptavidin–agarose beads, and examined by Western blot analysis using anti-DCCIN, anti-trkBECD, or anti-TAG-1. A single band was detected using anti-DCCIN (Fig. 3A) at the expected molecular weight of full-length DCC (∼180 kDa). The same band was detected using anti-DCCEX (data not shown). The addition of netrin-1 increased the amount of cell surface DCC in comparison with control (Fig. 3A). Under these conditions, the amount of either cell surface trkB or TAG-1 was not affected by the addition of netrin-1 (Fig. 3A).
Figure 3.

Netrin-1 increases DCC immunoreactivity at the cell surface. Dissociated commissural neurons were grown for 6 d in vitro before treatment for 15 min with 50 ng/ml netrin-1 or vehicle. A, Cell surface proteins were biotinylated and then isolated using streptavidin–agarose beads. The biotinylated proteins were analyzed by Western blot with antibodies directed against DCC, trkB, or TAG-1. B, Cells were fixed but not permeabilized, then immunostained with anti-DCCEX, anti-TAG-1, or anti-trkBECD, all against extracellular epitopes. Magnification, 100×. Scale bar, 25 μm.
The netrin-1-induced increase in cell surface DCC protein was then evaluated immunocytochemically. Commissural neurons were treated for 15 min with either 50 ng/ml netrin-1 or vehicle, fixed without permeabilization, and then immunostained with anti-DCCEX (Fig. 3B). Netrin-1 caused a significant increase in cell surface DCC immunoreactivity but had no effect on cell surface levels of TAG-1 or trkB (Fig. 3B; Table 1). Thus, netrin-1 selectively increased the amount of DCC on the neuronal surface.
Table 1.
DCC, TAG-1, and trkB immunofluorescence intensity (f.i./μm2)
|
|
Control |
Net |
FSK+Net |
SQ22536FSK+Net |
KT5720FSK+Net |
TeTxFSK+Net |
CHXFSK+Net |
|---|---|---|---|---|---|---|---|
| Neurites | |||||||
| DCC(EX) | 461 ± 80# | 1658 ± 102*# | 4699 ± 650* | 1470 ± 269*# | 1677 ± 201*# | 1236 ± 58*# | 5408 ± 612* |
| TAG-1 | 5456 ± 443 | 5123 ± 472 | 4681 ± 886 | 5345 ± 657 | 4993 ± 457 | 5234 ± 843 | 5033 ± 702 |
| trkB(ECD) | 6059 ± 962 | 5879 ± 934 | 6549 ± 425 | 6166 ± 420 | 5907 ± 637 | 6143 ± 762 | 5689 ± 678 |
| Growth cones | |||||||
| DCC(EX) | 239 ± 44# | 2034 ± 213*# | 6644 ± 728* | 1933 ± 275*# | 1433 ± 346*# | 1400 ± 423*# | 7623 ± 710* |
| TAG-1 |
7354 ± 266 |
7023 ± 354 |
7493 ± 455 |
8423 ± 1003 |
8439 ± 897 |
7799 ± 910 |
8067 ± 734 |
Levels of DCC, TAG-1, and trkB present at the cell surface were compared by quantitative immunofluorescence with antibodies raised against extracellular epitopes of these proteins. Results are expressed as the mean ± SEM of the neurite or growth cone surface fluorescence intensity (f.i.) (arbitrary units) (n = 6-14 per condition). Cells were treated with various inhibitors (1 mm SQ22536, 200 nm KT5720, 1.6 nm TeTx, or 100 μm CHX) or their respective vehicles for 15 min, after which 50 ng/ml netrin-1 or vehicle was added to the culture media for 15 min. *p < 0.01 versus control; #p < 0.01 versus 10 μm FSK plus 50 ng/ml netrin-1.
PKA activation stimulates DCC translocation to the plasma membrane
PKA influences the response of neuronal growth cones to netrin-1 (for review, see Song and Poo, 1999). Therefore, we determined whether PKA activation might influence the distribution of DCC. FSK activates adenylate cyclase, increases intracellular cAMP, and activates PKA (for review, see Nairn et al., 1985). Cells were treated with 10 μm FSK alone (Fig. 4A), 50 ng/ml netrin-1 alone (Fig. 3B), or 50 ng/ml netrin-1 in combination with 10 μm FSK (Fig. 4C) and then immunostained with anti-DCCEX to visualize DCC on the neuronal surface. FSK alone produced no change in DCC immunoreactivity (Fig. 4A,M) whereas netrin-1 (15 min) produced a modest increase (Fig. 4M,N). Substantially increased DCC immunoreactivity was detected in neurons treated with FSK and 50 ng/ml netrin-1 (Fig. 4C,M). Netrin-1 plus FSK did not increase cell surface immunoreactivity for TAG-1 (Fig. 4B,D,F,H,J,L; Table 1) or trkB (Table 1).
Figure 4.

DCC distribution is regulated by PKA and netrin-1. Dissociated commissural neurons, 6 d in vitro (A–L). Cells in C–L were treated with 50 ng/ml netrin-1; cells in A and B were not. Cells were exposed for 15 min to 10 μm FSK (A–L), 1 mm SQ 22536 (E,F), 200 nm KT5720 (G,H), 1.6 nm TeTx(I,J), or 100 μm CHX(K,L). After treatment, cultures were fixed without permeabilization and immunostained with anti-DCCEX (A, C, E, G, I, K) or anti-TAG-1 (B, D, F, H, J, L), both recognized extracellular epitopes. A–L, Cy2- or Cy3-conjugated secondary antibodies. Magnification, 100×. Scale bar, 25 μm. M, Quantification of DCC fluorescence intensity (mean ± SEM; n = 6–14 per condition). *p < 0.01 versus control or application of 10 μm FSK alone; #p < 0.01 versus 50 ng/ml netrin-1 in combination with 10 μm FSK. N, Effect of a 15 min application of netrin-1 alone on DCC immunofluorescence intensity. *p < 0.05 versus control. O, Western blot analysis of total cell extracts for phospho-CREB (P-CREB) and total CREB (∼43 kDa).
To investigate the mechanism regulating the increase in cell surface DCC, cultures of dissociated commissural neurons were exposed to different enzyme inhibitors 15 min before the addition of netrin-1, thus 30 min before the addition of FSK to the media. To confirm that FSK was acting by increasing adenylate cyclase activity, cells were treated with 1 mm SQ22536 (Fig. 4E,F), a specific inhibitor of adenylate cyclase (Fabbri et al., 1991; Goldsmith and Abrams, 1991; Tamaoki et al., 1993). SQ22536 blocked the increase in DCC surface immunoreactivity (Fig. 4E,M), consistent with the effect of FSK being attributable to adenylate cyclase activation. To confirm that cAMP produced by the adenylate cyclase was acting through PKA, commissural neurons were pretreated with 200 nm KT5720, a specific inhibitor of PKA (Kase et al., 1987). KT5720 blocked the increase in DCC surface immunoreactivity produced by FSK and netrin-1 (Fig. 4G,M). FSK-induced PKA activation was monitored by assaying phosphorylation of the PKA substrate CREB. Treatment of commissural neurons with FSK increased CREB phosphorylation, which was blocked by SQ22536 and KT5720 (Fig. 4O). These findings indicate that PKA activation is essential for the increase in cell surface DCC induced by FSK in the presence of netrin-1.
We then tested the hypothesis that recruitment from an intracellular store might contribute to the increase in plasma membrane DCC using TeTx, an inhibitor of exocytosis that acts by cleaving soluble vesicle-associated N-ethylmaleimide-sensitive factor attachment protein receptors (v-SNAREs) (Schiavo et al., 1992). TeTx (1.6 nm) blocked the FSK-induced increase in surface DCC immunoreactivity (Fig. 4I,M), consistent with the increase requiring exocytosis. The rapid increase in DCC protein on the neuronal surface, as early as 15 min after the addition of FSK and netrin-1, suggests that it is unlikely to be accounted for by increased transcription or translation of DCC mRNA. Consistent with this, a 15 min application of 100 μm CHX, a concentration sufficient to block protein synthesis (Twiss and Shooter, 1995), had no effect on the increase in cell surface DCC induced by FSK and netrin-1 (Fig. 4 K, M). Insertion of locally translated protein into the plasma membrane has been detected in axonal growth cones (Brittis et al., 2002). Furthermore, Campbell and Holt (2001) provide evidence that protein synthesis is required for netrin-1-dependent growth cone turning. Our results indicate that a pre-existing intracellular pool of DCC protein is present in the neuron, that activation of PKA promotes the insertion of DCC into the plasma membrane, and that this recruitment of DCC occurs through a protein synthesis-independent mechanism. Therefore, we conclude that the PKA-dependent increase in cell surface DCC is not the protein synthesis-sensitive step described by Campbell and Holt (2001).
DCC insertion into the growth cone plasma membrane
We then examined the effect of PKA activation on the distribution of the DCC protein on the surface of commissural neuron growth cones. Consistent with the findings presented above, treatment with FSK and netrin-1 increased DCC immunoreactivity on the surface of growth cones, netrin-1 alone produced a smaller increase, and FSK alone had no effect. FSK and netrin-1 had no effect on cell surface immunoreactivity for TAG-1 (Fig. 5; Table 1). Inhibition of adenylate cyclase (1 mm SQ22536) or PKA (200 nm KT5720) blocked the increase in DCC surface immunoreactivity induced by FSK and netrin-1 (Fig. 5), demonstrating that FSK produces this effect via the adenylate cyclase and activation of PKA. In contrast, inhibition of protein synthesis using 100 μm CHX did not affect the increased DCC on the growth cone surface (Fig. 5B). Application of 1.6 nm TeTx reduced the increase in cell surface DCC caused by FSK and netrin-1 to the level induced by netrin-1 alone (Fig. 5). These findings support the conclusion that cAMP elevation potentiates the translocation of DCC to the plasma membrane of neuronal growth cones.
Figure 5.
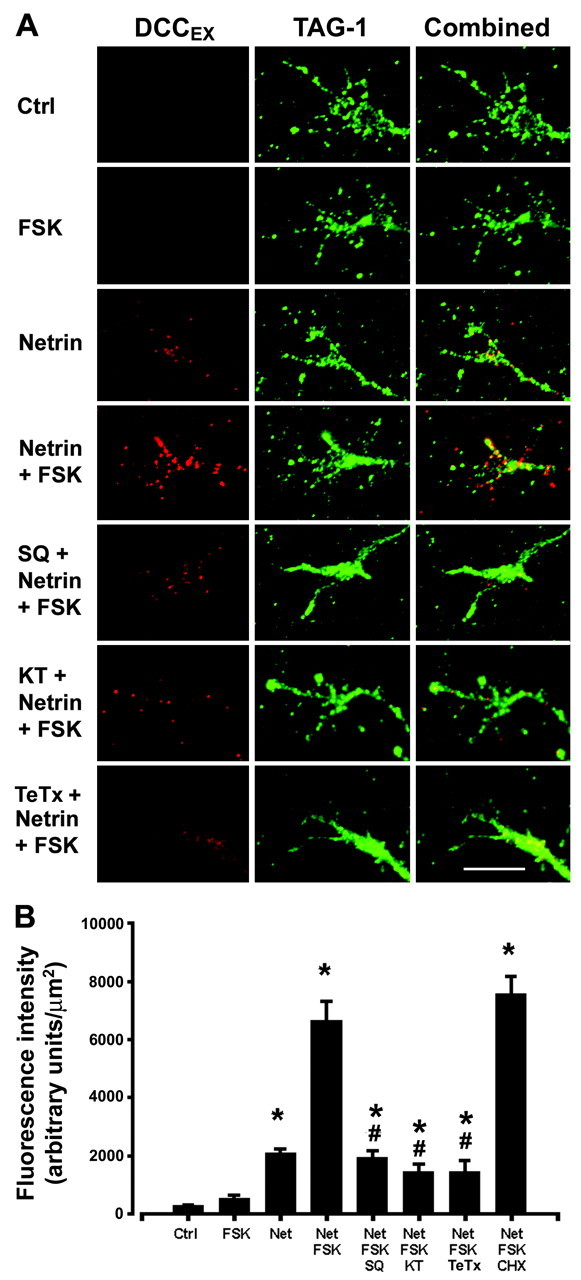
Distribution of DCC in growth cones is regulated by PKA and netrin-1. A, Dissociated commissural neurons were cultured for 2 d in vitro before treatment for 15 min with or without 50 ng/ml netrin-1. FSK (10 μm) was then added for 15 min in combination with 1 mm SQ22536, 200 nm KT5720, or 1.6 nm TeTx. Cells were fixed without permeabilization and immunostained with anti-DCCEX or anti-TAG-1 (Cy3- or Alexa 488-conjugated secondary antibodies). Magnification, 100×. Scale bar, 10 μm. B, Quantification of DCC fluorescence intensity. Values represent the mean ± SEM (n = 6–8 per condition). *p < 0.01 versus control or 10 μm FSK alone; #p < 0.01 versus netrin-1 (50 ng/ml) plus FSK (10 μm).
Netrin-1 does not activate PKA in commissural neurons and increases cell surface DCC by a PKA-independent mechanism
Immunocytochemical evidence obtained using cultured Xenopus retinal neurons suggests that netrin-1 elevates the concentration of cAMP in neurons (Hopker et al., 1999), raising the possibility that netrin-1 itself might promote DCC translocation by activating PKA. To test this hypothesis, cultures of embryonic rat spinal commissural neurons were treated with 50 ng/ml or 200 ng/ml netrin-1, or 10 μm FSK as a positive control, for either 5 or 15 min. Western blot analysis indicated that application of netrin-1 alone produced no detectable change in PKA-dependent phosphorylation of CREB, whereas the expected FSK-induced increase in phospho-CREB was readily detectable (Fig. 6A). Western blot analysis monitors global changes in CREB phosphorylation throughout the cell and may not detect localized changes in PKA activation. To determine whether netrin-1 might regulate cAMP concentration locally, we examined the level of cAMP in the growth cones of commissural neurons immunocytochemically. Consistent with the results of Western blot analysis, 10 μm FSK added to the media increased cAMP immunoreactivity in growth cones, whereas 50 ng/ml or 200 ng/ml netrin-1 did not (Fig. 6B). Furthermore, the increase in cell surface DCC triggered by the addition of netrin-1 alone was not blocked by inhibition of either the adenylate cyclase or PKA (Fig. 4N). These findings indicate that netrin-1 does not increase the concentration of cAMP or activate PKA in embryonic rat spinal commissural neurons and support the conclusion that the netrin-1-induced increase in DCC at the cell surface occurs through a PKA-independent mechanism.
Figure 6.

Netrin-1 does not activate PKA in embryonic rat spinal commissural neurons. Dissociated commissural neurons were cultured for 2 d in vitro (B) or 6 d in vitro (A) before treatment for 5 or 15 min with 50 or 200 ng/ml netrin-1 or 10 μm FSK. A, Western blot analysis of total cell extracts for phospho-CREB (P-CREB) and total CREB (∼43 kDa). The histograms illustrate quantification of the optical density of CREB phosphorylation. In B, cells were fixed, permeabilized, and immunostained with anti-cAMP (Alexa 546-conjugated secondary antibody). Magnification, 100×. Scale bar, 10 μm. The histograms illustrate quantification of cAMP fluorescence intensities. Values represent the mean ± SEM (n = 3 and 16 per condition, respectively, for A and B). *p < 0.01 versus control.
The adenosine A2b receptor activates the adenylate cyclase (for review, see Ralevic and Burnstock, 1998). Increasing the concentration of intracellular cAMP by activating A2b has been used to modulate the response to netrin-1 (Shewan et al., 2002). Furthermore, evidence has been provided that A2b is a receptor for netrin-1 (Corset et al., 2000). Therefore, we tested the hypothesis that activating A2b might enhance the response to netrin-1 in commissural neurons. Consistent with evidence indicating that A2b is not expressed by embryonic rat commissural neurons (Stein et al., 2001), we found that the adenosine receptor agonist NECA does not affect netrin-1-induced commissural axon outgrowth (supplemental Fig. 1; available at www.jneurosci.org), supporting the conclusion that A2b does not contribute to the response to netrin-1 in these cells.
PKA activation produces a netrin-1-dependent increase in cell surface DCC via a mechanism that requires exocytosis
The increased cell surface DCC detected immunocytochemically could be produced either by a selective increase in the amount of DCC protein on the cell surface, or by clustering DCC protein present more diffusely on the surface before treatment. To differentiate between these two possibilities, cell surface DCC protein was assessed directly by biotinylating cell surface proteins and quantifying the relative amount of DCC on the neuronal surface in different conditions. Dissociated commissural neurons were cultured for 6 d, and the cells were treated for 15 min with SQ22536, KT5720, or TeTx. Netrin-1 (50 ng/ml) was added to the culture media for 15 min, and cultures were then exposed to 10 μm FSK for 15 min. Cell surface proteins were then biotinylated, isolated using streptavidin–agarose beads, and examined by Western blot analysis using anti-DCCIN, anti-TAG-1, anti-NCAM and anti-trkBECD. A single ∼180 kDa band was detected by anti-DCCIN (Fig. 7). Analysis of biotinylated proteins indicated that netrin-1 in combination with FSK produced a 10-fold increase in the amount of cell surface DCC compared with control (Fig. 7). Pretreatment with SQ22536, KT5720, or TeTx before netrin-1 and FSK significantly reduced the level of cell surface DCC, when compared with netrin-1 plus FSK. Inhibition of protein synthesis with CHX did not affect the induced increase in cell surface DCC (data not shown). Under the same conditions, the amount of biotinylated trkB, NCAM, or TAG-1 was not affected by cAMP elevation. Nor did we detect a change in nonbiotinylated DCC protein, consistent with a relatively small amount of the total DCC being on the cell surface (Fig. 7)
Figure 7.
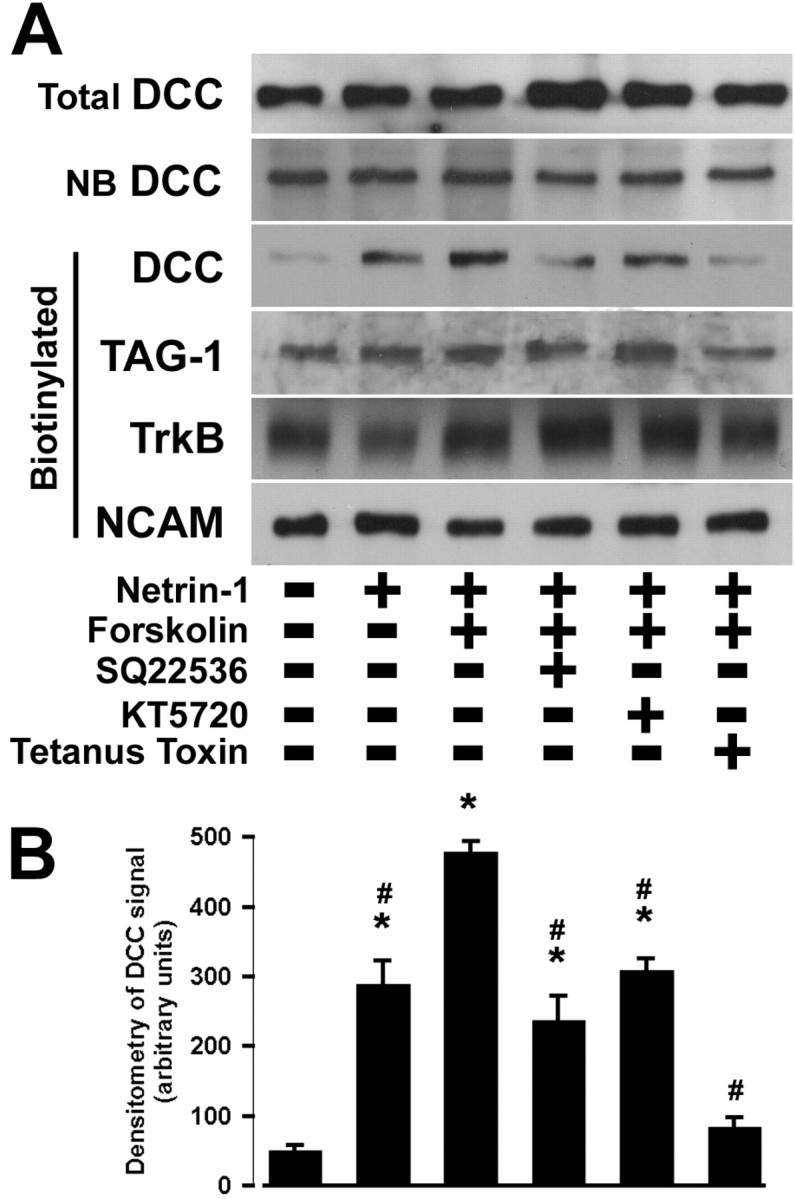
Netrin-1 and FSK increase cell surface DCC. Dissociated commissural neurons were cultured for 6 d in vitro before treatment for 15 min with or without 50 ng/ml netrin-1. FSK (10 μm) was then added for 15 min in combination with 1 mm SQ22536, 200 nm KT5720, or 1.6 nm TeTx. Cell surface proteins were biotinylated and isolated. A, Total, nonbiotinylated, and biotinylated proteins were analyzed by Western blot with antibodies directed against either DCC (∼180 kDa), TAG-1 (∼135 kDa), trkB (∼145 kDa), or NCAM (∼200 kDa). B, Quantification of the optical density of biotinylated DCC immunoreactivity. Values are the mean ± SEM (n = 4 per condition). *p < 0.01 versus control; #p < 0.01 versus 50 ng/ml netrin-1 in combination with 10 μm FSK.
PKA-dependent exocytosis promotes netrin-1-induced commissural axon outgrowth
Netrin-1 evokes commissural axon outgrowth from explants of embryonic dorsal spinal cord cultured in a three-dimensional collagen gel (Kennedy et al., 1994; Serafini et al., 1994). We tested the hypothesis that manipulation of PKA activation would cause DCC protein to be recruited to the surface of commissural axons and promote netrin-1-dependent axon outgrowth. Explants of E13 rat dorsal spinal cord were cultured in the presence of FSK (10 μm) alone or FSK (10 μm) and netrin-1 (50 ng/ml). At this concentration, netrin-1 alone evoked ∼30% of maximal commissural axon outgrowth (data not shown). After 16 hr of culture, FSK alone did not enhance axon outgrowth (Fig. 8B,J). In contrast, FSK (10 μm) plus netrin-1 (50 ng/ml) (Fig. 8D,J) produced a dramatic increase in axon outgrowth compared with explants exposed to netrin-1 alone (Fig. 8C,J). In all cases, extending axons express TAG-1, a marker for commissural axons (data not shown) (Dodd et al., 1988).
Figure 8.

PKA activation enhances netrin-1-dependent commissural axon outgrowth. A–I, E13 rat dorsal spinal cord axon outgrowth assays: control (A); 10 μm FSK (B); 50 ng/ml netrin-1 (C); 50 ng/ml netrin-1 plus 10 μm FSK (D); 50 ng/ml netrin-1, 10 μm FSK, and 1 mm SQ 22536 (E); 50 ng/ml netrin-1, 10 μm FSK, and 200 nm KT5720 (F); 50 ng/ml netrin-1, 10 μm FSK, and 1.6 nm TeTx (G); 50 ng/ml netrin-1, 10 μm FSK, and 30 ng/ml anti-DCCFB (H); 50 ng/ml netrin-1, 10 μm FSK, and 10 μg/ml anti-DCCFB (I). Magnification, 20×. Scale bar, 100 μm. J, Quantification of axon outgrowth shown in A–I. *p < 0.01 versus control; #p < 0.01 versus 50 ng/ml netrin-1 plus 10 μm FSK. K, Quantification of the effect of increasing concentrations of anti-DCCFB in the presence of netrin-1 and FSK. *p < 0.01 versus 50 ng/ml netrin-1 plus 10 μm FSK. J and K show the mean total axon bundle length per explant ± SEM for between 4 and 26 explants per condition.
To determine whether FSK acts via the adenylate cyclase and PKA, explants of dorsal spinal cord were exposed to different enzyme inhibitors 15 min before the addition of netrin-1, thus 30 min before the addition of FSK, and then cultured for an additional 16 hr. SQ22536 completely blocked the increase in axon outgrowth caused by FSK in the presence of netrin-1 (Fig. 8E,J). KT5720 blocked the effect of FSK, demonstrating that PKA activation is required to produce the netrin-1-dependent increase in axon outgrowth evoked by FSK (Fig. 8F,J). We then examined whether the increase in cell surface DCC requires v-SNARE-dependent exocytosis. Importantly, TeTx-induced cleavage of v-SNAREs does not block axon extension because the neuronal v-SNAREs that are required for axon outgrowth are insensitive to TeTx (Osen-Sand et al., 1996; Martinez-Arca et al., 2001). Treatment with 1.6 nm TeTx (16 hr) (Fig. 8G,J) reduced axon outgrowth to the level found in the presence of netrin-1 alone, consistent with the increased outgrowth caused by netrin-1 and FSK requiring exocytosis. Notably, the inhibitors used reduced outgrowth to the level evoked by netrin-1 alone, suggesting that outgrowth evoked by netrin-1 alone does not require TeTx-sensitive exocytosis.
Increased axon outgrowth evoked by FSK and netrin-1 requires DCC
The results described above suggest that FSK activates PKA, potentiating netrin-1-dependent outgrowth of commissural axons via a mechanism that requires exocytosis. To determine whether the increased axon outgrowth caused by netrin-1 and FSK required DCC, dorsal spinal cord explants were exposed to increasing concentrations of the DCC function blocking monoclonal antibody (anti-DCCFB; from 30 ng/ml to 10 μg/ml) 15 min before the addition of netrin-1, thus 30 min before the addition of FSK to the media, and then cultured for 16 hr. Anti-DCCFB has been reported to block netrin-1-dependent commissural axon outgrowth in vitro at a concentration of 10 μg/ml (Keino-Masu et al., 1996). In the presence of FSK and netrin-1, anti-DCCFB blocked axon outgrowth in a concentration-dependent manner (Fig. 8H,I,K). The same concentrations of nonimmune mouse IgG had no effect (data not shown), indicating that the increased netrin-1-dependent axon outgrowth induced by FSK requires DCC.
PKA modulates DCC-dependent axon extension to the ventral midline of the embryonic spinal cord
Our findings predict that cell surface levels of DCC are coregulated by netrin-1 and PKA. To determine whether this contributes to commissural axon extension to the floor plate in the embryonic spinal cord, we used a semi-intact explant preparation. Segments of E11 rat brachial spinal cords, approximately three somites long, were isolated and embedded in collagen (Fig. 9A,B), and the length of extending TAG-1-immunoreactive commissural axons were quantified. Commissural axons in control explants, cultured for 40 hr, followed their normal trajectory to the floor plate (Fig. 9A). Consistent with the results of assaying axon outgrowth into collagen (Fig. 8), activating PKA with FSK significantly increased the length of commissural axons extending within explanted spinal cords (Fig. 9A,C,D). Furthermore, inhibiting PKA with KT5720 significantly reduced axon extension to the floor plate, suggesting that endogenous PKA activity normally facilitates axon growth in the embryonic spinal cord. In contrast, inhibiting PKA with KT5720 does not reduce axon outgrowth into collagen below the level evoked by netrin-1 alone (Fig. 8), suggesting that the neuroepithelium may contain a PKA agonist that is not present when the axons grow into collagen. Anti-DCCFB (10 μg/ml) significantly reduced axon extension to the floor plate. Furthermore, application of KT5720 or 10 μg/ml anti-DCCFB to the explants 15 min before the addition of FSK significantly reduced the FSK-induced increase in axon extension (Fig. 9A,C,D). These findings indicate that the enhancement of commissural axon extension in the embryonic spinal cord caused by activating PKA requires cell surface DCC.
Figure 9.

PKA regulates axon extension to the ventral midline of the embryonic spinal cord in a DCC-dependent manner. A, Brachial segments of E11 rat dorsal spinal cords were embedded in collagen and cultured for 40 hr in the following conditions: control; 200 nm KT5720; 10 μg/ml anti-DCCFB; 10 μm FSK; 10 μg/ml anti-DCCFB + 10 μm FSK. TAG-1 immunoflorescence (black and white reversed image). Magnification, 10×. Scale bar, 100 μm. B, Diagram of an E11 spinal cord explant illustrating commissural neuron cell bodies in the dorsal (D) spinal cord extending an axon ventrally (V) to the floor plate (FP). RP, Roof plate; R, rostral; C, caudal. C, Quantification of axon bundle length (mean ± SEM for 183–548 axons per condition). D, Quantification of the percentage of axons reaching the floor plate (mean ± SEM for 6–10 explants per condition). *p < 0.01 versus control; #p < 0.01 versus 10 μm FSK.
Discussion
The findings reported here indicate that post-translational recruitment of DCC to the cell surface from an intracellular pool regulates the response of axons to netrin-1. Application of netrin-1 alone produced a modest increase in the amount of DCC on the neuronal surface. Activation of PKA coincident with theaddition of netrin-1 potentiated the insertion of DCC into the plasma membrane and increased axon outgrowth. Blocking DCC function significantly reduced the effect of PKA activation in each assay used. Furthermore, inhibiting PKA in explanted intact embryonic spinal cord assays reduced axon extension (Fig. 9), providing evidence that the embryonic spinal neuroepithelium may contain an endogenous PKA agonist. These results identify a novel modulatory role for PKA in the growth cone, regulating the presentation of DCC and thereby enhancing the extension of commissural axons in response to netrin-1.
Netrin-1 does not activate PKA in commissural neurons nor is PKA activation required for commissural axon outgrowth evoked by netrin-1
Previous studies performed using either Xenopus retinal neurons or spinal neurons grown in dispersed cell culture indicate that the intracellular level of cAMP plays a key role in determining whether a growth cone responds to netrin-1 as an attractant or a repellent (Ming et al., 1997; Hopker et al., 1999; Nishiyama et al., 2003). Low levels of intracellular cAMP correlate with a repellent response, whereas high levels of cAMP, and presumably activation of PKA, correlate with an attractant response. Netrin-1 itself has been reported to increase the concentration of intracellular cAMP in Xenopus retinal ganglion cell growth cones in vitro (Hopker et al., 1999). cAMP immunofluorescence in retinal ganglion cell growth cones supports this conclusion, but additional biochemical data were not provided. On the basis of these studies, models of netrin-1 signal transduction place activation of PKA directly downstream of DCC (Song and Poo, 1999; Nishiyama et al., 2003).
In contrast, our findings provide direct evidence that netrin-1 does not elevate intracellular cAMP or activate PKA in embryonic rat spinal commissural neurons. Furthermore, they indicate that activating PKA is not required for netrin-1-evoked axon outgrowth. This conclusion is based on the finding that application of netrin-1, while inhibiting adenylate cyclase (SQ22536) (Fig. 8) or PKA (KT5720) (Fig. 8), did not reduce axon outgrowth below the level produced by netrin-1 alone. The finding that PKA inhibition does not affect netrin-1-evoked commissural axon outgrowth (Fig. 8) also appears to be at odds with Ming et al. (1997) and Nishiyama et al. (2003), who reported that reduced levels of cAMP in cultured Xenopus spinal neurons causes growth cones to be repelled by netrin-1. Additional analysis will be required to determine whether these differences are attributable to the species, cell types, or methodologies used. However, based on our findings, we conclude that current models do not provide a widely generalizable description of the neuronal response to netrin-1.
We conclude that a major effect of activating PKA on netrin-1-induced commissural axon outgrowth is to potentiate translocation of DCC to the plasma membrane. We do not rule out a role for PKA exerting other effects on axon extension; however, our conclusion is supported by the finding that blocking DCC function dramatically reduced both the FSK-induced enhancement of netrin-1-dependent axon outgrowth into collagen (Fig. 8) and the FSK-induced enhancement of axon extension toward the floor plate in the explanted embryonic spinal cord (Fig. 9). These results indicate that a major component of the effect of FSK on netrin-1-evoked axon outgrowth requires DCC.
Interestingly, different mechanisms may underlie the increase in cell surface DCC caused by netrin-1 alone and the recruitment of DCC triggered by netrin-1 and activation of PKA. Notably, the increase induced by netrin-1 alone was not blocked by inhibiting PKA or blocking TeTx-sensitive exocytosis. In contrast, the increase in cell surface DCC triggered by netrin-1 and PKA activation was blocked by TeTx. Similarly, application of TeTx or inhibiting PKA did not affect commissural axon outgrowth in response to netrin-1 but reduced the PKA-induced increase in axon outgrowth to the level produced by netrin-1 alone (Fig. 8). These findings suggest that a TeTx-sensitive v-SNARE protein is required for PKA-dependent translocation of DCC but is not required for the increase in cell surface DCC caused by netrin-1 alone.
Specificity of DCC translocation
There are multiple examples of the elevation of intracellular cAMP causing the translocation of proteins from an intracellular vesicular store to the plasma membrane. These include transporters (Yao et al., 1996), ion pumps (Schwartz and Al Awqati, 1986), ion channels (Barres et al., 1989), and trophic factor receptors (Meyer-Franke et al., 1998). Here, both immunoflorescent and surface biotinylation analyses indicate a surprising level of specificity in the PKA-dependent increase in DCC at the cell surface. We hypothesize two mechanisms that may account for this specificity. First, commissural neurons may contain vesicles that specifically traffic DCC to the cell surface. A second, but not necessarily mutually exclusive, mechanism is suggested by the finding that increased cell surface DCC requires the presence of netrin-1. Activation of PKA alone produced no detectable increase in plasma membrane DCC (Figs. 4, 6), whereas PKA activation plus netrin-1 increased the amount of cell surface DCC and increased axon outgrowth. This suggests that netrin-1 is required to hold DCC at the plasma membrane. In this case, the vesicle bringing DCC to the cell surface may or may not exhibit specificity for DCC, but the presence of netrin-1 selects DCC and stabilizes it on the cell surface. Similarly, the PKA-independent increase in cell surface DCC produced by netrin-1 alone may be attributable to netrin-1-dependent stabilization of DCC on the cell surface and not netrin-1-induced DCC translocation.
Interestingly, this selection and cell surface capture model predicts that DCC would accumulate at the cell surface in regions of the cell in contact with extracellular netrin-1, a prediction that we are currently testing. We have previously reported that netrin-1 and DCC direct the organization of F-actin, causing Rac1- and Cdc42-dependent cell spreading and filopodia formation (Shekarabi and Kennedy, 2002). Together, these findings suggest that DCC will accumulate at the cell surface in regions corresponding to high concentrations of extracellular netrin-1, locally triggering filopodia formation and the extension of a leading edge, thereby directing axon outgrowth. Interestingly, local redistribution of receptors for guidance cues to the leading edge has been observed in directionally migrating lymphocytes and Dictyostelium (for review, see Manes et al., 2003), suggesting that this may be a general mechanism used by directionally migrating cells.
Recruitment of receptors to the cell surface: a post-translational mechanism regulating axon extension
Our findings support a model in which a post-translational mechanism plays a key role regulating the presentation of DCC. DCC is a member of a large family of type I transmembrane proteins containing IgG repeats and fibronectin type III repeats that includes Roundabout (Robo) and L1 (for review, see Brummendorf and Lemmon, 2001). Selective trafficking of such adhesion molecule-like receptors may be a widespread mechanism regulating the response of growth cones to extracellular cues that influence motility. For example, endoexocytic recycling regulates the distribution of L1 in growth cones (Kamiguchi and Lemmon, 2000). Furthermore, presentation of Robo on the cell surface determines whether an axon will cross the ventral midline of the embryonic CNS (Keleman et al., 2002).
Increasing the amount of UNC5 homolog expressed by cultured embryonic Xenopus spinal neurons causes axons that would normally be attracted to netrin-1 to be repelled (Hong et al., 1999). This study and genetic manipulation of UNC5 expression (Hamelin et al., 1993; Keleman and Dickson, 2001) indicate that changing the complement of netrin receptors expressed by a neuron can alter its response to netrin. Recently, Williams et al. (2003) provided evidence that PKC activation triggers the internalization of ectopically expressed UNC5H1 from neuronal growth cones and that this reduces the probability that growth cones will collapse in response to netrin-1. The mechanism underlying the ability of a neuron to switch its response to netrin-1 from attraction to repulsion remains unclear. We have not ruled out that PKA-regulated alterations in intracellular signal transduction may contribute to this; however, a straight-forward alternative is that growth cones change their response to netrin-1 based on the selective presentation of different classes of netrin receptors on the plasma membrane.
The role identified for PKA regulating cell surface presentation of receptors for axon guidance cues may extend beyond embryonic neural development. A decrease in the steady-state level of cAMP inside a neuron during maturation contributes to a decrease in the capacity of axons to regenerate in the adult mammalian CNS (Cai et al., 2001). Furthermore, PKA activation promotes regeneration of sensory axons in the CNS (Neumann et al., 2002; Qiu et al., 2002). The mechanisms underlying this change in neuronal response are not known, but the findings presented here raise the possibility that modulation of PKA activity may influence the ability of an axon to regenerate by regulating the complement of receptors presented by the growth cone.
Footnotes
This work was supported by grants to T.E.K. from the Canadian Institutes of Health Research (CIHR) and the Christopher Reeve Paralysis Foundation. J.-F.B. was supported by a CIHR postdoctoral fellowship, P.P.R. by a CIHR studentship, and T.E.K. by a CIHR Scholar award. We thank Adriana Di Polo, Colleen Manitt, Peter McPherson, and Wayne Sossin for comments on this manuscript.
Correspondence should be addressed to Dr. Timothy E. Kennedy, Centre for Neuronal Survival, Montreal Neurological Institute, McGill University, 3801 University Street, Montreal, Quebec, H3A 2B4 Canada. E-mail: timothy.kennedy@mcgill.ca.
P. P. Roux's present address: Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Laboratory for Human Reproduction and Reproductive Biology Building, Boston, MA 02115.
DOI:10.1523/JNEUROSCI.4934-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243040-11$15.00/0
References
- Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB (1997) The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 386: 838–842. [DOI] [PubMed] [Google Scholar]
- Barker PA, Shooter EM (1994) Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron 13: 203–215. [DOI] [PubMed] [Google Scholar]
- Barres BA, Chun LL, Corey DP (1989) Calcium current in cortical astrocytes: induction by cAMP and neurotransmitters and permissive effect of serum factors. J Neurosci 9: 3169–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brittis PA, Lu Q, Flanagan JG (2002) Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell 110: 223–235. [DOI] [PubMed] [Google Scholar]
- Brummendorf T, Lemmon V (2001) Immunoglobulin superfamily receptors: cis-interactions, intracellular adapters and alternative splicing regulate adhesion. Curr Opin Cell Biol 13: 611–618. [DOI] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT (2001) Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci 21: 4731–4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DS, Holt CE (2001) Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32: 1013–1026. [DOI] [PubMed] [Google Scholar]
- Corset V, Nguyen-Ba-Charvet KT, Forcet C, Moyse E, Chedotal A, Mehlen P (2000) Netrin-1-mediated axon outgrowth and cAMP production requires interaction with adenosine A2b receptor. Nature 407: 747–750. [DOI] [PubMed] [Google Scholar]
- de la Torre JR, Hopker VH, Ming GL, Poo MM, Tessier-Lavigne M, Hemmati-Brivanlou A, Holt CE (1997) Turning of retinal growth cones in a netrin-1 gradient mediated by the netrin receptor DCC. Neuron 19: 1211–1224. [DOI] [PubMed] [Google Scholar]
- Dodd J, Morton SB, Karagogeos D, Yamamoto M, Jessell TM (1988) Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1: 105–116. [DOI] [PubMed] [Google Scholar]
- Fabbri E, Brighenti L, Ottolenghi C (1991) Inhibition of adenylate cyclase of catfish and rat hepatocyte membranes by 9-(tetrahydro-2-furyl)adenine (SQ 22536). J Enzyme Inhib 5: 87–98. [DOI] [PubMed] [Google Scholar]
- Fazeli A, Dickinson SL, Hermiston ML, Tighe RV, Steen RG, Small CG, Stoeckli ET, Keino-Masu K, Masu M, Rayburn H, Simons J, Bronson RT, Gordon JI, Tessier-Lavigne M, Weinberg RA (1997) Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 386: 796–804. [DOI] [PubMed] [Google Scholar]
- Forcet C, Stein E, Pays L, Corset V, Llambi F, Tessier-Lavigne M, Mehlen P (2002) Netrin-1-mediated axon outgrowth requires deleted in colorectal cancer-dependent MAPK activation. Nature 417: 443–447. [DOI] [PubMed] [Google Scholar]
- Goldsmith BA, Abrams TW (1991) Reversal of synaptic depression by serotonin at Aplysia sensory neuron synapses involves activation of adenylyl cyclase. Proc Natl Acad Sci USA 88: 9021–9025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamelin M, Zhou Y, Su MW, Scott IM, Culotti JG (1993) Expression of the UNC-5 guidance receptor in the touch neurons of C. elegans steers their axons dorsally. Nature 364: 327–330. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D (1988) Immunoblotting. In: Antibodies: a laboratory manual (Harlow E, Lane D, eds), pp 471–510. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Hong K, Hinck L, Nishiyama M, Poo MM, Tessier-Lavigne M, Stein E (1999) A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell 97: 927–941. [DOI] [PubMed] [Google Scholar]
- Hopker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C (1999) Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature 401: 69–73. [DOI] [PubMed] [Google Scholar]
- Kamiguchi H, Lemmon V (2000) Recycling of the cell adhesion molecule L1 in axonal growth cones. J Neurosci 20: 3676–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M (1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142: 436–440. [DOI] [PubMed] [Google Scholar]
- Keino-Masu K, Masu M, Hinck L, Leonardo ED, Chan SS, Culotti JG, Tessier-Lavigne M (1996) Deleted in colorectal cancer (DCC) encodes a netrin receptor. Cell 87: 175–185. [DOI] [PubMed] [Google Scholar]
- Keleman K, Dickson BJ (2001) Short- and long-range repulsion by the Drosophila unc5 netrin receptor. Neuron 32: 605–617. [DOI] [PubMed] [Google Scholar]
- Keleman K, Rajagopalan S, Cleppien D, Teis D, Paiha K, Huber LA, Technau GM, Dickson BJ (2002) Comm sorts robo to control axon guidance at the Drosophila midline. Cell 110: 415–427. [DOI] [PubMed] [Google Scholar]
- Kennedy TE, Serafini T, de la Torre JR, Tessier-Lavigne M (1994) Netrins are diffusible chemotropic factors for commissural axons in the embryonic spinal cord. Cell 78: 425–435. [DOI] [PubMed] [Google Scholar]
- Leonardo ED, Hinck L, Masu M, Keino-Masu K, Ackerman SL, Tessier-Lavigne M (1997) Vertebrate homologues of C. elegans UNC-5 are candidate netrin receptors. Nature 386: 833–838. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Le Bivic A, Sargiacomo M, Rodriguez-Boulan E (1989) Steadystate distribution and biogenesis of endogenous Madin-Darby canine kidney glycoproteins: evidence for intracellular sorting and polarized cell surface delivery. J Cell Biol 109: 2117–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes S, Ana LR, Gomez-Mouton C, Martinez A (2003) From rafts to crafts: membrane asymmetry in moving cells. Trends Immunol 24: 320–326. [DOI] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouille P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, Galli T (2001) A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci 21: 3830–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz DC, Zheng H, Killeen MT, Krizus A, Culotti JG (2001) Multiple signaling mechanisms of the UNC-6/netrin receptors UNC-5 and UNC-40/DCC in vivo. Genetics 158: 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson Jr MG, Reichardt LF, Barres BA (1998) Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 21: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhardt JA, Caca K, Eckstrand BC, Hu G, Lengauer C, Banavali S, Look AT, Fearon ER (1999) Netrin-1: interaction with deleted in colorectal cancer (DCC) and alterations in brain tumors and neuroblastomas. Cell Growth Differ 10: 35–42. [PubMed] [Google Scholar]
- Ming GL, Song HJ, Berninger B, Holt CE, Tessier-Lavigne M, Poo MM (1997) cAMP-dependent growth cone guidance by netrin-1. Neuron 19: 1225–1235. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H, Berninger B, Inagaki N, Tessier-Lavigne M, Poo MM (1999) Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron 23: 139–148. [DOI] [PubMed] [Google Scholar]
- Ming GL, Henley J, Tessier-Lavigne M, Song H, Poo MM (2001) Electrical activity modulates growth cone guidance by diffusible factors. Neuron 29: 441–452. [DOI] [PubMed] [Google Scholar]
- Ming GL, Wong ST, Henley J, Yuan XB, Song HJ, Spitzer NC, Poo MM (2002) Adaptation in the chemotactic guidance of nerve growth cones. Nature 417: 411–418. [DOI] [PubMed] [Google Scholar]
- Nairn AC, Hemmings Jr HC, Greengard P (1985) Protein kinases in the brain. Annu Rev Biochem 54: 931–976. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI (2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34: 885–893. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hoshino A, Tsai L, Henley JR, Goshima Y, Tessier-Lavigne M, Poo MM, Hong K (2003) Cyclic AMP/GMP-dependent modulation of Ca(2+) channels sets the polarity of nerve growth-cone turning. Nature 423: 990–995. [DOI] [PubMed] [Google Scholar]
- Osen-Sand A, Staple JK, Naldi E, Schiavo G, Rossetto O, Petitpierre S, Malgaroli A, Montecucco C, Catsicas S (1996) Common and distinct fusion proteins in axonal growth and transmitter release. J Comp Neurol 367: 222–234. [DOI] [PubMed] [Google Scholar]
- Placzek M, Tessier-Lavigne M, Jessell T, Dodd J (1990) Orientation of commissural axons in vitro in response to a floor plate-derived chemoattractant. Development 110: 19–30. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hofmann PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cAMP. Neuron 34: 895–903. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G (1998) Receptors for purines and pyrimidines. Pharmacol Rev 50: 413–492. [PubMed] [Google Scholar]
- Reale MA, Hu G, Zafar AI, Getzenberg RH, Levine SM, Fearon ER (1994) Expression and alternative splicing of the deleted in colorectal cancer (DCC) gene in normal and malignant tissues. Cancer Res 54: 4493–4501. [PubMed] [Google Scholar]
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino dL, DasGupta BR, Montecucco C (1992) Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 359: 832–835. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Al Awqati Q (1986) Regulation of transepithelial H+ transport by exocytosis and endocytosis. Annu Rev Physiol 48: 153–161. [DOI] [PubMed] [Google Scholar]
- Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M (1994) The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78: 409–424. [DOI] [PubMed] [Google Scholar]
- Shekarabi M, Kennedy TE (2002) The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol Cell Neurosci 19: 1–17. [DOI] [PubMed] [Google Scholar]
- Shewan D, Dwivedy A, Anderson R, Holt CE (2002) Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat Neurosci 5: 955–962. [DOI] [PubMed] [Google Scholar]
- Shibata D, Reale MA, Lavin P, Silverman M, Fearon ER, Steele Jr G, Jessup JM, Loda M, Summerhayes IC (1996) The DCC protein and prognosis in colorectal cancer. N Engl J Med 335: 1727–1732. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Mirzayan C, Tessier-Lavigne M, Murakami F (1996) Guidance of circumferentially growing axons by netrin-dependent and -independent floor plate chemotropism in the vertebrate brain. Neuron 17: 1079–1088. [DOI] [PubMed] [Google Scholar]
- Song HJ, Poo MM (1999) Signal transduction underlying growth cone guidance by diffusible factors. Curr Opin Neurobiol 9: 355–363. [DOI] [PubMed] [Google Scholar]
- Stein E, Zou Y, Poo M, Tessier-Lavigne M (2001) Binding of DCC by netrin-1 to mediate axon guidance independent of adenosine A2B receptor activation. Science 291: 1976–1982. [DOI] [PubMed] [Google Scholar]
- Tamaoki J, Chiyotani A, Takeyama K, Yamauchi F, Tagaya E, Konno K (1993) Relaxation and inhibition of contractile response to electrical field stimulation by Beraprost sodium in canine airway smooth muscle. Prostaglandins 45: 363–373. [DOI] [PubMed] [Google Scholar]
- Tessier-Lavigne M, Placzek M, Lumsden AG, Dodd J, Jessell TM (1988) Chemotropic guidance of developing axons in the mammalian central nervous system. Nature 336: 775–778. [DOI] [PubMed] [Google Scholar]
- Twiss JL, Shooter EM (1995) Nerve growth factor promotes neurite regeneration in PC12 cells by translational control. J Neurochem 64: 550–557. [DOI] [PubMed] [Google Scholar]
- Williams ME, Wu SC, McKenna WL, Hinck L (2003) Surface expression of the netrin receptor UNC5H1 is regulated through a protein kinase C-interacting protein/protein kinase-dependent mechanism. J Neurosci 23: 11279–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Karam SM, Ramilo M, Rong Q, Thibodeau A, Forte JG (1996) Stimulation of gastric acid secretion by cAMP in a novel alpha-toxin-permeabilized gland model. Am J Physiol 271: C61–C73. [DOI] [PubMed] [Google Scholar]
- Yu TW, Bargmann CI (2001) Dynamic regulation of axon guidance. Nat Neurosci 4[Supp 1]: 1169–1176. [DOI] [PubMed] [Google Scholar]