

Abstract
Increasing data suggest that impairments of cellular plasticity underlie the pathophysiology of bipolar disorder. In this context, it is noteworthy that AMPA glutamate receptor trafficking regulates synaptic plasticity, effects mediated by signaling cascades, which are targets for antimanic agents. The present studies were undertaken to determine whether two clinically effective, but structurally highly dissimilar, antimanic agents lithium and valproate regulate synaptic expression of AMPA receptor subunit glutamate receptor 1 (GluR1). Chronic (but not acute) treatment of rats with therapeutically relevant concentrations of lithium or valproate reduced hippocampal synaptosomal GluR1 levels. The reduction in synaptic GluR1 by lithium and valproate was attributable to a reduction of surface GluR1 distribution onto the neuronal membrane as demonstrated by three independent assays in cultured hippocampal neurons. Furthermore, these agents induced a decrease in GluR1 phosphorylation at a specific PKA site (GluR1p845), which is known to be critical for AMPA receptor insertion. Sp-cAMP treatment reversed the attenuation of phosphorylation by lithium and valproate and also brought GluR1 back to the surface, suggesting that phosphorylation of GluR1p845 is involved in the mechanism of GluR1 surface attenuation. In addition, GluR1p845 phosphorylation also was attenuated in hippocampus from lithium- or valproate-treated animals in vivo. In contrast, imipramine, an antidepressant that can trigger manic episodes, increased synaptic expression of GluR1 in hippocampus in vivo. These studies suggest that regulation of glutamatergically mediated synaptic plasticity may play a role in the treatment of bipolar disorder and raise the possibility that agents more directly affecting synaptic GluR1 may represent novel therapies for this devastating illness.
Keywords: lithium, valproate, antidepressant, bipolar disorder, GluR1, phosphorylation, PKA
Introduction
Bipolar disorder is a common, severe, often life-threatening illness (Goodwin and Jamison, 1990). Despite the devastating impact that bipolar disorder has on the lives of millions worldwide, there is still a dearth of knowledge concerning its underlying etiology and pathophysiology. The brain systems that traditionally have received the greatest attention in neurobiological studies of bipolar disorder have been the monoaminergic neurotransmitter systems, which are distributed extensively throughout the network of limbic, striatal, and prefrontal cortical neuronal circuits (Drevets, 2000, 2001; Manji et al., 2001; Nestler et al., 2002a). However, increasing recent data suggest that impairments of cellular plasticity also may underlie the pathophysiology of severe mood disorders. These observations have raised the possibility that effective treatments may exert effects on systems known to play critical roles in neuronal plasticity, such as the glutamatergic system (Manji and Lenox, 2000; Manji et al., 2001; D'Sa and Duman, 2002; Nestler et al., 2002a; Young, 2002).
Somewhat surprisingly, the potential role of the glutamatergic system in the pathophysiology and treatment of mood disorders has begun to be investigated in earnest only recently (Krystal et al., 2002; Coyle and Duman, 2003; Moghaddam and Wolf, 2003). Glutamate, the major excitatory neurotransmitter in the CNS, regulates numerous physiological functions in the mammalian CNS, most notably various forms of plasticity, and represents a major neurotransmitter system in the circuitry thought to sub-serve many of the symptoms of severe, recurrent mood disorders (Drevets, 2001). Three major classes of ionotropic glutamate receptors are expressed throughout the mammalian CNS, including AMPA, kainate, and NMDA receptors. AMPA receptors mediate the majority of excitatory synaptic transmission in the CNS; the AMPA R channel is composed of the combination of glutamate receptor 1 (GluR1), GluR2, GluR3, and GluR4 subunits.
Modification of the levels of synaptic AMPA receptors, in particular by receptor subunit trafficking, insertion, and internalization, is a critically important mechanism for regulating postsynaptic responsiveness at many different synapses (Malinow and Malenka, 2002). AMPA receptor subunit GluR1 trafficking is regulated by protein kinase A (PKA), Ca2+/calmodulin-dependent protein kinase II (CaMKII), and protein kinase C (PKC) via phosphorylation of specific sites on GluR1 (Lee et al., 1998; Malinow and Malenka, 2002; Esteban et al., 2003). Phosphorylation of the receptor subunits regulates not only the intrinsic channel properties of the receptor but also the interaction of the receptor with associated proteins that modulate the membrane trafficking and synaptic targeting of the receptors (Malinow and Malenka, 2002).
The possibility that AMPA receptor trafficking also may be involved in the pathophysiology of neuropsychiatric disorders is suggested by recent studies showing that the ability of drugs of abuse to elevate levels of the AMPA GluR1 receptors in the ventral tegmental area is crucial for the development of behavioral sensitization (Carlezon and Nestler, 2002). More recently, it has been demonstrated that both the in vivo administration of drugs of abuse with different molecular mechanisms of action and acute stress increase in strength at excitatory synapses on midbrain dopamine neurons; these results have led to the postulate that AMPA-mediated plasticity changes at excitatory synapses on dopamine neurons may be a key neural adaptation contributing to addiction (Saal et al., 2003).
In the present series of experiments we sought to determine whether two structurally highly dissimilar antimanic agents, lithium and valproate, exert effects on AMPA receptor trafficking. Because lithium and valproate require days to weeks to exert their therapeutic effects, it is widely believed that adaptive changes in intracellular signaling and/or cellular physiology underlie the beneficial effects; moreover, these two agents have been shown to exert robust effects on the very same signaling pathways known to regulate AMPA receptor trafficking. We found that these anti-manic agents, when administered in therapeutically relevant paradigms, attenuate AMPA receptor distribution at synapses. In dramatic contrast, an antidepressant drug (which is known to be capable of inducing mania) exerted opposite effects on GluR1 localization. This attenuation on GluR1 receptor synaptic receptors by antimanic agents may play an important role in the therapeutic effects of lithium and valproate, suggesting avenues for the development of novel therapeutics.
Materials and Methods
Animals. All animal treatments, procedures, and care were approved by the National Institute of Mental Health Animal Care and Use Committee and followed the Guide for the Care and Use of Laboratory Animals (ISBN 0-309-05377-3). Male Wistar Kyoto rats 7-8 weeks of age from Harlan (Indianapolis, IN) were housed two to three per cage in a 12 hr light/dark cycle with ad libitum access to water and food. Rat chow was custom-produced by BioServe (Frenchtown, NJ). Chows containing drugs and control chow were identical with the exception of the added drug and were produced at both a low and high concentration for each drug, with concentrations of 1.2 and 2.4 gm/kg lithium carbonate and 10 and 20 gm/kg sodium valproate. These doses of lithium and valproate were used previously by our group and were found to lead to therapeutic serum drug levels (Yuan et al., 1999). For the in vivo treatment studies we mimicked the clinical situation by treating the animals with a lower dose for a short period of time (to acclimatize them and reduce side effects), followed by an additional, longer period of time at the full dose. Thus, for the chronic treatment studies the rats were treated with half-dose chow for 5-7 d to prevent adverse effects, followed by 3 weeks of full-dose treatment. For the short-term treatment studies the rats were treated with low (half) dose chow for 2 d, followed by 3-4 d of full-dose treatment. Only animals with drug levels within the therapeutic concentrations were used for additional studies; in general, ∼80% of animals achieved therapeutic levels. The number of animals used for each assay is indicated in the figure legends and was generally 6-12 animals/group. Serum blood levels of the animals used for additional analysis were lithium, 0.80 ± 0.13 mEq/l (long term), and valproate, 80.6 ± 8.8 μg/ml (long term); and lithium, 0.702 ± 0.126 mEq/l (short term), and valproate, 58.65 ± 23.0 μg/ml (short term).
For the imipramine treatment studies the animals were injected with imipramine (10 mg/kg in 0.3 ml of saline, twice daily, i.p.) or saline for 4 weeks.
All rats were decapitated during the morning hours. Trunk blood was collected for analysis of drug levels. Drug serum levels were performed by Medtox Scientific (St. Paul, MN), and ∼80% of animals were found to be within the therapeutic range (only these were used for subsequent biochemical analyses). Animals were killed (and the tissue subsequently processed) by alternating control and treated animals, with separate control groups for each drug. Hippocampi were dissected immediately after decapitation. Brain specimens then were frozen rapidly in liquid nitrogen and stored at -80°C until additional analyses.
Synaptosomal preparation and Western blot analysis. Both hippocampi from one animal were used to prepare the synaptosomes, using the differential and discontinuous Ficoll gradient centrifugation method (Pozzo-Miller et al., 1999). Briefly, each group of hippocampal tissue was homogenized in 3 ml of cold Syn buffer (300 mm mannitol/1 mm EDTA, pH 7.4). Aliquots of the crude homogenates were taken to determine the levels of synaptic proteins in the whole hippocampus. The remaining homogenates were centrifuged twice at 5000 × g for 10 min. The supernatants were centrifuged at 15,000 × g for 30 min. The pellets then were resuspended in Syn buffer, loaded on Ficoll gradient tubes, and centrifuged at 32,000 × g at 4°C for 90 min. The proteins in the interfaces between 8 and 12% and between 12 and 16% Ficoll gradients were collected, diluted in Syn buffer in a ratio of 1:4, and then centrifuged for 20 min at 15,000 × g. Pellets were resuspended in 500 μl of 1× P buffer [containing (in mm): 5.4 KCl, 0.8 MgSO4, 5.5 glucose, 50 HEPES, 130 choline chloride, 1 BSA, plus 0.01% CHAPS]. Synaptosomal proteins were solubilized with 300 μl of solubilization buffer (150 mm NaCl, 10 mm HEPES, and 1.5% CHAPS), stirred at 4°C for 1.5 hr, and additionally centrifuged at 12,000 × g for 1 hr to remove any insoluble impurities. Proteins from the crude homogenates (in Syn buffer) were solubilized by the same protocol after the addition of a concentrated solubilization buffer. Protein concentration was determined by using the BCA assay (Pierce, Rockford, IL); the presence of mannitol in the crude homogenates was not found to affect the linearity of this assay significantly. Equal amounts of proteins from either homogenized hippocampus (3 μg) or hippocampal synaptosomes (1 μg) were separated by 10% SDS gradient gel electrophoresis (Invitrogen, San Diego, CA), transferred to 0.45 μm pore size polyvinylidene difluoride membranes (Millipore, Bedford, MA), and immunoblotted with anti-GluR1 (1:2000; Chemicon, Temecula, CA), anti-phospho-GluR1p845 (1:100; Upstate Biotechnology, Lake Placid, NY), anti-phospho-GluR1p831, anti-GluR2/3, anti-synaptobrevin, anti-postsynaptic density 95 (PSD95) (all at 1:1000; Chemicon), anti-synaptophysin(1:5000; Chemicon), anti-phospho-synapsin I (1:200; for site 1, a PKA site; a generous gift from Paul Greengard's laboratory, New York, NY), and anti-NR1 (1:200; Chemicon) antibodies. Horseradish peroxidase-conjugated anti-rabbit and anti-mouse IgG antibodies [Amersham Biosciences (Arlington Heights, IL) and Vector Laboratories (Burlingame, CA)] were the secondary antibodies. The immunoreactive bands were visualized by enhanced chemiluminescence (ECL+, Amersham Biosciences) and exposed to Kodak BioMax or Biolight film (Rochester, NY). The ECL signal intensities were quantified by using the Kodak Imaging System, based on standard curves of these synaptic proteins. All data were analyzed by Student's t test and presented as the mean ± SE (n = number of the animals).
Primary hippocampal culture preparations. Cultures of hippocampal neurons were prepared according to established procedures with minor modifications (Du et al., 2000). Briefly, whole hippocampi were dissected from embryonic day 18 (E18) rats, dissociated in Ca2+ and Mg2+-free HBSS containing 0.125% trypsin for 15 min, triturated in DMEM (Invitrogen)/10% fetal bovine serum, and plated at 0.2 million cells/well in 12-well plates. Cells were grown at 37°C, 5% CO2/95% humidity, first in 10% fetal bovine serum/DMEM and then 1 d later switched to serum-free medium Neurobasal plus B27 (B27NB, Invitrogen). Cultures were grown in serum-free medium for 8-10 d before the start of experiments, and the medium was changed every 3 d. Fresh medium was applied 24 hr before each experiment. These cultures yielded virtually pure neurons (data not shown).
Surface biotinylation and Western blot analysis of GluR1 and phospho-GluR1. Surface GluR1 receptors were detected by a biotinylation assay, followed by Western blot analysis that used either a GluR1 antibody (1:200; Chemicon) or a pan-cadherin antibody (1:500; Sigma, St. Louis, MO). At the end of valproate or lithium treatment for the time indicated, ice-cold PBS (with calcium and magnesium, pH 7.4; Invitrogen) was added to the cultures to prevent receptor internalization. After three washes with ice-cold PBS the cells were incubated in sulfo-NHS-LC-biotin (0.25 mg/ml in cold PBS; Pierce) for 30 min. The surface biotinylation was stopped by removal of the above solution and incubation in 10 mm ice-cold glycine in PBS for 20 min. Cells then were washed three times with cold PBS and lysed by RIPA buffer, which contained (in mm) 20 HEPES, pH 7.4, 100 NaCl, 1 EGTA, 1 Na-orthovanadate, 50 NaF, 1 mm 4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride, plus 1% NP-40, 1% deoxycholate, 0.1% SDS, 10 μg/ml leupeptin, 1 μg/ml aprotinin. Biotinylated proteins were precipitated with 100 μl of ImmunoPure Immobilized Streptavidin (Pierce). Biotinylated proteins were separated on 10% SDS-PAGE gel and transferred to nitrocellulose membrane. The membranes were probed with a polyclonal anti-GluR1 antibody (1:200; Chemicon) or an anti-pan-cadherin antibody (1:200; Sigma), followed by peroxidase-conjugated goat anti-rabbit IgG (1:3000; Vector Laboratories). Immunoreactive bands were visualized by ECL (Amersham Biosciences). The ECL signal intensities were quantified by the Kodak Imaging System.
To verify that only a specific protein population, membrane proteins, was biotinylated, we performed silver staining of the total protein gel and Western blot analysis with avidin-conjugated peroxidase for the biotinylated proteins. Briefly, after biotinylation the hippocampal neurons were harvested, and equal amounts of proteins were separated by electrophoresis. The gel was stained by Simply Blue SafeStain (Invitrogen) to show the total protein pattern. The proteins also were transferred onto nitrocellulose membrane and blotted by avidin-peroxidase to show the biotinylated protein pattern (supplemental Fig. 1, available at www.jneurosci.org/cgi/content/full/24/29/6578/DC1). Additionally, experiments also were performed to determine the biotinylation of the total hippocampal protein. The hippocampal neurons were harvested in lysis buffer and centrifuged at 14,000 × g for 10 min to discard debris. Then 50 μg of protein in PBS with magnesium and calcium was incubated with 500 ng of NHS-LC-biotin for 2 hr at 4°C. Equal amounts of proteins were separated by 10% electrophoresis. The protein gel was stained by a silver staining kit (Bio-Rad, Hercules, CA) according to the manufacturer's instructions. The same proteins were transferred onto nitrocellulose membranes and stained by streptavidin conjugated with horseradish peroxidase (HRP-streptavidin; 1:10,000; Chemicon). All of the protein profiles stained by silver staining were biotinylated. Because HRP-streptavidin staining is very sensitive, proteins that could not be shown by silver staining also were biotinylated and recognized by HRP-streptavidin (supplemental Fig. 2, available at www.jneurosci.org/cgi/content/full/24/29/6578/DC1).
To measure the total (surface and internalized) amount of GluR1, we simply harvested cultured hippocampal neurons by RIPA buffer and processed them for Western blot. For phospho-GluR1, after lithium and valproate treatment equal amounts of cell homogenates (15 μg) were separated by electrophoresis. Western blot analysis was performed with anti-phospho-GluR1 (Ser845; 1:100; Upstate Biotechnology) or anti-phospho-GluR1 (Ser831; 1:100) antibodies (Upstate Biotechnology). Quantitation for each experimental condition was based on three to six independent experiments (samples); each was repeated at least two to three times. The results were pooled, averaged, and presented as the mean ± SE.
GluR1 surface staining. The rat hippocampal cultures were treated with lithium or valproate at 37°C. Then the cells were blocked with 0.5% BSA in B27NB medium for 10 min at 37°C, followed by treatment with rabbit anti-GluR1 N-terminal antibody (Oncogene Research, San Diego, CA) in 0.1% BSA in B27NB (1:10) for 15 min. After being washed with 0.1% BSA in B27NB, the cells were incubated with Cy3-goat anti-rabbit antibody (1:30; Jackson ImmunoResearch, West Grove, PA) for 15 min at 37°C, washed with B27NB three times, and fixed with 4% paraformaldehyde in PBS for 1 hr on ice before mounting with Antifade mounting medium. Z-stack images were acquired by 510-Meta confocal microscope under exactly the same conditions. To acquire high-quality images for quantification, we took only five confocal images randomly from each slice, mounted with Antifade mounting medium. The samples were assigned a blinded code. The quantitation was performed by a technician who was completely blind to the experiments. The mean fluorescence of the dendrites ∼55 μm in length (starting point was one cell body in length apart from the neuronal cell body) was measured from 12 neurons in each treated group. ANOVA was used for statistical analysis.
Fluorescent double immunostaining. Cultured rat hippocampal neurons were treated with lithium (Li; 1.0 mm) or valproate (VPA; 1.0 mm) for 1 or 4 d and fixed in 4% paraformaldehyde in PBS for 1 hr on ice. Then the cells were blocked with 10% normal goat serum, 1% bovine serum albumin (BSA), and 0.4% Triton X-100 in PBS for 1 hr. After being incubated in rabbit anti-GluR1 (1:40; Chemicon) and mouse anti-synaptotagmin (1:100; Chemicon) at 4°C overnight, the cells were washed extensively in PBS. The secondary antibodies were FITC-conjugated anti-mouse (for multiple staining, 1:50; Jackson ImmunoResearch) and Cy3-conjugated anti-rabbit (for multiple staining, 1:150; Jackson ImmunoResearch) antibodies. After extensive washing, the cells were mounted onto slides with Antifade mounting medium (Molecular Probes, Eugene, OR). Z-stack imaging was acquired by using a 510-Meta confocal microscope under exactly the same conditions. The samples were assigned a blinded code. The quantitation was performed by a technician who was completely blind to the experiments. Neurons, which were viewed under the confocal microscope, were photographed randomly; only five images were taken from each slice to ensure that the condition of slices for each group was the same. The longest dendrite was picked from each neuron treated in each group for quantification, and the average length of dendrites from each cell used for quantification had no significant difference among the control, lithium-, and valproate-treated groups. Fluorescent intensity of GluR1 (red) at each individual synapse on the longest dendrite of the neuron was determined by 510-Meta software; data were analyzed by using ANOVA.
PKA enzyme assay. PKA activity was determined by using previously described methods (Tartaglia et al., 2001). In brief, hippocampi from chronic lithium- or valproate-treated rats (4 weeks), which were kept frozen at -70°C, were sonicated in homogenization buffer [50 mm Tris-Cl, pH 7.5, containing (in mm) 2 dithiothreitol, 2 EDTA, 1 EGTA, 50 KF, plus 50 nm okadaic acid, Na-orthovanadate, and NP-40 as well as 50 μm 4-(2-aminoethyl)-benzenesulfonylfluoride hydrochloride, 5 mg/ml leupeptin, aprotinin, chymostatin, pepstatin A] and spun in the Eppendorf 5810R centrifuge (Madison, WI) for 20 min at 4°C at 20,000 × g; the clear homogenate was used for protein determination and PKA enzymatic assay experiments.
PKA activity of brain samples prepared from the hippocampus of mice was measured at 30°C for 5 min in a mixture (50 μl) containing 30 mm Tris-Cl, pH 7.5, 6 mm MgCl2, 120 μm [γ-32P]ATP, 1.0 mg/ml bovine serum albumin, 40 μm PKA substrate peptide (PGRQRRHTLPANEFRC), and 1 μg protein of tissue extract. Reactions were stopped by placing the tubes at 99°C for 3 min and being allowed to cool for 10 min. Tubes were centrifuged with the Eppendorf Centrifuge 5415D for 3 min; 5 μl of reaction mixture was blotted on 4 × 4 cm Whatman P81 chromatography paper (Maidstone, UK). Blotted papers were allowed to dry for 30 min and then were washed in a 7% phosphoric acid solution three times for 10 min each. The papers were placed on aluminum foil and allowed to dry overnight. 32P-labeled peptide substrates blotted on chromatography paper were measured in a scintillation counter. All reactions were run in duplicate. PKA activities were converted to picomoles of 32P incorporated into peptides per microgram of protein per minute.
Results
Chronic treatment with the structurally dissimilar antimanic agents lithium and valproate reduces AMPA receptor subunit GluR1 synaptic expression in vivo
To determine whether chronic treatment with structurally dissimilar antimanic agents altered synaptic AMPA GluR1 levels, we treated rats with lithium or valproate for 4 weeks, achieving therapeutically relevant drug concentrations. GluR1 receptor expression in the synaptosomal preparations from the hippocampus was reduced significantly in the lithium- or valproate-treated rats (Fig. 1A,B,I). Synaptophysin, used as a presynaptic protein marker, and PSD95, used as a postsynaptic marker to control for protein loading, remained unchanged in synaptosomal preparations of three groups (Fig. 1C-F). Another presynaptic marker, synaptobrevin, also remained unchanged among these three groups in hippocampal synaptosomal preparation (data not shown). As a quality control measure the fold-enrichment of both a presynaptic and postsynaptic marker was evaluated in five independent experiments. GluR1 levels were enriched approximately fourfold (3.82 ± 1.24-fold; n = 5) in the synaptosomal preparation compared with the tissue original homogenates in a pool of control animals from five independent synaptic preparations. A presynaptic marker synaptobrevin-2 showed a very similar fold-enrichment in synaptosomal preparation (4.01 ± 0.69-fold; n = 5), suggesting that both the presynaptic marker synaptobrevin-2 and postsynaptic marker GluR1 are solubilized in the 1.5% CHAPS-soluble pool.
Figure 1.

AMPA receptor subunit GluR1 was attenuated in synaptosomal preparation from long-term lithium- and valproate-treated animals. A, Quantification of GluR1 content in hippocampal synaptosomes from lithium-treated (n = 10) and control; (n = 12) animals (t test; p < 0.01). B, GluR1 protein content in hippocampal synaptosomes from valproate-treated (n = 11) and control (n = 12) animals (t test; p < 0.05). C, Quantification of synaptophysin protein in hippocampal synaptosomal preparation from lithium-treated (n = 5) and control (n = 6) animals. D, Synaptophysin protein levels in hippocampal synaptosomal preparation from valproate-treated (n = 8) and control (n = 7) animals. E, PSD95 protein levels in synaptosomal preparation from lithium-treated (n = 11) and control (n = 12) animals. F, PSD95 protein levels in synaptosomal preparation from valproate-treated (n = 12) and control (n = 12) animals. G, Total GluR1 expression levels in hippocampal tissue homogenates from lithium-treated (n = 9) and control (n = 9) animals. H, Total GluR1 expression levels in hippocampal tissue homogenates from valproate-treated (n = 5) and control (n = 9) animals. I, Samples of Western blot analysis of hippocampal synaptosomal preparation from lithium-treated (L) or valproate-treated (V) animals with anti-GluR1, anti-synaptophysin (SYP), or anti-PSD95 antibodies. J, NMDA receptor NR1 levels remained unchanged in synaptosomal preparation from lithium-treated (n = 10) or valproate-treated (n = 12) animals compared with control animals (n = 10). K, AMPA GluR2/3 receptors also were attenuated in hippocampal synaptosomal preparations from lithium- and valproate-treated animals (control, n = 7; lithium, n = 7; valproate, n = 6). ANOVA; *p < 0.05. L, GluR1 and synaptophysin (SYP) levels in synaptosomal preparation from short-term (5 d) lithiumtreated (n = 7), valproate-treated (n = 8), and control (n = 7) animals.
Chronic treatment with the structurally dissimilar antimanic agents lithium and valproate does not affect total AMPA receptor subunit GluR1 expression in vivo
One possibility for the reduction in synaptosomal GluR1 levels is a decrease in total GluR1 protein expression. However, Western blot analysis of GluR1 in tissue homogenates from hippocampus of chronically treated rats indicated that there was no significant change in the total amount of GluR1 levels (Fig. 1G,H) either after chronic lithium or after chronic valproate treatment. We therefore postulated that lithium or valproate might exert selective effects on GluR1 synaptic expression, which involves AMPA receptor trafficking. This possible mechanism was examined additionally in cultured hippocampal neurons (see below).
AMPA receptor GluR2/3 levels also are attenuated in synaptosomal preparations from lithium- and valproate-treated animals after chronic administration; however, NMDA receptor subunit NR1 levels at synapses remain unchanged
Previous studies have shown that three major subunits of AMPA receptors, GluR1, GluR2, and GluR3, are all localized in the hippocampus. Thus, it is possible that the effects of GluR1 could be compensated by alterations in GluR2/3 synaptic localization. Therefore, we sought to determine whether GluR2/3 levels were altered in synaptosomal preparation from lithium- and valproate-treated animals after chronic administration, using an anti-GluR2/3 antibody. The data showed that GluR2/3 levels also were attenuated significantly in synaptosomal preparations from lithium- or valproate-treated animals by 29.6 or 20.9%, respectively (Fig. 1K). Synaptophysin was used as the loading control. In summary, the three major AMPA receptor subunits GluR1, GluR2, and GluR3 were reduced at synapses after lithium or valproate treatment in vivo (Fig. 1K).
We next sought to investigate the specificity of the effects on AMPA receptors and undertook studies to determine whether the major NMDA receptor subunit, NR1, also was reduced at synapses after chronic lithium or valproate treatment. Western blot analysis with anti-NR1 antibody revealed that NR1 levels at synapses remained unchanged in synaptosomal preparations after chronic lithium or valproate treatment (Fig. 1J).
Acute treatment with the structurally dissimilar antimanic agents lithium and valproate does not alter AMPA receptor subunit GluR1 synaptic expression in vivo
To ascribe potential therapeutic relevance, we also undertook short-term treatment studies (at a time point when clinical effects generally are not observed). To learn whether short-term lithium or valproate treatment has an effect on GluR1 synaptic localization, we determined GluR1 levels in hippocampal synaptosomal preparations from short-term-treated animals by Western blot analysis. Synaptic GluR1 levels were not changed significantly after short-term (5 d) lithium or valproate treatment (Fig. 1L). Synaptophysin was used as a loading control and also was unaltered similarly after short-term lithium or valproate treatment (Fig. 1L).
Lithium and VPA treatments attenuate surface expression and synaptic localization of GluR1 in cultured hippocampal neurons
To investigate in more detail the mechanisms by which lithium or valproate selectively reduced synaptosomal GluR1 on AMPA receptor levels and trafficking, we established hippocampal neuronal cultures. Three independent assays, biotinylation, surface GluR1 immunostaining, and GluR1/synaptotagmin double staining, were used to determine the surface and synaptic localization of GluR1.
Hippocampal neuronal cultures were prepared from E18 embryos to yield a pure neuronal population and were cultured for 8-10 d to allow for synaptic neuronal connections. Therapeutically relevant concentrations of lithium and valproate were applied to cultured hippocampal neurons, and surface GluR1 levels were determined by biotinylation assay. Both lithium and valproate significantly attenuated GluR1 surface expression in a dose- and time-dependent manner. In the dose-response experiments the hippocampal neurons were treated with lithium and valproate with various concentrations (0.3, 1.0, 2.0 mm) for 4 d (Fig. 2A). Surface GluR1 levels were normalized to a neuronal surface protein marker pan-cadherin. Surface GluR1 levels were reduced significantly in 1.0 or 2.0 mm lithium- or valproate-treated groups (Fig. 2A,B), suggesting that concentrations approximating the therapeutic antimanic concentrations (∼1.0 mm of both agents) were required for these effects. Concentrations that were considered subtherapeutic (0.3 mm) resulted in a slight decrease. Therapeutic doses of lithium or valproate (1.0 mm) produced significant decreases, with a maximum reduction of 43 and 53%, respectively (Fig. 2B,D,F). Similar to their clinical effects, the reductions in surface GluR1 required more prolonged exposure and were not seen with shorter-term (6 hr) administration (Fig. 2D,F). The time frame differs markedly from that seen with activity-dependent regular of AMPA receptor trafficking, which occurs in minutes, suggesting that complex second messenger cascades are involved (addressed further below) (Lin et al., 2000). The reduction in surface GluR1 was maintained through 4 and even 7 d (data not shown) of lithium and valproate treatment, suggesting that it is a sustained effect.
Figure 2.

Surface GluR1 on cultured hippocampal neurons was decreased after lithium and valproate treatment within a therapeutic range in a dose- and time-dependent manner. Hippocampalprimary culture neurons were prepared from E18 Sprague Dawley embryos. After 10 d of culturing in B27 Neurobasal media, the neurons were treated with lithium or valproate with the dose indicated and for various durations. Surface proteins of the neurons were labeled with biotin, and then the cells were harvested with lysis buffer. Biotinylated surface proteins were precipitated by immobilized avidin and analyzed by Western blot analysis with anti-GluR1 or anti-pan-cadherin (surface protein control) antibodies. Data were analyzed by the Kodak Imaging System and pooled from three independent experiments. A, Dose dependency of lithium and valproate effect on surface GluR1 after 4 d of treatment. Con, Control. B, Dose-response curve for surface GluR1 expression after lithium and valproate treatment for 4 d. Pan-cadherin was used as a loading control (n = 2, n = 4; *p < 0.05). C, Lithium (1.0 mm) effects on surface GluR1 after 1 or 4 d of treatment. D, Quantification of the time course on lithium effect of GluR1 on neuronal surface (n = 3; n as indicated on the bars; t test; *p < 0.05). E, Valproate (1.0 mm) effect on GluR1 surface expression after 1 or 4 d of treatment. F, Quantification for the effect of valproate on GluR1 surface expression (n = 3; n as indicated on the bars; t test; *p < 0.05). C, Control; L, lithium; V, valproate.
To demonstrate that only a specific protein population, membrane protein, was biotinylated, we performed Simply Blue (Invitrogen) staining of the total protein gel and Western blot analysis with avidin-conjugated peroxidase for the biotinylated proteins (supplemental Fig. 1, available at www.jneurosci.org/cgi/content/full/24/29/6578/DC1).
To confirm the biochemical finding further, we immunostained live hippocampal neurons after lithium and valproate treatment with an anti-GluR1 antibody, which recognizes the GluR1 N-terminal on its extracellular domain. Previous studies have shown that this antibody does not trigger internalization of GluR1 receptors after binding to the receptor under similar conditions (Liao et al., 2001). In Figure 3A, the orthogonal picture shows that the GluR1 labeling was localized mainly on the neuronal surface, as indicated by the arrows. After lithium and valproate treatment the fluorescent intensity of GluR1 patches on the dendrites of the neuron was reduced, as indicated by the arrows, demonstrating a reduction in GluR1 staining on the neuronal surface (Fig. 3A). Using 510-Meta software, we performed quantification analysis to determine the mean fluorescence in 55 μm length of dendrites from 10 neuronal images. As demonstrated in Figure 3B, the mean fluorescence per 55 μm of dendrites was decreased significantly in lithium- and valproate-treated neurons, by 28.4 and 29.2%, respectively.
Figure 3.
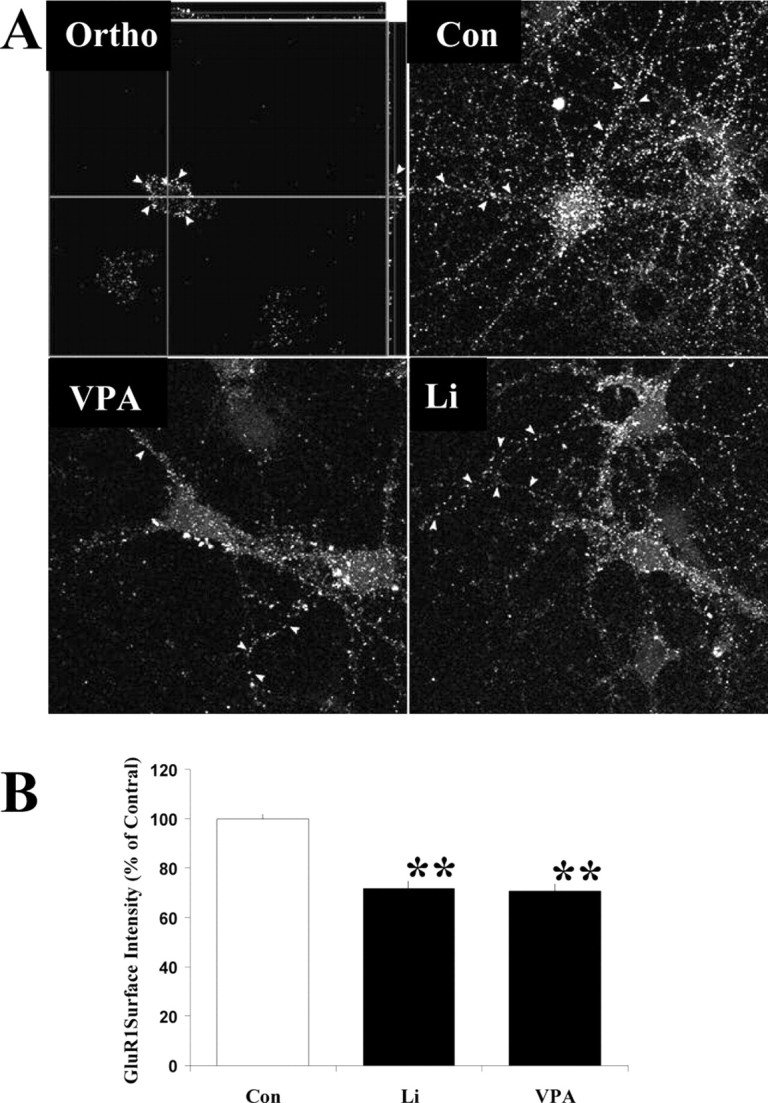
A, Surface staining with anti-GluR1 N-terminal antibody after lithium and valproate treatment in cultured hippocampal neurons. Hippocampal neurons were cultured for 10 d, followed by treatment with Li (1.0 mm) or VPA (1.0 mm) for an additional 1 d. The surface of living neurons was labeled with anti-GluR1 (against N-terminal epitope) antibody and subsequently with Cy3-anti-rabbit IgG. Z-stack images were acquired with a 510-Meta confocal microscopy under exactly the same setup for different experimental groups. Three-dimensional images were reconstructed with 510-Meta software. The orthogonal picture (Ortho) indicates that GluR1 stainings are at the neuronal surface. B, Quantification of surface GluR1 in hippocampal neurons after lithium and valproate treatment. The mean fluorescent intensity on the longest dendrite was determined (n = 10 for each group; ANOVA; **p < 0.01). This experiment was repeated. Con, Control.
To determine whether this attenuation is attributable to a decrease in total GluR1 expression, we performed Western blot analysis of lithium- or valproate-treated hippocampal neuronal lysate with anti-GluR1 antibody. Actin was used as a loading control. Total GluR1 levels remained unchanged after lithium or valproate treatment for 1 or 4 d (Fig. 4). Increasingly, as is the case for the treatment of other complex illnesses like epilepsy, combinations of treatments are being used to treat bipolar disorder. We therefore next investigated the effects of the combination of therapeutic doses of both agents. Treatment of hippocampal cultures with a combination of both lithium and valproate produced additive effects, resulting in ∼65% reduction in surface GluR1 expression. The cultured hippocampal neurons underwent routine visual inspection microscopically and showed no signs of toxicity secondary to medication exposure (Fig. 5).
Figure 4.

GluR1 total protein expression remained unchanged after lithium or valproate treatment in cultured hippocampal neurons. Hippocampal neurons were treated with lithium (1.0 mm) or valproate (1.0 mm) for 1 or 4 d. Equal amounts of proteins were applied for Western blot analysis by anti-GluR1 antibody. The loading of the protein was corrected by actin. Con, Control.
Figure 5.

Treatment with both lithium and valproate had an additive effect on the surface GluR1 expression of hippocampal neurons. After being cultured for 10 d, hippocampal neurons were treated with lithium only, valproate only, and lithium plus valproate for 4 d. Surface GluR1 was determined by biotinylation assay. Samples were pooled from three independent experiments, with n of 9-12 (ANOVA; #,**p < 0.05). Con, Control.
GluR1 immunostaining at synapses is attenuated after lithium and valproate treatment in cultured hippocampal neurons
Because GluR1 receptors at synapses are important for glutamate synaptic transmission, additionally we specifically quantitated GluR1 levels at synapses by double-immunostaining of GluR1 and synaptotagmin. Synaptotagmin puncta were used as a crude indicator of synaptic specialization. After 4 d treatment with lithium (1.0 mm) or valproate (1.0 mm) fluorescent intensities of GluR1 (red) and synaptotagmin (green) were determined from individual synapses (Fig. 6A,B). All of the synapses on the longest dendrite from seven neuronal images for each treatment (∼150-190 synapses in each experimental condition) were counted for fluorescent intensity (Fig. 6B). After 1 d of treatment with lithium or valproate the GluR1 fluorescent intensity at synapses remained unchanged, although the surface GluR1 previously was decreased. After 4 d of treatment the GluR1 intensity at synapses was reduced significantly in lithium- and valproate-treated neurons, by 48.8 and 40.3%, respectively. Together, these data suggest that the mood stabilizer-induced decreased surface expression of GluR1 brings about changes in the synaptic expression of GluR1 in a time-dependent manner.
Figure 6.
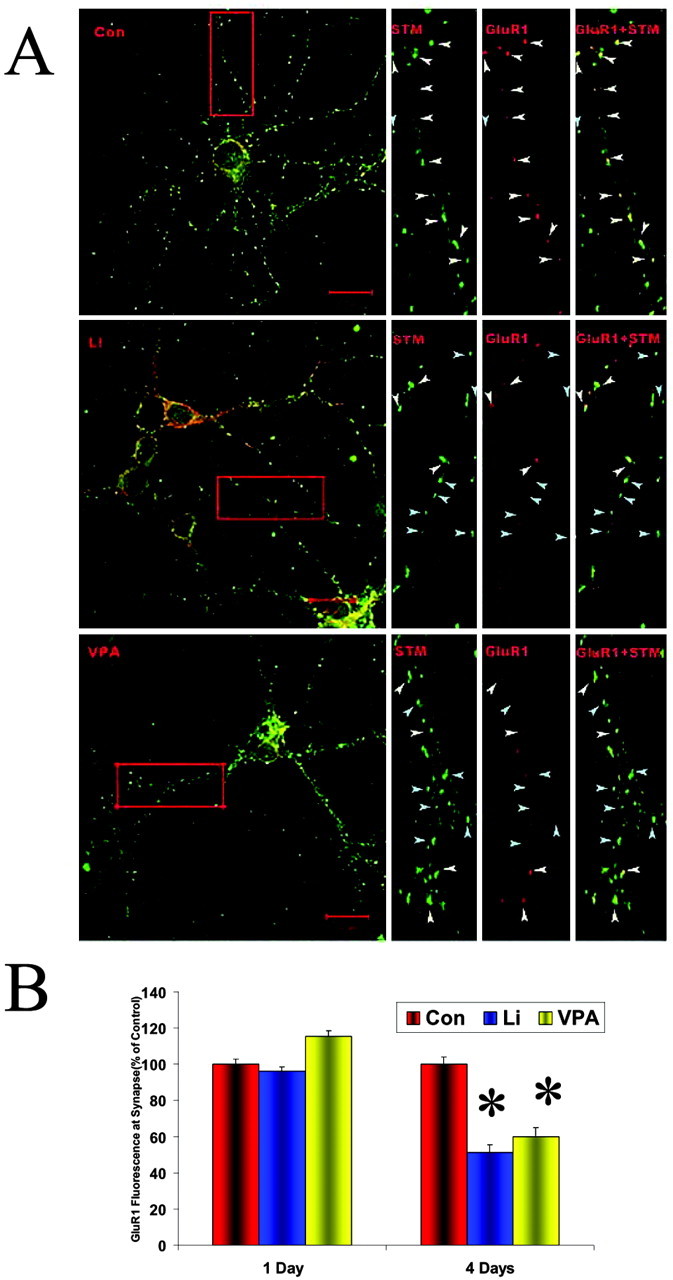
Double immunostaining of GluR1 and synaptotagmin indicated that GluR1 was attenuated at synapses. A, Double immunostaining of GluR1 and synaptotagmin in hippocampal neurons treated with lithium or valproate for 4 d. After lithium (1.0 mm) or valproate (1.0 mm) treatment for 4 d, hippocampal neurons were double-stained with anti-GluR1 (red) and anti-synaptotagmin (STM; green) antibodies. Z-stack images were acquired by using a confocal microscope under the same setup for lithium-treated (Li) or valproate-treated (VPA) and untreated (Con) groups. Three-dimensional images were reconstructed by 510-Meta software. Portions of dendrite (red rectangle) were taken from the neurons for a close observation on their synapses. The GluR1-positive synapses are indicated with white arrows, and GluR1-negative synapses are indicated with blue arrows. B, Quantification of GluR1 (red) fluorescent intensity at the synapses. GluR1 fluorescent intensities were quantified at the individual synapses from the longest dendrite from seven neuronal images for each condition. The average length of the longest dendrite in each condition was not significantly different. In total, ∼150-190 synapses were measured for each condition (ANOVA; *p < 0.01). This experiment was repeated. Con, Control.
GluR1 phosphorylation at the PKA site plays an important role in lithium- or valproate-induced regulation of surface expression of GluR1
Phosphorylation and dephosphorylation of GluR1 plays an important role for AMPA glutamate receptor insertion and internalization (Ehlers, 2000; Lee et al., 2000; Malinow and Malenka, 2002). Previous studies have demonstrated that phosphorylation of the PKA site (GluR1p845) and/or the Ca MKII/PKC site (GluR1p831) are the key regulators for synaptic distribution of GluR1. Therefore, we next investigated the effects of lithium and valproate on the phosphorylation of GluR1. After 4 d treatments with lithium (1.0 mm) or valproate (1.0 mm) equal amounts of hippocampal proteins were analyzed by Western blot analysis with anti-phospho-GluR1p845 and anti-phospho-GluR1p831 antibodies. Phosphorylation of GluR1 at the PKA site (p845) was attenuated significantly after lithium and valproate treatment by 52.0 and 31.0%, respectively (Fig. 7A,C). Western blot analysis of GluR1 also was performed with stripped membrane, and total GluR1 level remain unchanged. However, phosphorylation of GluR1 at CaMKII/PKC site was not altered by lithium or valproate treatment (Fig. 7B,C), suggesting that lithium and valproate regulates GluR1 phosphorylation in a site-specific manner.
Figure 7.

Phosphorylation of GluR1 at PKA site was attenuated significantly by lithium or valproate treatment in cultured hippocampal neurons. A, Phosphorylation of GluR1 at p845 (PKA site) after lithium (1.0 mm) or valproate (1.0 mm) treatment for 4 d in cultured hippocampal neurons. The membranes were stripped and reprobed with anti-GluR1 antibody. B, Phosphorylation of GluR1 at p831 (CaMKII site) after lithium (1.0 mm) or valproate (1.0 mm) treatment for 4 d. The same blot was stripped and reprobed with anti-GluR1 antibody. C, Quantification of GluR1 phosphorylation at p845 and p831 sites after lithium or valproate treatment for 4 d. Results were pooled from two to three independent experiments. The number for each condition is indicated on the bars (ANOVA; *p < 0.05). Con, Control.
To determine whether this attenuation of phosphorylation on GluR1p845 correlates with the surface localization of GluR1, we stimulated the phosphorylation of GluR1 on the p845 site with Sp-cAMP, a membrane-permeable specific PKA activator. The surface GluR1 levels subsequently were investigated to determine whether reversing lithium and valproate effects on phosphorylation of GluR1 might bring GluR1 back to the neuronal surface. As depicted in Figure 8, Sp-cAMP significantly enhanced the phosphorylation of GluR1 on the PKA site in all control (138%), lithium-treated (125%), and valproate-treated (93%) groups in comparison to the original 100, 61, and 63%, respectively. Furthermore, Sp-cAMP treatment also resulted in GluR1 insertion onto the membrane such that surface GluR1 levels were increased significantly to reach 134% in control, 107% in lithium-treated, and 112% in valproate-treated groups in comparison to the original 100, 66, and 68%, respectively.
Figure 8.

Sp-cAMP enhanced GluR1 phosphorylation on the PKA site in lithium- or valproate-treated neurons and also resulted in an increase in GluR1 surface expression. Hippocampal neurons were cultured for 8 d in B27 plus Neurobasal medium and treated with lithium (1.0 mm) or valproate (1.0 mm) for an additional 4 d. Sp-cAMP (50 μm) was applied to the cultured hippocampal neurons for 30 min. The surface proteins were labeled with sulfo-NHS-LC-biotin, and the proteins were harvested with lysis buffer. Equal amounts of proteins were analyzed by Western blot with anti-phospho-GluR1p845 and anti-actin antibody. The blots were stripped and reprobed with anti-GluR1 antibody. A, Sp-cAMP significantly enhanced phosphorylation of GluR1 at the p845 site in all control, lithium-, and valproate-treated groups. B, Sp-cAMP treatment also reversed the effects of lithium and valproate on GluR1 surface expression. Data were pooled from two to three independent experiments (n = 5-8; t test; **p < 0.01 and *p < 0.05). Con, Control.
GluR1 phosphorylation both at the PKA site and PKA basal activity are attenuated after chronic administration of lithium or valproate in vivo
To determine whether GluR1 phosphorylation was altered in vivo after lithium or valproate treatment, we investigated phosphorylation of GluR1 at p845 or p831 in hippocampal homogenates from chronically lithium- or valproate-treated animals by Western blot analysis with anti-phospho-GluR1p845 and anti-phospho-GluR1p831 antibodies (Fig. 9A). The data have shown that GluR1 phosphorylation at the p845 site was attenuated by 30.3 and 31.0%, respectively. To determine the specificity of effects (i.e., that phosphorylation of all PKA substrates is not similarly reduced), we investigated the effects of chronic lithium or valproate on the PKA site of synapsin I (site 1), another major synaptic protein. We found that chronic treatment with either drug was not associated with a significant reduction in hippocampal synapsin I phosphorylation at the PKA site (data not shown).
Figure 9.

Both GluR1 phosphorylation at the PKA site and PKA autonomous activity were reduced in lithium- and valproate-treated animals in hippocampus. A, B, Phosphorylation of GluR1 in hippocampus from lithium (L) or valproate (V) chronically treated animals. C, Control. Rats were treated with lithium or valproate for 4 weeks; hippocampal tissues were isolated and homogenated. Equal amounts of homogenate proteins were separated by gel electrophoresis and analyzed with anti-GluR1p845, anti-GluR1p831, and anti-actin antibodies. The bands were analyzed by the Kodak Imaging System (n = 8 for all three groups; ANOVA; *p < 0.05). C, PKA activity in hippocampus from lithium or valproate chronically treated animals. Protein samples were prepared from one hippocampus of lithium- or valproate-treated animals. Equal amounts of proteins were assayed for PKA activity (n = 8 for all three groups; ANOVA; *p < 0.05).
One of the possible mechanisms for the attenuation of GluR1 phosphorylation at the PKA site is that basal PKA activity was decreased in the hippocampus after lithium or valproate treatment. Therefore, we measured the PKA activity in hippocampal homogenates from lithium- or valproate-treated animals. Indeed, basal levels of PKA activity in tissue homogenates from both lithium- and valproate-treated animals were reduced significantly (Fig. 9B).
The antidepressant agent imipramine (which is capable of inducing mania) increases surface GluR1 levels
The two structurally dissimilar antimanic agents lithium and valproate robustly decrease synaptic GluR1 expression, effects that previously have been associated with reduced AMPA receptor synaptic strength. We therefore next sought to determine whether a promanic agent might produce opposite effects. It is well established that tricyclic antidepressants can precipitate manic episodes in individuals with bipolar disorder (Nolen and Bloemkolk, 2000); therefore, we used the mixed serotonin/norepinephrine reuptake inhibitor antidepressant imipramine. Indeed, as predicted, chronic treatment of rats with imipramine produced increases in synaptosomal GluR1 levels, whereas synaptophysin levels in the same preparations remained unchanged (Fig. 10).
Figure 10.

Antidepressant imipramine (Im) induces an increase in synaptic GluR1 in vivo. Rats were treated with imipramine (10 mg/kg, twice daily, i.p.) for 4 weeks; hippocampal tissues were isolated, and synaptosome preparations were obtained by using a Ficoll gradient assay. A, B, Equal amounts of proteins were separated by gel electrophoresis and analyzed with anti-GluR1 antibodies and anti-synaptophysin antibodies. The bands were analyzed by the Kodak Imaging System (n = 8; t test; *p < 0.05). Con, Control.
Discussion
In this study we have demonstrated for the first time that AMPA GluR1 receptor trafficking may play an important role in the adaptive synaptic plasticity underlying the treatment of bipolar disorder. Thus, we found that the structurally highly dissimilar antimanic agents lithium and valproate have a common effect on downregulating AMPA GluR1 synaptic expression in the hippocampus after prolonged treatment with therapeutically relevant concentrations as assessed both in vitro and in vivo. In cultured hippocampal neurons lithium and valproate attenuated surface GluR1 expression after chronic (days) treatment. Additionally, we found that this effect on GluR1 surface expression may be attributable to a reduction of GluR1 phosphorylation at the PKA (GluR1p845) site. Furthermore, rather than compensating for GluR1 reductions, GluR2/3 levels also were reduced at synapses, suggesting a coordinated regulation of the major AMPA receptor subtypes. However, NR1 levels at synapses remain unchanged after mood stabilizer treatment, suggesting an AMPA-specific mechanism. Further supporting the therapeutic relevance of the finding, we found that an agent that provokes mania, namely the antidepressant imipramine, has an opposite effect because it upregulates hippocampal GluR1. Notably, a similar upregulation of GluR1 receptors also has been reported with psychostimulants, which also are capable of inducing manic episodes (Carlezon and Nestler, 2002).
Because chronic administration of mood stabilizers brings about numerous biochemical effects, our laboratory (Manji and Lenox, 1999, 2000) and others (Coyle and Duman, 2003) have established several criteria that findings should meet to maximize the likelihood of their therapeutic importance. First, this effect of mood stabilizers on GluR1 is a common effect of the structurally highly dissimilar antimanic agents lithium, which is a monovalent cation, and valproate, which is an eight-carbon branched fatty acid. Second, this attenuation of synaptic GluR1 by lithium and valproate occurs in the hippocampus, a brain region known to be involved in critical affective neuronal circuits. Third, this effect of lithium and valproate on synaptic GluR1 occurs at therapeutic concentrations both in vivo and in vitro. Fourth, similar to the clinical therapeutic effects, the changes in GluR1 were observed only after chronic (and not acute) administration. Fifth, all three types of AMPA receptors are attenuated at synapses after lithium or valproate treatment, suggesting that the GluR1 attenuation at synapses will not be compensated by other subtypes of AMPA receptors. Finally, the effects are specific for antimanic agents, because the promanic antidepressant produced opposite effects. Although it is impossible to determine whether synaptic GluR1 attenuation occurs in the human brain in vivo, our experimental conditions attempted to mimic this situation as closely as possible.
Further supporting our data are the recent studies that show that AMPA receptor antagonists attenuate several “manic-like” behaviors produced by amphetamine administration. Thus, AMPA antagonists have been demonstrated to attenuate psychostimulant-induced development or expression of sensitization and hedonic behavior without affecting spontaneous locomotion; additionally, some studies have demonstrated that AMPA receptor antagonists reduce amphetamine/cocaine-induced hyperactivity (Willins et al., 1992; Burns et al., 1994; Li et al., 1997; Mead and Stephens, 1998; Hotsenpiller et al., 2001; Backstrom and Hyytia, 2003). The need to use caution in the appropriate application of animal models to complex neuropsychiatric disorders has been well articulated, and in fact it is un-likely we will ever develop rodent models that display the full range of symptomatology as clinically expressed in man (Nestler et al., 2002b; Einat et al., 2003; McClung and Nestler, 2003). However, one current model of mania, which has been used extensively and has reasonable heuristic value in the studies of mood disorders, involves the use of psychostimulants in appropriate paradigms. Thus, psychostimulants like amphetamine and cocaine are known to induce manic-like symptoms in healthy volunteers and trigger frank manic episodes in individuals with bipolar disorder (Goodwin and Jamison, 1990). Thus, the best-established animals of mania use the administration of amphetamine or cocaine to produce hyperactivity, risk-taking behavior, and increased hedonic drive, all very important facets of the human clinical condition of mania. Moreover, these psychostimulant-induced behavioral changes are attenuated by the administration of chronic lithium in a therapeutically relevant time frame. Thus, the fact that AMPA receptor antagonists are capable of attenuating psychostimulant-induced sensitization, hyperactivity, and hedonic behavior (Willins et al., 1992; Burns et al., 1994; Li et al., 1997; Mead and Stephens, 1998; Schmidt, 1998; Hotsenpiller et al., 2001; Backstrom and Hyytia, 2003) provides compelling behavioral support for our contention that AMPA receptors play important roles in regulating affective behavior.
As discussed previously, in striking contrast to the effects seen with the anti-manic agents lithium and valproate, we found that the chronic administration of the antidepressant imipramine, which is capable of triggering manic episodes, in susceptible individuals (Goodwin and Jamison, 1990) increased hippocampal synaptic expression of GluR1. Very recent studies from other laboratories also have demonstrated that chronic administration of antidepressants enhances membrane expression of GluR1 as well as phosphorylation of GluR1 at the PKA site (p845) and the CaMKII/PKC site (p831)(Martinez-Turrillas et al., 2002; Li et al., 2003). Furthermore, it is noteworthy that AMPA-potentiating agents reportedly have efficacy in preclinical models of depression (Li et al., 2001). An elegant series of studies recently has provided insights into how dopamine receptors, which are activated during psychostimulant administration, might influence glutamate-dependent forms of synaptic plasticity that are increasingly recognized as important for the long-term behavioral effects of these drugs (Carlezon and Nestler, 2002; Chao et al., 2002). These studies have demonstrated that surface GluR1 labeling on processes of medium spiny neurons and interneurons was increased by brief incubation with a D1 agonist. Although these studies were designed to investigate the role of GluR1 in mediating the effects of psychostimulants in the context of drug abuse, it is noteworthy that many of the symptoms of mania resemble the effects of psychostimulants (e.g., locomotor hyperactivity, racing thoughts, reduced sleep, psychosis, increased hedonic drive). Taken together, the biochemical and behavioral studies investigating the effects of antimanic (lithium and valproate) and promanic (antidepressants, cocaine, amphetamine) agents on GluR1 strongly suggest that AMPA receptor trafficking may represent an important mediator of the pathogenesis and treatment of certain facets of bipolar disorder.
Our mechanistic studies suggested that GluR1 phosphorylation at the PKA site plays an important role in lithium- or valproate-induced regulation of surface expression of GluR1. Previous studies have demonstrated that phosphorylation of the PKA site (GluR1p845) and/or the CaMKII/PKC site (GluR1p831) is the key regulator for synaptic distribution of GluR1. Therefore, we next investigated the effects of lithium and valproate on the phosphorylation of GluR1. We found that phosphorylation of GluR1 at the PKA site (p845) was attenuated significantly after lithium and valproate treatment by 52.0 and 31.0%, respectively; in contrast, phosphorylation of GluR1 at CaMKII/PKC site was not altered by lithium or valproate treatment (Fig. 7B,C), suggesting that lithium and valproate regulate GluR1 phosphorylation in a site-specific manner. To determine whether this attenuation of phosphorylation on GluR1p845 correlates with the surface localization of GluR1, we stimulated the phosphorylation of GluR1 on the p845 site with Sp-cAMP, a membrane-permeable specific PKA activator. The surface GluR1 levels subsequently were investigated to determine whether reversing lithium and valproate effects on phosphorylation of GluR1 might bring GluR1 back to the neuronal surface. We found that Sp-cAMP reversed the lithium- or VPA-induced reductions in phosphorylation of GluR1 on the PKA site and normalized surface GluR1 levels.
Adding additional support to our contention that the antimanic agents bring about these effects via PKA, we found that not only GluR1 phosphorylation at the PKA site but also PKA activity were attenuated after chronic administration of lithium or valproate in vivo. To determine that these effects were relatively specific for GluR1s and that not all PKA phosphorylation was altered similarly, we investigated the effects of these agents on another major synaptic PKA target, synapsin I (Kao et al., 2002). We found that, in contrast to GluR1 receptors, 4 weeks of treatment with lithium or VPA did not reduce PKA phosphorylation of synapsin I. Additionally, we examined three other proteins, including the protein PSD95 as well as the presynaptic proteins synaptophysin and synaptobrevin. We found that chronic treatment with the mood stabilizers did not alter these proteins significantly, suggesting that GluR receptors are important and relatively selective synaptic targets for the chronic actions of mood stabilizers.
It is now well established that regulation of synaptic GluR1 trafficking is a complex, multifaceted process. We propose that the attenuation of synaptic GluR1 by mood stabilizers is a two step process whereby (1) insertion of GluR1 onto neuronal surface is governed by phosphorylation of GluR1 on the PKA site (GluR1p845) and (2) GluR1 moving into synapses is regulated by the activation of CaMKII (Hayashi et al., 2000; Lee et al., 2000). Lithium and valproate attenuate only phosphorylation of PKA site, resulting in a decrease in surface GluR1, which is the resource of synaptic GluR1. Consistent with the effects observed in the present study, previous in vivo and in vitro studies have shown that chronic lithium and valproate treatments attenuate adenylyl cyclase activity via distinct mechanisms (Gould and Manji, 2002; Gould et al., 2004). Earlier work suggests that there is a significant delay between an attenuation of GluR1 surface expression and a decrease in GluR1 at functional synapses in cultured cortical neurons (Wolf et al., 2003). The time course of lithium and valproate effects on surface and synaptic GluR1 is in agreement with these findings. Although requiring chronic treatment, surface GluR1 levels were reduced at a time point when the number of GluR1-positive synapses was unchanged; the downregulation of synaptic GluR1 was detected only after more prolonged treatment. These findings are compatible with the delayed onset and long-term efficacy of lithium and valproate.
Both lithium and valproate attenuate the phosphorylation of GluR1 on its PKA site after chronic administration in vitro and in vivo. Several lines of evidence support activity-dependent endocytosis of the AMPA receptor, and two mechanisms have been proposed for AMPA receptor internalization: AMPA-dependent and AMPA-independent mechanisms. Synaptic activity (induced by picrotoxin) can stimulate AMPA receptor endocytosis in an NMDA receptor-independent manner (Ehlers, 2000; Lin et al., 2000). In contrast, insulin can induce AMPA receptor internalization by a clathrin-dependent mechanism independent of AMPA release (Man et al., 2000). The internalization of the receptor usually is in response to extracellular stimuli and occurs in an acute manner. Because we observed that the phosphorylation of GluR1 at the PKA site is attenuated by lithium and valproate, we propose that this decrease in surface expression might be caused by reduced insertion of GluR1 receptors onto the membrane. However, as elucidated previously, this is a highly complex multi-step process, and additional studies will be necessary to delineate the precise mechanisms by which mood stabilizers exert their effects on GluR trafficking.
In conclusion, we have shown that AMPA GluR1 receptor trafficking may play a critical role in the treatment of mania. Now it is well established that glutamatergic neurotransmission plays a critical role in regulating various forms of plasticity; the regulation of synaptic AMPA receptors thus has the potential to contribute to the communication of critical circuits involved in affective functioning and buffering. The mechanisms by which glutamate receptors are actively recruited to synapses have long intrigued the neuroscience community; the results presented here suggest that they may also play important roles in the pathophysiology and treatment of complex neuropsychiatric disorders. This progress holds much promise for the development of novel therapeutics for the long-term treatment of severe refractory mood disorders and for improving the lives of millions.
Footnotes
We acknowledge the support of the Intramural Research Program of the National Institute of Mental Heath and the Stanley Medical Research Institute. We also thank Drs. Kazutoshi Nakazawa and Jeffrey Diamond for invaluable discussions and important recommendations.
Correspondence should be addressed to Dr. Husseini K. Manji, Chief, Laboratory of Molecular Pathophysiology, National Institute of Mental Health, 49 Convent Drive MSC 4405, Building 49, B1EE16, Bethesda, MD 20892. E-mail: manjih@intra.nimh.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246578-12$15.00/0
References
- Backstrom P, Hyytia P (2003) Attenuation of cocaine-seeking behaviour by the AMPA/kainate receptor antagonist CNQX in rats. Psychopharmacology (Berl) 166: 69-76. [DOI] [PubMed] [Google Scholar]
- Burns LH, Everitt BJ, Kelley AE, Robbins TW (1994) Glutamate-dopamine interactions in the ventral striatum: role in locomotor activity and responding with conditioned reinforcement. Psychopharmacology (Berl) 115: 516-528. [DOI] [PubMed] [Google Scholar]
- Carlezon Jr WA, Nestler EJ (2002) Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci 25: 610-615. [DOI] [PubMed] [Google Scholar]
- Chao SZ, Ariano MA, Peterson DA, Wolf ME (2002) D1 dopamine receptor stimulation increases GluR1 surface expression in nucleus accumbens neurons. J Neurochem 83: 704-712. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Duman RS (2003) Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 38: 157-160. [DOI] [PubMed] [Google Scholar]
- Drevets WC (2000) Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813-829. [DOI] [PubMed] [Google Scholar]
- Drevets WC (2001) Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Curr Opin Neurobiol 11: 240-249. [DOI] [PubMed] [Google Scholar]
- D'Sa C, Duman R (2002) Antidepressants and neuroplasticity. Bipolar Disord 4: 183-194. [DOI] [PubMed] [Google Scholar]
- Du J, Feng L, Yang F, Lu B (2000) Activity- and Ca2+-dependent modulation of surface expression of brain-derived neurotrophic factor receptors in hippocampal neurons. J Cell Biol 150: 1423-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G (2003) The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23: 7311-7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban JA, Shi SH, Wilson C, Nuriya M, Huganir RL, Malinow R (2003) PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat Neurosci 6: 136-143. [DOI] [PubMed] [Google Scholar]
- Goodwin FK, Jamison KR (1990) Manic-depressive illness. New York: Oxford UP.
- Gould TD, Manji HK (2002) The Wnt signaling pathway in bipolar disorder. Neuroscientist 8: 497-511. [DOI] [PubMed] [Google Scholar]
- Gould TD, Chen G, Manji HK (2004) In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29: 32-38. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262-2267. [DOI] [PubMed] [Google Scholar]
- Hotsenpiller G, Giorgetti M, Wolf ME (2001) Alterations in behaviour and glutamate transmission following presentation of stimuli previously associated with cocaine exposure. Eur J Neurosci 14: 1843-1855. [DOI] [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P (2002) A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci 5: 431-437. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, Epperson CN, Goddard A, Mason GF (2002) Glutamate and GABA systems as targets for novel antidepressant and mood-stabilizing treatments. Mol Psychiatry 7[Suppl 1]: S71-S80. [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21: 1151-1162. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955-959. [DOI] [PubMed] [Google Scholar]
- Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P (2001) Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology 40: 1028-1033. [DOI] [PubMed] [Google Scholar]
- Li X, Witkin JM, Need AB, Skolnick P (2003) Enhancement of antidepressant potency by a potentiator of AMPA receptors. Cell Mol Neurobiol 23: 419-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vartanian AJ, White FJ, Xue CJ, Wolf ME (1997) Effects of the AMPA receptor antagonist NBQX on the development and expression of behavioral sensitization to cocaine and amphetamine. Psychopharmacology (Berl) 134: 266-276. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R (2001) Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci 21: 6008-6017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M (2000) Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 3: 1282-1290. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT (2000) Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25: 649-662. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH (1999) Ziskind-Somerfeld research award. Protein kinase C signaling in the brain: molecular transduction of mood stabilization in the treatment of manic-depressive illness. Biol Psychiatry 46: 1328-1351. [DOI] [PubMed] [Google Scholar]
- Manji HK, Lenox RH (2000) The nature of bipolar disorder. J Clin Psychiatry 61[Suppl 13]: 42-57. [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS (2001) The cellular neurobiology of depression. Nat Med 7: 541-547. [DOI] [PubMed] [Google Scholar]
- Martinez-Turrillas R, Frechilla D, Del Rio J (2002) Chronic antidepressant treatment increases the membrane expression of AMPA receptors in rat hippocampus. Neuropharmacology 43: 1230-1237. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ (2003) Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat Neurosci 6: 1208-1215. [DOI] [PubMed] [Google Scholar]
- Mead AN, Stephens DN (1998) AMPA receptors are involved in the expression of amphetamine-induced behavioural sensitization, but not in the expression of amphetamine-induced conditioned activity in mice. Neuropharmacology 37: 1131-1138. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Wolf ME (2003) Glutamate and disorders of cognition and motivation. Ann NY Acad Sci 1003: 1-481.14684431 [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002a) Neurobiology of depression. Neuron 34: 13-25. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Gould E, Manji H, Buncan M, Duman RS, Greshenfeld HK, Hen R, Koester S, Lederhendler I, Meaney M, Robbins T, Winsky L, Zalcman S (2002b) Preclinical models: status of basic research in depression. Biol Psychiatry 52: 503-528. [DOI] [PubMed] [Google Scholar]
- Nolen WA, Bloemkolk D (2000) Treatment of bipolar depression, a review of the literature and a suggestion for an algorithm. Neuropsychobiology 42[Suppl 1]: 11-17. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Gottschalk W, Zhang L, McDermott K, Du J, Gopalakrishnan R, Oho C, Sheng ZH, Lu B (1999) Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knock-out mice. J Neurosci 19: 4972-4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577-582. [DOI] [PubMed] [Google Scholar]
- Schmidt WJ (1998) Dopamine-glutamate interactions in the basal ganglia. Amino Acids 14: 5-10. [DOI] [PubMed] [Google Scholar]
- Tartaglia N, Du J, Tyler WJ, Neale E, Pozzo-Miller L, Lu B (2001) Protein synthesis-dependent and -independent regulation of hippocampal synapses by brain-derived neurotrophic factor. J Biol Chem 276: 37585-37593. [DOI] [PubMed] [Google Scholar]
- Willins DL, Wallace LJ, Miller DD, Uretsky NJ (1992) α-Amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor antagonists in the nucleus accumbens and ventral pallidum decrease the hypermotility response to psychostimulant drugs. J Pharmacol Exp Ther 260: 1145-1151. [PubMed] [Google Scholar]
- Wolf ME, Mangiavacchi S, Sun X (2003) Mechanisims by which dopamine receptors may influence synaptic plasticity. Ann NY Acad Sci 1003: 241-249. [DOI] [PubMed] [Google Scholar]
- Young LT (2002) Neuroprotective effects of antidepressant and mood stabilizing drugs. J Psychiatry Neurosci 27: 8-9. [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Chen G, Manji HK (1999) Lithium activates the c-Jun NH2-terminal kinases in vitro and in the CNS in vivo J Neurochem 73: 2299-2309. [DOI] [PubMed] [Google Scholar]