

Abstract
Manic-depressive illness has been conceptualized as a neurochemical illness. However, brain imaging and postmortem studies reveal gray-matter reductions, as well as neuronal and glial atrophy and loss in discrete brain regions of manic-depressive patients. The roles of such cerebral morphological deficits in the neuropathophysiology and therapeutic mechanisms of manic-depressive illness are unknown. Valproate (2-propylpentanoate) is a commonly used mood stabilizer. The ERK (extracellular signal-regulated kinase) pathway is used by neurotrophic factors to regulate neurogenesis, neurite outgrowth, and neuronal survival. We found that chronic treatment of rats with valproate increased levels of activated phospho-ERK44/42 in neurons of the anterior cingulate, a region in which we found valproate-induced increases in expression of an ERK pathway-regulated gene, bcl-2. Valproate time and concentration dependently increased activated phospho-ERK44/42 and phospho-RSK1 (ribosomal S6 kinase 1) levels in cultured cortical cells. These increases were attenuated by Raf and MEK (mitogen-activated protein kinase/ERK kinase) inhibitors. Although valproate affects the functions of GSK-3 (glycogen synthase kinase-3) and histone deacetylase (HDAC), its effects on the ERK pathway were not fully mimicked by selective inhibitors of GSK-3 or HDAC. Similar to neurotrophic factors, valproate enhanced ERK pathway-dependent cortical neuronal growth. Valproate also promoted neural stem cell proliferation-maturation (neurogenesis), demonstrated by bromodeoxyuridine (BrdU) incorporation and double staining of BrdU with nestin, Tuj1, or the neuronal nuclei marker NeuN (neuronal-specific nuclear protein). Chronic treatment with valproate enhanced neurogenesis in the dentate gyrus of the hippocampus. Together, these data demonstrate that valproate activates the ERK pathway and induces ERK pathway-mediated neurotrophic actions. This cascade of events provides a potential mechanism whereby mood stabilizers alleviate cerebral morphometric deficits associated with manic-depressive illness.
Keywords: valproate, ERK, neurite outgrowth, neurogenesis, mania, mood disorders
Introduction
Manic-depressive illness (also known as bipolar disorder) has been conceptualized traditionally as a neurochemical illness involving imbalances in certain neurotransmitter systems (Goodwin and Jamison, 1990). However, recent brain-imaging and postmortem morphometric studies reveal regional reductions in CNS volumes, as well as reductions in neuropil and in numbers of glia and neurons in discrete brain regions of manic-depressive patients (Harrison, 2002; Coyle and Duman, 2003). A volume reduction of ∼40% of subgenual prefrontal cortex gray matter has been observed in familial bipolar patients (Drevets et al., 1997). Initial results suggest that such reductions may be prevented or reversed when patients are treated with mood stabilizers (Drevets et al., 1997; Drevets, 2000). This hypothesis is supported by our finding that chronic treatment with lithium, a classic antimanic mood stabilizer, increases cerebral gray-matter volumes and N-acetyl aspartate (NAA) (a neuronal viability marker) levels of patients with mood disorders (Moore et al., 2000a,b). The cause(s) of these volumetric reductions, the interrelationship of these reductions and occurrences of mania or depression, and the mechanisms by which mood stabilizers reverse the reductions and improve clinical symptoms are unknown.
Valproate (2-propylpentanoate) is an anticonvulsant and antimanic mood stabilizer (Bowden et al., 1994). Valproate-induced modulation of GABA levels in the brain may underlie its anticonvulsive action (Gould et al., 2002). However, the antimanic mechanism of valproate is primarily unknown (Gould et al., 2002). Neurotrophic factors activate the Ras-Raf-MEK [MAP (mitogen-activated protein) kinase/ERK (extracellular signal-regulated kinase) kinase] pathway and, through this pathway, produce cellular neurotrophic actions, including neurite growth, regeneration, and neurogenesis (Kaplan and Miller, 2000; Marinissen and Gutkind, 2001; Weeber and Sweatt, 2002; Huang and Reichardt, 2003). The ERK pathway has also been shown to be involved in long-term potentiation, long-term depression, learning and memory, cognition, and neurogenesis (Kaplan and Miller, 2000; Marinissen and Gutkind, 2001; Weeber and Sweatt, 2002; Huang and Reichardt, 2003). Recently, we found that chronic treatment of rats with valproate or lithium activates the ERK pathway. The activation is demonstrated by increases in phospho-ERK44/42, phospho-RSK1 (ribosomal S6 kinase 1) (ERK substrate), phospho-CREB (cAMP response element-binding protein), phospho-BAD [Bcl-2 (B-cell lymphoma protein 2)-associated death protein] (RSK substrates), BDNF, and Bcl-2 (CREB-regulated gene products) in total homogenates of rat prefrontal cortex and hippocampus (Chen et al., 1999a; Yuan et al., 2001; Einat et al., 2003). Treatment of rats with an inhibitor of MEK blocks activation of ERK44/42 and induces mood disorder-related behavioral deficits (Einat et al., 2003). These data suggest that activation of the ERK pathway is a therapeutically relevant action of antimanic mood stabilizers.
On the basis of known effects of mood stabilizers, functions of the ERK pathway, and known neurobiology of bipolar disorder, we postulate that mood stabilizers may activate the ERK pathway and produce morphological actions similar to those of neurotrophic factors in CNS cells. We also postulate that such actions play an important role in the prevention or recovery of brain structural deficits observed in manic-depressive patients and thus play a fundamental role in the prophylaxis of recurrent mood episodes.
Materials and Methods
Reagents. Sodium valproate, bromodeoxyuridine (BrdU), poly-D-lysine, dimethylsulfoxide (DMSO), trichostatin A (TSA), protease inhibitor mixture, and phosphatase inhibitor mixtures I and II were from Sigma-Aldrich (St. Louis, MO). Raf inhibitor I (5-iodo-3-[(3,5-dibromo-4-hydroxyphenyl)methylene]-2-indolinone), ZM336372 (N-[5-(3-dimethylaminobenzamido)-2-methylphenyl]4-hydroxybenzamide), PD98059 (2′-amino-3′-methoxyflavone), U0126 (1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene), SB202190 (4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole), and GSK-3 (glycogen synthase kinase-3) inhibitor II (2-thio(3-iodobenzyl-5-(1-pyridyl)-[1,3,4]-oxadiazole) were from Calbiochem (La Jolla, CA). All cell culture products were from Invitrogen (Carlsbad, CA) unless specified otherwise. All antibodies were from Cell Signaling Technology (Beverly, MA) unless specified otherwise.
Animals and animal treatments. All animal experiments were approved by the National Institutes of Health Animal Care and Use Committee in accordance with National Institutes of Health guidelines on the care and use of animals. Animal treatment was performed using our previously reported protocol (Einat et al., 2003). Male Wistar rats (150-200 gm) were housed four to five per cage with water and food available ad libitum and were maintained under a 12 hr light/dark cycle. After a 1 week accommodation period, the rats were fed either a regular or a sodium valproate-supplemented (20 gm/kg) chow. Treatment with the sodium valproate-supplemented chow routinely yields blood levels of valproic acid close to or within the human therapeutic window (50-150 mg/l or 0.35-1.04 mm) (McElroy and Keck, 1995). Rats were killed between 8:00 A.M and 12:00 P.M. after 4 weeks of treatment. Blood samples were collected to detect valproic acid levels. Blood valproic acid levels were 42 ± 6 mg/l. This level may not reflect the average daily level of valproic acid because the half-life of valproic acid is <0.5 hr in rats (Yoshioka et al., 2000; Stout et al., 2001).
For the in vivo neurogenesis studies, male C57BL/6 mice (20-25 gm) were injected with 300 mg/kg BrdU, a dosage selected on the basis of a previous report (Cameron and McKay, 2001). To study the potential effects of valproate on proliferation of progenitor cells, mice first were fed either regular or sodium valproate-supplemented (20 gm/kg) chows for 6 weeks and then administered a single injection (intraperitoneally) of BrdU and killed 2 hr after the injection. To further study the potential effects of valproate on survival, differentiation, and proliferation of progenitor cells, mice were first injected with BrdU and then fed either regular or sodium valproate-supplemented (20 gm/kg) chows for 6 weeks. The pooled blood valproic acid level of treated mice was 79 mg/l.
Given that valproate exposure causes neural developmental defects, we examined the brains of adult rats and mice treated with valproate chow and did not find marked anatomical alterations.
Immunohistochemistry of phospho-ERK44/42 and ERK44/42. After treatments, rats were deeply anesthetized and perfused via the ascending aorta with saline, followed by 0.1 m phosphate buffer, pH 7.4, containing 4% paraformaldehyde. The brains were removed and postfixed in the same fixative overnight at 4°C. After immersion in 0.1 m PBS containing 20% sucrose for 48 hr (4°C), the brains were rapidly frozen and stored at -75°C. Serial sections (30 μm) were cut coronally through the entire cerebrum. Immunostaining was performed using the free-floating method with mouse monoclonal anti-phospho-ERK44/42 (Thr202/Tyr204) IgG (1:600). The immunoreaction product was visualized using the avidin-biotin complex method (Hsu and Raine, 1981) with the Vectastain Elite ABC Peroxidase kit (Vector Laboratories, Burlingame, CA). Immunostaining of phospho-ERK44/42 was blocked by phospho-ERK44/42-blocking peptide to verify phospho-ERK44/42 staining specificity. Two investigators that were blind to the treatment codes of the slices independently conducted the comparisons of valproate and control samples.
Primary cortical cell culture and treatments. Embryonic day 18 (E18) cortical cells were isolated from embryonic brain and cultured in a humidified atmosphere of 95% air-5% CO2 at 37°C in Neurobasal medium plus B27 supplement and antibiotics for 8 d in vitro (DIV 8). More than 95% of cells in culture expressed neuronal markers. Cells were then treated with basic fibroblast growth factor (bFGF), neurotrophin-3 (NT-3), valproate, protein kinase inhibitors, TSA, or DMSO (0.1%; vehicle control for kinase inhibitors and TSA) for times and concentrations indicated in the figures and legends. To observe neurite growth-regeneration, cell proliferation, and neurogenesis, a gap was created in the middle of cultured dishes or slides using a 100 μl pipette tip. Cultures were then treated with reagents indicated in the figures. Gaps were monitored for signs of growth. Digital pictures of cultures were taken using a Nikon (Tokyo, Japan) Eclipse microscope equipped with a Nikon TS100 camera. For the observation of cell proliferation and neurogenesis, cortical cells were incubated with BrdU (50 μg/ml) for 6 hr and treated with different reagents as indicated in the figures.
Immunoblot analysis. Immunoblotting was conducted as described previously (Yuan et al., 2001). In brief, the cells were washed with PBS and homogenized by brief sonication in an extraction buffer containing 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, protease inhibitor mixture, and phosphatase inhibitor mixtures I and II. Homogenates were centrifuged at 14,000 × g for 15 sec to remove undissolved debris. Immunoblotting was performed using protein amounts demonstrated to be within the linear range for the detection system. The antibodies for immunoblotting were diluted according to the recommendations of the manufacturer. The resultant primary-secondary immunocomplex was subsequently detected with an ECL kit (Amersham Biosciences, Piscataway, NJ). Quantitation of the immunoblots was performed by densitometric scanning of exposed film using a Kodak Image Analysis (Eastman Kodak, Rochester, NY) system.
Confocal immunocytochemistry for cell proliferation and neurogenesis in cultured cortical cells. Cells were fixed in 70% cold alcohol for 30 min, washed three times with PBS, and permeabilized with 0.4% Triton X-100 in PBS for 30 min. After washing, the cultures were incubated in 2 m HCl for 10 min and with Na2O4B7 for another 10 min. Cultures were washed three times again with PBS. Cultures were incubated with primary antibodies in blocking solution (1% BSA in PBS) at 4°C overnight. Cells were washed three times in PBS and incubated in secondary antibody in blocking solution at room temperature for 2 hr. Antibodies were as follows: rat monoclonal anti-BrdU (1:100; Accurate, Westbury, NY); mouse monoclonal anti-unique β tubulin (TuJ1) (1:500; Babco, Richmond, CA); mouse monoclonal anti-nestin (1:1000; BD PharMingen, San Diego, CA); mouse monoclonal anti-NeuN (neuronal-specific nuclear protein) (1:200; Chemicon, Temecula, CA); and rabbit polyclonal anti-GFAP (1: 500; Dako, Carpinteria, CA), and FITC- or rhodamine-conjugated secondary antibodies (1:200; Jackson ImmunoResearch, West Grove, PA). 4′,6′-Diamidino-2-phenylindole (DAPI) (10 μg/ml; Molecular Probes, Eugene, OR) was used to counterstain nuclei in some cases. Fluorescence was detected with a Zeiss (Thornwood, NY) LSM510 Meta multiphoton system at excitation/emission wavelengths of 535/565 nm (rhodamine, red), 488/505 nm (FITC, green), and 745/400 nm (DAPI, blue). Controls were prepared by omitting or preabsorbing the primary antibody or omitting the secondary antibody. ProLong Antifade Kit (Molecular Probes) was used to mount the slides.
Confocal immunocytochemistry for neurogenesis in adult mice hippocampi. Mouse brain was initially processed as described above for the rat brain immunohistochemical experiments. Neurogenesis was examined using our previously reported method with some modifications (Chen et al., 2000). In brief, stereological principles were followed to avoid potential biases. Serial coronal sections (30 μm) were cut through the entire anteroposterior extension of the hippocampi. Every first, second, and third section from each series was collected separately. The set of second sections were used for BrdU and NeuN immunofluorescence stainings. Sections were processed as described for primary cortical cell culture and examined with a Zeiss LSM510 Meta multiphoton system. BrdU-positive cells were counted within and one-cell-wide below the granule cell layer. Only profiles of neuronal nuclei with a complete nuclear contour were included. In total, eight sections from each mouse were counted.
Statistical analysis. Statistical analyses were performed by ANOVA, followed by Fisher's PLSD or Scheffe's tests. Unpaired t tests were used for comparisons of two groups. p < 0.05 was considered significant. Data are expressed as the means ± SE.
Results
Valproate increases immunoreactivity of activated phospho-ERK44/42 in cells of anterior cingulate
First, we investigated whether valproate activates the ERK pathway in a specific population or region of CNS cells. Rats were chronically treated with valproate in a clinically relevant regimen. Immunohistochemical staining revealed that valproate treatment did not significantly alter total ERK44/42 immunoreactivity (Fig. 1A) but markedly increased activated phospho-ERK44/42 immunoreactivity in anterior cingulate cells with neuronal characteristics (Fig. 1B). The processes and nuclei were heavily stained. This staining pattern is known to be associated with alterations in synaptic function and gene expression (Kaplan and Miller, 2000; Marinissen and Gutkind, 2001; Weeber and Sweatt, 2002; Huang and Reichardt, 2003). Interestingly, it is the same region in which we found valproate-induced expression of bcl-2 (Chen et al., 1999a), a gene regulated by the ERK pathway (Riccio et al., 1999). Other cortical and subcortical regions were also examined; however, detection of potential changes in those regions may require more sensitive quantifications.
Figure 1.
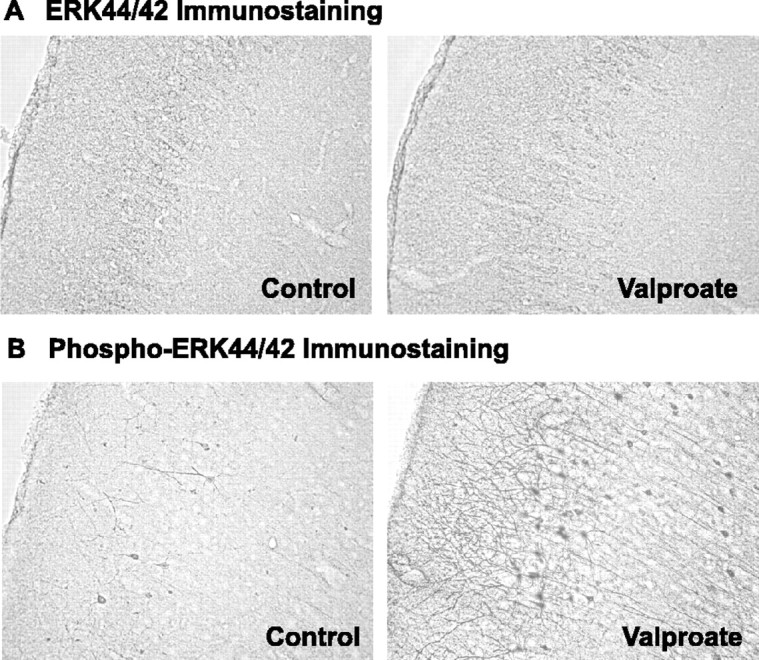
Increases in prefrontal cortical phospho-ERK44/42, but not total ERK44/42, immunoreactivities by chronic valproate treatment. Rats were fed regular or sodium valproate-supplemented (20 gm/kg) chows for 4 weeks, yielding a mean serum valproic acid concentration of 42 mg/l. Immunostaining of the brain slices was performed. Valproate treatment markedly increased intensities of phospho-ERK44/42 (B), but not of total ERK44/42 (A), in prefrontal cortical cells with neuronal characteristics. Two investigators, blind to the treatments, independently examined 10 sets of paired samples and reported clear increases in phospho-ERK44/42 staining intensities in eight sets.
Valproate activates the ERK pathway in cultured cortical cells in a time- and concentration-dependent matter
To confirm that valproate activates the ERK pathway in cortical neurons and to understand the potential neurobiological significance of valproate-induced ERK pathway activation, we conducted experiments in cultured E18, DIV 8 cortical cells using neurotrophic factors as a comparison. The activation of the ERK pathway was monitored by measuring levels of activated phospho-ERK44/42 and phospho-RSK1. As expected, incubation of cultured cortical cells with neurotrophic factors bFGF and NT-3 resulted in rapid increases of phospho-ERK44/42 and phospho-RSK1 levels (Fig. 2A-E). In the same system, therapeutic concentrations of valproate increased phospho-ERK44/42 and phopsho-RSK1 levels in concentration- (Fig. 3A,B) and time-dependent manners (Fig. 3C,D). Neurotrophic factor- and valproate-induced increases in phospho-ERKs or phospho-RSK1 are not caused by changes in total ERK44/42 or RSK1 proteins (Figs. 2F,3A,C). Neurotrophic factor-induced increases in both phospho-ERKs and phospho-RSK1 rapidly subsided (Fig. 2A-E), whereas valproate-induced increases were more sustained (Fig. 3C,D). A possible explanation for the difference is that neurotrophic factors desensitize trophic pathways through receptor downregulation (Frank et al., 1996). Valproate may not directly bind to receptor tyrosine kinase(s) and therefore may not desensitize the system.
Figure 2.

Time-dependent activation of the ERK pathway by bFGF, NT-3, or in combination. Rat cortical cells isolated from E18 embryos were cultured in vitro for 8 d, at which time the culture reached confluence. The cells were then treated with bFGF and NT-3. Immunoblotting of phospho-ERK44/42, phospho-RSK1, total ERK44/42, and total RSK1 was conducted. bFGF (10 ng/ml), NT-3 (20 ng/ml), or a combination of both time dependently increased phospho-ERK44/42 (A-C) and phospho-RSK1 levels (D, E) but not total ERK44/42 and total RSK1 (F). Bar graphs (B, C, E) depict densitometric results representing mean ± SE of three or more sets of samples immunoblotted in duplicates as presented in the figure (A, D, F).
Figure 3.

Concentration- and time-dependent activation of the ERK pathway and accumulation of acetylated histone-3 by valproate (VPA) in cortical cells. Rat cortical cells isolated from E18 embryos were cultured in vitro for 8 d, at which time the culture reached confluence. The cells were then treated with sodium valproate. Immunoblotting of phospho-ERK44/42, phospho-RSK1, total ERK44/42, total RSK1, or acetyl-H3 was conducted. Valproate concentration (48 hr) (A, B) and time (0.8 mm) (C, D) dependently increased phospho-ERK44/42, phospho-RSK1, and acetyl-H3 levels are shown. Bar graphs (B, D) depict densitometric results representing mean ± SE of three or more sets of samples immunoblotted in duplicates as presented in A and C. †F(5, 18) = 9.974, p = 0.001 for phosho-ERK44; F(5,18) = 2.892, p = 0.0435 for phospho-ERK42; F(5,18) = 11.404, p < 0.0001 for phospho-RSK1; F(5,18) = 10.224, p < 0.0001 for acetyl-H3. *p < 0.05 compared with 8, 24, 48, 72, and 96 hr valproate-treated cells. #p < 0.05 compared with valproate-treated cells.
Valproate-induced activation of ERK and RSK1 requires MEK and Raf
MEK is the immediate upstream activating kinase of ERK in this pathway. MEK inhibitors U0126 and PD98059 attenuated the magnitudes of valproate-induced increases in phospho-ERK44/42 and phospho-RSK1 (Fig. 4A,B). Raf is the immediate upstream kinase of MEK in the pathway. Raf1 inhibitor I lowered the magnitude of valproate-induced increases in phospho-ERK44/42 and phospho-RSK1 (Fig. 4C,D). Another Raf inhibitor, ZM336372, produced similar effects, but it was less effective (Fig. 4C,D). The Raf1 inhibitor I and MEK inhibitor data are consistent with our previous observations that a dominant-negative Raf mutant and PD98059 block valproate-induced expression of a reporter gene driven by Elk1, an ERK-regulated transcription factor in human neuroblastoma cells (Yuan et al., 2001). Both data sets demonstrate that valproate-induced ERK pathway activation requires Raf and MEK.
Figure 4.

Involvement of ERK pathway components in valproate-(VPA) induced ERK pathway activation. E18, DIV 8 cortical cells obtained as described in Figures 2 and 3 were treated with valproate (0.8 mm) in the absence or presence of indicated inhibitors for 2 d. Immunoblotting was conducted as described in Figures 2 and 3. MEK inhibitors [PD98059 (50 μm) and U0126 (10 μm)] (A, B) and Raf inhibitors [Raf inhibitor I (1 μm) and ZM336372 (10 μm)] (C, D) attenuated valproate-induced increases in phospho-ERK44/42 and phospho-RSK1. U0126 and Raf1 inhibitor I attenuated basal phospho-ERK44/42 and phospho-RSK1 (A-D). Bar graphs (B, D) depict densitometric results representing mean ± SE from three or more sets of samples immunoblotted in duplicates on two gels as presented in A and C. *p < 0.05 compared with cells treated with DMSO alone.
Valproate enhances ERK pathway-dependent neuronal growth
The effects of valproate on the ERK pathway suggest that valproate may function as neurotrophic factors do in promoting neuronal growth and cortical damage recovery. To test this premise, we removed a strip of cortical cells in the middle of confluent cortical cultures (E18, DIV 8) and observed neurite growth into the created gap for 5 d. Neurites and cells reappeared faster in the gaps of cultures treated with neurotrophic factors or valproate than controls (Fig. 5). The MEK inhibitor blocked the reappearance of neurites and cells (Fig. 5), indicating that ERK pathway activation is required for cortical neuronal growth.
Figure 5.

Induction of neurite growth and cell reemergence by neurotrophic factors and valproate (VPA). For observation of cortical cell regeneration, gaps (indicated by arrows; bars indicate the edges of the gaps) were created by removing strips of cells from the middle of dishes or culture slides containing E18, DIV 8 confluent cortical cells as described in Figures 2 and 3. After 2 d, neurite growth and cell reemergence were more pronounced in cultures treated with bFGF (10 ng/ml), NT-3 (20 ng/ml), bFGF plus NT-3, or valproate (0.8 mm) (left) than controls. MEK inhibitor PD98059 (50 μm) blocked growth in valproate-treated and nontreated cultures (left). Similar results were also obtained in three or more sets of samples. Neurite lengths were traced in three sets of samples. Two day valproate (0.8 mm) treatment significantly increased neurite lengths in the gaps (right). *p < 0.05 compared with controls.
Valproate promotes cortical neurogenesis in primary culture
Unexpectedly, we observed reemergence of cells in the created gaps. To characterize these cells, we treated the cultures with BrdU (cell proliferation marker) and stained the cultures with BrdU antibody and DAPI (nuclear staining dye) to monitor cell migration and proliferation. Some DAPI-positive cells were also BrdU positive (Fig. 6A,B). Some BrdU-positive cells appeared to be undergoing mitosis (Fig. 6A). Thus, the reemerged cells either migrated from surrounding areas or were born in situ. Furthermore, we found cells positive for both BrdU and nestin (neural progenitor cell marker) (Fig. 6A), cells positive for both BrdU and TuJ1 (immature neuron marker) (Fig. 6B), and cells positive for both BrdU and NeuN (mature neuron marker) (Fig. 6C). These series of observations indicate the presence of neural stem cells undergoing neurogenesis. Valproate or a combination of bFGF and NT-3 significantly increased numbers of both BrdU- and NeuN-positive cells (Fig. 6), demonstrating that both treatments promoted neurogenesis. The data are consistent with reports showing that neurotrophic factors promote neurogenesis through the ERK pathway in E14 cortical cells (Ghosh and Greenberg, 1995; Menard et al., 2002; Barnabe-Heider and Miller, 2003).
Figure 6.

Induction of neurogenesis by neurotrophic factors and valproate in cortical cells. The cortical cell cultures were obtained and handled as described in Figure 5. To monitor cell proliferation, cultures were treated with BrdU (50 μg/ml) for 6 hr after creating gaps and treated with reagents as described in Figure 5 for 1, 2, or 5 d. The cultures were processed for staining of nuclei with DAPI (blue) and for staining of antigens of BrdU (red), nestin (green), TuJ1 (green), or NeuN (green). Images were obtained using a Zeiss LSM510 Meta multiphoton system. DAPI-positive cells, DAPI plus BrdU-positive cells, and cells undergoing mitosis were present (A, B), suggesting that cells in the gaps migrated from surrounding regions or were born de novo. BrdU plus nestin-(A), BrdU plus TuJ1-(B), or BrdU plus NeuN-(C) positive cells were present, suggesting the existence of neural stem cell neurogenesis. Valproate and the combination of bFGF and NT-3 significantly increased the numbers of both BrdU- and NeuN-positive cells, indicating that neurotrophic factor and valproate treatments promoted neurogenesis (right). *p < 0.05 compared with controls. Scale bars, 10 μm..
Valproate promotes hippocampal neurogenesis in adult mice
Cortical neurogenesis has been observed in adult animals by some (Gould et al., 1999, 2001), but not all (Kornack and Rakic, 2001), investigators. Therefore, we first examined BrdU-positive cells in cortical regions. Although we found BrdU-positive cells in cortical regions, we failed to identify cells that were positive for both BrdU and NeuN (data not shown). To address the potential effects of valproate on the proliferation of hippocampal progenitor cells, we chronically treated mice with valproate for 6 weeks, followed by a single BrdU injection. The mice were killed 2 hr after the BrdU injection. There were BrdU-positive cells in every animal but not in every slice. The difference in proliferating hippocampal cell numbers between control and valproate-treated mice was not statistically significant (control, 100.0 ± 20.8%; valproate, 142.3 ± 54.7%; F(1,10) = 0.523; p > 0.05). Finally, we administered a single BrdU injection, followed by valproate treatment for 6 weeks. In this paradigm, the majority of BrdU-positive cells within the granular layer of the dentate gyrus were also NeuN positive (Fig. 7A). These results are consistent with previous reports (Chen et al., 2000). Six week valproate treatment after single BrdU injection significantly increased numbers of BrdU-positive cells in the dentate gyrus (Fig. 7B,C), suggesting that valproate promotes neurogenesis.
Figure 7.

Induction of hippocampal neurogenesis by chronic valproate treatment in adult mice. Male C57BL/6 (25-30 gm) mice were treated first with a single injection of BrdU (300 mg/kg) and then fed valproate-containing chow (20 gm/kg) for 6 weeks. The potential effect of chronic valproate on hippocampal neurogenesis was investigated according to standard stereological techniques. Each treatment group contained six mice. Eight serial sections from each mouse were immunostained with antibodies against either BrdU (red) alone or BrdU and NeuN (green). The sections were examined using a Zeiss LSM510 Meta multiphoton system. Double staining of BrdU and NeuN revealed that the majority of the BrdU-positive cells in the dentate gyrus also stained positively for NeuN antibody (A). There were greater numbers of BrdU-positive cells in the section from the valproate-treated mouse (B). The difference in the numbers of BrdU-positive cells between the two groups was statistically significant (C). *p < 0.05 compared with controls. Scale bars, 10 μm.
Roles of p38 kinase and GSK-3 in valproate-induced activation of the ERK pathway in cortical cells
ERKs and p38 kinase belong to the MAP kinase superfamily. To test the selectivity of the effects of valproate on MAP kinases, we used the p38 kinase inhibitor SB202190 and found that it lowered phospho-ERK44/42 and phospho-RSK1 levels but did not significantly alter the magnitude of valproate-induced increases in phospho-ERK44/42 and phospho-RSK1 levels (Fig. 8A,B).
Figure 8.

Involvements of other signaling proteins in valproate-(VPA) induced ERK pathway activation. E18, DIV8 cortical cells obtained as described in Figures 2, 3, 4 were treated with valproate (0.8 mm) in the absence or presence of indicated inhibitors for 2d. Immunoblotting was conducted as described in Figures 2, 3, 4. p38 inhibitor [SB202190(2 μm)] lowered basal levels but not the magnitudes of valproate-induced increases in levels of phospho-ERK44/42 and phopspho-RSK1 (A, B). Selective GSK-3 inhibitor [GSK-3 inhibitor II (20 μm)] neither induced increases in basal levels of phospho-ERK44/42 and phospho-RSK1 nor altered valproate-induced increases in levels of phospho-ERK44/42 and phospho-RSK1 (A, B). Valproate and TSA induced increases in acetyl-H3 levels (C, E). The effects of valproate and TSA (at low concentration) on acetyl-H3 accumulation appeared to be additive (C, E). TSA alone appeared to elevate levels of phospho-ERK44/42 and phospho-RSK1, but the increases were not concentration dependent (C, D). A high concentration of TSA (300 nm) attenuated valproate-induced increases in phospho-ERK44/42 and phopspho-RSK1 levels (C, D). Bar graphs (B, D, E) depict densitometric results representing mean ± SE of three or more sets of samples immunoblotted in duplicates on two gels as presented in A and C. †p < 0.05, with versus without VPA. *p < 0.05, with versus without VPA. #p < 0.05, TSA-nonVPA-treated cells versus nontreated cells.
It is suggested that valproate inhibits GSK-3 either directly (Chen et al., 1999b; Grimes and Jope, 2001) or indirectly (Gould and Manji, 2002; Gould et al., 2004). GSK-3 is a Wnt pathway enzyme that has been postulated to play a role in mood disorders and neurodegenerative diseases (Gould and Manji, 2002; Jope and Bijur, 2002; Phiel et al., 2003). A selective GSK-3 inhibitor (GSK-3 inhibitor II) did not significantly increase phospho-ERK44/42 levels as would be expected if valproate produced its effects on ERK primarily through inhibition of GSK-3 (Fig. 8A,B). The inhibitor lowered basal phospho-RSK1 level but did not significantly alter the magnitudes of valproate-induced increases in phospho-ERK44/42 and phospho-RSK1 levels (Fig. 8A,B). These data do not support a critical role for GSK-3 in valproate-induced ERK pathway activation.
Role of histone deacetylase in valproate-induced activation of the ERK pathway
Valproate is an inhibitor of HDAC (Phiel et al., 2001). It rapidly and concentration dependently induced accumulation of acetylated histone-3 (acetyl-H3) in cortical cells (Fig. 3A-D). TSA, a potent HDAC inhibitor, concentration dependently induced accumulation of acetyl-H3 (Fig. 8C,E). TSA induced nonsignificant increases in phospho-ERK44/42 levels (Fig. 8C,D). TSA at a low concentration induced a significant increase in phospho-RSK1 levels, yet this increase plateaued at a higher TSA concentration (Fig. 8C,D). These data indicate that TSA produces a limited effect on ERK pathway activation that does not parallel its effect on acetyl-H3 accumulation (Fig. 8C,E). TSA at a low concentration induced additive effects on valproate-induced acetyl-H3 accumulation (Fig. 8C,E) but not on valproate-induced increases in phospho-ERK44/42 levels (Fig. 8C,D). TSA at a high concentration significantly attenuated valproate-induced increases in phospho-ERK44/42 and phospho-RSK1 levels (Fig. 8C,D). These data indicate that valproate-induced ERK pathway activation is unlikely mediated solely by its effect on HDAC.
Discussion
Mood stabilizers produce neurotrophic actions in CNS cells
We observed intensified immunostaining of activated phospho-ERK44/42 in neuronal processes and soma of anterior cingulate cortical cells after chronic valproate treatment (Fig. 1), suggesting that valproate activates the ERK pathway in cortical neurons. We also demonstrated that valproate time and concentration dependently increased phospho-ERK44/42 and phospho-RSK1 levels in cultured cortical neurons (Fig. 3). Neurotrophic factors activated the ERK pathway in the same cortical cell-culture system (Fig. 2). Valproate-induced activations of ERK44/42 and RSK1 required MEK and Raf (Fig. 4) as did neurotrophic factor-induced activations (Kaplan and Miller, 2000; Marinissen and Gutkind, 2001; Huang and Reichardt, 2003). Similar to neurotrophic factors, valproate promoted neurite growth and cell reemergence in the created gaps in an ERK pathway-dependent manner (Fig. 5). The reemerged cells were positive for progenitor cell marker nestin, immature neuron marker TuJ1, and mature neuron marker NeuN (Fig. 6), indicating that these cells had undergone neurogenesis in culture. Both valproate and neurotrophic factors increased numbers of BrdU- and NeuN-positive cells, demonstrating that both treatments promoted cortical neurogenesis (Fig. 6). The data indicate that valproate produces neurotrophic actions in CNS cells.
Although there is no direct evidence, existing data suggest that lithium produces neurotrophic actions in cortical cells. For instance, lithium treatment increases cortical levels of activated phospho-ERK44/42 and phospho-RSK1 (Einat et al., 2003) and enhances functions of ERK pathway-regulated transcription factors CREB (Ozaki and Chuang, 1997; Grimes and Jope, 2001; Einat et al., 2003) and AP-1 (activator protein-1) (Ozaki and Chuang, 1997; Asghari et al., 1998; Chen et al., 1998; Yuan et al., 1998). Lithium also induces cortical expressions of bcl-2 (Chen et al., 1999a) and bdnf (Fukumoto et al., 2001; Hashimoto et al., 2002; Einat et al., 2003), ERK pathway-regulated genes (Riccio et al., 1999; Weeber and Sweatt, 2002).
Valproate promotes hippocampal neurogenesis in adult mice
Neurotrophic factors and the ERK pathway have been suggested to play roles in hippocampal neurogenesis (Collazo et al., 1992). This is also supported by the finding that adult BDNF+/- mice exhibit lower proliferation and survival rates of hippocampal progenitor cells (Lee et al., 2002). Administration of corticosterone reduces levels of phospho-ERKs, phospho-CREB, and BDNF, as well as the rate of neurogenesis in the dentate gyrus (Yu et al., 2004). In addition to the frontal cortex, we found that mood stabilizers lithium and valproate also activate the ERK pathway and increase BDNF in the hippocampus. We also demonstrated that lithium promotes hippocampal neurogenesis in adult animals (Chen et al., 2000). In the present study, we found that chronic valproate treatment resulted in a significant increase in numbers of BrdU-positive cells in the dentate gyrus (Fig. 7). The data suggest that a common action of the two mood stabilizers is enhanced hippocampal neurogenesis, possibly through an ERK pathway-mediated mechanism.
Dysfunction of neurotrophic signaling in the CNS is a pathogenic factor of bipolar mood disorder
A Val66Met BDNF polymorphism is associated with reductions in activity-dependent BDNF secretion in cultured cells and low levels of NAA in human hippocampus (Egan et al., 2003). Two independent studies suggest that the Val66Met polymorphism is associated with high risk for bipolar disorder (Neves-Pereira et al., 2002; Sklar et al., 2002). Lack of neurotrophic support triggers axonal and dendritic withdrawal and collapse and apoptosis in nervous systems (Kaplan and Miller, 2000; Huang and Reichardt, 2003). In concert with the roles of neurotrophic factors and the genetic association of the BDNF polymorphism with bipolar risk, brain-imaging and postmortem studies reveal volume reductions of cerebral gray matter and atrophy-loss of neurons and glial cells in discrete brain regions of bipolar patients (Harrison, 2002; Coyle and Duman, 2003). Collectively, clinical evidence supports a hypothesis that neurotrophic signaling pathway dysfunction is a pathogenic factor of bipolar disorder. This dysfunction is targeted by mood-stabilizing treatment with valproate (Figs. 1, 3, 5, 6) (Chen et al., 1999a; Einat et al., 2003) or lithium (Ozaki and Chuang, 1997; Asghari et al., 1998; Chen et al., 1998, 1999a; Yuan et al., 1998; Fukumoto et al., 2001; Grimes and Jope, 2001; Hashimoto et al., 2002; Einat et al., 2003).
Induction of neurotrophic or neurotrophic-related actions is a common effect of mood stabilizers and antidepressants
Although mood stabilizers and antidepressants possess unique therapeutic profiles, they produce some comparable neurotrophic or neurotrophic-related actions. These actions include induction of BDNF expression (Nibuya et al., 1995, 1996; Fukumoto et al., 2001; Hashimoto et al., 2002; Altar et al., 2003; Einat et al., 2003), activation of CREB (Nibuya et al., 1996; Chen et al., 1997; Ozaki and Chuang, 1997; Thome et al., 2000; Grimes and Jope, 2001; Einat et al., 2003), increases in numbers and lengths of neuronal processes (Vaidya et al., 1999; Yuan et al., 2001; Williams et al., 2002; Cui et al., 2003) (Fig. 5), and enhancement of neurogenesis (Chen et al., 2000; Malberg et al., 2000; Santarelli et al., 2003) (Figs. 5, 6). Electroconvulsive therapy (ECT) is a very effective treatment for depression (UK ECT Review Group, 2003), an effective treatment for mania (Mukherjee et al., 1994) and a maintenance therapy to prevent mood episode relapses (Russell et al., 2003). The electroconvulsive shock (ECS) paradigm is a modified version of ECT used to study the mechanisms of clinical actions of ECT in animal models. Animal studies show that ECS activates ERKs (Baraban et al., 1993; Kang et al., 1994; Bhat et al., 1998), increases BDNF expression (Nibuya et al., 1995; Newton et al., 2003; Altar et al., 2004), and promotes hippocampal neurogenesis (Madsen et al., 2000, 2003; Scott et al., 2000; Hellsten et al., 2002).
Both bipolar disorder and unipolar depression are associated with brain volumetric reductions and neuropathological findings of decreased neuronal and/or glial cell volumes and densities (Drevets et al., 1997; Harrison, 2002; Coyle and Duman, 2003). Therefore, neurotrophic or neurotrophic-related actions may be beneficial for both disorders. The therapeutic profiles and the known biological actions of mood stabilizers and antidepressants suggest that each of these two classes of mood-modulating agents produce two major categories of therapeutically relevant actions. The first consists of neurotrophic or neurotrophic-related actions, which are fundamental for recovery of structural and morphometric deficits. This set of actions is common to both classes of agents. The other major sets of actions are distinct to their own class, alleviating either mania or depression.
Potential initial mechanism(s) by which valproate activates the ERK pathway
GSK-3 and HDAC are two known targets of valproate. However, we failed to obtain conclusive evidence to support the notion that valproate activates the ERK pathway by inhibiting GSK-3 or HDAC (Fig. 8). Lithium lowers myo-inositol levels in the brain through inhibition of inositol phosphatases (Berridge, 1989; Gould et al., 2002). Studies have suggested that valproate may also lower inositol levels in brain (O'Donnell et al., 2003) by blocking de novo inositol synthesis from glucose (Vaden et al., 2001; Ju et al., 2004). Inositol depletion affects synthesis of phosphatidylinositol (Gould et al., 2002), a molecule used by multiple signaling pathways [such as PKC and PI3K phosphoinositide 3-kinase)] pathways that crosstalk with the ERK pathway (Gould et al., 2002). Valproate can enter cells and incorporate into phospholipids (Siafaka-Kapadai et al., 1998). Some of these phospholipids and their derivatives are potent activators of the ERK pathway (Abdel-Latif, 2001; Andresen et al., 2002; Yart et al., 2002). Presently, the precise initial mechanism by which valproate activates the ERK pathway is unknown.
Valproate as an alternative or complementary neurotrophic therapeutic agent
Neurotrophic factors and their analogs have been tested for their effectiveness in treatments of a variety of diseases of the nervous system (Apfel, 2002). Valproate is a widely used neurologic and psychiatric drug. Our data demonstrate that valproate produces neurotrophic actions (Figs. 3, 5, 6) in CNS cells. In addition, valproate-induced ERK pathway activations were more sustained compared with those produced by neurotrophic factors (Figs. 2, 3). Recent animal and cell-culture studies demonstrate that valproate enhances axonal regeneration and neuronal survival against a variety of insults (Cui et al., 2003; Dou et al., 2003; Jeong et al., 2003). Valproate may be an appropriate alternative or complementary neurotrophic treatment for brain trauma, ischemia, and neurodegenerative diseases (Loy and Tariot, 2002).
Footnotes
This work was supported by the Intramural Research Program of the National Institute of Mental Health, the Theodore and Vada Stanley Foundation, and the National Alliance for Research on Schizophrenia and Depression.
Correspondence should be addressed to Guang Chen, Laboratory of Molecular Pathophysiology, National Institute of Mental Health, National Institutes of Health, Building 49, Room B1EE16, 49 Convent Drive, MSC 4405, Bethesda, MD 20892-4405. E-mail: cheng@intra.nimh.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246590-10$15.00/0
References
- Abdel-Latif AA (2001) Cross talk between cyclic nucleotides and polyphosphoinositide hydrolysis, protein kinases, and contraction in smooth muscle. Exp Biol Med 226: 153-163. [DOI] [PubMed] [Google Scholar]
- Altar CA, Whitehead RE, Chen R, Wortwein G, Madsen TM (2003) Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry 54: 703-709. [DOI] [PubMed] [Google Scholar]
- Altar CA, Laeng P, Jurata LW, Brockman JA, Lemire A, Bullard J, Bukhman YV, Young TA, Charles V, Palfreyman MG (2004) Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J Neurosci 24: 2667-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen BT, Rizzo MA, Shome K, Romero G (2002) The role of phosphatidic acid in the regulation of the Ras/MEK/Erk signaling cascade. FEBS Lett 531: 65-68. [DOI] [PubMed] [Google Scholar]
- Apfel SC (2002) Is the therapeutic application of neurotrophic factors dead? Ann Neurol 51: 8-11. [DOI] [PubMed] [Google Scholar]
- Asghari V, Wang JF, Reiach JS, Young LT (1998) Differential effects of mood stabilizers on Fos/Jun proteins and AP-1 DNA binding activity in human neuroblastoma SH-SY5Y cells. Brain Res Mol Brain Res 58: 95-102. [DOI] [PubMed] [Google Scholar]
- Baraban JM, Fiore RS, Sanghera JS, Paddon HB, Pelech SL (1993) Identification of p42 mitogen-activated protein kinase as a tyrosine kinase substrate activated by maximal electroconvulsive shock in hippocampus. J Neurochem 60: 330-336. [DOI] [PubMed] [Google Scholar]
- Barnabe-Heider F, Miller FD (2003) Endogenously produced neurotrophins regulate survival and differentiation of cortical progenitors via distinct signaling pathways. J Neurosci 23: 5149-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1989) The Albert Lasker Medical Awards. Inositol trisphosphate, calcium, lithium, and cell signaling. JAMA 262: 1834-1841. [PubMed] [Google Scholar]
- Bhat RV, Engber TM, Finn JP, Koury EJ, Contreras PC, Miller MS, Dionne CA, Walton KM (1998) Region-specific targets of p42/p44MAPK signaling in rat brain. J Neurochem 70: 558-571. [DOI] [PubMed] [Google Scholar]
- Bowden CL, Brugger AM, Swann AC, Calabrese JR, Janicak PG, Petty F, Dilsaver SC, Davis JM, Rush AJ, Small JG, Garza-Trevino ES, Risch SC, Goodnick PJ, Morris DD (1994) Efficacy of divalproex vs lithium and placebo in the treatment of mania. The Depakote Mania Study Group. JAMA 271: 918-924. [PubMed] [Google Scholar]
- Cameron HA, McKay RD (2001) Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol 435: 406-417. [DOI] [PubMed] [Google Scholar]
- Chen G, Yuan P, Hawver DB, Potter WZ, Manji HK (1997) Increase in AP-1 transcription factor DNA binding activity by valproic acid. Neuropsychopharmacology 16: 238-245. [DOI] [PubMed] [Google Scholar]
- Chen G, Yuan PX, Jiang YM, Huang LD, Manji HK (1998) Lithium increases tyrosine hydroxylase levels both in vivo and in vitro. J Neurochem 70: 1768-1771. [DOI] [PubMed] [Google Scholar]
- Chen G, Zeng WZ, Yuan PX, Huang LD, Jiang YM, Zhao ZH, Manji HK (1999a) The mood-stabilizing agents lithium and valproate robustly increase the levels of the neuroprotective protein bcl-2 in the CNS. J Neurochem 72: 879-882. [DOI] [PubMed] [Google Scholar]
- Chen G, Huang LD, Jiang YM, Manji HK (1999b) The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J Neurochem 72: 1327-1330. [DOI] [PubMed] [Google Scholar]
- Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji HK (2000) Enhancement of hippocampal neurogenesis by lithium. J Neurochem 75: 1729-1734. [DOI] [PubMed] [Google Scholar]
- Collazo D, Takahashi H, McKay RD (1992) Cellular targets and trophic functions of neurotrophin-3 in the developing rat hippocampus. Neuron 9: 643-656. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Duman RS (2003) Finding the intracellular signaling pathways affected by mood disorder treatments. Neuron 38: 157-160. [DOI] [PubMed] [Google Scholar]
- Cui SS, Yang CP, Bowen RC, Bai O, Li XM, Jiang W, Zhang X (2003) Valproic acid enhances axonal regeneration and recovery of motor function after sciatic nerve axotomy in adult rats. Brain Res 975: 229-236. [DOI] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE (2003) Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci 23: 9162-9170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drevets WC (2000) Neuroimaging studies of mood disorders. Biol Psychiatry 48: 813-829. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M, Raichle ME (1997) Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386: 824-827. [DOI] [PubMed] [Google Scholar]
- Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR (2003) The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112: 257-269. [DOI] [PubMed] [Google Scholar]
- Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G (2003) The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23: 7311-7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank L, Ventimiglia R, Anderson K, Lindsay RM, Rudge JS (1996) BDNF down-regulates neurotrophin responsiveness, TrkB protein and TrkB mRNA levels in cultured rat hippocampal neurons. Eur J Neurosci 8: 1220-1230. [DOI] [PubMed] [Google Scholar]
- Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S (2001) Chronic lithium treatment increases the expression of brain-derived neurotrophic factor in the rat brain. Psychopharmacology (Berl) 158: 100-106. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME (1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89-103. [DOI] [PubMed] [Google Scholar]
- Goodwin FK, Jamison KR (1990) Manic-depressive illness. New York: Oxford UP.
- Gould E, Reeves AJ, Graziano MS, Gross CG (1999) Neurogenesis in the neocortex of adult primates. Science 286: 548-552. [DOI] [PubMed] [Google Scholar]
- Gould E, Vail N, Wagers M, Gross CG (2001) Adult-generated hippocampal and neocortical neurons in macaques have a transient existence. Proc Natl Acad Sci USA 98: 10910-10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TD, Manji HK (2002) The Wnt signaling pathway in bipolar disorder. The Neuroscientist 8: 497-511. [DOI] [PubMed] [Google Scholar]
- Gould TD, Chen G, Manji HK (2002) Mood stabilizer psychopharmacology. Clin Neurosci Res 2: 193-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TD, Chen G, Manji HK (2004) In vivo evidence in the brain for lithium inhibition of glycogen synthase kinase-3. Neuropsychopharmacology 29: 32-38. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS (2001) CREB DNA binding activity is inhibited by glycogen synthase kinase-3 beta and facilitated by lithium. J Neurochem 78: 1219-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison PJ (2002) The neuropathology of primary mood disorder. Brain 125: 1428-1449. [DOI] [PubMed] [Google Scholar]
- Hashimoto R, Takei N, Shimazu K, Christ L, Lu B, Chuang DM (2002) Lithium induces brain-derived neurotrophic factor and activates TrkB in rodent cortical neurons: an essential step for neuroprotection against glutamate excitotoxicity. Neuropharmacology 43: 1173-1179. [DOI] [PubMed] [Google Scholar]
- Hellsten J, Wennstrom M, Mohapel P, Ekdahl CT, Bengzon J, Tingstrom A (2002) Electroconvulsive seizures increase hippocampal neurogenesis after chronic corticosterone treatment. Eur J Neurosci 16: 283-290. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L (1981) Protein A, avidin, and biotin in immunohistochemistry. J Histochem Cytochem 29: 1349-1353. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003) TRK receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609-642. [DOI] [PubMed] [Google Scholar]
- Jeong MR, Hashimoto R, Senatorov VV, Fujimaki K, Ren M, Lee MS, Chuang DM (2003) Valproic acid, a mood stabilizer and anticonvulsant, protects rat cerebral cortical neurons from spontaneous cell death: a role of histone deacetylase inhibition. FEBS Lett 542: 74-78. [DOI] [PubMed] [Google Scholar]
- Jope RS, Bijur GN (2002) Mood stabilizers, glycogen synthase kinase-3beta and cell survival. Mol Psychiatry 7 [Suppl 1]: S35-S45. [DOI] [PubMed] [Google Scholar]
- Ju S, Shaltiel G, Shamir A, Agam G, Greenberg ML (2004) Human 1D-myoinositol 3-phosphate synthase is functional in yeast. J Biol Chem 279: 21759-21765. [DOI] [PubMed] [Google Scholar]
- Kang UG, Hong KS, Jung HY, Kim YS, Seong YS, Yang YC, Park JB (1994) Activation and tyrosine phosphorylation of 44-kDa mitogen-activated protein kinase (MAPK) induced by electroconvulsive shock in rat hippocampus. J Neurochem 63: 1979-1982. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Kornack DR, Rakic P (2001) Cell proliferation without neurogenesis in adult primate neocortex. Science 294: 2127-2130. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP (2002) Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem 82: 1367-1375. [DOI] [PubMed] [Google Scholar]
- Loy R, Tariot PN (2002) Neuroprotective properties of valproate: potential benefit for AD and tauopathies. J Mol Neurosci 19: 303-307. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A (2000) Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry 47: 1043-1049. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Newton SS, Eaton ME, Russell DS, Duman RS (2003) Chronic electroconvulsive seizure up-regulates beta-catenin expression in rat hippocampus: role in adult neurogenesis. Biol Psychiatry 54: 1006-1014. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104-9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinissen MJ, Gutkind JS (2001) G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci 22: 368-376. [DOI] [PubMed] [Google Scholar]
- McElroy SL, Keck Jr PE (1995) Antiepileptic drugs. In: Textbook of psychopharmacology (Schatzberg AF, Nemeroff CB, eds), pp 351-375. Washington, DC: American Psychiatric.
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD (2002) An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron 36: 597-610. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB, Faulk MW, Koch S, Glitz DA, Jolkovsky L, Manji HK (2000a) Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2's neurotrophic effects? Biol Psychiatry 48: 1-8. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK, Menji HK (2000b) Lithium-induced increase in human brain grey matter. Lancet 356: 1241-1242. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Sackeim HA, Schnur DB (1994) Electroconvulsive therapy of acute manic episodes: a review of 50 years' experience. Am J Psychiatry 151: 169-176. [DOI] [PubMed] [Google Scholar]
- Neves-Pereira M, Mundo E, Muglia P, King N, Macciardi F, Kennedy JL (2002) The brain-derived neurotrophic factor gene confers susceptibility to bipolar disorder: evidence from a family-based association study. Am J Hum Genet 71: 651-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton SS, Collier EF, Hunsberger J, Adams D, Terwilliger R, Selvanayagam E, Duman RS (2003) Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J Neurosci 23: 10841-10851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS (1995) Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci 15: 7539-7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibuya M, Nestler EJ, Duman RS (1996) Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 16: 2365-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell T, Rotzinger S, Nakashima TT, Hanstock CC, Ulrich M, Silver-stone PH (2003) Chronic lithium and sodium valproate both decrease the concentration of myoinositol and increase the concentration of inositol monophosphates in rat brain. Eur Neuropsychopharmacol 13: 199-207. [DOI] [PubMed] [Google Scholar]
- Ozaki N, Chuang DM (1997) Lithium increases transcription factor binding to AP-1 and cyclic AMP-responsive element in cultured neurons and rat brain. J Neurochem 69: 2336-2344. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001) Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276: 36734-36741. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423: 435-439. [DOI] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD (1999) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358-2361. [DOI] [PubMed] [Google Scholar]
- Russell JC, Rasmussen KG, O'Connor MK, Copeman CA, Ryan DA, Rummans TA (2003) Long-term maintenance ECT: a retrospective review of efficacy and cognitive outcome. J ECT 19: 4-9. [DOI] [PubMed] [Google Scholar]
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301: 805-809. [DOI] [PubMed] [Google Scholar]
- Scott BW, Wojtowicz JM, Burnham WM (2000) Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp Neurol 165: 231-236. [DOI] [PubMed] [Google Scholar]
- Siafaka-Kapadai A, Patiris M, Bowden C, Javors M (1998) Incorporation of [3H]valproic acid into lipids in GT1-7 neurons. Biochem Pharmacol 56: 207-212. [DOI] [PubMed] [Google Scholar]
- Sklar P, Gabriel SB, McInnis MG, Bennett P, Lim YM, Tsan G, Schaffner S, Kirov G, Jones I, Owen M, Craddock N, DePaulo JR, Lander ES (2002) Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry 7: 579-593. [DOI] [PubMed] [Google Scholar]
- Stout SC, Owens MJ, Lindsey KP, Knight DL, Nemeroff CB (2001) Effects of sodium valproate on corticotropin-releasing factor systems in rat brain. Neuropsychopharmacology 24: 624-631. [DOI] [PubMed] [Google Scholar]
- Thome J, Sakai N, Shin K, Steffen C, Zhang YJ, Impey S, Storm D, Duman RS (2000) cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci 20: 4030-4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK ECT Review Group (2003) Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet 361: 799-808. [DOI] [PubMed] [Google Scholar]
- Vaden DL, Ding D, Peterson B, Greenberg ML (2001) Lithium and valproate decrease inositol mass and increase expression of the yeast INO1 and INO2 genes for inositol biosynthesis. J Biol Chem 276: 15466-15471. [DOI] [PubMed] [Google Scholar]
- Vaidya VA, Siuciak JA, Du F, Duman RS (1999) Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience 89: 157-166. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Sweatt JD (2002) Molecular neurobiology of human cognition. Neuron 33: 845-848. [DOI] [PubMed] [Google Scholar]
- Williams RS, Cheng L, Mudge AW, Harwood AJ (2002) A common mechanism of action for three mood-stabilizing drugs. Nature 417: 292-295. [DOI] [PubMed] [Google Scholar]
- Yart A, Chap H, Raynal P (2002) Phosphoinositide 3-kinases in lysophosphatidic acid signaling: regulation and cross-talk with the Ras/mitogen-activated protein kinase pathway. Biochim Biophys Acta 1582: 107-111. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Ida S, Yokota M, Nishimoto A, Shibata S, Sugawara A, Takiguchi Y (2000) Effects of lithium on the pharmacokinetics of valproate in rats. J Pharm Pharmacol 52: 297-301. [DOI] [PubMed] [Google Scholar]
- Yu IT, Lee SH, Lee YS, Son H (2004) Differential effects of corticosterone and dexamethasone on hippocampal neurogenesis in vitro. Biochem Biophys Res Commun 317: 484-490. [DOI] [PubMed] [Google Scholar]
- Yuan PX, Chen G, Huang LD, Manji HK (1998) Lithium stimulates gene expression through the AP-1 transcription factor pathway. Brain Res Mol Brain Res 58: 225-230. [DOI] [PubMed] [Google Scholar]
- Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G (2001) The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem 276: 31674-31683. [DOI] [PubMed] [Google Scholar]