

Abstract
The efficacy of excitatory transmission in the brain depends to a large extent on synaptic AMPA receptors, hence the importance of understanding the delivery and recycling of the receptors at the synaptic sites. Here we report a novel regulation of the AMPA receptor transport by a PDZ (postsynaptic density-95/Drosophila disc large tumor suppressor zona occludens 1) and LIM (Lin11/rat Isl-1/Mec3) domain-containing protein, RIL (reversion-induced LIM protein). We show that RIL binds to the AMPA glutamate receptor subunit GluR-A C-terminal peptide via its LIM domain and to α-actinin via its PDZ domain. RIL is enriched in the postsynaptic density fraction isolated from rat forebrain, strongly localizes to dendritic spines in cultured neurons, and coprecipitates, together with α-actinin, in a protein complex isolated by immunoprecipitation of AMPA receptors from forebrain synaptosomes. Functionally, in heterologous cells, RIL links AMPA receptors to the α-actinin/actin cytoskeleton, an effect that appears to apply selectively to the endosomal surface-internalized population of the receptors. In cultured neurons, an overexpression of recombinant RIL increases the accumulation of AMPA receptors in dendritic spines, both at the total level, as assessed by immunodetection of endogenous GluR-A-containing receptors, and at the synaptic surface, as assessed by recording of miniature EPSCs. Our results thus indicate that RIL directs the transport of GluR-A-containing AMPA receptors to and/or within dendritic spines, in anα-actinin/actin-dependent manner, and that such trafficking function promotes the synaptic accumulation of the receptors.
Keywords: AMPA receptor, transport, dendritic spine, α-actinin/actin cytoskeleton, PDZ domain, LIM domain
Introduction
AMPA receptors mediate the fast excitatory transmission in the mammalian CNS. Accumulating evidence indicates that regulation of the number of synaptic AMPA receptors underlies some aspects of glutamatergic synaptic plasticity, a process believed to be essential for CNS development, learning, and memory (Sheng and Lee, 2001; Malinow and Malenka, 2002; Song and Huganir, 2002). Activity-induced decrease of synaptic AMPA receptors was shown to participate in long-term depression (LTD) of glutamatergic synaptic transmission in hippocampal neurons and in cerebellar Purkinje cells (Carroll et al., 1999; Heynen et al., 2000; Matsuda et al., 2000; Wang and Linden, 2000; Lee et al., 2002). Conversely, NMDA receptor-dependent long-term potentiation (LTP) in CA1 hippocampal neurons is mediated at least in part by an increase of AMPA receptors at the synaptic surface membrane (Shi et al., 1999; Heynen et al., 2000; Lu et al., 2001; Passafaro et al., 2001; Shi et al., 2001).
AMPA receptors are heterotetrameric complexes formed by combinations of four glutamate receptor (GluR) subunits, termed GluR-A through GluR-D (or GluR1-GluR4) (Boulter et al., 1990; Keinanen et al., 1990). Principal neurons in the hippocampus express the GluR-A, GluR-B, and GluR-C subunits, which form two major receptor populations, the GluR-A/GluR-B and GluR-B/GluR-C complexes (Wenthold et al., 1996). In the adult brain, activity-dependent synaptic insertion of GluR-A/GluR-B receptors, which is regulated by the GluR-A intracellular C-terminal domain, contributes to lasting increases of synaptic strength during LTP in CA1 neurons in vitro, as well as to sensory-evoked synaptic plasticity in cortical neurons in vivo (Shi et al., 2001; Takahashi et al., 2003).
In the present study, we report a novel molecular mechanism that may link the transport of GluR-A-containing AMPA receptors to actin cytoskeleton. We show that a PDZ [PSD -95 (postsynaptic density-95)/Drosophila disc large tumor suppressor zona occludens 1)] and LIM (Lin11/rat Isl-1/Mec3) domain-containing protein termed RIL (for reversion-induced LIM protein) (Kiess et al., 1995) binds via its LIM domain to the last 10 amino residues of the GluR-A C-terminal peptide and via its PDZ domain to the carboxyl region of α-actinin, a member of the spectrin/dystrophin family of actin-cross-linking proteins (Vandekerckhove, 1990). Filamentous actin is the principal cytoskeleton of dendritic spines (Fifkova and Delay, 1982; Matus et al., 1982) and is known to be critical for maintaining AMPA receptor synaptic function. Acute disruption of postsynaptic actin inhibits the induction of LTP, whereas prolonged treatment with actin-depolymerizing agents reduces the basal level of AMPA receptor-mediated transmission, resulting from the decreased steady-state synaptic dwell time of the receptors (Allison et al., 1998; Kim and Lisman, 1999; Zhou et al., 2001). These findings imply that actin cytoskeleton plays distinct roles during activity-driven insertion and during stabilization of the receptors at the postsynaptic membrane. A role in the synaptic stabilization of the receptors was previously proposed for the 4.1N protein that links AMPA receptors to the spectrin/actin cytoskeleton (Shen et al., 2000). Our results suggest that RIL links the GluRA-containing AMPA receptors to the α-actinin/actin cytoskeleton, spatially directing the synaptic trafficking of the receptors to and/or within the dendritic spine compartment toward an insertion at the postsynaptic membrane.
Materials and Methods
Yeast two-hybrid system. The yeast two-hybrid screening of adult rat brain cDNA library (Clontech, Cambridge, UK; pACT2 vector) was performed using HF7c yeast strain according to the Yeast Protocols Handbook (PT3024-1; Clontech). The bait constructs were subcloned in the DNA binding domain vector pGBT9. The efficiency of screening with pGBT9-R-B/R-At10 bait was 5.1 × 106 and 1.0 × 106 with pGBT9-PDZ bait. Yeast-mating assays for GluR-A and RIL binding were performed using yeast strains HF7c and Y187, with pGBT9 and pACT2 vectors. The assays for RIL and α-actinin binding were performed with pGAD10 and pBHA vectors.
Generation of anti-RIL antibody. Anti-RIL polyclonal serum against the C-terminal RIL aa 317-330 peptide CDVVAVYPNAKVEL was generated at AnaSpec (San Jose, CA). Affinity purification was done by peptide-coupled SulfoLink gel (Pierce,Rockford, IL) column: 20 ml of serum, diluted 1:10 in 4°C cold PBS, was run over the column overnight at 4°C. After wash with PBS, 1 ml fractions were eluted with glycine, pH 2.5, and immediately neutralized with 1/10 volume of 1 m Tris, pH 8.0. Anti-RIL antibody (Ab)-positive fractions were selected by Western blotting of FlagRIL-expressing African green monkey kidney fibroblast cells (COS)1 lysate.
Heterologous cell experiments. COS1 or human embryonic kidney cells were transfected by the calcium phosphate method. For immunoprecipitations (IPs), cells were lysed after 48 hr in buffer A [0.5% Triton X-100, 150 mm NaCl, 1 mm Na3V04, 25 mm HEPES, pH 7.6; Boehringer Complete protease inhibitors (Boehringer Mannheim, Indianapolis, IN)]. For each IP, 300 μl of high speed supernatant lysate, at 1.5 mg per ml, was precleared with 20 μl of Protein-G agarose (1 hr) (Santa Cruz Biotechnology, Santa Cruz, CA), followed by incubation with 6 μg of α-Flag antibody (4 hr) (Sigma, St. Louis, MO). Immunoprecipitated proteins collected on protein G agarose were eluted by SDS-PAGE sample buffer. For immunocytochemistry, cells were fixed after 48 hr by 4% paraformaldehyde (PFA) (room temperature) or 100% methanol (-20°C), with the exception of internalization assays, where cells were first incubated with α-Myc antibody at 4°C for 30 min, extensively washed with PBS, and either directly fixed by PFA or returned to 37°C for an internalization period and then fixed. Stainings were done with the following primary antibodies: α-GluR1, α-GluR2/3 (Chemicon, Temecula, CA), α-calnexin, α-lysosome-associated membrane protein 1 (Lamp1) (Stressgene Biotechnologies, Victoria, British Columbia, Canada), α-early endosome antigen 1 (EEA1) (BD Biosciences, Franklin Lake, NJ), α-human transferrin receptor (TfR) (Zymed, San Francisco, CA), α-Flag (Sigma), α-Myc (9-E10; Santa Cruz Biotechnology), and secondary antibodies conjugated with FITC, Texas Red, Cy3, or Cy5 (Jackson ImmunoResearch, West Grove, PA). Actin was visualized by phalloidin-tetramethylrhodamine isothiocyanate (TRITC) (Sigma).
For glutathione S-transferase (GST) pull-downs, GST and GST-R-A proteins were expressed by baculovirus expression (BacPak6, Clontech) in Sf9 cells, cultured in TMN-FH medium (Invitrogen, San Diego, CA) with 10% FCS at 27°C, and harvested in buffer containing 20 mm HEPES, pH 7.2, 150 mm NaCl, 1 mm EGTA, 1% Triton X-100, and protease inhibitors (buffer B). An equal amount of protein bound on glutathione Sepharose was used for pull-downs with COS1-expressed FlagRIL and FlagRIL deletion mutants in buffer containing 20 mm Tris-HCl, pH 8.5, 150 mm NaCl, 1 mm EGTA, 1% Triton X-100, 0.1% SDS, and protease inhibitors (buffer C). GST, GST-RIL, and GST-RIL deletion proteins were produced in bacteria using the pGEX system (Amersham Biosciences, Arlington Heights, IL). COS1 cells expressing α-actinin2 were lysed in buffer C. High-speed supernatant of adult rat brain lysate was prepared in buffer B by differential centrifugation at 3000 × g and 100,000 × g. GST-bound precipitates were eluted in SDS-PAGE sample buffer. Western blotting was performed with antibodies already listed above and the α-α-actinin2 antibody (Wyszynski et al., 1998).
Brain fractionation, coimmunoprecipitation, and neuronal cultures. Brain protein fractionation was done from adult Wistar rats (Carlin et al., 1980) with modifications as described (Srivastava et al., 1998). Antibodies for Western blotting (next to those listed already) were as follows: α-NR1 (Chemicon) and α-synaptophysin (Zymed). Hippocampal primary cultures were prepared from embryonic day 18 Sprague Dawley tissue (Osten et al., 2000) and cultured for 2-3 weeks in Neurobasal medium supplemented with B27 (Invitrogen) (Brewer et al., 1993). Organotypic slice cultures were prepared as described (Stoppini et al., 1991). For coimmunoprecipitations, P2 fraction obtained from rat brain lysate was solubilized with 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and cleared by ultracentrifugation. The extract (input) was applied to protein A Sepharose conjugated with anti-GluR2/3 antibody or normal rabbit IgG. After column wash, bound proteins were eluted with competitive peptide, which is the antigen for GluR2/3 antibody. Input and elution fractions were analyzed by Western blotting.
Sindbis virus-based expression, immunostaining of neuronal cultures, and confocal microscopy. Attenuated Sindbis virus-based expression vector (Dryga et al., 1997), plasmid SINrep(nsp2S), was used with the helper plasmid DH(26S)5′tRNA (Kim et al., 2004) to express recombinant proteins in hippocampal primary or organotypic slice cultures. The virus was prepared as described previously (Kim et al., 2004). Infected primary neurons either were fixed with 4% PFA in PBS containing 0.12 m sucrose (30 min at room temperature) and permeabilized with 0.2% Triton X-100 in PBS or were incubated with anti-Myc antibody under living conditions, as described previously (Osten et al., 1998), followed by PFA fixation. The primary antibodies used were as follows (next to those listed already): α-RIL (Cuppen et al., 1998) and α-SV2 (Buckley and Kelly, 1985). These were maintained by the University of Iowa, Department of Biological Sciences). All images were acquired on a Leica TCS NT microscope. Deconvolution was done with the Huygens 2 software as described previously (Osten et al., 2000).
For quantitation of spine enrichment, regions of interest (ROI) of dendritic spine heads and of neighboring dendrites were selected by the ImageJ program (ROI manager), first on a confocal maximal projection image of a recombinant protein (e.g., EGFPRIL) dendritic distribution, and the values of mean immunofluorescence were calculated. The same ROI mask was then applied to the corresponding image of endogenous protein distribution (e.g., anti-α-actinin2/3 immunostaining). For quantitation of co-distribution of EGFPRIL and total GluR-A, spine and shaft ROIs were first selected based on EGFPRIL distribution, and the same mask was applied to anti-GluR-A immunostaining (fixed and permeabilized conditions), detecting total GluR-A expression.
Electrophysiology. Recordings of miniature EPSCs (mEPSCs) were done from primary rat hippocampal neurons [14 d in vitro (14DIV)] or CA1 pyramidal neurons in organotypic slice cultures (7DIV; prepared from postnatal day 7 rats). The recording chamber was continuously perfused with (in mm): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2, 25 glucose, 2 CaCl2, 0.001 TTX, 0.01 bicuculline methiodide, 0.03 d-AP5, pH 7 (gassed with 95% O2/5% CO2). To enhance mEPSC frequency in the organotypic slice cultures, 150 mm sucrose was added to the extracellular solution after whole-cell configuration was established. Only one recording was performed per slice. The patch pipettes were filled with (in mm): 125 Cs-gluconate, 20 CsCl, 10 NaCl, 10 HEPES, 0.2 EGTA, 4 MgATP, 0.3 NaGTP, pH 7.25 (305 mOsm). Pipette resistances were 5-6 mΩ. The series resistance was monitored throughout the recordings. mEPSCs were recorded at room temperature (22°C), at -70 mV using an EPC-9 amplifier (HEKA Elektronik, Lambrecht, Germany), and were stored on tape. Recordings were analyzed off-line (filtering, 3 kHz; sampling, 5 kHz) using an event detection program (kindly provided by Prof. Ulrich Misgeld, Institute of Physiology, University of Heidelberg). For event detection, the trigger level was set at 15 pA (three times higher than the baseline noise), and events were individually selected if the rise time was <2 msec. The average 20-80% rise time of the mEPSC events in EGFpRIL, noninfected, EGFPRILΔPDZ, and EGFPRILΔLIM cells was as follows (mean ± SD): 0.84 ± 0.11, 0.85 ± 0.1, 0.85 ± 0.1, and 0.7 ± 0.1 in primary cultures, and 0.8 ± 0.2 and 0.81 ± 0.17 in organotypic slice cultures. The average frequency of the mEPSC events in EGFPRIL, noninfected, EGFPRILΔPDZ, and EGFPRILΔLIM cells was as follows (in Hertz; mean ± SD): 0.36 ± 0.3, 0.14 ± 0.19, 0.05 ± 0.06, and 0.19 ± 0.19 in primary cultures, and 0.31 ± 0.14 and 0.16 ± 0.13 in organotypic slice cultures. The total numbers of analyzed events in EGFPRIL, noninfected, EGFPRILΔPDZ, and EGFPRILΔLIM cells were 1156, 462, 188, and 1106 in primary cultures, and 890 and 813 in organotypic slice cultures. Data were compared by one-way ANOVA test and unpaired two-tailed Student's t test.
Results
GluR-A C-terminal domain interacts, via its 10 C-terminal residue motif, with the LIM domain of RIL
To identify proteins that interact with the GluR-A C-terminal domain, we screened an adult rat brain cDNA library using a chimeric peptide, R-B/R-At10, as a bait in the yeast two-hybrid system. This bait consisted of the membrane-proximal 40 amino acids of the GluR-B C-terminal tail fused to the terminal 10 amino acids of the GluR-A sequence (Fig. 1a). This fusion protein circumvented a decreased growth rate observed in yeast transfected with a bait vector expressing the full GluR-A C-terminal peptide (data not shown). Of 34 positive clones, 3 encoded partially overlapping open reading frames (ORFs) of the RIL gene, which codes for a 330 amino acid protein containing two recognizable protein-protein interaction motifs, an N-terminal PDZ domain and a C-terminal LIM domain. Of the three RIL clones isolated, one contained the full ORF, whereas the two remaining clones showed either partial or complete lack of the N-terminal PDZ domain coding sequence (RILΔ1-59 and RILΔ1-130, respectively) (Fig. 1a). As judged by the intensity of β-galactosidase (β-gal) signal, RILΔ1-130 showed the strongest binding (Fig. 1a). To examine the specificity of RIL interaction with the GluR-A 10 amino residue peptide, we first tested the binding of RIL ORF or RILΔ1-130 with the GluR-B portion of the chimeric bait lacking the GluR-A sequence, R-BΔ10, as well as with an unrelated bait construct and with an empty-vector pGBT9 GAL4 DNA-binding domain. As determined by yeast mating assays, both RIL ORF and RILΔ1-130 interacted only with the original R-B/R-At10 bait, indicating that the GluR-A C-terminal 10 residues are required for RIL binding (Fig. 1b). Next, we examined the specificity of the interaction with respect to the GluR-A PDZ-binding motif, amino residues -TGL. Bait R-B/R-At10(T887A), containing an alanine substitution of the critical threonine-887 that mutates the PDZ-binding motif to -AGL (Hayashi et al., 2000; Passafaro et al., 2001), was able to bind to both RIL ORF and RILΔ1-130 (Fig. 1b). These data indicate different sequence requirements for PDZ and LIM domain-mediated interactions at the GluR-A C-terminal peptide. In addition, dual alanine substitution within the GluR-A-binding sequence, bait R-B/R-At10(P883A,T887A), but not a substitution of the proline-883 alone, bait R-B/RAt10(P883A), resulted in a near loss of binding with both RIL ORF and RILΔ1-130 (Fig. 1b).
Figure 1.

GluR-A C-terminal 10 residues constitute a binding motif for the LIM domain of RIL. a, Top, Representation of the chimeric bait comprising the proximal 40 amino residues of the GluR-B C-terminal domain (R-B) fused to the C-terminal 10 residues of GluR-A (R-At10); Gal4 BD, Gal4 DNA-binding domain. Bottom, Representation of RIL and the isolated RIL clones (numbers = amino residues).β-Gal activity for the corresponding RIL clones is indicated: +++ = strong, ++ = good, += visible blue color. b, Yeast-mating assays were performed between RIL ORF or RILΔ1-130 and the bait constructs: R-B/R-At10, R-BΔ10 containing only the 40 residues of the GluR-B C-terminal domain, dendrin#2 (Herb et al., 1997) (unrelated bait), and an empty bait vector, pGBT9. β-Gal activity is indicated as in a: - = nondetected. Matings with mutation-containing baits were assayed by liquid β-gal measurements and normalized to the R-B/R-At10 values with RIL ORF and RILΔ1-130, respectively (n = 2). c, GluR-A but not GluR-B coimmunoprecipitates with FlagRIL. COS1 cells, transfected with plasmid DNA as indicated below the panels, were lysed and immunoprecipitated with α-Flag antibody (panels labeled IP α-Flag). Input panels show 5% of the protein used for the precipitations. Antibodies that used Western blotting are indicated on the right. d, RIL binding with GST fusion protein containing the GluR-A C-terminal domain (GST-R-A). GST and GST-R-A were used in pull-down assays with FlagRIL and FlagRIL-deletion constructs graphed as in Figure 1a; aa borders: FlagRIL-PDZ = 1-115; FlagRIL-PDZ+L = 1-255; FlagRIL-L = 96-255; FlagRIL-L+LIM = 96-330; FlagRIL-LIM = 238-330. Input panels show 5% of the protein used for the pull-downs. L, Linker region.
To confirm our yeast interaction data in a mammalian expression system, we first tested whether RIL binds to GluR-A when coexpressed in COS1 cells. As shown in Figure 1c, RIL with an N-terminal Flag epitope, FlagRIL, indeed coimmunoprecipitated GluR-A from COS1 lysates. Under the same conditions, FlagRIL failed to bind with GluR-B (Fig. 1c), which supports the specificity of RIL interaction with GluR-A. In the second set of experiments, a GST fusion protein containing the GluR-A C-terminal peptide, GST-R-A, was used in pull-down assays with recombinant FlagRIL and FlagRIL deletion mutants. GST-R-A, but not GST alone, bound with FlagRIL as well as with RIL containing the linker region and the C-terminal LIM domain but lacking the N-terminal PDZ domain, FlagRIL-L+LIM (Fig. 1d). In contrast, all RIL deletion mutants lacking the LIM domain failed to bind to GST-R-A, confirming that the LIM domain is required for the interaction with GluR-A (Fig. 1d) (the PDZ+L construct bound very weakly to GST-R-A, and it is not clear whether this interaction is specific). The LIM domain itself, FlagRIL-LIM, was not sufficient to mediate the binding, suggesting that the flanking sequence is also required, possibly for proper protein folding of the LIM domain (Fig. 1d).
The PDZ domain of RIL interacts with the carboxyl PDZ-binding motif of α-actinin
To search for potential RIL PDZ domain-interacting proteins, we screened an adult rat brain cDNA library using the RIL N-terminal 110 residues as bait in the yeast two-hybrid system. Of 28 positive clones, 4 encoded the actin-binding protein α-actinin, 3 encoded theα-actinin4, and 1 encoded theα-actinin2 isoforms (Fig. 2a). The RIL interaction site on α-actinin2 was mapped to the extreme carboxyl region, which terminates with amino acids -SDL. This sequence is a typical class I PDZ binding motif and is conserved among all four α-actinin isoforms, α-actinin1-4 (Fig. 2b); the NMDA receptor subunit NR1 C-terminal bait, shown previously to bind to the internal spectrin-like repeats ofα-actinin2 (Wyszynski et al., 1997), was used as a control for functional expression of the SDL-lacking constructs.
Figure 2.

RIL PDZ domain binds to the carboxyl -SDL motif of α-actinin. a, Top, Representation of α-actinin. ABD, Actin-binding domain; SD, spectrin-like repeat; EF, Ca2+-binding EF hand motif. Bottom, A single α-actinin2 and three α-actinin4 clones isolated by screening with the RIL PDZ domain as bait (numbers = amino residues; slashed lines indicate that the clones were only partially sequenced). β-Gal activity is indicated on the right. b, Deletion constructs of α-actinin2, represented as gray horizontal bars with indicated amino acid borders, were tested for their binding activity with the RIL PDZ domain or with the NR1 C-terminal domain that served as a control, shown previously to interact with the α-actinin spectrin-like repeats (Wyszynski et al., 1997). β-Gal activity is indicated on the right. c, GST, GST-RIL, and GST-RIL deletion constructs containing the indicated portions of RIL (same borders as for the FlagRIL truncations in Fig. 1d) were used in pull-down assays with heterologous α-actinin2 (top panel) or with endogenous brain α-actinin2 (bottom panel). Input = 15% of the lysates used for the pull-downs. Western blotting was done with anti-α-actinin2 antibody.
To confirm the yeast-based data, we performed pull-down assays with GST fusion protein containing the full RIL molecule, GST-RIL ORF, or various RIL deletions. As shown in Figure 2c, GST-RIL ORF as well as GST-RIL deletion mutants containing the PDZ domain, GST-RIL-PDZ and GST-RIL-PDZ+L, bound recombinant as well as endogenous α-actinin2 from brain homogenate. We conclude that RIL is a bifunctional protein that binds to the C-terminal tails of the GluR-A subunit and α-actinin.
Endogenous RIL is enriched at excitatory synapses and interacts with AMPA receptors
RIL mRNA is expressed most prominently in the adult rat brain, heart, and lung, and at lower levels in other tissues (Kiess et al., 1995; Vallenius et al., 2004). To study RIL protein expression, we generated a polyclonal antibody against the RIL C-terminal 14 amino acids. As shown in Figure 3a, anti-RIL serum recognized recombinant FlagRIL, and an affinity-purified anti-RIL antibody detected a single band of the predicted molecular size, 35.5 kDa, in the rat forebrain lysate (Fig. 3a). RIL was found to be expressed in the brain, in hippocampal and cortical lysates, from early postnatal stages until adulthood (data not shown). Subcellular fractionation of the forebrain lysate showed RIL enriched in the PSD fraction, to a similar degree as GluR-A, but less than NR1 (Fig. 3b). In addition, the postsynaptic expression of RIL was further confirmed by immunostainings of hippocampal primary cultures with a previously characterized RIL-specific antibody raised against the C-terminal portion of the molecule (Cuppen et al., 1998); our anti-peptide-raised antibody did not show a specific signal for immunocytochemistry and thus could not be used. As shown in Figure 3c, RIL immunoreactivity was concentrated in dendritic spines and showed partial colocalization with the presynaptic vesicular protein SV2 (Buckley and Kelly, 1985), confirming that in hippocampal principal neurons, RIL is localized mainly to the postsynaptic sites of synapses.
Figure 3.

Endogenous RIL is postsynaptically enriched and interacts with AMPA receptors and α-actinin. a, Specificity of anti-RIL antibody. Lanes 1-3, Lysates from COS1 cells transfected with FlagRIL-expressing plasmid were Western blotted with anti-Flag Ab (1), preimmune serum (2), and anti-RIL serum (3). Lane 4, Whole-cell forebrain lysate Western blotted with affinity-purified anti-RIL antibody. Molecular weight markers in kilodaltons are on the right. b, Whole-cell forebrain homogenate (hom.), synaptosomal (synapt.), and PSD fractions were probed with affinity-purified anti-RIL antibody (top panel) and with anti-GluR-A, NR1, and synaptophysin (synapt.) Ab, as indicated on the right. c, Hippocampal primary cultures (14DIV) were fixed and stained with anti-RIL Ab [characterized previously in Cuppen et al. (1998)] (red channel) and anti-SV2 Ab (Buckley and Kelly, 1985) (green channel). Right panels were enlarged from the framed area in the left and middle panels; arrows point to examples of RIL distribution in spine-like structures along the distal dendrite, showing partial overlap with anti-SV2 staining of presynaptic terminals. The images are maximal projections of a stack of 0.08 μm separated confocal z-sections, processed by deconvolution for enhanced resolution. Scale bar, 5 μm. d, Solubilized rat brain P2 fraction was applied to protein-A Sepharose column conjugated with either anti-GluR-B/C antibody (GluR-B/C col.) or normal rabbit IgG (IgG col.). After extensive washing, bound proteins were eluted with the GluR-B/C antigen peptide, and the elution fractions 2 and 3 (lanes 2 and 3) were analyzed by Western blotting with antibodies as indicated on the right. Note that α-actinin2 bound weakly to a control normal IgG-conjugated column; however, it bound at only 18% of the signal compared with the GluR-B/C antibody-conjugated column. Input lane is 1% of total protein used for the immunoprecipitations.
To examine whether native RIL binds with AMPA receptors, we isolated AMPA receptor complexes from synaptosomal lysate using a protein A Sepharose column conjugated with anti-GluR-B/C antibody (we used the anti-GluR-B/C antibody because the anti-GluR-A antibody and RIL recognize the same region of the GluR-A C-terminal peptide and thus may compete for binding). Western blotting of fractions eluted from the column by excess of the GluRB/C antigen peptide revealed that RIL was indeed complexed with GluR-A/GluR-B receptors (Fig. 3d). In addition, we also detected α-actinin2 in the GluR-A and RIL-containing fractions, suggesting that all three proteins can bind simultaneously (Fig. 3d).
EGFPRIL increases the abundance of recombinant GluR-A receptors in endosomal compartments in heterologous cells
Protein-protein interactions mediated by the C-terminal domains of the AMPA receptor subunits regulate different aspects of the receptor trafficking (Malinow and Malenka, 2002; Song and Huganir, 2002). We examined whether RIL affects the transport of GluR-A in COS1 cells, a heterologous system in which we demonstrated RIL binding to GluR-A by coimmunoprecipitation (Fig. 1c). We analyzed the GluR-A steady-state distribution, alone or cotransfected with RIL, in the following trafficking compartments: the endoplasmatic reticulum (ER; the site of synthesis and maturation of transmembrane proteins), early and recycling endosomes (the transport pathway for internalized proteins sorted for reinsertion at the surface membrane), and lysosomes (the site of protein degradation). GluR-A expressed alone was concentrated in the ER, identified by staining against the ER-resident protein calnexin (Fig. 4a). In addition, GluR-A weakly accumulated in the perinuclear recycling endosomes, colabeled with antibody against TfR (Fig. 4b). GluR-A was not accumulated detectably in early endosomes, labeled with antibody against EEA1 (data not shown), or in lysosomes, identified by staining against Lamp1 (data not shown). RIL expressed alone, either FlagRIL (data not shown) or EGFPRIL (Fig. 4c), colocalized with phalloidin-labeled actin cytoskeleton, suggesting that RIL is targeted to the actin filaments via its interaction with α-actinin [a similar distribution of recombinant RIL was reported previously in Cuppen et al. (2000) and Vallenius et al. (2004); expression of recombinant RIL did not change the overall distribution of F-actin compared with nontransfected cells; no appreciable amount of endogenous RIL was detected in COS1 cells by anti-RIL immunostaining or Western blot; data not shown]. EGFPRIL lacking the LIM domain, EGFPRILΔLIM, still localized efficiently to F-actin, whereas EGFPRIL lacking the PDZ domain, EGFPRILΔPDZ, showed only a diffuse cytoplasmic distribution (data not shown) (Fig. 5b,c) (in cotransfection with GluRA). This shows that the PDZ domain is required for targeting of RIL to actin cytoskeleton in COS1 cells.
Figure 4.
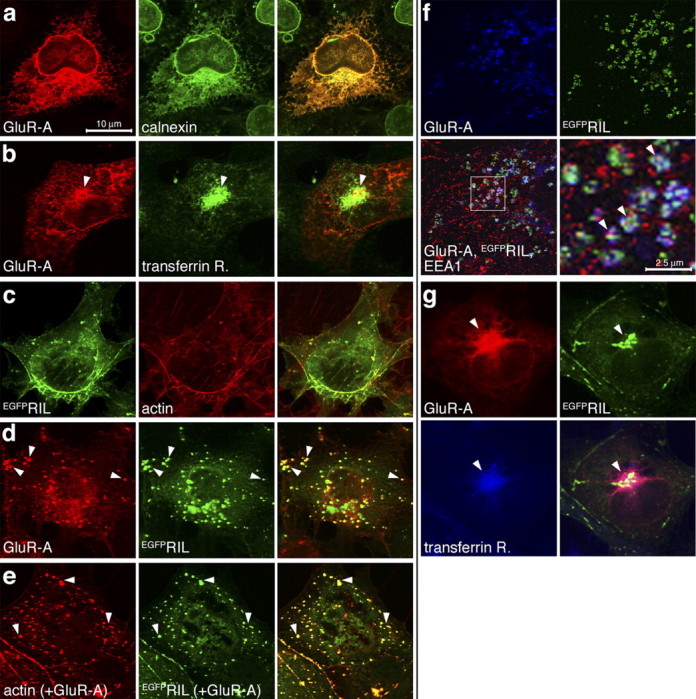
Colocalization of GluR-A and EGFPRIL in endosomes in COS1 cells. a, COS1 cells were transfected with GluR-A-expressing plasmid and stained with anti-GluR-A and anti-calnexin antibodies. Calnexin is an ER-resident protein. b, Cells were transfected as in a and stained with anti-GluR-A and anti-TfR antibodies. Arrows mark GluR-A in TfR-labeled recycling endosomes. c, Cells were transfected with EGFPRIL-expressing plasmid and labeled with TRITC-phalloidin identifying actin cytoskeleton. d, Cells were transfected with GluR-A and EGFPRIL plasmids and stained with anti-GluR-A antibody. Arrows mark colocalization of both proteins in small vesicular-like structures. e, Cells were transfected as in d and labeled with TRITC-phalloidin. Arrows point to examples of punctate colocalization of EGFPRIL and phalloidin-labeled actin resembling the EGFPRIL and GluR-A colocalization in early endosomes (d, f). f, Cells were transfected as in d and stained with anti-GluR-A (blue channel) and anti-EEA1 (red channel) antibodies. Arrows mark colocalization of GluR-A and EGFPRIL in vesicular-like structures that also contain EEA1. g, Cells were transfected as in d and stained with anti-GluR-A (red channel) and anti-TfR (blue channel) antibodies. Arrows mark colocalization of GluR-A and EGFPRIL in TfR-containing recycling endosomes. All images in a-d and g are maximal projections of a stack of 0.3 μm separated confocal z-sections through the entire cell thickness; images in e and f are single z-confocal sections. Scale bars: a-g, 10 μm; f, 2.5 μm.
Figure 5.
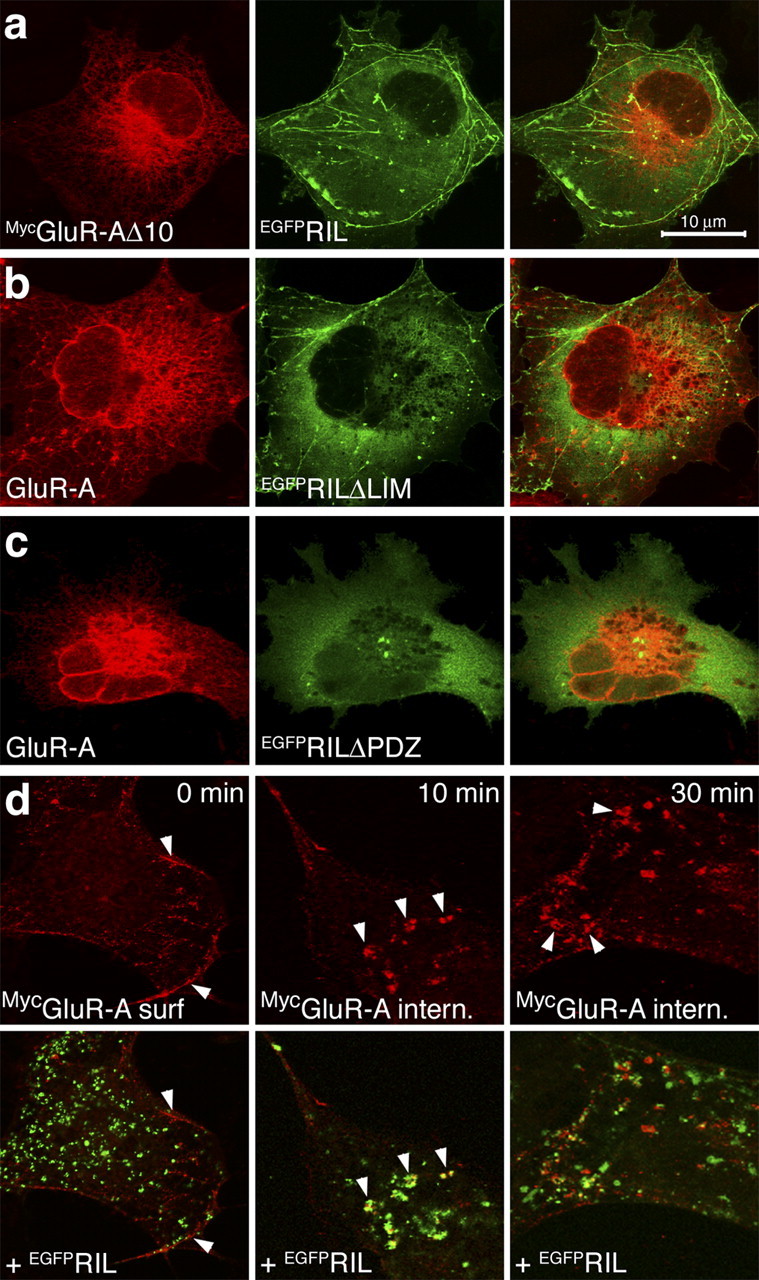
EGFPRIL-GluR-A colocalization in early endosomes requires EGFPRIL binding with both GluR-A and α-actinin. a, Cells were transfected with MycGluR-AΔ10 and EGFPRIL plasmids and labeled with anti-Myc antibody. b, c, Cells were transfected with GluR-A and EGFPRILΔLIM (b) or EGFPRILΔPDZ (c) plasmids and labeled with anti-GluR-A antibody. d, Cells transfected with MycGluR-A and EGFPRIL plasmids were assayed for colocalization of surface-internalized MycGluR-A with EGFPRIL. Live cells were incubated with anti-Myc Ab at 4°C to label surface-expressed receptors and then either fixed (panels 0 min) or returned to 37°C for internalization periods of 10 or 30 min. Top panels show labeled MycGluR-A receptors; bottom panels show corresponding overlays with EGFPRIL distribution. Arrows at time 0 min point to surface-expressed MycGluR-A at the edges of the cell; arrows at time 10 and 30 min point to colocalization of internalized MycGluR-A with EGFPRIL. Images in a-c are maximal projections of z-sections through the entire cell thickness; images in d are single z-confocal sections taken near the cell membrane. Scale bar: (in a) a-d, 10 μm.
Coexpression of GluR-A and EGFPRIL resulted in redistribution of both proteins into small vesicular-like structures that were identified as early endosomes by anti-EEA1 staining (Fig. 4d,f). Furthermore, we also observed a similar redistribution of F-actin in cells transfected with EGFPRIL and GluR-A, resulting in punctate actin-EGFPRIL colocalization (Fig. 4e) (a comparable result was also observed for coexpression of GluR-A and FlagRIL; data not shown). These data imply that the accumulation of GluR-A and EGFPRIL in early endosomes may involve formation of GluR-A-EGFPRIL-α-actinin/actin protein complexes (see more below). In addition, in a small population of cells, GluR-A and EGFPRIL accumulated in the perinuclear region corresponding to recycling endosomes colabeled with antibody against TfR (Fig. 4g). In contrast, GluR-A and EGFPRIL showed no detectable colocalization in Lamp1-labeled lysosomes (data not shown). Similarly, the rate of degradation of GluR-A in COS1 cells, as determined by S35Cys/Met pulse chase, was not different if the receptor subunit was expressed alone or coexpressed with EGFPRIL (data not shown).
To examine whether RIL colocalization with GluR-A requires RIL interaction with both the receptor subunit and α-actinin, we coexpressed GluR-A and RIL mutants lacking the corresponding protein-protein interaction motifs. As shown in Figure 5a, no colocalization was observed between MycGluR-AΔ10, a GluR-A form lacking the last 10 amino residues of the C-terminal domain, and EGFPRIL. At the same time, both EGFPRILΔLIM and EGFPRILΔPDZ failed to induce colocalization with GluR-A in early endosomes (Fig. 5b,c) (in some cases we observed a weak recruitment of EGFPRILΔPDZ to GluR-A localized in the perinuclear recycling endosomal region). These data indicate that the GluR-A C-terminal 10 residues are essential for its colocalization with RIL and that the RIL PDZ domain is required for the accumulation of GluR-A in early endosomes. In addition, coexpression of EGFPRIL with either GluR-B or GluR6 subunits failed to show any colocalization, further confirming the specificity for the GluR-A subunit (data not shown).
Next, we wanted to confirm the finding that RIL and GluR-A co-distribute to early endosomes. For this purpose, we examined whether RIL colocalizes with GluR-A receptors that entered the early endosomal compartment from the cell surface, after antibody-induced receptor internalization. Cells expressing the N-terminally tagged MycGluR-A and EGFPRIL were first incubated at 4°C with anti-Myc antibody to selectively label surface-inserted MycGluR-A receptors. After extensive washing, the cells were either fixed or returned to 37°C for 10 or 30 min. As shown in Figure 5d (time, 0 min), surface-expressed MycGluR-A did not show any significant colocalization with EGFPRIL. In contrast, newly internalized MycGluR-A strongly colocalized with EGFPRIL in endosomal compartments in the proximity of the cell membrane, after both 10 and 30 min internalization periods (Fig. 5d). We conclude that EGFPRIL is recruited to surface-internalized endosomal population of the MycGluR-A receptors.
EGFPRIL is targeted via its PDZ domain to dendritic spines in hippocampal primary neurons
To examine RIL function in neurons, we first characterized the subcellular distribution of EGFPRIL, EGFPRILΔPDZ, and EGFPRILΔLIM expressed from an attenuated Sindbis virus vector, SINrep(nsp2S726) (Kim et al., 2004), in hippocampal cultured neurons. As shown in Figure 6, a and e, EGFPRIL was enriched in numerous but not all spine-like structures along dendritic branches. Assuming that EGFPRIL is targeted to spines by binding to the α-actinin-actin complex, the variability of EGFPRIL enrichment in spine-like protrusions may reflect varying levels of endogenous actin and α-actinin at these sites. To test this, we first selected a set of spine-like protrusions with obvious EGFPRIL enrichment and measured the ratio of spine/shaft fluorescence (spine enrichment) for EGFPRIL as well as for actin and α-actinin2/3 (see Materials and Methods). We found that EGFPRIL was enriched approximately threefold in the selected spine-like protuberances, and these also showed 3.5-fold enrichment of actin and α-actinin 2/3 (Fig. 6i) (EGFPRIL enriched). Next we selected spine-like protrusions with apparent lack of EGFPRIL enrichment and found that these showed only ∼1.5-fold enrichment of actin and α-actinin 2/3 (Fig. 6i)(EGFPRIL equal). This shows that EGFPRIL is preferentially enriched at sites containing larger amounts of actin and α-actinin. The same distribution into α-actinin2/3 and actin-rich sites was also observed for EGFPRILΔLIM, whereas EGFPRILΔPDZ as well as free EGFP were distributed equally between spine-like protrusions and dendritic shafts (Fig. 6). In addition, we also compared the spine enrichment of actin and α-actinin2/3 without selecting for EGFPRIL or EGFPRILΔLIM distribution. In this analysis we found that the overall actin and α-actinin2/3 distribution was similar for all four constructs expressed: actin enrichment (mean ± SEM) was 250.3 ± 20.9, 231.2 ± 20.09, 221.9 ± 15.6, and 242.3 ± 8.7, and α-actinin2/3 enrichment (mean ± SEM) was 265.7 ± 40.1, 289.4 ± 36.6, 250 ± 17.7, and 292 ± 21.5 in cells expressing EGFPRIL, EGFPRILΔLIM, EGFPRILΔPDZ, and EGFP, respectively (at least 9 dendritic branches and in total 200 spines were analyzed per each construct). This suggests that expression of EGFPRIL or EGFPRILΔLIM alone did not alter the distribution of endogenous actin or α-actinin in the infected neurons. We thus conclude that EGFPRIL is targeted efficiently to spines via its PDZ domain-mediated interaction with the α-actinin-actin cytoskeletal complex.
Figure 6.
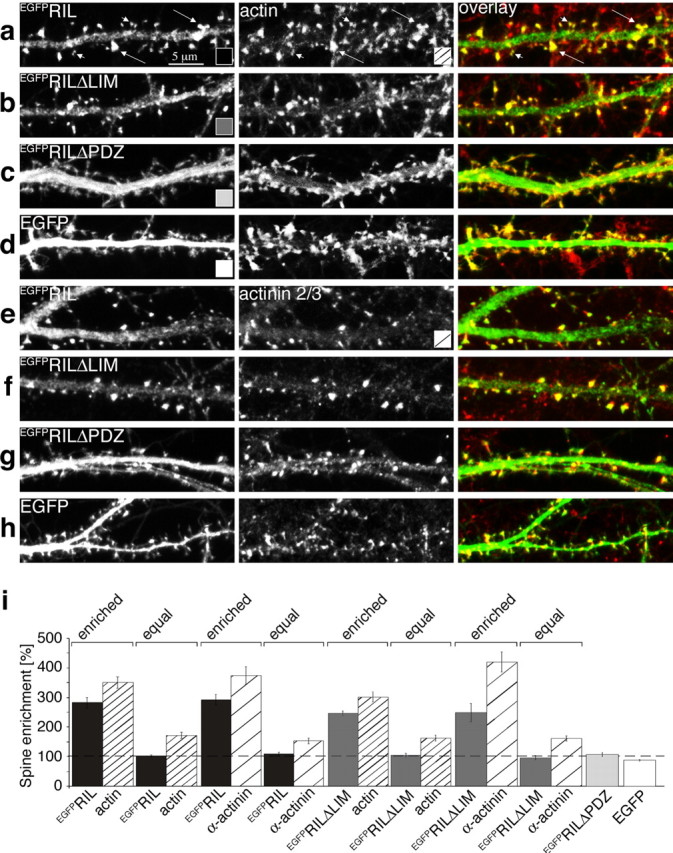
EGFPRIL is targeted via its PDZ domain to α-actinin/actin-rich spines. Hippocampal primary cultures (14DIV) were infected with Sindbis virus expressing EGFPRIL (a), (e), EGFPRILΔLIM (b), (f), EGFPRILΔPDZ (c), (g) or EGFP (d), (h). After 24 hr, neurons were fixed and stained with TRITC-conjugated phalloidin to label F-actin (red channel in overlay; a-d) or with anti-α-actinin2/3 antibody (red channel in overlay; e-h). Notice that EGFPRIL and EGFPRILΔLIM show highly enriched distribution in a number of spine-like protrusions along dendritic shafts (in a, long arrows point to examples of high EGFPRIL and corresponding high actin-content spine-like structures; short arrows point to low EGFPRIL and actin content). The color- and dash-coding in the bottom right corner of the panels indicates the type of heterologous protein expression or immunostaining for the bar graph quantitation below. i, Quantitation of spine enrichment: the ratio of fluorescence between spine-like protrusion and neighboring dendritic shaft. Comparison of spine enrichment for actin and α-actinin 2/3 (as indicated under the bars) in spine-like protrusions selected for either clear enrichment of EGFPRIL (enriched) or for equal distribution of EGFPRIL (equal), and for clear enrichment of EGFPRILΔLIM (enriched) or for equal distribution of EGFPRILΔLIM (equal). In contrast, both EGFPRILΔPDZ and EGFP were equally distributed between dendritic shafts and spines. Measured values (mean ± SEM): EGFPRIL-enriched = 282.7 ± 18.1/actin = 351.1 ± 19.9, n = 10 dendritic branches, 212 spines; EGFPRIL-equal = 102.8 ± 3.9/actin = 172.1 ± 10.4, n = 10 dendritic branches, 135 spines; EGFPRIL-enriched = 194.2 ± 18.2/α-actinin2/3 = 377.1 ± 30.2, n = 10 dendritic branches, 258 spines; EGFPRIL-equal = 109.2 ± 6.8, α-actinin2/3 = 154.3 ± 8.7, n = 10 dendritic branches, 185 spines; EGFPRILΔLIM-enriched = 244.8 ± 8.2/actin = 300.5 ± 16.3, n = 8 dendritic branches, 178 spines; EGFPRILΔLIM-equal = 104.4 ± 6.8/actin = 161.8 ± 9.6, n = 8 dendritic branches, 136 spines; EGFPRILΔLIM-enriched = 248.1 ± 30.9/α-actin in 2/3 = 418.2 ± 34.2, n = 9 dendritic branches, 154 spines; EGFPRILΔLIM-equal = 95.8 ± 7.1/actin = 160.6 ± 8.6, n = 9 dendritic branches, 155 spines; EGFPRILΔPDZ = 111.3 ± 7.6, n = 9 dendritic branches, 198 spines; EGFP = 91.6 ± 3.2, n = 10 dendritic branches, 251 spines.
EGFPRIL increases the abundance of GluR-A-containing receptors in dendritic spines in hippocampal primary neurons
Next, we examined EGFPRIL distribution in primary cultured neurons with respect to spine localization of endogenous AMPA receptors. In this set of experiments, analysis of immunostaining against the GluR-A subunit revealed that GluR-A-containing AMPA receptors were 2.4-fold enriched in dendritic spines selected on the basis of high EGFPRIL expression and 1.6-fold enriched at sites with equal spine/shaft EGFPRIL distribution (Fig. 7a,e). In comparison, cells expressing the EGFPRILΔLIM construct showed overall lower synaptic enrichment for the GluR-A-containing AMPA receptors, with only 1.6-fold enrichment at spines selected for high EGFPRILΔLIM expression and 1.3-fold enrichment at sites with equal spine/shaft EGFPRILΔLIM distribution (Fig. 7b,e). Furthermore, analysis of GluRA spine/shaft ratio distribution without selecting for EGFPRIL or EGFPRILΔLIM enrichment revealed that neurons expressing EGFPRIL had overall higher spine accumulation of GluR-A in comparison with cells expressing EGFPRILΔLIM, EGFPRILΔPDZ, or only EGFP (Fig. 7f). In summary, we conclude that expression of EGFPRIL enhances the total accumulation of GluR-A-containing AMPA receptors in dendritic spines.
Figure 7.

EGFPRIL enhances synaptic accumulation of GluR-A-containing AMPA receptors. Hippocampal primary cultures (14DIV) were infected with Sindbis virus expressing EGFPRIL (a), EGFPRILΔLIM (b), EGFPRILΔPDZ (c), or EGFP (d). After 24 hr, neurons were fixed and immunostained against GluR-A (red channel in overlay; a-d). In a, long arrows point to examples of high EGFPRIL and corresponding high GluR-A-content spine-like structures (short arrows point to low EGFPRIL and GluR-A content). The color- and dash-coding in the bottom right corner of the panels indicates the type of heterologous protein expression or immunostaining for the bar graph quantitation below. e, Quantitation of spine enrichment for GluR-A (as indicated under the bars) in spine-like protrusions selected for either clear enrichment of EGFPRIL (enriched) or for equal distribution of EGFPRIL (equal), and for clear enrichment of EGFPRILΔLIM (enriched) or for equal distribution of EGFPRILΔLIM (equal). Both EGFPRILΔPDZ and EGFP were equally distributed between shafts and spines. Measured values (mean ± SEM): EGFPRIL-enriched = 235.2 ± 9.6/GluR-A = 241.6 ± 10.0, n = 16 dendritic branches, 277 spines; EGFPRIL-equal = 100.1 ± 2.2/GluR-A = 157.5 ± 9.6, n = 15 dendritic branches, 255 spines; EGFPRILΔLIM-enriched = 226.6 ± 13.6/GluR-A = 163.7 ± 6.4, n = 12 dendritic branches, 221 spines; EGFPRILΔLIM-equal = 101.3 ± 1.6/GluR-A = 131.2 ± 5.0, n = 12 dendritic branches, 202 spines; EGFPRILΔPDZ = 112.1 ± 5.0, n = 11 dendritic branches, 293 spines; EGFP = 94.4 ± 4.7, n = 12 dendritic branches, 263 spines. f, GluR-A spine enrichment in cells with expression of heterologous proteins as indicated under the bar graph (mean ± SEM): 197.3 ± 12.4, 147.5 ± 5.2, 157.1 ± 8.9, and 171.7 ± 7.3 for cells expressing EGFPRIL, EGFPRILΔLIM, EGFPRILΔPDZ, and EGFP, respectively. Statistical significance: ANOVA (df = 3, F = 5.0), p = 0.003; Student's t test (unpaired, 2-tailed); GluR-A in cells with EGFPRIL versus EGFPRILΔLIM: p = 0.001; EGFPRIL versus EGFPRILΔPDZ: p = 0.016; EGFPRIL versus noninfected: p = 0.042.
EGFPRIL enhances AMPA receptor-mediated synaptic currents
To examine selectively the effects of EGFPRIL on synaptic surface-expressed AMPA receptors, we analyzed AMPA receptor-mediated mEPSCs in noninfected cells and in neurons expressing EGFPRIL or EGFPRIL deletion mutants. These experiments showed that the average peak mEPSC amplitude recorded from EGFPRIL-expressing neurons was increased by ∼20% compared with noninfected neurons or neurons expressing either EGFPRILΔPDZ or EGFPRILΔLIM (Fig. 8a,b) (the mEPSC rise time was the same among all conditions; the frequency showed a trend toward higher values in EGFPRIL-expressing cells; see Materials and Methods). These data suggest that overexpression of EGFPRIL, but not of EGFPRILΔPDZ or EGFPRILΔLIM, in primary hippocampal neurons moderately but significantly increases the overall number of endogenous AMPA receptors at synaptic sites. Furthermore, we obtained an essentially identical result, relative to control noninfected cells, when EGFPRIL was expressed in CA1 neurons in hippocampal organotypic slices, an alternative in vitro hippocampal preparation. As shown in Figure 8, c and d, the mean mEPSC amplitude in EGFPRIL-expressing CA1 neurons was again increased significantly, by ∼25%, compared with noninfected neurons (the rise time of the mEPSCs was the same; see Materials and Methods). These data thus confirm that overexpression of EGFPRIL increases AMPA receptor-mediated synaptic currents in cultured CA1 neurons. We conclude that increased expression of EGFPRIL in hippocampal pyramidal neurons results in an overall increased accumulation of GluR-A-containing AMPA receptors in dendritic spines, including the population of surface-expressed synaptic receptors.
Figure 8.

Overexpression of EGFPRIL increases AMPA receptor-mediated mEPSCs in hippocampal cultured neurons. a, Representative recordings (left) and averaged events (right) from EGFPRIL, EGFPRILΔPDZ, EGFPRILΔLIM-expressing, and noninfected neurons of hippocampal primary cultures, as indicated. b, Average current amplitudes (mean ± SEM) recorded from neurons expressing EGFPRIL (121.5 ± 5.2%; 25.6 ± 1.1 pA; n = 7),EGFPRILΔPDZ (100.2 ± 4.7%; 21.1 ± 1.0 pA; n = 4), and EGFPRILΔLIM (99.6 ± 7.5; 21.0 ± 1.6; n = 11) relative to control noninfected cells (100 ± 7.5%; 21.0 ± 1.6 pA; n = 7). Statistical significance: ANOVA (df = 3, F = 3.2), p = 0.036; Student's t test (unpaired, 2-tailed) EGFPRIL versus EGFPRILΔPDZ: p = 0.027; EGFPRIL versus EGFPRILΔLIM: p = 0.04;EGFPRIL versus noninfected: p = 0.035. c, Representative recordings (left) and averaged events (right) from EGFPRIL-expressing and noninfected hippocampal CA1 neurons in organotypic slices, as indicated. d, Average current amplitudes (mean ± SEM) recorded from CA1 neurons expressing EGFPRIL (125.1 ± 6.0%; 26.5 ± 1.3 pA; n = 6) relative to control noninfected cells (100 ± 4.1%; 21.2 ± 0.9 pA; n = 7). Statistical significance: Student's t test (unpaired, 2-tailed): p = 0.006.
Discussion
We have identified protein-protein interactions that may link the transport of AMPA receptors to the actin cytoskeleton of dendritic spines: the PDZ-LIM protein RIL binds via its PDZ domain to the actin-binding protein α-actinin and via its LIM domain to the carboxyl region of the GluR-A C-terminal domain. RIL is a member of a family of cytoplasmic proteins containing one N-terminal PDZ domain and one or three C-terminal LIM domains (Bach, 2000). Common characteristics among these proteins include PDZ domain-mediated association with actin-binding proteinsα-actinin or tropomyosin and a broad range of interactions mediated by their LIM domains. These proteins are thus proposed to function as adaptors linking transport of transmembrane and cytosolic proteins to the actin cytoskeleton, typically in muscle and epithelial cells where they are prominently expressed (Bach, 2000). In contrast, RIL is the only member of the PDZ-LIM protein family with highest expression in brain (Kiess et al., 1995).
LIM domain-mediated binding of RIL to GluR-A
The LIM domain, an ∼50-residue motif formed by two tandemly repeated zinc fingers, functions as a protein-protein interaction module in various proteins (Bach, 2000). The RIL LIM domain was shown previously to bind to the RIL PDZ domain and to the second and fourth PDZ domains of the protein tyrosine phosphatase PTP-BL (basophil-like) (Cuppen et al., 1998) (we have confirmed the RIL LIM to PDZ interaction in yeast-mating assays; data not shown). The RIL LIM to PDZ domain binding can presumably occur intermolecularly, allowing RIL to homo-oligomerize, or intramolecularly. Supporting the intramolecular folding, RIL constructs containing the linker region and the LIM domain but lacking the PDZ domain appeared to bind more efficiently to the GluR-A C-terminal motif than the full RIL molecule in vitro (Fig. 1). RIL thus may be able to undergo conformational switching from a “closed” LIM-PDZ-bound state to an “open” state in which RIL binds to other interacting partners. Similar folding-mediated regulation of function was described previously for the ezrin, radixin, and moesin proteins that link actin filaments to the cytoplasmic membrane: both the N-terminal membrane-binding and C-terminal actin-binding domains are masked by intramolecular interactions (Pufall and Graves, 2002).
The RIL LIM domain interaction site on GluR-A comprises the 10 carboxyl residues, including the class I PDZ domain-binding motif. Mutation of the GluR-A threonine-877, T887A, is predicted to result in a loss of PDZ domain-mediated binding (Shi et al., 1999; Passafaro et al., 2001), yet the bait R-B/R-At10(T887A) still bound with RIL. This indicates that the PDZ and LIM domains recognize distinct motifs within the GluR-A C-terminal peptide. A comparison of known interacting partners of RIL-related LIM domains so far fails to identify a common LIM domain-binding motif. For example, the second and third LIM domain of the single PDZ- and three LIM-domain protein Enigma bind to the intracellular C-terminal domains of the insulin receptor and the receptor tyrosine kinase Ret, respectively, and these interactions require Tyr- and Pro-based motifs (Wu and Gill, 1994; Wu et al., 1996); however, each of the three Enigma LIM domains also binds to the N-terminal portions of protein kinase C isoforms α, β, and ζ, without a clear common binding motif found in these sequences (Kuroda et al., 1996). Thus it appears that LIM domains may recognize a wide range of protein-protein interacting motifs within the C-terminal peptides of transmembrane receptors as well as coding sequences of soluble signaling molecules.
PDZ domain-mediated binding of RIL to α-actinin/actin cytoskeleton
The molecular structure of α-actinin consists of an N-terminal actin-binding domain, four spectrin-like repeats forming the central rod of the molecule, and two Ca2+-binding EF hands in the C terminus (Baron et al., 1987) (Fig. 2a). The spectrin-like repeats mediate α-actinin anti-parallel homodimerization, which allows α-actinin to cross-link actin fibers. In addition, the central repeats serve as a docking site for the binding of a number of proteins via different motifs, including the PDZ domains of RIL-related ALP (actinin-associated LIM protein) and CLP-36 (C-terminal LIM protein 36), as well as the intracellular C-terminal domains of several transmembrane proteins, such as the β1 and β2 integrins, adhesion molecules ICAM and Ep-CAM, and the NMDA receptor subunits NR1 and NR2B (Djinovic-Carugo et al., 2002). Recently, an interaction between a recombinant RIL and α-actinin, mediated by the RIL PDZ-domain binding to theα-actinin spectrin-like repeats, was described in epithelial cells (Vallenius et al., 2004). In contrast, we showed here that in yeast-mating assays the RIL PDZ domain selectively binds to the -SDL class I PDZ-binding motif ofα-actinin1-4. Thus it appears possible that RIL may associate with either the spectrin repeats or the C-terminal residues on the α-actinin molecule. Functionally, we showed that the PDZ-domain-based binding targets RIL to dendritic spines in hippocampal neurons, an effect that likely mediates the observed RIL enrichment at the excitatory postsynaptic side.
EGFPRIL-mediated regulation of recombinant GluR-A receptors in COS1 cells
After internalization, surface-expressed proteins are first targeted to early endosomes where they are sorted to be delivered back to the plasma membrane via recycling endosomes or targeted to late endosomes and lysosomes for degradation (Gruenberg and Maxfield, 1995). We found that although GluR-A expressed alone in COS1 cells was localized mainly to ER and EGFPRIL to actin cytoskeleton, coexpression of the two proteins resulted in their prominent colocalization in early endosomes and, to a lesser extent, in recycling endosomes. Importantly, the accumulation of GluR-A in early endosomes required EGFPRIL binding with both GluR-A and α-actinin.
Recently, RIL overexpression in osteosarcoma U2OS cells was reported to change the dynamics of actin in stress fibers, with high formation and collapse of new fibers (Vallenius et al., 2004). Although we did not observe obvious changes in actin distribution after expression of RIL alone, the endosomal GluR-A-RIL colocalization included a partial redistribution of actin into a punctate pattern overlapping with EGFPRIL. Together, these data imply that RIL can act as an actin-based anchor for GluR-A-containing AMPA receptors undergoing endosomal sorting and that such an RIL-based link of the receptors to actin may affect, in addition, the arrangement of the actin cytoskeleton itself.
EGFPRIL-mediated regulation of endogenous AMPA receptors in cultured neurons
In the first set of experiments examining RIL effect on endogenous AMPA receptors in hippocampal cultured neurons, we showed that expression of EGFPRIL, but not of EGFPRILΔPDZ, EGFPRILΔLIM, or EGFP, resulted in an increased accumulation of GluR-A-containing AMPA receptors in dendritic spines, as examined by anti-GluR-A immunostaining. Because EGFPRIL is targeted to spines by its PDZ domain, these data suggest that RIL may act to increase the content of the GluR-A-containing AMPA receptors in dendritic spines by linking the receptors to the α-actinin/actin cytoskeleton.
In addition, we also showed that expression of EGFPRIL, but not of EGFPRILΔPDZ, EGFPRILΔLIM, or EGFP, resulted in ∼25% potentiation of endogenous AMPA receptor-mediated mEPSCs in hippocampal primary cultures and in organotypic slice cultures. These data support the finding that increased levels of EGFPRIL in dendritic spines induce a larger accumulation of GluR-A-containing AMPA receptors, including the functional surface-expressed receptor pool.
Functional significance of RIL for AMPA receptor transport
Here we present evidence that RIL regulates, in an α-actinin/actin-dependent manner, trafficking of AMPA receptors in dendritic spines. Our experiments show that increased levels of EGFPRIL translate to increased levels of AMPA receptors within dendritic spines as well as at the synaptic surface. In principal, such an effect can be achieved by RIL-based recruitment of extrasynaptic receptors to the dendritic spine compartment and/or by limiting the spine exit of the receptors undergoing endosomal synaptic recycling. In both cases, RIL appears to provide an α-actinin/actin-dependent spatially directive regulation for the transport of GluR-A-containing AMPA receptors in dendritic spines, ultimately promoting the transport and/or recycling of the receptors toward insertion at the postsynaptic membrane.
Footnotes
This study was supported in part by Deutsche Forschungsgemeinschaft Grant Os 191/1-1 to P.O. We thank Andrea Migala, Carmen Grosskurth, Judith Muller, Sabine Gruenewald, Günter Giese, and Horst Grosskurth for excellent technical assistance and Sondra Schlesinger for Sindbis(nsp2S726) plasmid and helpful discussions.
Correspondence should be addressed to Pavel Osten, Max Planck Institute for Medical Research, Department of Molecular Neurobiology, Jahnstrasse 29, 69120 Heidelberg, Germany. E-mail: posten@mpimf-heidelberg.mpg.de.
A. Rozov's present address: University of Heidelberg, Interdisciplinary Center for Neurosciences, 69120 Heidelberg, Germany.
J. Kim's present address: National Institutes of Health, Bethesda, MD 20892-9495.
T. W. Schulz's present address: Responsif GmbH, Schallershofer Strasse 84, D-91056 Erlangen, Germany.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248584-11$15.00/0
T.W.S., T.N., P.L., and V.P. contributed equally to this work.
References
- Allison DW, Gelfand VI, Spector I, Craig AM (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18: 2423-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach I (2000) The LIM domain: regulation by association. Mech Dev 91: 5-17. [DOI] [PubMed] [Google Scholar]
- Baron MD, Davison MD, Jones P, Critchley DR (1987) The structure and function of alpha-actinin. Biochem Soc Trans 15: 796-798. [DOI] [PubMed] [Google Scholar]
- Boulter J, Hollmann M, O'Shea-Greenfield A, Hartley M, Deneris E, Maron C, Heinemann S (1990) Molecular cloning and functional expression of glutamate receptor subunit genes. Science 249: 1033-1037. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Buckley K, Kelly RB (1985) Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol 100: 1284-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol 86: 831-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Lissin DV, von Zastrow M, Nicoll RA, Malenka RC (1999) Rapid redistribution of glutamate receptors contributes to long-term depression in hippocampal cultures. Nat Neurosci 2: 454-460. [DOI] [PubMed] [Google Scholar]
- Cuppen E, Gerrits H, Pepers B, Wieringa B, Hendriks W (1998) PDZ motifs in PTP-BL and RIL bind to internal protein segments in the LIM domain protein RIL. Mol Biol Cell 3: 671-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuppen E, van Ham M, Wansink DG, de Leeuw A, Wieringa B, Hendriks W (2000) The zyxin-related protein TRIP6 interacts with PDZ motifs in the adaptor protein RIL and the protein tyrosine phosphatase PTP-BL. Eur J Cell Biol 79: 283-293. [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Gautel M, Ylanne J, Young P (2002) The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett 513: 119-123. [DOI] [PubMed] [Google Scholar]
- Dryga SA, Dryga OA, Schlesinger S (1997) Identification of mutations in a Sindbis virus variant able to establish persistent infection in BHK cells: the importance of a mutation in the nsP2 gene. Virology 228: 74-83. [DOI] [PubMed] [Google Scholar]
- Fifkova E, Delay RJ (1982) Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol 95: 345-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Maxfield FR (1995) Membrane transport in the endocytic pathway. Curr Opin Cell Biol 7: 552-563. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R (2000) Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287: 2262-2267. [DOI] [PubMed] [Google Scholar]
- Herb A, Wisden W, Catania MV, Marechal D, Dresse A, Seeburg PH (1997) Prominent dendritic localization in forebrain neurons of a novel mRNA and its product, dendrin. Mol Cell Neurosci 8: 367-374. [DOI] [PubMed] [Google Scholar]
- Heynen AJ, Quinlan EM, Bae DC, Bear MF (2000) Bidirectional, activity-dependent regulation of glutamate receptors in the adult hippocampus in vivo. Neuron 28: 527-536. [DOI] [PubMed] [Google Scholar]
- Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH (1990) A family of AMPA-selective glutamate receptors. Science 249: 556-560. [DOI] [PubMed] [Google Scholar]
- Kiess M, Scharm B, Aguzzi A, Hajnal A, Klemenz R, Schwarte-Waldhoff I, Schafer R (1995) Expression of ril, a novel LIM domain gene, is down-regulated in Hras-transformed cells and restored in phenotypic revertants. Oncogene 10: 61-68. [PubMed] [Google Scholar]
- Kim CH, Lisman JE (1999) A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci 19: 4314-4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Dittgen T, Nimmerjahn A, Waters J, Pawlak V, Helmchen F, Schlesinger S, Seeburg PH, Osten P (2004) Sindbis vector SINrep(nsP2S726): a tool for rapid heterologous expression with attenuated cytotoxicity in neurons. J Neurosci Methods 133: 81-90. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Tokunaga C, Kiyohara Y, Higuchi O, Konishi H, Mizuno K, Gill GN, Kikkawa U (1996) Protein-protein interaction of zinc finger LIM domains with protein kinase C. J Biol Chem 271: 31029-31032. [DOI] [PubMed] [Google Scholar]
- Lee SH, Liu L, Wang YT, Sheng M (2002) Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36: 661-674. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29: 243-254. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103-126. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Launey T, Mikawa S, Hirai H (2000) Disruption of AMPA receptor GluR2 clusters following long-term depression induction in cerebellar Purkinje neurons. EMBO J 19: 2765-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K (1982) High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci USA 79: 7590-7594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osten P, Srivastava S, Inman GJ, Vilim FS, Khatri L, Lee LM, States BA, Einheber S, Milner TA, Hanson PI, Ziff EB (1998) The AMPA receptor GluR2 C-terminus can mediate a reversible, ATP-dependent interaction with NSF and a- and b-SNAPs. Neuron 21: 99-110. [DOI] [PubMed] [Google Scholar]
- Osten P, Khatri L, Perez JL, Kohr G, Giese G, Daly C, Schulz TW, Wensky A, Lee LM, Ziff EB (2000) Mutagenesis reveals a role for ABP/GRIP binding to GluR2 in synaptic surface accumulation of the AMPA receptor. Neuron 27: 313-325. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4: 917-926. [DOI] [PubMed] [Google Scholar]
- Pufall MA, Graves BJ (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu Rev Cell Dev Biol 18: 421-462. [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL (2000) Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci 20: 7932-7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Lee SH (2001) AMPA receptor trafficking and the control of synaptic transmission. Cell 105: 825-828. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105: 331-343. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R (1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811-1816. [DOI] [PubMed] [Google Scholar]
- Song I, Huganir RL (2002) Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci 25: 578-588. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Osten P, Vilim FS, Khatri L, Inman G, States B, Daly C, DeSouza S, Abagyan R, Valtschanoff JG, Weinberg RJ, Ziff EB (1998) Novel anchorage of GluR2/3 to the postsynaptic density by the AMPA receptor-binding protein ABP. Neuron 21: 581-591. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R (2003) Experience strengthening transmission by driving AMPA receptors into synapses. Science 299: 1585-1588. [DOI] [PubMed] [Google Scholar]
- Vallenius T, Scharm B, Vesikansa A, Luukko K, Schafer R, Makela TP (2004) The PDZ-LIM protein RIL modulates actin stress fiber turnover and enhances the association of alpha-actinin with F-actin. Exp Cell Res 293: 117-128. [DOI] [PubMed] [Google Scholar]
- Vandekerckhove J (1990) Actin-binding proteins. Curr Opin Cell Biol 2: 41-50. [DOI] [PubMed] [Google Scholar]
- Wang YT, Linden DJ (2000) Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 25: 635-647. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS (1996) Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci 16: 1982-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Durick K, Songyang Z, Cantley LC, Taylor SS, Gill GN (1996) Specificity of LIM domain interactions with receptor tyrosine kinases. J Biol Chem 271: 15934-15941. [DOI] [PubMed] [Google Scholar]
- Wu RY, Gill GN (1994) LIM domain recognition of a tyrosine-containing tight turn. J Biol Chem 269: 25085-25090. [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M (1997) Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature 385: 439-442. [DOI] [PubMed] [Google Scholar]
- Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M (1998) Differential regional expression and ultrastructural localization of α-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci 18: 1383-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA (2001) Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci USA 98: 1261-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]