

Abstract
Microglia are the resident macrophage population of the CNS and are considered its major immunocompetent elements. They are activated by any type of brain pathology and can migrate to the lesion site. The chemokine CXCL10 is expressed in neurons in response to brain injury and is a signaling candidate for activating microglia and directing them to the lesion site. We recently identified CXCR3, the corresponding receptor for CXCL10, in microglia and demonstrated that this receptor system controls microglial migration. We have now tested the impact of CXCR3 signaling on cellular responses after entorhinal cortex lesion. In wild-type mice, microglia migrate within the first 3 d after lesion into the zone of axonal degeneration, where 8 d after lesion denervated dendrites of interneurons are subsequently lost. In contrast, the recruitment of microglia was impaired in CXCR3 knock-out mice, and, strikingly, denervated distal dendrites were maintained in zones of axonal degeneration. No differences between wild-type and knock-out mice were observed after facial nerve axotomy, as a lesion model for assessing microglial proliferation. This shows that CXCR3 signaling is crucial in microglia recruitment but not proliferation, and this recruitment is an essential element for neuronal reorganization.
Keywords: microglia, migration, chemokine, receptor, CXCL10, CXCR3, dendritic loss, entorhinal cortex lesion, facial nerve lesion
Introduction
Microglia, the major immunocompetent cells of the CNS (Gehrmann et al., 1995; Benveniste, 1997), play a key role in brain pathology. During virtually any type of lesion, microglial cells are activated and transform in a multistep process from a ramified morphology into an amoeboid macrophage- or dendritic cell-like (immune) phenotype (Kreutzberg, 1996; Streit et al., 1999; Fischer and Reichmann, 2001; Santambrogio et al., 2001). Microglial cells detect neuronal injury at a very early stage, and activation of microglial cells is limited to those anatomical regions occupied by the injured neurons. It is thus reasonable to hypothesize that neurons provide signals that regulate microglial activation. Chemokines are known to play a crucial role in the regulation of migratory behavior and activation of immune cells (Mackay, 2001). One of the chemokines, CXCL10 [previously known as interferon-inducible protein of 10 kDa (IP-10)], is strongly expressed in many neurodegenerative diseases (Riemer et al., 2000; Xia et al., 2000) and after entorhinal lesion (Babcock et al. 2003). The specific receptor for CXCL10 in microglia is CXCR3 (Biber et al., 2001, 2002; Rappert et al., 2002). To study the impact of CXCL10 and CXCR3 signaling on microglial response, we used two well characterized brain lesion models with different microglial reactions: the facial nerve axotomy model (Blinzinger and Kreutzberg, 1968) and entorhinal cortex lesion (Lynch et al., 1972). The fibers from the entorhinal cortex form the cortical input to the hippocampus. Axotomy of this tract causes a rapid astroglial and microglial reaction in the deafferentiated middle and outer molecular layers of the dentate gyrus, whereas the inner molecular layer is not affected (Gall et al., 1979; Gehrmann et al., 1991; Jensen et al., 1994, 1999; Bechmann and Nitsch, 1997; Hailer et al., 1999). This reaction involves morphological and functional changes, such as phagocytic activity and a loss of ramification (Bechmann and Nitsch, 1997). Additionally, the number of microglial cells in the molecular layer increases, whereas in adjacent hippocampal subfields, the number of microglia decreases, indicating migration of microglial cells into the zone of axonal degeneration.
Facial nerve axonal injury leads to an organized sequence of morphological and metabolic changes in and around the affected motoneurons (Blinzinger and Kreutzberg, 1968; Kreutzberg, 1996). Microglial cells adjacent to the transected motoneurons show a rapid increase in cell adhesion molecules, receptors for microglial mitogens, and cytoskeletal proteins. Within the first 48 hr after lesion, these activated microglia displace synaptic input (Raivich et al., 1994). Within 7 d after axotomy, microglial cells increase in number by four-fold to six-fold, but this increase is attributable to vigorous proliferation rather than recruitment of microglia from regions apart from the retrogradely degenerating neurons or blood-derived monocytic cells (Blinzinger and Kreutzberg, 1968; Raivich et al., 1994). We used these two models in CXCR3 knock-out mice to determine the impact of CXCL10 and CXCR3 signaling on microglial response.
Materials and Methods
Animals and surgical procedures
All operations were performed on 8- to 12-week-old mice. Generation of the homozygotes of the CXCR3 knock-out line has been described previously (Hancock et al., 2000). The CXCR3 knock-out animals have a C57Bl/6 background and were bred at local animal facilities. C57Bl/6 animals were obtained from Charles River (Hannover, Germany) and used as a control strain. The animals were kept under standardized conditions at constant temperature and controlled lighting and had ad libitum access to food and water. The animal experiments and care protocols were approved by the Landesamt für Arbeitsschutz (Berlin, Germany) (facial nerve axotomy, G0086/98; entorhinal cortex lesion, G0155/00).
For facial nerve axotomy, animals were anesthetized by 50 mg/kg sodium pentobarbital (Sanofi, Hannover, Germany). The right facial nerve was transected at the level of the stylomastoid foramen, whereas the contralateral side was left intact. Animals were killed 3 d after axotomy.
For unilateral entorhinal cortex lesion, the animals were anesthetized with a mixture consisting of 20% ketamine (CuraMED GmbH) and 8% Rompun (Bayer, Wuppertal, Germany) dissolved in 0.9% NaCl. The animals were fixed in a stereotaxic apparatus (Kopf Instruments) and the following coordinates measured from λ were used for lesions: anteroposterior, 0.4 mm; lateral, 1.2 mm; and dorsoventral, down to the base of the skull. The monopolar blade of the surgical needle was lowered only once to the base of the skull. For sham operations, the skull was opened along the coordinates described above, but no lesion was performed. Animals were killed 6, 12, or 24 hr or 1.5, 2, 3, 8, or 31 d after axotomy.
Immunocytochemistry
All animals were deeply anesthetized with the ketamine mixture (for composition, see above) and perfused transcardially with phosphate buffer (PB) followed by a solution of 4% paraformaldehyde in 0.1 m PB, pH 7.4. Brains were removed and postfixed overnight at 4°C in the same fixative and subsequently rinsed in cold PB. Sagittal sections (40 μm) of brainstem containing facial nerve nuclei or coronal sections (50 μm) of the hippocampus were cut with a vibratome (VT 1000 S; Leica, Nussloch, Germany; or Vibratome 1000; Shandon-Lipshaw, Pittsburgh, PA). In addition, 15 μm cryosections of brainstem containing facial nerve nuclei and hippocampal sections were also used for immunohistochemistry.
Immunocytochemical procedures were performed as described before (Deller et al., 1995; Nolte et al., 2001). Briefly, free-floating vibratome sections or cryosections were permeabilized with 0.1% Triton X-100 in PB for 30 min. Endogenous peroxidases were blocked by 0.3% H2O2 for 5 min at room temperature. Sections were incubated in blocking solution [5% bovine serum albumin, 5% normal goat serum (NGS), and 0.1% Triton X-100 in 0.1 m PB] for 10 min, followed by overnight incubation at 4°C with primary antibodies in a dilution buffer containing 0.1% Triton X-100 and 1% NGS in 0.1 m PB.
Both anti-mouse CD11b (Mac-1; Leinco Technologies, St. Louis, MO) for the detection of macrophages and microglial cells and the anti-human proliferation marker Ki-67 (PharMingen, San Diego, CA) were diluted 1:100 in blocking solution. In some experiments, lectin from Lycopersicon esculentum (coupled to Alexa Fluor 594; BioTez Berlin-Buch, Berlin, Germany) in a dilution of 1:100 was used for the detection of macrophages and microglial cells. To label astrocytes, sections were stained for glial fibrillary acidic protein (GFAP; Dako Diagnostika GmbH, Hamburg, Germany; 1:400); neurons were stained for neuronal nuclei (NeuN; Chemicon Europe Ltd.; 1:1000). An antiserum against activated caspase 3 (R&D Systems, Wiesbaden-Nordenstadt, Germany; 1:1000) was used to stain apoptotic cells. An antibody against the CXCR3 ligand CXCL10 (PeproTech, London, UK; 1:500) was used to show the expression of this ligand after lesion. Biotinylated secondary anti-rabbit or anti-rat IgG (Linaris Biologische Produkte GmbH, Wertheim, Germany) was applied at a dilution of 1:250 for 2 hr at room temperature. Antibodies were then visualized with avidin-biotin-peroxidase-complex (ABC-Elite; Linaris Biologische Produkte GmbH) and diaminobenzidine (Sigma, Deisenhofen, Germany) following the supplier's instructions. For fluorescence labeling, Fluor (FITC) AffiniPure donkey anti-mouse IgG [heavy and light chain (H+L)] secondary antibody (1: 250) and Cy3-conjugated AffiniPure goat anti-rabbit IgG (H+L) secondary antibody (1:500; Jackson ImmunoResearch, West Grove, PA) were used. Sections were washed in 0.1 m PB, mounted on slides, dehydrated through a graded series of ethanol, cleared in xylene, and coverslipped.
Microscopy
The sections were analyzed by a conventional microscope (Axioskop; Zeiss, Oberkochen, Germany) equipped with epifluorescence illumination. The morphology of lectin-labeled microglial cells and macrophages was visualized by a confocal laser scanning microscope (CLSM) equipped with a 488 nm argon-ion laser (Molecular Dynamics, Sunnyvale CA) mounted on an upright microscope (Axioskop). Red fluorescence of Alexa was excited at 517 nm (beam splitter, 535 nm) and detected with a 570 nm long-pass filter. Serial sections of CLSM images were stored and processed on an Indigo workstation using the program Imagespace.
Comparison of microglial recruitment after entorhinal cortex lesion
For blind analysis, samples were prepared with one cross section of the wild-type mouse and one cross section of the knock-out mouse showing the same hippocampal region. The cross sections were randomly placed next to each other by a person not involved in the histological evaluation. The task of the blind investigator was to state in which of the two sections microglial recruitment to the denervated molecular layers and morphologic activation were more pronounced. On decoding the samples, it became evident that all four investigators uniformly judged the sections from the knock-out animal as exhibiting less migration and less morphologic transformation than the matching cross section from the wild-type animals. Most importantly, in the outer molecular layer of the knock-out animals, only a few microglia could be observed, whereas in the wild-type animals, this layer was full of microglia.
Quantification of microglia and astrocyte numbers in the molecular layers after entorhinal cortex lesion
As a parameter for quantifying this obvious difference, the number of microglia was counted in the inner, middle, and outer molecular layers of the lesioned and the contralateral hippocampus by blind investigators in digital pictures (taken with a 20× objective) of three wild-type and three knock-out mice. Five Mac-1-stained slices from matching hippocampal regions of each animal were used for this analysis. Although there is no clear border between the molecular layers in Mac-1-stained sections of unlesioned animals, the middle molecular layer can be easily identified after entorhinal cortex lesion because of the strong microglial reactions in this zone of axonal degeneration (Jensen et al. 1999) (Figs. 1 A, 2C). The sum of the number of microglial cells within the middle and outer molecular layers was divided by the number of Mac-1-positive cells in the inner molecular layer of each slice. The mean of this ratio was then calculated and statistically analyzed with a two-tailed Student's t test. The same procedure was performed in GFAP-stained slices for the calculation of astrocyte distribution.
Figure 1.
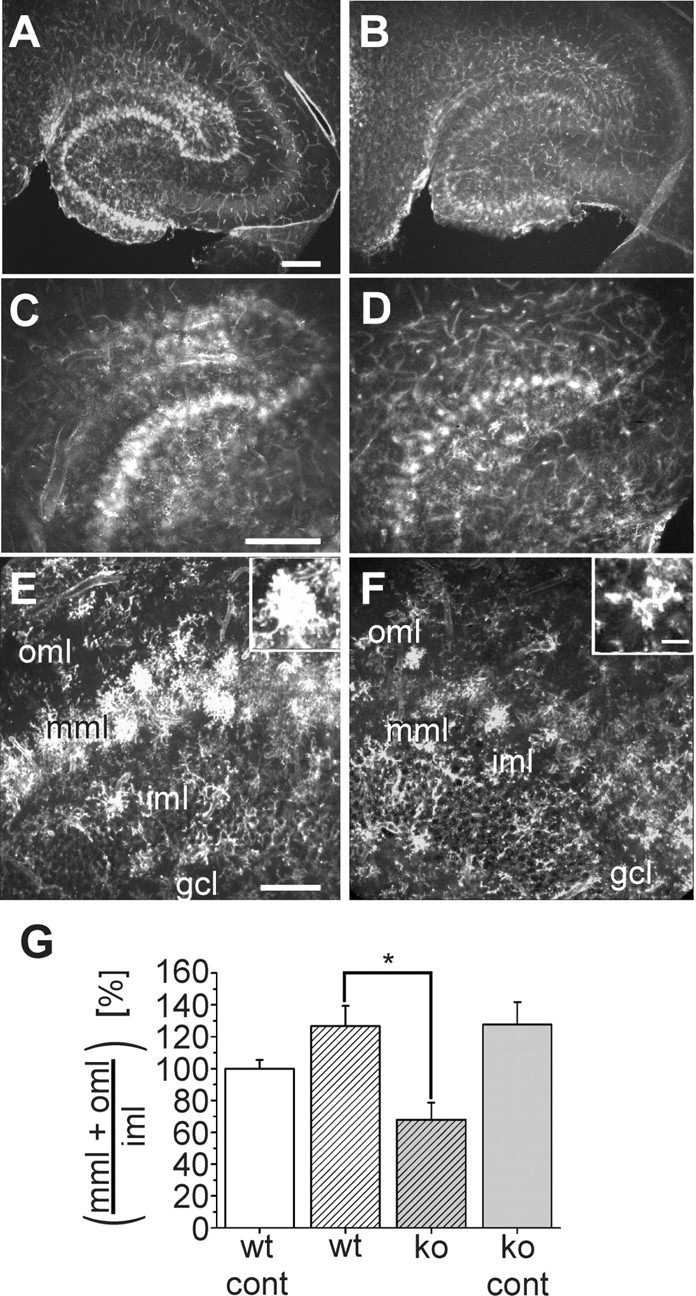
Distribution and morphology of Alexa Fluor 594-coupled lectin-positive microglia in the hippocampus of wild-type and CXCR3 knock-out mice 3 d after entorhinal cortex lesion. A-F, Horizontal vibratome sections of the temporal hippocampal formation were stained with Alexa Fluor 594-coupled lectin 3 d after entorhinal cortex lesion. A, B, Overviews of the lesioned hippocampus of wild-type animals (A) and CXCR3 knock-out animals (B). C, D, Magnified areas from A, B, respectively. E, F, Areas from the dentate granule cell layer (gcl), inner molecular layer (iml), middle molecular layer (mml), and outer molecular layer (oml) in more detail. In wild-type animals, lectin-stained microglial cells are visible in the oml and mml. In CXCR3 knock-out animals, microglial cells are restricted to the mml. Note the different morphologies of the microglial cells in CXCR3 knock-out and wild-type animals, as shown in the insets. Scale bars: A-D, 250 μm; E, F, 100 μm. G, Analysis of the number of microglial cells in slices from three wild-type and knock-out mice on the contralateral (cont) and lesioned sides. The number of microglia in CXCR3 knock-out (ko) animals on both sides and in wild-type (wt) animals on the lesioned side was normalized to the number of microglia in the wild-type animals on the contralateral side and presented as percentage of wild type ± SEM. *p < 0.05.
Figure 2.
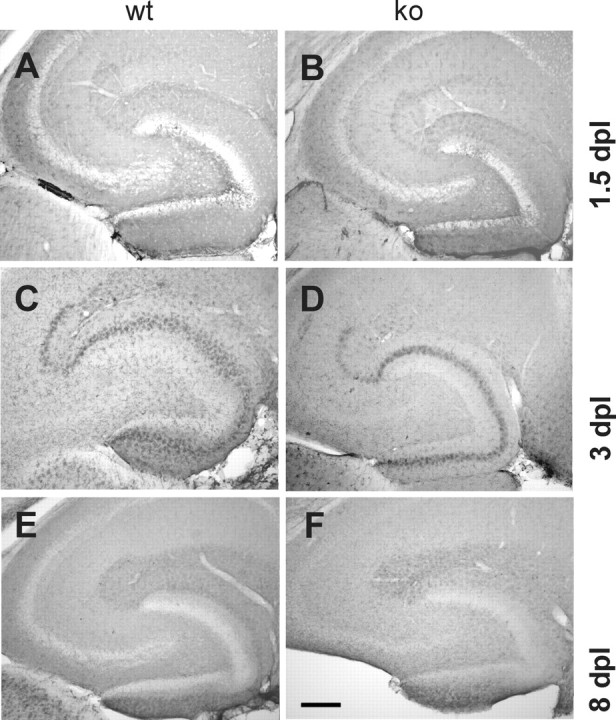
Time course of microglial activation in the hippocampus in wild-type and CXCR3 knock-out animals after entorhinal cortex lesion. Distribution and morphology of Mac-1-positive microglia were compared at 1.5, 3, and 8 d postlesion (dpl) in wild-type (wt; left column) and CXCR3 knock-out (ko; right column) animals. A, B, At 1.5 dpl, Mac-1 immunoreactivity is increased in zones of axonal degeneration in both wild-type and knock-out animals. The immunoreactive band, appearing in the middle and, to a lesser extent, outer molecular layers, is more pronounced in wild-type animals. C, D, At 3 dpl, the difference in distribution and intensity of Mac-1 immunolabeling between wild-type and knock-out animals is clearly visible. This difference allowed blind investigators to distinguish cross sections of wild-type from knock-out mice. E, F, At 8 dpl, Mac-1 immunoreactivity in the middle and outer molecular layers has decreased. Differences between knock-out and wild-type animals can no longer be distinguished by immunolabeling. Scale bar, 250 μm.
Quantification of Ki-67-positive cells
Photographs were taken at 20× magnification using a Magnafire digital camera (Intas, Göttingen, Germany). The Ki-67 labeled cells in the axotomized facial nucleus and dentate gyrus after entorhinal cortex lesion of CXCR3 knock-out mice (n = 3) and C57Bl/6 wild-type mice (n = 3). At least nine sections per animal and four randomly selected regions in each section were analyzed. All statistical analyses were performed using a two-tailed Student's t test and presented as mean ± SEM.
Analysis of dendrite length
The length of the dendrites was measured by a blind investigator using a custom-made pixel-based program. Using a 10× objective, a digital picture of the dentate gyrus was taken. The molecular layers of the dentate gyrus, including all dendrites in this area, were outlined. The program calculated the quantity of the outlined area (pixels). With the aid of a cursor, a person labeled the dendrites, and the program calculated the length of the dendrites (micrometers). Because we analyzed more than one slice per animal (on average, nine slices per animal), the mean of the length of all slices per animal was calculated. Finally, a ratio of dendrites per area was calculated and statistically analyzed.
Reverse transcription-PCR for the CXCR3 ligands CXCL9, CXCL10, and CXCL11
For reverse transcription (RT)-PCR analysis, brains were removed 6, 12, 24, and 48 hr after entorhinal cortex lesion and immediately frozen in liquid nitrogen. After separation of the two hemispheres, the lesioned site as well as its contralateral nonlesioned control were dissected. The tissue sections were homogenized in 2 ml of ice-cold guanidinium isothiocyanate/mercaptoethanol buffer, and total RNA was extracted with slight modifications according to the method of Chomczynski and Sacchi (1987).
Reverse transcription. One microgram of total RNA was transcribed into cDNA as described (Biber et al., 1997). Potential contamination by genomic DNA was checked by running the reactions (35 cycles) without reverse transcriptase and using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers in subsequent PCR amplifications. Only RNA samples that showed no bands after that procedure were used for further investigation.
PCR. Two microliters of the RT reaction were used in subsequent PCR amplification as described (Biber et al., 1997). The oligonucleotide sequence used to amplify the 347 bp fragment of CXCL10 was 5′-AAGTGCTGCCGTCATTTTCT-3′ (forward primer) and 5′-GAGGCTCTCTGCTGTCCATC-3′ (backward primer) according to the published sequence (GenBank accession number, AF227743) at 56°C with 30 cycles. The primer sequence for the 302 bp fragment of CXCL9 was 5′-GCACGATCCACTACAAATCC-3′ (forward primer) and 5′-ATTCAGGGTGCTTGTTGGTA-3′ (backward primer) (GenBank accession number, BC003343), and the primer sequence for the 262 bp fragment of CXCL11 was 5′-ATTACCAGGCTGCAGAACTTT-3′ (forward primer) (GenBank accession number, BC025903) and 5′-TCTTCCTAGATGTTCGTGTGC-3′ (backward primer). The cycler number was 34, and the annealing temperature was 58°C for both primers. The housekeeping gene (GAPDH) was primed for the 346 bp fragment with 5′-CATCCTGCACCACCAACTGCTTAG-3′ (forward primer) and 5′-GCCTGCTTCACCACCTTCTTGATG-3′ (backward primer) at 60°C with 28 cycles. All oligonucleotides were obtained from GenSet Oligos (Proligo, Paris, France). Cloning into pCR2.1 (Invitrogen, Breda, The Netherlands) and subsequent sequencing checked the identity of all PCR products.
Results
Microglial migration into the projection area of the lesioned perforant path is impaired in CXCR3 knock-out animals
Transection of the perforant path was used as a model of anterograde (Wallerian) axonal degeneration in a morphologically well circumscribed area, the middle molecular layer (i.e., the termination zone of the medial perforant path) and outer molecular layer (i.e., the termination zone of the lateral perforant path) of the dentate gyrus (Jensen et al., 1994; Bechmann and Nitsch, 1997; Hailer et al., 1999; Jensen et al., 1999; Bechmann et al., 2000). In accordance with a previous study in adult mice (Jensen et al., 1999), the microglial reaction is clearly potentiated and accelerated in the middle compared with the outer molecular layer, probably reflecting different fiber density and myelination (Figs. 1A-F, 2C,D).
Microglia were labeled using Mac-1 antibodies or Alexa Fluor 594-coupled lectin 1.5, 3, and 8 d after lesion. Under normal conditions, the microglia are evenly distributed throughout the molecular layers (data not shown) (Wirenfeld et al., 2003). In wild-type animals (n = 10), a broad band of labeled microglial cells appeared in the projection areas of the lesioned fibers at 3 d after lesion, with an accumulation in the middle being more pronounced as anticipated, compared with the outer molecular layer (Figs. 1A-F, 2C,D) (Jensen et al., 1994; Bechmann and Nitsch, 1997). The microglia had transformed from their resting, ramified morphology into an activated cell, which shows fewer ramified processes and an increase in the cell body size or “bushy” phenotypes (Fig. 1, left column) (Gehrmann et al., 1995; Kreutzberg, 1996; Benveniste, 1997; Streit et al., 1999). The hilus and inner molecular layer clearly contained fewer Mac-1-positive cells compared with the middle molecular layer, suggesting microglia migration from these adjacent regions into the denervated areas (Jensen et al., 1994).
In CXCR3 knock-out animals (n = 8), the increase in microglia reactivity 3 d after lesion appeared to be more restricted to the middle molecular layer, with fewer microglia in the outer molecular layer (Fig. 1, right column). Overall, microglial cells were less densely distributed in zones of degeneration, and, in contrast to the wild type, individual labeled cells could be easily recognized. These cells did not display the morphological characteristics of activated microglia as seen in the wild type and still retained their ramified morphology. When compared with the ramified microglia of the unlesioned control side, however, they appeared to have an increase in cell body size and a decrease in ramification of distal branches (data not shown). On the basis of these differences in microglial distribution and morphology in wild-type versus knock-out animals, four blind investigators could uniformly distinguish cross sections of the wild-type from knock-out animals in three independent experiments and at least 30 randomly paired samples.
As described above, in addition to differences in microglial morphology, another obvious difference between CXCR3 knock-out and wild-type animals after entorhinal cortex lesion is the distribution of microglia along the molecular layers. To quantify this altered recruitment of microglia to zones of axonal degeneration, microglia were counted by blind investigators. A ratio was calculated from the sum of the number of microglia in the denervated middle and outer molecular layers divided by the number of cells within the nonaffected inner molecular layer. The same was performed for the unlesioned contralateral side. The ratio of microglial cells in CXCR knock-out mice (n = 3, five slices per animal) was normalized to the ratio of microglial cells found on the unlesioned control side in wild-type animals (n = 3, five slices per animal). As shown in Figure 1G, on the lesioned side of CXCR3 knock-out animals (23.84 ± 3.81 cells), the calculated ratio differs significantly from the ratio in lesioned wild-type animals (44.45 ± 4.49 cells) as well as the contralateral side in wild-type animals (35.10 ± 1.90 cells) and knock-out animals (44.81 ± 4.96 cells) (p < 0.05). No significant difference could be found between the unlesioned contralateral sides of wild-type and CXCR3 knock-out animals.
The difference in microglial activation between CXCR3 knock-out and wild-type animals is most evident 3 d after lesion
To monitor the impact of CXCR3 deficiency on microglial activation and migration after entorhinal cortex lesion, we analyzed Mac-1-stained sections 1.5, 3, and 8 d after entorhinal cortex lesion (Fig. 2) as well as after sham operation. At 1.5 d after lesion, Mac-1 immunoreactivity was clearly increased in the hippocampus of both CXCR3 knock-out (n = 4) and wild-type (n = 4) animals when compared with the sham-operated animals (n = 3) and the contralateral unlesioned hemisphere (Fig. 2A,B). At 3 d after lesion, the spreading migration of cells into the denervated layers was clearly more pronounced than 36 hr before (Fig. 2C,D). In comparison with sham-operated animals, there was an obvious increase in Mac-1 immunolabeling in wild-type and knock-out animals. However, at 1.5 and 3 d after lesion, blind investigators could distinguish cross sections of CXCR3 knock-out mice from those of wild-type animals based on the morphology and distribution of Mac-1 immunostaining. In accordance with a previous study (Bechmann et al., 2000), apoptosis of glial cells was not evident either in wild-type or in knock-out animals using activated caspase 3 immunostaining, suggesting that knocking out the CXCR3 receptor did not enhance early apoptotic signals (data not shown).
At 8 d after lesion, Mac-1 expression still appeared to be enhanced in the lesioned hippocampus compared with sham-operated animals, indicating that activated microglia are still present. However, blind investigators could no longer distinguish cross sections of the wild-type mice (n = 7) from cross sections of the knock-out mice (n = 8) (Fig. 2E,F) (see Fig. 4, bottom panels).
Figure 4.
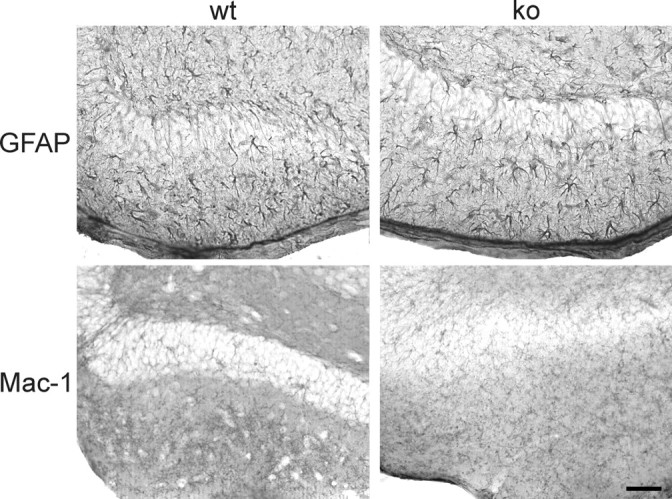
Astrocyte activation 8 d after entorhinal cortex lesion does not differ in wild-type and CXCR3 knock-out mice. Horizontal vibratome sections of hippocampus from wild-type mice (wt; left column) and CXCR3 knock-out mice (ko; right column) show GFAP-positive astrocytes (top panels) and Mac-1-positive microglia (bottom panels). No difference in the astrocytic activation could be observed between knock-out and wild-type animals. As described in Figure 2, microglial activation has already ceased at 8 days after lesion. Scale bar, 100 μm.
In sham-operated animals, there was a slight increase in Mac-1 immunoreactivity at the side of operation compared with the contralateral side. These changes were clearly less pronounced than in lesioned animals. It was not possible to distinguish cross sections from sham-operated wild-type and CXCR3 knock-out animals (data not shown).
The lesion-induced dendritic retraction of interneurons is prevented in CXCR3 knock-out animals
In the dentate gyrus, two subpopulations of GABAergic interneurons (basket and chandelier cells) express the calcium-binding protein parvalbumin. Their long dendrites extend to the stratum lacunosum-moleculare and to the outer molecular layer of the dentate gyrus (Kosaka et al., 1987; Sloviter, 1989; Nitsch et al., 1990; Gulyas et al., 1993), where they receive synaptic input from entorhinal fibers (Ruth et al., 1982; Zipp et al., 1989). Lesion of the perforant path thus denervates the distal part of their dendrites, resulting in significant and long-lasting loss of these denervated segments (Nitsch and Frotscher, 1991, 1993) (Fig. 3B) (n = 7). A decrease in dendrites was not observed before 8 d after entorhinal cortex lesion but persisted for 55 d without further reduction (Nitsch and Frotscher, 1991). We therefore chose 8 d to examine possible differences between CXCR3 knock-out and wild-type mice. In fact, analysis of the length of the dendrites (area covered by dendritic profiles in pixels) in the molecular layer of the dentate gyrus of wild-type animals performed by blind investigators revealed significant (p < 0.01) differences between lesioned and sham-operated animals (15.21 ± 1.91 vs 24.61 ± 3.06 μm/pixel; n = 4) (Fig. 3D).
Figure 3.

Parvalbumin immunostaining in the dentate gyrus 8 and 31 d after entorhinal cortex lesion. A-C, Horizontal vibratome sections of hippocampus from wild-type (wt; A, B) and CXCR3 knock-out (ko; C) mice showing parvalbumin-positive dendrites at 8 d postlesion (dpl). The density of parvalbumin-positive dendrites in wild-type animals (B) has markedly decreased when compared with CXCR3 knock-out animals (C) and sham-operated animals (A). Only a few parvalbumin-positive dendrites are visible in the middle and outer molecular layers of the fascia dentata of wild-type animals. Scale bar, 125 μm. D, E, Quantitative analysis of dendrite length 8 d (D) and 31 d (E) after entorhinal cortex lesion. D, Comparison of dendrite length 8 d after entorhinal cortex lesion revealed a significant difference between sham-operated and lesioned wild-type animals as well as between lesioned wild-type and CXCR3 knock-out mice (p < 0.01).E, Thirty-one days after entorhinal cortex lesion, the dendrite length of lesioned wild-type animals is significantly reduced compared with lesioned CXCR3 knock-out mice. A significant reduction in dendrite length in lesioned CXCR3 knock-out animals compared with the unlesioned contralateral side of CXCR3 knock-out animals (cont) also occurs (p < 0.05). ECL, Entorhinal cortex lesion; GCL, granular cell layer; ML, molecular layer.
In the CXCR3 receptor knock-out mice (n = 8), there was a striking difference from the wild-type mice at 8 d after lesion: the number of parvalbuminergic dendrites was not visibly reduced compared with sham-operated animals (n = 4) (Fig. 3A,C), indicating their maintenance on the entorhinal cortex attributable to CXCR3 knock-out. In fact, the dendritic length of CXCR3 knock-out mice after the entorhinal cortex (24.63 ± 1.58 μm/pixel) was significantly higher (p < 0.01) compared with wild-type animals (Fig. 3D) but, as anticipated, not in comparison with sham-operated knock-out animals (26.73 ± 0.49 μm/pixel). There was no difference between sham-operated wild-type and sham-operated knock-out mice. Thus, diminished microglial recruitment is associated with diminished dendritic loss in CXCR3 knock-out mice.
At 31 d after entorhinal cortex lesion, a significant (p < 0.05) decrease in dendrite length (7.11 ± 3.18 μm/pixel) was still evident in the ipsilateral dentate gyrus of wild-type animals (n = 2) compared with the nonlesioned contralateral side (25.87 ± 2.10 μm/pixel) (Fig. 3E, wt cont). Compared with 8 d after lesion, there was an additional slight reduction, yet this difference was not significant. Interestingly, the dendritic length was also significantly reduced on the ipsilateral side (16.85 ± 1.10 μm/pixel) compared with the contralateral unlesioned side (25.82 ± 1.83 μm/pixel) (Fig. 3E, ko cont) in CXCR3 knock-out mice (n = 5) at 31 d after entorhinal cortex lesion. Nevertheless, there are still significantly more dendritic profiles in the knock-out compared with the wild-type mice.
Astrocyte activation is not visibly altered in CXCR3 knock-out and wild-type animals
Microglial activation after entorhinal cortex lesion is followed, after a delay, by astrocytic hypertrophy and enhanced GFAP expression (Rose et al., 1976; Gage et al., 1988; Poirier et al., 1991; Steward et al., 1993; Jucker et al., 1995). Astrocytes can influence microglial activation (Becher et al., 2000), and they also express the chemokine receptor CXCR3 (Biber et al., 2002). Therefore, we examined astrocytic activation in CXCR3 knock-out versus wild-type animals 3 and 8 d after entorhinal cortex lesion. At neither 3 d after lesion (data not shown) nor 8 d after lesion (Fig. 4) were differences in the distribution or morphology of individual GFAP-positive cells evident to blind investigators. As for microglia, the ratio of the sum of the number of astrocytes in the middle and outer molecular layers divided by the number of cells within the inner molecular layer was calculated and subsequently normalized to the wild-type unlesioned contralateral side. The ratios revealed no differences in the distribution of GFAP-positive cells along the molecular layers (data not shown) (n = 3, three slices per animal; wild-type unlesioned side, 32.7 ± 4.6 cells; lesioned side, 33.17 ± 4.15 cells; CXCR3 knock-out unlesioned side, 33.26 ± 1.12 cells; lesioned side, 31.48 ± 4.65 cells). Thus, the astrocytic response to axonal degeneration is apparently not influenced by knocking out the CXCR3 receptor.
The CXCR3 ligand CXCL10 is induced in wild-type and knock-out animals
To investigate the induction of the CXCR3 ligand CXCL10, RTPCR and immunohistochemical analyses were performed. Because we were interested in early signals possibly provided by neurons in response to the lesion, different time points (6, 12, 24, and 48 hr) of CXCL10 mRNA expression after entorhinal cortex lesion were chosen. The results of RT-PCR clearly indicated an early induction of CXCL10 mRNA expression in the lesioned hemisphere at 6 and 12 hr after entorhinal cortex lesion (Fig. 5A) (n = 4). At 24 and 48 hr after entorhinal cortex lesion, CXCL10 mRNA induction was still detectable but less pronounced (data not shown). This is in line with the finding of Babcock et al. (2003), who demonstrated CXCL10 RNA expression in the lesioned hippocampus 24 hr after entorhinal cortex lesion. Immunohistochemical experiments corroborated these results at the protein level, because CXCL10-positive cells were found in the entorhinal cortex and the dentate gyrus ipsilateral to the lesion at 24 hr (Fig. 5B). Interestingly, CXCL10-positive staining was restricted to neuronal cells of the entorhinal cortex next to the lesion site and to a lesser extent of the dentate gyrus (Fig. 5C). No evident differences in the induction of CXCL10 mRNA and protein expression between CXCR3 knock-out (n = 4) and wild-type (n = 4) mice could be observed by blind investigators (Fig. 5). Thus, the observed differences in microglial activation and migration between wild-type and knock-out mice were apparently not attributable to differential expression of the CXCR3 ligand CXCL10. The expression of CXCL9 and CXCL11 as two other known ligands for CXCR3 was not detected using RT-PCR. This emphasizes the specificity of CXCL10 in this model.
Figure 5.

Expression of the CXCR3 ligand CXCL10 after entorhinal cortex lesion. A, RT-PCR analysis revealed an induction of CXCL10 mRNA expression in caudal forebrain tissue of wild-type (wt) and CXCR3 knock-out (ko) animals. As shown in the top panel, CXCL10 mRNA expression was induced in the ipsilateral hemisphere (LS) of the brains of both wild-type and CXCR3 knock-out mice 6 and 12 hr after lesion. Weaker CXCL10 mRNA expression was found on the contralateral side (CS). The bottom panel shows the results obtained with GAPDH house keeping gene primers to verify the quality of the cDNAs used in the experiment. Similar results were obtained in three independent experiments. The numbers of cycles used in PCR were 30 for CXCL10 and 28 for GAPDH; neg, PCR-negative control; MM, molecular weight marker; the highlighted band is 500 bp. B, Twenty-four hours after lesion, immunohistochemical staining showed numerous CXCL10-positive neuron-like cells in the entorhinal cortex next to the lesion (ipsilateral), as indicated by arrows. In contrast, no CXCL10-positive cells were observed in the contralateral entorhinal cortex. Blinded studies demonstrated no differences in CXCL10 immunohistochemistry between wild-type and CXCR3 knock-out animals. Similar results were obtained in three independent experiments. Scale bar, 100 μm. C, The double staining against CXCL10 and the neuronal marker NeuN clearly indicates that the CXCL10-positive cells are neurons.
Microglial response in the axotomized facial nucleus is not altered in CXCR3 knock-out mice: proliferation is unchanged in this model and after entorhinal cortex lesion
Transection of the facial nerve in adult mice leads to axonal loss of the motoneurons in the facial nucleus and to a pronounced microglial reaction around the axotomized motoneurons (for review, see Kreutzberg, 1996). Microglial cells are quickly activated, as indicated by morphological transformation, onset of proliferation, and the release of cytokines and chemokines (Raivich et al., 1994; Boucsein et al., 2000). We examined whether proliferation and the morphological transformation of microglia were altered in CXCR3 knock-out mice (n = 3) 3 d after axotomy compared with the wild-type mice (n = 3). Proliferating cells were labeled with the cell proliferation-associated nuclear antigen Ki-67, which is expressed in all active parts of the cell cycle (Gerdes et al., 1984). The density of Ki-67-positive cells was then evaluated microscopically. The time point chosen corresponds to the time point after axotomy when microglial proliferation has reached the maximum in the mouse (Raivich et al., 1994). At the same time, there were no Ki67-positive microglia in the facial nucleus on the control (unlesioned) side. The prominent microglial response induced by facial nerve axotomy 3 d after lesion in CXCR3 knockout mice could not be distinguished from that in the wild-type mice (Fig. 6A-D). Proliferating microglia could not be detected in the unlesioned contralateral facial nucleus either in the wild-type or in the CXCR3 knock-out animals.
Figure 6.
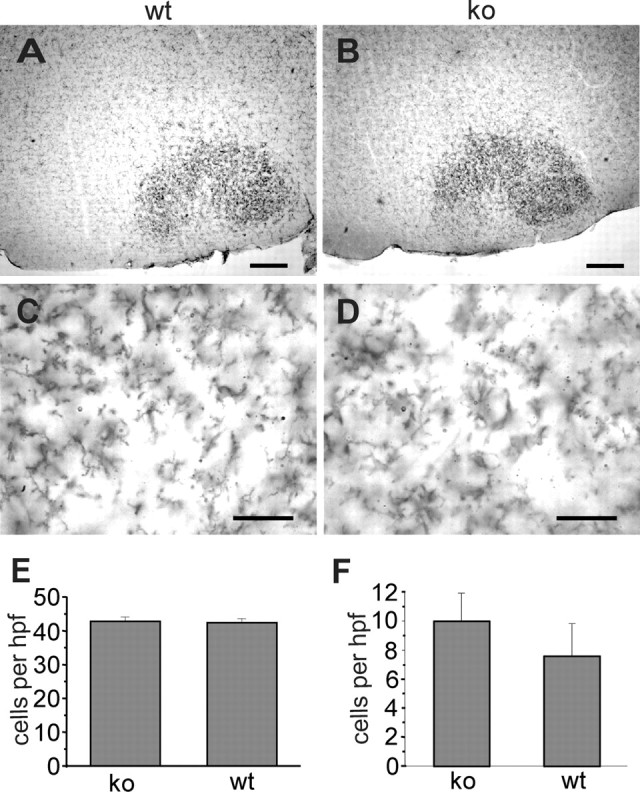
Morphology of microglia in the axotomized facial nucleus and comparison of cell proliferation after facial nerve axotomy and entorhinal cortex lesion. A-D, Wild-type (wt; A, C) and CXCR3 knock-out (ko; B, D) mice were stained for Mac-1 immunohistochemistry 3 d after facial nerve axotomy as described. Differences in density and morphology of facial nucleus microglia could not be observed (compare A with B, C with D). Scale bars: A, B, 250 μm; C, D, 50 μm. E, F, Quantification of lesion-induced proliferation: consecutive sections of axotomized facial nuclei (E) or deafferentiated hippocampus (F) were labeled with Ki67 antibodies. Ki67-positive nuclei were quantified as described. In both models, there was no significant difference between wild-type and knock-out animals in the density of Ki67-positive cells. Cell numbers are represented as mean ± SEM per high power field (hpf) and are derived from three animals per group.
Microglial activation is most apparent at day 3 after facial nerve axotomy and entorhinal cortex lesion (Raivich et al., 1994; Hailer et al., 1999). Proliferating cell counts in the facial nucleus 3 d after axotomy (Fig. 6E) and in the dentate gyrus 3 d after entorhinal cortex lesion (Fig. 6F) did not differ between wild-type (n = 4) and CXCR3 knock-out (n = 4) animals, indicating that CXCR3 signaling does not influence the proliferative activity of microglial cells.
Discussion
To test the effect of CXCR3 signaling on microglial migration and proliferation, two established models were used: one in which microglia migrate to zones of axonal degeneration, the entorhinal cortex lesion (Lynch et al., 1972; Peterson et al., 1994; Buzsaki et al., 1995; Deller et al., 1995; Frotscher et al., 1997); and another, in which local microglial cells predominantly increase by proliferation, namely, the facial nerve lesion (Blinzinger and Kreutzberg, 1968). In this set of experiments, using a CXCR3 knock-out mouse, we studied the relevance of CXCR3 signaling in vivo because our previous in vitro experiments suggested an important role for this chemokine in the control of microglial migration (Biber et al., 2001, 2002; Rappert et al., 2002). Indeed, we found profound differences in the entorhinal lesion model, whereas microglial behavior in the facial nucleus was not apparently affected by the loss of CXCR3 signaling. This is compatible with our studies in cell culture, which show that CXCR3 signaling controls the migration but not the proliferation of microglia (Biber et al., 2001; Rappert et al., 2002).
Entorhinal fibers form synaptic connections with the distal dendrites of parvalbumin-positive GABAergic interneurons in the middle and outer molecular layers of the dentate gyrus. In response to lesion of the entorhinal fibers, these interneurons lose the denervated distal segments of their dendrites within 8 d after lesion (Nitsch and Frotscher, 1991, 1993). Therefore, it has been suggested that the maintenance of these dendrites requires input from the entorhinal cortex (Zipp et al., 1989; Nitsch and Frotscher, 1991; Kiess et al., 1996). Our data show that the recruitment of microglia via CXCR3 signaling is involved in the fate of denervated dendrites because the lack of recruited microglia in knock-out mice correlates to the maintenance of dendrites. The activation process of microglial cells appears to proceed through a series of steps, which could end in phagocytotic microglial cells (Raivich et al., 1999). In the white matter, phagocytosis and the degradation of myelin is a slow processes, and we have shown before that the number of phagocytotic microglia increases until 8 d after entorhinal cortex lesion (Bechmann and Nitsch, 1997), the time point at which we have here demonstrated the difference in dendritic length between CXCR3 knock-out and wild-type mice. This could explain the time delay between the most obvious difference between CXCR3 knock-out and wild-type animals at 3 d after lesion and the retraction of the dendrites at 8 d after lesion. It is conceivable that microglia are involved in the observed dendritic loss by phagocytosing the denervated segments. Using organotypic entorhinohippocampal slice cultures, we have already shown that downregulation of microglial phagocytosis also preserves denervated dendrites after entorhinal cortex lesion (Eyupoglu et al., 2003).
Moreover, significantly more dendrites are found in CXCR3 knock-out animals compared with wild-type animals 31 d after lesion. Interestingly, the number of dendrites on the lesioned side in knock-out animals is significantly decreased compared with the unlesioned contralateral side. In addition, there is a noticeable additional decrease in the number of dendrites in wild-type animals 31 d after lesion compared with 8 d after lesion. One explanation might be that some microglia are still activated and phagocytose the dendrites in wild-type as well as knock-out animals at this late phase after lesion. It is also known that astrocytes can phagocytose neuronal debris, and we have demonstrated in this work that astrocytes in both groups of animals become activated after lesion. We also cannot eliminate the possibility that the missing signaling input could lead in the late phase to dendrite retraction. Nevertheless, in knock-out animals, there are still more dendrites than in wild-type animals. The extent to which such dendrite protection is functionally relevant remains to be seen.
It has been described that neurons can also express CXCR3 (Xia et al., 2000). However, CXCR3-positive cells were found only in the inner molecular layer and granule cells of the dentate gyrus (Xia et al., 2000), which clearly does not reflect the distribution of the parvalbumin-positive neurons studied here. Thus, although we cannot exclude a direct effect of the lack of CXCR3 in neurons, our data most likely reflect the function of CXCR3 in microglia.
Although astrocytes are also involved in the removal of debris after entorhinal cortex lesion (Bechmann and Nitsch, 1997), their response was not visibly altered in the CXCR3 knock-out mice. This is in line with experiments in which we found no effects of ligands of the CXCR3 receptor on electrophysiologic characteristics of astrocytes (A. Rappert and H. Kettenmann, unpublished observations). The role of astrocytic recruitment and phagocytosis of dendrites thus cannot be evaluated in CXCR3 knock-out mice but may become clear in future ultrastructural studies.
We have previously demonstrated that microglia express the chemokine receptor CXCR3, which triggers chemotaxis in vitro (Biber et al., 2001, 2002; Rappert et al., 2002). Here we describe an impaired migration of microglia into zones of axonal degeneration after entorhinal lesion in CXCR3 knock-out mice. In lesioned wild-type mice, the number of activated microglia increases in the denervated middle and outer molecular layers, whereas their density decreases in the hilus (Jensen et al., 1994). In contrast, in lesioned CXCR3 knock-out mice, the accumulation, particularly in the outer molecular layer, and concurrent depletion in the inner molecular layer and hilus were not as apparent. Moreover, it was evident that in the knock-out animals, microglial cells do not acquire a highly amoeboid morphology but instead show a more ramified morphology. The basal morphology and distribution of microglia in wild-type and CXCR3 knock-out mice are comparable, suggesting that the difference in the microglia distribution is a consequence of a microglial activation process. Because we observed no difference in proliferative activity between wild-type and knock-out animals using Ki-67 immunohistochemistry, we conclude that the accumulation of microglia in the molecular layer of the dentate gyrus is indeed attributable to migration, and CXCR3 is important in the migration process in vivo.
In vitro studies have shown that stimulation with CXCL10-induced calcium influx and electrophysiological responses is crucial for chemotaxis of cultured microglia, indicating that CXCL10 is an important chemokine controlling microglial properties (Biber et al., 2001, 2002; Rappert et al., 2002). Here we demonstrate an apparently equal induction of CXCL10 mRNA and protein expression in both wild-type and CXCR3 knock-out mice, indicating that the chemokine is produced even when its target is not present. Thus, the difference in microglial activation between wild-type and CXCR3 knock-out animals cannot be attributed solely to divergent expression levels of the CXCR3 ligand CXCL10. It therefore appears that lack of CXCR3 cannot be substituted by alternate receptors, indicating that CXCR3 may be the only receptor for CXCL10-mediated microglial recruitment. Generally, it seems that the CXCL10 level in the brain under pathological conditions correlates with the development of neurological symptoms (Kolb et al., 1999; Fife et al. 2001). Under pathological conditions, CXCL10 is produced by astrocytes, microglia, and neurons (Ransohoff et al., 1993; Vanguri and Farber, 1994; Wang et al., 1998). After entorhinal cortex lesion, we found CXCL10-positive neurons (Fig. 5C) in the hippocampal formation in both in CXCR3 knock-out and wild-type animals but no CXCL10-positive astrocytes or microglia. The expression of other CXCR3 ligands such as CXCL9 and CXCL11 could not be detected in the unlesioned and lesioned mice (data not shown), indicating the specificity of CXCL10 in this model. These findings support our hypothesis that lesioned neurons express CXCL10, which leads to the migration of microglia, a response that is certainly inhibited in CXCR3 knock-out mice.
Taken together, our data show that microglial recruitment to zones of axonal degeneration is hindered in CXCR3 knock-out mice, and this hampered microglial recruitment is accompanied by the maintenance of denervated dendrites. Parvalbumin-positive neurons are preferentially vulnerable in Alzheimer's disease (Inaguma et al., 1992; Brady and Mufson, 1997). Because of the early effect on the entorhinal cortex (Braak and Braak, 1991), transneuronal dendritic alterations similar to what has been observed after entorhinal cortex lesion are prominent features of this disease (Einstein et al., 1994). It is evident that parvalbumin-positive neurons degenerate or lose their dendrites mainly in the areas where entorhinal input is lost. Our data point to the possibility that the initial signal is not a loss of this input itself but rather a result of activated microglial cells recruited to the zones of axonal degeneration. It is, however, unclear whether this preservation of dendrites in the knock-out animals is functionally relevant, e.g., in that the protected distal dendrites are reinnervated by sprouting fibers after entorhinal cortex lesion.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Graduiertenkolleg, Schwerpunktprogramm, and Dutch-German collaboration grant).
Correspondence should be addressed to Helmut Kettenmann, Department of Cellular Neuroscience, Max-Delbrück-Center for Molecular Medicine, Robert-Rössle-Strasse 10, 13092 Berlin, Germany. E-mail: hketten@mdc-berlin.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248500-10$15.00/0
A.R. and I.B. contributed equally to this work.
References
- Babcock AA, Kuziel WA, Rivest S, Owens T (2003) Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 23: 7922-7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becher B, Prat A, Antel JP (2000) Brain-immune connection: immunoregulatory properties of CNS-resident cells. Glia 29: 293-304. [PubMed] [Google Scholar]
- Bechmann I, Nitsch R (1997) Astrocytes and microglial cells incorporate degenerating fibers following entorhinal lesion: a light, confocal, and electron microscopical study using a phagocytosis-dependent labeling technique. Glia 20: 145-154. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Lassau S, Steiner B, Mor G, Gimsa U, Nitsch R (2000) Reactive astrocytes upregulate Fas (CD95) and Fas ligand (CD95L) expression but do not undergo programmed cell death during the course of anterograde degeneration. Glia 32: 25-41. [DOI] [PubMed] [Google Scholar]
- Benveniste EN (1997) Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med 75: 165-173. [DOI] [PubMed] [Google Scholar]
- Biber K, Klotz KN, Berger M, Gebicke-Haerter PJ, van Calker D (1997) Adenosine A1 receptor-mediated activation of phospholipase C in cultured astrocytes depends on the level of receptor expression. J Neurosci 17: 4956-4964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Sauter A, Brouwer N, Copray SC, Boddeke HWGM (2001) Ischemia-induced neuronal expression of the microglia attracting chemokine secondary lymphoid-tissue chemokine (SLC). Glia 34: 121-133. [DOI] [PubMed] [Google Scholar]
- Biber K, Dijkstra I, Trebst C, De Groot CJ, Ransohoff RM, Boddeke HW (2002) Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience 112: 487-497. [DOI] [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G (1968) Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 85: 145-157. [DOI] [PubMed] [Google Scholar]
- Boucsein C, Kettenmann H, Nolte C (2000) Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci 12: 2049-2058. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 82: 239-259. [DOI] [PubMed] [Google Scholar]
- Brady DR, Mufson EJ (1997) Parvalbumin-immunoreactive neurons in the hippocampal formation of Alzheimer's diseased brain. Neuroscience 80: 1113-1125. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Penttonen M, Bragin A, Nadasdy Z, Chrobak JJ (1995) Possible physiological role of the perforant path-CA1 projection. Hippocampus 5: 141-146. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Deller T, Frotscher M, Nitsch R (1995) Morphological evidence for the sprouting of inhibitory commissural fibers in response to the lesion of excitatory entorhinal input to the rat dentate gyrus. J Neurosci 15: 6868-6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein G, Buranosky R, Crain BJ (1994) Dendritic pathology of granule cells in Alzheimer's disease is unrelated to neuritic plaques. J Neurosci 14: 5077-5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyupoglu IY, Bechmann I, Nitsch R (2003) Modification of microglia function protects from lesion-induced neuronal alterations and promotes sprouting in the hippocampus. FASEB J 17: 1110-1111. [DOI] [PubMed] [Google Scholar]
- Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD, Karpus WJ (2001) CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol 166: 7617-7624. [DOI] [PubMed] [Google Scholar]
- Fischer HG, Reichmann G (2001) Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol 166: 2717-2726. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Heimrich B, Deller T (1997) Sprouting in the hippocampus is layer-specific Trends Neurosci 20: 218-223. [DOI] [PubMed] [Google Scholar]
- Gage FH, Olejniczak P, Armstrong DM (1988) Astrocytes are important for sprouting in the septohippocampal circuit. Exp Neurol 102: 2-13. [DOI] [PubMed] [Google Scholar]
- Gall C, Rose G, Lynch G (1979) Proliferative and migratory activity of glial cells in the partially deafferented hippocampus. J Comp Neurol 183: 539-549. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Schoen SW, Kreutzberg GW (1991) Lesion of the rat entorhinal cortex leads to a rapid microglial reaction in the dentate gyrus: a light and electron microscopical study. Acta Neuropathol (Berl) 82: 442-455. [DOI] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Rev 20: 269-287. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H (1984) Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 133: 1710-1715. [PubMed] [Google Scholar]
- Gulyas AI, Miles R, Sik A, Toth K, Tamamaki N, Freund TF (1993) Hippocampal pyramidal cells excite inhibitory neurons through a single release site. Nature 366: 683-687. [DOI] [PubMed] [Google Scholar]
- Hailer NP, Grampp A, Nitsch R (1999) Proliferation of microglia and astrocytes in the dentate gyrus following entorhinal cortex lesion: a quantitative bromodeoxyuridine-labelling study. Eur J Neurosci 11: 3359-3364. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Lu B, Gao W, Csizmadia V, Faia K, King JA, Smiley ST, Ling M, Gerard NP, Gerard C (2000) Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 192: 1515-1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaguma Y, Shinohara H, Inagaki T, Kato K (1992) Immunoreactive parvalbumin concentrations in parahippocampal gyrus decrease in patients with Alzheimer's disease. J Neurol Sci 110: 57-61. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Gonzalez B, Castellano B, Zimmer J (1994) Microglial and astroglial reactions to anterograde axonal degeneration: a histochemical and immunocytochemical study of the adult rat fascia dentata after entorhinal perforant path lesions. Exp Brain Res 98: 245-260. [DOI] [PubMed] [Google Scholar]
- Jensen MB, Hegelund IV, Poulsen FR, Owens T, Zimmer J, Finsen B (1999) Microglial reactivity correlates to the density and the myelination of the anterogradely degenerating axons and terminals following perforant path denervation of the mouse fascia dentata. Neuroscience 93: 507-518. [DOI] [PubMed] [Google Scholar]
- Jucker M, Mondadori C, Mohajeri H, Bartsch U, Schachner M (1995) Transient upregulation of NCAM mRNA in astrocytes in response to entorhinal cortex lesions and ischemia. Brain Res Mol Brain Res 28: 149-156. [DOI] [PubMed] [Google Scholar]
- Kiess J, Buzsaki G, Morrow JS, Glantz SB, Leranth C (1996) Entorhinal cortical innervation of parvalbumin-containing neurons (basket and chandelier cells) in the rat Ammons' horn. Hippocampus 6: 239-246. [DOI] [PubMed] [Google Scholar]
- Kolb SA, Sporer B, Lahrtz F, Koedel U, Pfister HW, Fontana A (1999) Identification of a T cell chemotactic factor in the cerebrospinal fluid of HIV-1-infected individuals as interferon-gamma inducible protein 10. J Neuroimmunol 93: 172-181. [DOI] [PubMed] [Google Scholar]
- Kosaka T, Katsumaru H, Hama K, Wu JY, Heizmann CW (1987) GABAergic neurons containing the Ca2+-binding protein parvalbumin in the rat hippocampus and dentate gyrus. Brain Res 419: 119-130. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19: 312-318. [DOI] [PubMed] [Google Scholar]
- Lynch G, Matthews DA, Mosko S, Parks T, Cotman C (1972) Induced acetylcholinesterase-rich layer in rat dentate gyrus following entorhinal lesions. Brain Res 42: 311-318. [DOI] [PubMed] [Google Scholar]
- Mackay CR (2001) Chemokines: immunology's high impact factors. Nat Immunol 2: 95-101. [DOI] [PubMed] [Google Scholar]
- Nitsch R, Soriano E, Frotscher M (1990) The parvalbumin-containing nonpyramidal neurons in the rat hippocampus. Anat Embryol (Berl) 181: 413-425. [DOI] [PubMed] [Google Scholar]
- Nitsch R, Frotscher M (1991) Maintenance of peripheral dendrites of GABAergic neurons requires specific input. Brain Res 554: 304-307. [DOI] [PubMed] [Google Scholar]
- Nitsch R, Frotscher M (1993) Transneuronal changes in dendrites of GABAergic parvalbumin-containing neurons of the rat fascia dentata following entorhinal lesion. Hippocampus 3: 481-490. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H (2001) GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia 33: 72-86. [PubMed] [Google Scholar]
- Peterson DA, Lucidi-Phillipi CA, Eagle KL, Gage FH (1994) Perforant path damage results in progressive neuronal death and somal atrophy in layer II of entorhinal cortex and functional impairment with increasing post-damage age. J Neurosci 14: 6872-6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier J, Hess M, May PC, Finch CE (1991) Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res 11: 97-106. [DOI] [PubMed] [Google Scholar]
- Raivich G, Moreno-Flores MT, Moller JC, Kreutzberg GW (1994) Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur J Neurosci 6: 1615-1618. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CU, Werner A, Jones LL, Kreutzberg GW (1999) Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Rev 30: 77-105. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, Estes ML, Thomas DM, Tuohy VK (1993) Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J 7: 592-600. [DOI] [PubMed] [Google Scholar]
- Rappert A, Biber K, Nolte C, Lipp M, Schubel A, Lu B, Gerard NP, Gerard C, Boddeke HWGM, Kettenmann H (2002) Secondary lymphoid tissue chemokine (CCL21) activates CXCR3 to trigger a Cl- current and chemotaxis in murine microglia. J Immunol 168: 3221-3226. [DOI] [PubMed] [Google Scholar]
- Riemer C, Queck I, Simon D, Kurth R, Baier M (2000) Identification of up-regulated genes in scrapie-infected brain tissue. J Virol 74: 10245-10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose G, Lynch G, Cotman CW (1976) Hypertrophy and redistribution of astrocytes in the deafferented dentate gyrus. Brain Res Bull 1: 87-92. [DOI] [PubMed] [Google Scholar]
- Ruth RE, Collier TJ, Routtenberg A (1982) Topography between the entorhinal cortex and the dentate septotemporal axis in rats: I. Medial and intermediate entorhinal projecting cells. J Comp Neurol 209: 69-78. [DOI] [PubMed] [Google Scholar]
- Santambrogio L, Belyanskaya SL, Fischer FR, Cipriani B, Brosnan CF, Ricciardi-Castagnoli P, Stern LJ, Strominger JL, Riese R (2001) Developmental plasticity of CNS microglia. Proc Natl Acad Sci USA 98: 6295-6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS (1989) Calcium-binding protein (calbindin-D28k) and parvalbumin immunocytochemistry: localization in the rat hippocampus with specific reference to the selective vulnerability of hippocampal neurons to seizure activity. J Comp Neurol 280: 183-196. [DOI] [PubMed] [Google Scholar]
- Steward O, Kelley MS, Torre ER (1993) The process of reinnervation in the dentate gyrus of adult rats: temporal relationship between changes in the levels of glial fibrillary acidic protein (GFAP) and GFAP mRNA in reactive astrocytes. Exp Neurol 124: 167-183. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA (1999) Reactive microgliosis. Prog Neurobiol 57: 563-581. [DOI] [PubMed] [Google Scholar]
- Vanguri P, Farber JM (1994) IFN and virus-inducible expression of an immediate early gene, crg-2/IP-10, and a delayed gene, I-A alpha in astrocytes and microglia. J Immunol 152: 1411-1418. [PubMed] [Google Scholar]
- Wang X, Ellison JA, Siren AL, Lysko PG, Yue TL, Barone FC, Shatzman A, Feuerstein GZ (1998) Prolonged expression of interferon-inducible protein-10 in ischemic cortex after permanent occlusion of the middle cerebral artery in rat. J Neurochem 71: 1194-1204. [DOI] [PubMed] [Google Scholar]
- Wirenfeld M, Dalmau I, Finsen B (2003) Estimation of absolute microglial cell numbers in mouse fascia dentata using unbiased and efficient stereological cell counting principles. Glia 44: 129-139. [DOI] [PubMed] [Google Scholar]
- Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT (2000) Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer's disease. J Neuroimmunol 108: 227-235. [DOI] [PubMed] [Google Scholar]
- Zipp F, Nitsch R, Soriano E, Frotscher M (1989) Entorhinal fibers form synaptic contacts on parvalbumin-immunoreactive neurons in the rat fascia dentata. Brain Res 495: 161-166. [DOI] [PubMed] [Google Scholar]