

Abstract
We studied the contribution of L-type Ca2+ channels to action potential-evoked Ca2+ influx in dendritic spines of CA1 pyramidal neurons and the modulation of these channels by the β2 adrenergic receptor. Backpropagating action potentials (bAPs) (three at 50 Hz) were evoked by brief somatic current injections, and Ca2+ transients were recorded in proximal basal dendrites and associated spines. The R- and T-type Ca2+ channel blocker NiCl2 (100 μm) significantly reduced Ca2+ transients in both spines and their parent dendrites (∼50%), suggesting that these channels are the major source of bAP-evoked Ca2+ influx in these structures. The L-type Ca2+ channel blockers nimodipine and nifedipine (both 10 μm) reduced spine Ca2+ transients by ∼10%, whereas the L-type Ca2+ channel activators FPL 64176 (2,5-dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylic acid methylester) and Bay K 8644 ((±)-1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)-phenyl]-3-pyridine carboxylic acid methyl ester) (both 10 μm) significantly enhanced the spine Ca2+ transients by 40-50%. Activation of β2 adrenergic receptors with salbutamol (40 μm) or formoterol (5 μm) resulted in significant enhancements of the spine (40-50%) but not dendritic Ca2+ transients. This increase was prevented when L-type Ca2+ channels were blocked with nimodipine (10 μm) or when cAMP-dependent protein kinase A (PKA) was inhibited with KT5720 (3 μm), Rp-cAMPS (Rp-adenosine cyclic 3′,5′-phosphorothioate) (100 μm), or PKI (100 μm). The above data suggest that L-type Ca2+ channels are functionally present in dendritic spines of CA1 pyramidal neurons, contribute to spine Ca2+ influx, and can be modulated by the β2 adrenergic receptor through PKA in a highly compartmentalized manner.
Keywords: dendritic spine, CA1, calcium, L-type, adrenergic, hippocampus, imaging
Introduction
Spines are protrusions that emanate from the dendrites of the majority of excitatory neurons and receive most of the synapses in the CNS. Since Ramon y Cajal (1891, 1894) first described dendritic spines, electron microscopy (Harris and Stevens, 1989; Harris et al., 1992; Sorra and Harris, 2000) as well as confocal and two-photon microscopy (Yuste and Denk, 1995; Denk et al., 1996; Kovalchuk et al., 2000; Sabatini and Svoboda, 2000; Sabatini et al., 2002) have contributed greatly to our understanding of these neuronal structures. The source and kinetics of spine Ca2+ signals have important consequences for the exact nature of synaptic plasticity (Cormier et al., 2001; Wang et al., 2000; Holthoff et al., 2002; Nishiyama et al., 2000). It is crucial, therefore, to map the distribution of the various Ca2+ sources in neurons, including the localization and functional presence of voltage-dependent Ca2+ channels (VDCCs) in spines and their parent dendrites. The functional presence of several types of VDCCs in spines has been mapped (Yuste et al., 1999; Kovalchuk et al., 2000; Sabatini and Svoboda, 2000). However, less is known about the functional presence of L-type VDCCs. Although L-type VDCCs localize to both dendrites and spines (Hell et al., 1993; Davare et al., 2001; Obermair et al., 2004), there is no evidence for a direct contribution of these channels to spine Ca2+ influx (Yasuda et al., 2003). The importance of L-type Ca2+ channels is underscored by the fact that they contribute to resting Ca2+ levels in dendrites (Magee et al., 1996), are involved in excitability (Moyer et al., 1992) and synaptic plasticity (Christie et al., 1997; Cavus and Teyler, 1998; Evers et al., 2002), and also couple postsynaptic Ca2+ influx to gene transcription (Murphy et al., 1991; Graef et al., 1999).
We used confocal imaging to examine the contribution of L-type Ca2+ VDCCs to spine Ca2+ transients evoked by brief bursts of backpropagating action potentials (bAPs). We show that the L-type Ca2+ channel blockers nimodipine and nifedipine reduce, and accordingly, the L-type Ca2+ channel activators Bay K 8644 ((±)-1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)-phenyl]-3-pyridine carboxylic acid methyl ester) and FPL 64176 (2,5-dimethyl-4-[2-(phenylmethyl)benzoyl]-1H-pyrrole-3-carboxylic acid methylester) enhance bAP-evoked spine Ca2+ transients. To further validate the presence of L-type VDCCs in spines, we used the coupling of β2 adrenergic receptors (β2-ARs) to a specific isoform of L-type Ca2+ channel, CaV1.2 or class C, which localizes to dendritic spines (Hell et al., 1993; Davare et al., 2001). We demonstrate that activation of β2 adrenergic receptors leads to a preferential enhancement of bAP-evoked Ca2+ transients in spines, but not their parent dendrites, and that this enhancement is prevented when L-type Ca2+ channels are blocked. We also show that blocking cAMP-dependent protein (PKA) occludes the enhancement of bAP-evoked spine Ca2+ transients during β2-AR activation. Our data thus suggest that L-type VDCCs are functionally present in dendritic spines and can be modulated in a highly compartmentalized manner, adding to a growing body of evidence demonstrating spine-restricted neuromodulation of VDCCs.
Materials and Methods
Slice preparation. Rat hippocampal slices were obtained from the brains of 4- to 6-week-old Sprague Dawley rats and prepared in accordance with the guidelines of the National Institutes of Health, as approved by the animal care and use committee of Baylor College of Medicine. Animals were anesthetized with a combination anesthetic containing ketamine, xylazine, and acepromazine (100, 20, and 10 mg/ml, respectively). Before removing the brain, an ice-cold solution (2-4°C; containing in mm: 2.5 KCl, 1.25 Na2H2PO4, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 110 choline chloride, 1.3 ascorbate, and 3 pyruvate) was perfused transcardially (through the left ventricle) to clear out blood cells and quickly cool down the brain. Hippocampal slices (350 μm) were cut with a Vibratome 1000+ (Ted Pella, St. Louis, MO) in the same perfusion solution. Slices were kept at 35°C for 0.5 hr in artificial CSF (ACSF) (containing in mm: 125 NaCl, 2.5 KCl, 1.25 Na2H2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, and 10 dextrose) and then allowed to adjust to room temperature before use. All solutions were continuously bubbled with 95% O2-5% CO2, and slices were superfused with ACSF during all experiments. Recordings were performed at 33-35°C.
Electrophysiology and optical recording. Whole-cell patch-clamp recordings were made from CA1 pyramidal neurons 30-40 μm below the surface of the brain slice, using an Axopatch 200A patch-clamp amplifier (Axon Instruments, Union City, CA). Pipette resistance was 2-6 MΩ, and series resistance was 10-20 MΩ. Series resistance was not compensated. The average resting potential of the neurons was -61.2 ± 0.2 mV. Direct current was injected into the cells to hold the membrane potential at -65 mV. Patch pipettes were filled with an internal solution (in mm: 135 KMeSO4, 10 HEPES, 8 NaCl, 2 Mg2GTP, and 0.3 NaGTP) that included 75 μm of the medium-affinity Ca2+ indicator Fluo-5F (Kd of 2.3 μm; Molecular Probes, Eugene, OR) to record Ca2+ transients and 75 μm of the fluorescent label Alexa Fluor 555 (Molecular Probes) to image fine neuronal structures (Fig. 1 A). The use of Alexa Fluor 555 to visualize dendritic spines was necessary because of the low fluorescence intensity of Fluo-5F at resting Ca2+ levels. After establishing whole-cell mode, both dyes were allowed to diffuse into the dendrites for ∼20 min before recordings were started. Neurons were visualized with an E600FN upright microscope (Nikon, Melville, NY) equipped with a 60×, 1.0 numerical aperture water immersion objective (Nikon), differential interference contrast infrared optics (Nikon), and an infrared-sensitive camera (IR-1000; Dage-MTI, Michigan City, IN). Readily identifiable spines on proximal basal dendrites were imaged (Fig. 1 B) using a PCM2000 confocal scan head (Nikon). Functional and structural imaging was performed using single-photon excitation using both an argon (laser line, 488 nm) and a helium-neon laser (laser line, 543 nm) to excite Fluo-5F and Alexa Fluor 555, respectively. Collected epifluorescence was separated using a polychroic mirror (488/543/633; Chroma, Rockingham, VT). A 515/30 bandpass filter was used for green emission, and a 565 long-pass filter was used for red emission (Chroma), which was detected by two fiber-coupled photomultiplier tubes (R928; Hamamatsu, Hamamatsu City, Japan). Ca2+ transients were acquired in line-scan mode using a large pinhole (50 μm) with a temporal resolution of 2.5 msec per line (Fig. 1C). Structural data were acquired with a small pinhole (20 μm), and image stacks were deconvolved using Huygens Essential (SVI, Hilversum, The Netherlands) using an iterative maximum likelihood estimation blind deconvolution algorithm. Image data that was reconstructed three-dimensionally using Amira (TGS, San Diego, CA) revealed that the spines that we recorded from lay in a plane approximately parallel to that of the dendrite and that no spines were protruding perpendicular to this plane where the line scan intersected the parent dendrite.
Figure 1.

Imaging of Ca2+ transients in individual dendritic spines. A, Maximum image projection of a CA1 pyramidal neuron. Cells were loaded with both Alexa Fluor 555 and Fluo 5F (both 75 μm) through the patch pipette and imaged with a confocal microscope. B, Close-up of the boxed region in A. Spines from proximal dendrites (visualized with Alexa-Fluor 555, ∼50 μm from soma) were selected for all measurements. The dashed scan line indicates the points from which fluorescence was sampled over time. C, Fluorescence changes (measured with Fluo 5F) from structures intersected by the scan line in response to three 50 Hz bAPs initiated at the soma. D, Ca2+ transients of spine (top) and parent dendrite (bottom), calculated from the boxed regions in C and expressed as ΔF/F × 100%. The shaded areas under the transients depict the Ca2+transient integrals. These were calculated from the start of the first bAP until 75 msec after the end of the last bAP and used to compare control and drug conditions. E, Action potentials evoked by brief current injections (3 msec, ∼1 nA) and measured at the soma.
In some experiments, the following drugs were added to the ACSF (in μm): 10 nimodipine (Bayer, Wuppertal, Germany), 10 nifedipine (Tocris Cookson, Ballwin, MO), 10 Bay K 8644 (Alamone Labs, Jerusalem, Israel), 10 FPL 641776 (Sigma, St. Louis, MO), 100 NiCl2 (Sigma), 40 salbutamol (Sigma), 5 formoterol (Tocris Cookson), 3 KT5720 (Calbiochem, La Jolla, CA), 100 Rp-cAMPS (Rp-adenosine cyclic 3′,5′-phosphorothioate) (Biomol, Plymouth Meeting, PA), and 100 PKI (Biomol). Most drugs were bath perfused for ∼6 min. PKI and Rp-cAMPS were included in the patch pipette. Care was taken to keep light-sensitive drugs in the dark. Ca2+ transients are shown as fractional changes of fluorescence, ΔF/F, calculated as ((F - Fprestimulus)/Fprestimulus) × 100%. Three to five trials were averaged, and a running average of 2 points was applied to the data to obtain the traces shown (Fig. 1 D). The maximal Ca2+ transient amplitude was defined as the peak amplitude of the Ca2+ transient during the last bAP in the burst stimulus. The integrals of the ΔF/F transients from the start of the first stimulus until 75 msec after the end of the last (third) stimulus were calculated to compare control and drug conditions (Fig. 1 D, hatched lines). Ca2+ transient integrals enabled us to measure small reductions or enhancements of the Ca2+ signals in spines and their parent dendrites and proved a method less sensitive to the noise inherent in measuring from very small neuronal structures. When applying multiple drugs, we calculated the percentage change of the Ca2+ transient integral by dividing the integral 6 min after start of wash in of the second drug to 6 min after wash in of the first drug. bAPs (three at 50 Hz) were elicited using brief current injections (∼1 nA, 3 msec) at the soma (Fig. 1 E). Ca2+ transients measured at the soma did not show any saturation of the indicator because the fractional change in fluorescence was unchanged with each action potential. Laser power was adjusted to give minimal photo-damage for the duration of the measurements. Estimated power delivered to the preparation was ∼ 500 μW.
Statistics. Paired t tests for comparison of two means (α = 0.05) were used to test for differences between control and drug conditions and to compare spines and their parent dendrites. For comparison between groups with different sample sizes, we used a two-sample t test assuming unequal variances. Notation of p values is as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Differences were considered significant for p < 0.05. Data are displayed as mean ± SEM.
Results
Amplitudes and kinetics of bAP-evoked Ca2+ transients in spines and dendrites of CA1 pyramidal neurons
Recordings were made from large-diameter spines (∼0.5 μm) of proximal basal dendrites (46 ± 3 μm from soma). Larger-diameter spines are associated with developed postsynaptic densities (Harris et al., 1992; Spacek and Harris, 1997) and are also believed to be the most stable and mature dendritic spines in vivo (Trachtenberg et al., 2002). For the bAP-evoked Ca2+ signals recorded from these proximal spines (Fig. 2A), no correlation was found between amplitude and distance from the soma (Fig. 2B). A linear fit through the data points gave R2 values for basal spines and dendrites of 0.004 and 0.001, an indirect assessment that no measurable decrement of the action potentials occurred over the distances investigated. We hypothesized that successful propagation of the bAPs should maximize the chances of opening all present VDCCs.
Figure 2.

Amplitudes and kinetics of bAP-evoked spine Ca2+ transients. A, Left, Maximum image projection of CA1 pyramidal neuron basal dendrites and close-up of dendritic spines. Right, Ca2+ transients displayed as ΔF/F × 100%, recorded from a single spine (gray trace) and its parent dendrite (black trace) shown on the left. B, Normalized maximal Ca2+ transient amplitudes of spines and parent dendrites (Dend.) did not change with distance from the soma (spines, R2 = 0.004; dendrites, R2 = 0.001). C, Ca2+ transient amplitudes of basal spines were significantly different from parent dendrites during the first, but not the last, bAP of a burst stimulus (first bAP, n = 61 pairs, *p < 0.05; last bAP, NS, p = 0.16). D, Ca2+ transients evoked by either one or three bAPs (overlaid) with the corresponding somatic action potentials. E, Ca2+ transient decay times of bAP-evoked Ca2+ transients in spines and their parent dendrites. Spines had significantly faster decay times than parent dendrites with either a single bAP or a burst of three bAPs (single bAP, n = 41 pairs, *p < 0.05; three bAPs, n = 61 pairs, ***p < 0.001).
With the Ca2+ indicator used, we observed large-amplitude changes of fluorescence (Fig. 2C). Spines and their parent dendrites had similar Ca2+ transient amplitudes during the first bAP (spines, 436 ± 24% ΔF/F, n = 61; dendrites, 403 ± 20% ΔF/F, n = 57) and the last bAP (spines, 688 ± 40% ΔF/F, n = 61; dendrites, 655 ± 35% ΔF/F, n = 57). Ca2+ transient amplitudes were significantly different during the first bAP (*p < 0.05) but not the last bAP (NS, p = 0.16) when amplitudes of spines were compared with those of parent dendrites. We also observed fast decay kinetics of the bAP-evoked Ca2+ transients (Fig. 2D). Two different types of decay kinetics have been described previously associated with bAP-evoked spine Ca2+ transients, depending on the diameter of the parent dendrite (Holthoff et al., 2002). Thin dendrites displayed single-exponential decays, whereas thick dendrites displayed double-exponential decays. All Ca2+ transients were recorded from spines of small-diameter dendrites (basal, < 1 μm) and could be well fit with single-exponential decays, in agreement with previous findings from spines of similarly thin dendrites (Sabatini et al., 2002). Ca2+ transient decay times were significantly faster in spines than their parent dendrites (Fig. 2E), using both a single bAP (spines, 33 ± 3 msec, n = 41; dendrites, 40 ± 6 msec, n = 38; *p < 0.05) or a brief burst of bAPs (spines, 39 ± 2 msec, n = 61; dendrites, 44 ± 2 msec, n = 57; ***p < 0.001). Although Ca2+ transient decay times in spines remained significantly faster than dendrites during a burst, the spine decay times were increased with respect to those during a single bAP (*p < 0.05).
In summary, we found basal spines to have bAP-evoked Ca2+ transients with similar amplitudes and significantly faster decay kinetics compared with their parent dendrites during both a single bAP and a brief burst of bAPs.
Contribution of R- and T-type VDCCs to bAP-evoked Ca2+ transients in spines and parent dendrites
Spine and dendritic Ca2+ transients evoked by brief bursts of bAPs were studied in the presence of NiCl2 (100 μm), which selectively blocks both R- and T-type VDCCs at low concentrations (Christie et al., 1995, 1997). Representative traces under control conditions and during bath application of NiCl2 are shown in Figure 3A. Ca2+ transient amplitudes (Fig. 3B) were significantly reduced in both spines and their parent dendrites during the first bAP (spines, 64 ± 5%, n = 6, ***p < 0.001; dendrites, 65 ± 9%, n = 6, ***p < 0.001) and last (third) bAP (spines, 60 ± 8%, n = 6, ***p < 0.001; dendrites, 59 ± 8%, n = 6, ***p < 0.001). Ca2+ transient integrals, calculated as described in Materials and Methods, before and during application of 100 μm NiCl2 were also compared (Fig. 3C). Ca2+ transient integrals were significantly reduced in spines and their parent dendrites (spines, 49 ± 5%, n = 6, ***p < 0.001; dendrites, 51 ± 7%, n = 6, ***p < 0.001). Ca2+ transient decay times were increased after the block with NiCl2 (Fig. 3D). Decay times increased nonsignificantly in spines and significantly in their parent dendrites (spines, 122 ± 16%, n = 6, NS, p = 0.2; dendrites, 122 ± 8%, n = 6, *p < 0.05).
Figure 3.

Blockade of R- and T-type Ca2+ channels with NiCl2 significantly reduces bAP-evoked spine Ca2+ transients. A, Representative Ca2+ transients of a basal spine and its parent dendrite under control conditions and during bath application of the R- and T-type Ca2+ channel blocker NiCl2 (100 μm). B, Ca2+ transient amplitudes were significantly reduced in spines and their parent dendrites during the first bAP (spines, n = 6, ***p < 0.001; dendrites, n = 6, ***p < 0.001) and last bAP of the train stimulus (spines, ***p < 0.001; dendrites, ***p < 0.001). C, Ca2+ transient integrals (see Materials and Methods) were significantly reduced in spines and their parent dendrites in the presence of NiCl2 (spines, n = 6, ***p < 0.001; dendrites, n = 6, ***p < 0.001). D, Ca2+ transient decay times shown as percentage of control. In the presence of 100 μm NiCl2, decay times of spines increased nonsignificantly, whereas those of dendrites increased significantly (spines, n = 6, NS, p = 0.2; dendrites, n = 6, *p < 0.05).
Together, these data suggest that R- and possibly T-type VDCCs contribute a major fraction to bAP-evoked Ca2+ transients but are not necessarily the sole source of bAP-evoked Ca2+ influx in spines and dendrites of CA1 pyramidal neurons.
Contribution of L-type VDCCs to bAP-evoked Ca2+ transients in spines and parent dendrites
We were particularly interested in measuring the contribution of L-type VDCCs to bAP-evoked spine Ca2+ transients. Although their anatomical presence in spines has been confirmed (Davare et al., 2001, Obermair et al., 2004), so far no data support a direct contribution of L-type VDCCs to spine Ca2+ influx (Yasuda et al., 2003). We studied burst-evoked Ca2+ transients in more mature rats (4-6 weeks) than those of similar studies (1-2 weeks), in view of reported changes in the number and distribution of VDCCs during early development (Tanaka et al., 1995; Shankar et al., 1998; Isomura and Kato, 1999). L-type Ca2+ channel blockers and activators were used to probe for the presence of L-type VDCCs in spines and their parent dendrites.
Two dihydropyridine (DHP) L-type VDCC blockers, nimodipine and nifedipine (both 10 μm), were used to selectively block L-type Ca2+ channels. The effect of these drugs on spine and dendritic Ca2+ transients was examined (Fig. 4A). In the presence of nifedipine, a significant reduction was observed of the spine and dendritic Ca2+ transient amplitudes (Fig. 4B) during the first bAP (spines, 87 ± 5%, n = 5, *p < 0.05; parent dendrites, 80 ± 6%, n = 5, *p < 0.05) but not the last bAP (spines, 89 ± 6%, n = 5, NS, p = 0.10; dendrites, 91 ± 6%, n = 5, NS, p = 0.15). With nimodipine, Ca2+ transient amplitudes were reduced significantly in spines and parent dendrites during the first bAP (spines, 93 ± 4%, n = 10, *p < 0.05; dendrites, 84 ± 7%, n = 10, *p < 0.05) and the last bAP of a burst (spines, 85 ± 3%, n = 10, **p < 0.01; dendrites, 91 ± 6%, n = 10, *p < 0.05). For quantitative comparisons, we referred to the less noise-sensitive Ca2+ transient integral (Fig. 4C). In the presence of nifedipine, Ca2+ transient integrals were significantly reduced in both spines and their parent dendrites (spines, 87 ± 5%, n = 5, *p < 0.05; dendrites, 87 ± 6%, n = 5, *p < 0.05). With nimodipine, Ca2+ transient integrals were significantly reduced in spines and their parent dendrites (spines, 87 ± 3%, n = 10, *p < 0.05; dendrites, 83 ± 6%, n = 10, *p < 0.05). Given that R- and L-type VDCCs could be blocked independently, we tested the effect of blocking these channels simultaneously. Coapplication of NiCl2 (100 μm) and nimodipine (10 μm) resulted in a somewhat stronger reduction of the Ca2+ transient integrals in both spines and their parent dendrites (spines, 33 ± 12%, n = 4, **p < 0.01; dendrites, 37 ± 13%, n = 4, **p < 0.01). However, spine and dendritic Ca2+ transient integrals did not differ significantly from those in the presence of NiCl2 alone (spines, NS, p = 0.15; dendrites, NS, p = 0.20). Ca2+ transient decay times were not significantly affected when L-type VDCCs were blocked (Fig. 4D). In the presence of nifedipine, Ca2+ transient decay times nonsignificantly increased in spines and their parent dendrites (spines, 109 ± 7%, n = 5, NS, p = 0.15; dendrites, 110 ± 15%, n = 5, NS, p = 0.27). Likewise, with nimodipine, spine decay times were nonsignificantly increased (spines, 109 ± 6%, NS, p = 0.09; dendrites, 102 ± 6%, NS, p = 0.36).
Figure 4.

Blockade of L-type Ca2+ channels with nimodipine or nifedipine significantly reduces bAP-evoked spine Ca2+ transients. A, Typical Ca2+ transients with and without nimodipine (Nim.; 10 μm) or nifedipine (Nif.; 10 μm). B, Ca2+ transient peak amplitudes were significantly reduced with nifedipine and nimodipine during the first bAP (nifedipine spines, n = 5, *p < 0.05; nifedipine dendrites, n = 5, *p < 0.05; nimodipine spines, n = 10, *p < 0.05; nimodipine dendrites, n = 10, *p < 0.05) and with nimodipine, but not nifedipine, during the last bAP (nifedipine spines, NS, p = 0.10; nifedipine dendrites, NS, p = 0.15; nimodipine spines, **p < 0.01; nimodipine dendrites, *p < 0.05). C,Ca2+ transient integrals shown as percentage of control in the presence of nimodipine, nifedipine, and both nimodipine and NiCl2 (100 μm). With nifedipine or nimodipine, the Ca2+ transient integrals were reduced significantly in both spines (nifedipine, n = 5, *p < 0.05; nimodipine, n = 10, *p < 0.05) and their parent dendrites (nifedipine, n = 5, *p < 0.05; nimodipine, n = 10, *p < 0.05). Coapplication of NiCl2 with nimodipine resulted in a significant reduction of the spine and dendritic integrals (spines, n = 4, **p < 0.01; dendrites, n = 4, **p < 0.01), which was nonsignificantly different from bath application of NiCl2 by itself. C,Ca2+ transient decay times, shown as percentage of control. With either nimodipine or nifedipine, Ca2+ transient decay times were nonsignificantly changed in spines (nimodipine, n = 10, NS, p = 0.09, nifedipine, n = 5, NS, p = 0.15) or their parent dendrites (nimodipine, n = 10, NS, p = 0.36; nifedipine, n = 5, NS, p = 0.27).
To further validate the contribution of L-type Ca2+ VDCCs to the bAP-evoked Ca2+ transients in spines, we tested whether spine Ca2+ transients could be enhanced by L-type Ca2+ channel activators. Figure 5A shows the spine and dendritic Ca2+ transients before and during application of the DHP L-type VDCC activator Bay K 8644 and the benzoylpyrrole FPL 64176. Ca2+ transient amplitudes (Fig. 5B) of spines increased significantly from first to last bAP of the burst stimulus with both Bay K 8644 (*p < 0.05) and FPL 64176 (**p < 0.01). Seven of eight spine-dendrite pairs showed an increase with Bay K 8644. Of these seven pairs, peak amplitudes of bAP-evoked Ca2+ transients in spines and their parent dendrites were nonsignificantly increased with Bay K 8644 during the first bAP (spines, 102 ± 10%, n = 7, NS, p = 0.41; dendrites, 117 ± 11%, n = 7, NS, p = 0.19) and significantly increased in spines but not parent dendrites during the last bAP (spines, 127 ± 9%, n = 7, *p < 0.05; dendrites, 104 ± 4%, n = 7, NS, p = 0.06). In the presence of FPL 64176, five of six spines showed an increase of the spine Ca2+ transients. Of these five pairs, Ca2+ amplitudes were nonsignificantly increased in spines and their parent dendrites during the first bAP (spines, 123 ± 14%, n = 5, NS, p = 0.09; dendrites, 100 ± 8%, n = 5, NS, p = 0.35) and significantly increased in spines and parent dendrites during the last bAP (spines, 145 ± 9%, n = 5, *p < 0.05; dendrites, 115 ± 4%, n = 5, *p < 0.05). With both FPL 64176 and Bay K 8644 (both 10 μm), we observed a significant increase of the spine Ca2+ transient integrals (Fig. 5C). Spine Ca2+ transient integrals were significantly different from parent dendrites with Bay K 8644 (**p < 0.01). Furthermore, with Bay K 8644, Ca2+ transient integrals increased significantly in both spines (146 ± 12%, n = 7, **p < 0.01) and their parent dendrites (124 ± 9%, n = 7, *p < 0.05). Bath application of FPL 64176 resulted in Ca2+ transient integrals of spines that were also significantly increased from those of their parent dendrites (**p < 0.01). In the presence of FPL 64176, Ca2+ transient integrals were increased significantly in spines but not their parent dendrites (spines, 146 ± 11%, n = 5, **p < 0.01; dendrites, 114 ± 6%, n = 5, NS, p = 0.06). Comparison of dendritic Ca2+ transient integrals in the presence of either Bay K or FPL 64176, on the other hand, showed no significant differences (NS, p = 0.18). The discrepancy between spines and parent dendrites suggests that there was little difference in the propagation of the APs in the basal dendrites in the presence of these drugs. Ca2+ transient decay times were increased with both L-type Ca2+ channel activators (Fig. 5D). With Bay K 8644, spine decay times increased nonsignificantly in spines and significantly in their parent dendrites (spines, 145 ± 25%, n = 7, NS, p = 0.06; dendrites, 129 ± 13%, n = 7; *p < 0.05). Using FPL 64176 to activate L-type VDCCs, both spine and dendritic decay times increased significantly with respect to control (spines, 133 ± 10%, n = 5, *p < 0.05; dendrites, 131 ± 12%, n = 5, *p < 0.05).
Figure 5.

Activation of L-type Ca2+ channels with Bay K 8644 or FPL 64176 significantly enhances bAP-evoked Ca2+ transients in spines. A, Ca2+ transients under control conditions and during bath application of the L-type channel activators Bay K 8644 (Bay K; 10 μm) or FPL 64176 (FPL; 10 μm). B, Ca2+ transient peak amplitudes showed a significant increase in spines relative to their parent dendrites during the last bAP of a burst with both Bay K 8644 (n = 7 pairs, *p < 0.05) and FPL 64176 (n = 5 pairs, *p < 0.05). Peak amplitudes were increased nonsignificantly with Bay K 8644 and FPL 64176 during the first bAP (Bay K 8644 spines, n = 7, NS, p = 0.41; Bay K 8644 dendrites, n = 7, NS, p = 0.19; FPL 64176 spines, n = 5, NS, p = 0.09; FPL 64176 dendrites, n = 5, NS, p = 0.35) and significantly in spines with Bay K 8644 and FPL 64176 during the last bAP of a burst (Bay K 8644 spines, *p < 0.05; Bay K 8644 dendrites, NS, p = 0.06; FPL 64176 spines, *p < 0.05; FPL 64176 dendrites, *p < 0.05). C, Ca2+ transient integrals shown as percentage of control in the presence of Bay K 8644 and FPL 64176. Ca2+ transient integrals increased significantly with Bay K 8644 in spines (n = 7, **p < 0.01) and parent dendrites (n = 7, *p < 0.05), whereas it increased only in spines (n = 5, **p < 0.01) but not parent dendrites (n = 5, NS, p = 0.06) with FPL 64176. With both activators, Ca2+ transient integrals in spines were significantly more enhanced than in their parent dendrites (both **p < 0.01). D, Ca2+ transient decay times as percentage of control for Bay K 8644 and FPL 64176. Ca2+ transients decay times were enhanced in spines (Bay K 8644, NS, p = 0.06; FPL 64176, *p < 0.05) and their parent dendrites (Bay K 8644, *p < 0.05; FPL 64176, *p < 0.05).
Together, these data suggest that L-type Ca2+ channels are functionally present in basal spines and could contribute to Ca2+ increases in these structures during brief bursts of bAPs.
Facilitation of L-type VDCCs in spines by activation of the β2 adrenergic receptor
A macro-molecular complex localized to dendritic spines of CA1 pyramidal neurons was described recently, which provides a direct functional coupling between the β2-ARs and class C (CaV1.2) L-type Ca2+ channels (Davare et al., 2001; Laporte et al., 2001). This suggests that noradrenergic afferents could selectively modulate VDCC-dependent Ca2+ increases in spines. We hypothesized, therefore, that stimulation of β2-ARs should lead to Ca2+ increases restricted to dendritic spines and that such increases would most likely be mediated by L-type Ca2+ channels. To test this hypothesis, and to further validate the functional presence of L-type VDCCs in dendritic spines, we activated β2-ARs with the specific agonists salbutamol and formoterol and compared bAP-evoked Ca2+ transients in spines and their parent dendrites. Spine and dendritic Ca2+ transients under control conditions and with either salbutamol (40 μm) or formoterol (5 μm) are shown in Figure 6A. In the presence of either salbutamol or formoterol, peak amplitudes of spine Ca2+ transients were significantly different from dendritic peak amplitudes (Fig. 6B) during the last peak of the burst-evoked transients (salbutamol, *p < 0.05; formoterol, *p < 0.05). Activation of β2-ARs with salbutamol resulted in an increase of the spine Ca2+ transients in 11 of 12 spine-dendrite pairs. Data are presented for these 11 pairs below. With salbutamol, Ca2+ transient amplitudes were nonsignificantly changed in spines and their parent dendrites during the first bAP (spines, 116 ± 12%, n = 11, NS, p = 0.06; dendrites, 89 ± 6%, n = 11, NS, p = 0.09). During the last bAP, Ca2+ transient amplitudes increased significantly in spines but not their parent dendrites (spines, 134 ± 7%, n = 11, *p < 0.05; dendrites, 112 ± 10%, n = 11, NS, p = 0.33). With formoterol, Ca2+ transient amplitudes increased nonsignificantly during the first bAP in both spines and their parent dendrites (spines, 123.8 ± 11%, n = 4, NS, p = 0.054; dendrites, 102 ± 14%, n = 4, NS, p = 0.35). During the last bAP, peak amplitudes were non-significantly increased in both spines and parent dendrites (spines, 131 ± 16%, n = 4, NS, p = 0.07; dendrites, 92 ± 6%, n = 4, NS, p = 0.13). However, in the presence of formoterol, Ca2+ transient amplitudes during the last bAP were significantly different in spines when compared with their parent dendrites (n = 4, *p < 0.05). We observed an increasingly pronounced enhancement of the spine but not dendritic Ca2+ transient integrals during sequential wash in of salbutamol (Fig. 6C). Spine Ca2+ transient integrals were significantly enhanced after wash in of either salbutamol or formoterol (Fig. 6D). When salbutamol was bath applied, spine, but not dendritic, Ca2+ transient integrals were significantly increased relative to control (spines, 147 ± 8%, n = 11, **p < 0.01; dendrites, 112 ± 6%, n = 11, NS, p = 0.20). When β2-ARs were activated with formoterol (5 μm), all spines showed an increase of the Ca2+ transient integral, whereas the dendritic Ca2+ transient integral increased nonsignificantly (spines, 147 ± 12%, n = 4, *p < 0.05; dendrites, 104 ± 12%, n = 4, NS, p = 0.38). Spine and dendritic Ca2+ transient integrals were significantly different from each other after activation of β2-ARs with both drugs (salbutamol, **p < 0.01; formoterol, *p < 0.05). Furthermore, with either salbutamol or formoterol, spine Ca2+ transient decay times were significantly enhanced relative to Ca2+ transient decay times in parent dendrites (Fig. 6E) (salbutmol, *p < 0.05; formeterol, *p < 0.05). With salbutamol, spine decay times were increased significantly, whereas dendritic decay times were increased nonsignificantly (spines, 144 ± 13%, n = 11, *p < 0.05; dendrites, 109 ± 10%, n = 11, NS, p = 0.22). Similarly, with formoterol, spine decay times were increased significantly, whereas dendritic decay times were nonsignificantly increased (spines, 139 ± 13%, n = 4, *p < 0.05; dendrites, 109 ± 6%, n = 4, NS, p = 0.11).
Figure 6.
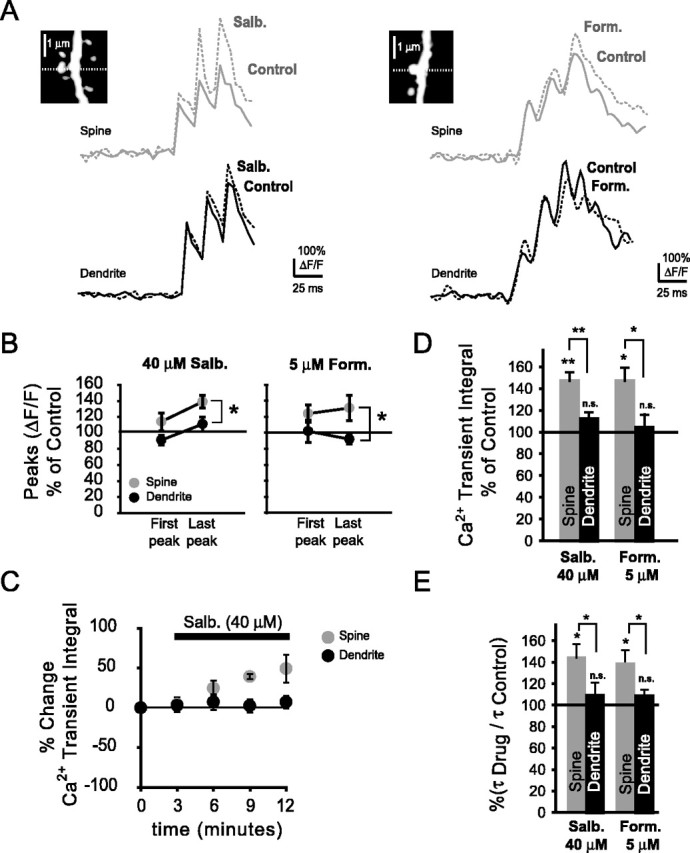
Activation of β2-ARs results in a preferential enhancement of spine Ca2+ transients. A, Ca2+ transients under control conditions and during wash in of the β2-AR activators salbutamol (salbutamol; 40 μm) or formoterol (Form.; 5 μm). Insets, Spine and parent dendrites from which Ca2+ transients were obtained (dashed line indicates position of the line scan). B, Ca2+ transient amplitudes in spines and their parent dendrites during the first and last bAP of the burst-evoked transients. During the last bAP, Ca2+ transients in spines were significantly different from those in their parent dendrites in the presence of either salbutamol or formoterol (salbutamol, n = 11 pairs, *p < 0.05; formoterol, n = 4 pairs, *p < 0.05). With salbutamol, peak amplitudes changed nonsignificantly during the first bAP (spines, n = 11, NS, p = 0.06; dendrites, n = 11, NS, p = 0.09) and significantly in spines, but not parent dendrites, during the last bAP (spines, n = 11, NS, p < 0.05; dendrites, n = 11, NS, p = 0.33). With formoterol, peak amplitudes changed nonsignificantly during the first bAP (spines, n = 4, NS, p = 0.05; dendrites, n = 4, NS, p = 0.35) and the last bAP (spines, n = 4, NS, p = 0.07; dendrites, n = 4, NS, p = 0.13). C, Time course of the Ca2+ transient integrals in spines and parent dendrites during bath application of salbutamol. D, Percentage change of the Ca2+ transient integrals in spines and their parent dendrites during β2-AR activation with salbutamol and formoterol. With salbutamol or formoterol, spine Ca2+ transient integrals were significantly enhanced in spines (salbutamol, n = 11, **p < 0.01; formoterol, n = 4, *p < 0.05) but not parent dendrites (salbutamol, n = 11, NS, p = 0.2; formoterol, n = 4, NS, p = 0.38). Ca2+ transient integrals in spines were significantly different from their parent dendrites with either salbutamol or formoterol (salbutamol, n = 11 pairs, **p < 0.01; formoterol, n = 4 pairs, *p < 0.05). E, Ratio of spine and dendritic Ca2+ transient decay times before and after drug application. Decay times were significantly enhanced in spines (salbutamol, n = 11, *p < 0.05; formoterol, n = 4, *p < 0.05) but not their parent dendrites (salbutamol, n = 11, NS, p = 0.22; formoterol, n = 4, NS, p = 0.11).
These data suggest that spine Ca2+ transients are selectively enhanced after activation of the β2 adrenergic receptor.
To study whether the spine-specific increase was mediated by L-type Ca2+ channels, we activated β2-ARs in the presence of nimodipine (Fig. 7A). Spine and dendritic Ca2+ transient amplitudes in the presence of both salbutamol (40 μm) and nimodipine (10 μm) were nonsignificantly changed relative to amplitudes with nimodipine alone (Fig. 7B). Ca2+ transient amplitudes changed nonsignificantly in spines and their parent dendrites during the first bAP of a burst (spines, 116 ± 11%, n = 4, NS, p = 0.08; dendrites, 100 ± 15%, n = 4, NS, p = 0.34). Equally, during the last bAP, Ca2+ transient amplitudes were nonsignificantly changed (spines, 100 ± 10%, NS, p = 0.38; dendrites, 95 ± 19%, NS, p = 0.17). Sequential wash in of nimodipine, followed by salbutamol, caused an expected small reduction of the spine and dendritic Ca2+ transient integrals (Fig. 7C), with no significant change of spine and dendritic Ca2+ transient integrals after wash in of salbutamol. During activation of β2-ARs with salbutamol and additional block of L-type VDCCs with nimodipine, Ca2+ transient integrals were nonsignificantly changed in both spines and their parent dendrites (spines, 109 ± 9%, n = 4, NS, p = 0.38; dendrites, 99 ± 8%, n = 4, NS, p = 0.13) (Fig. 7D). Furthermore, Ca2+ transient integrals in spines were not significantly different with respect to their parent dendrites (NS, p = 0.09). Ca2+ transient decay times were nonsignificantly increased in spines and their parent dendrites when L-type VDCCs were blocked, but β2-ARs were activated with salbutamol (Fig. 7E) (spines, 114 ± 11%, NS, p = 0.14; dendrites, 106 ± 14%, NS, p = 0.34).
Figure 7.
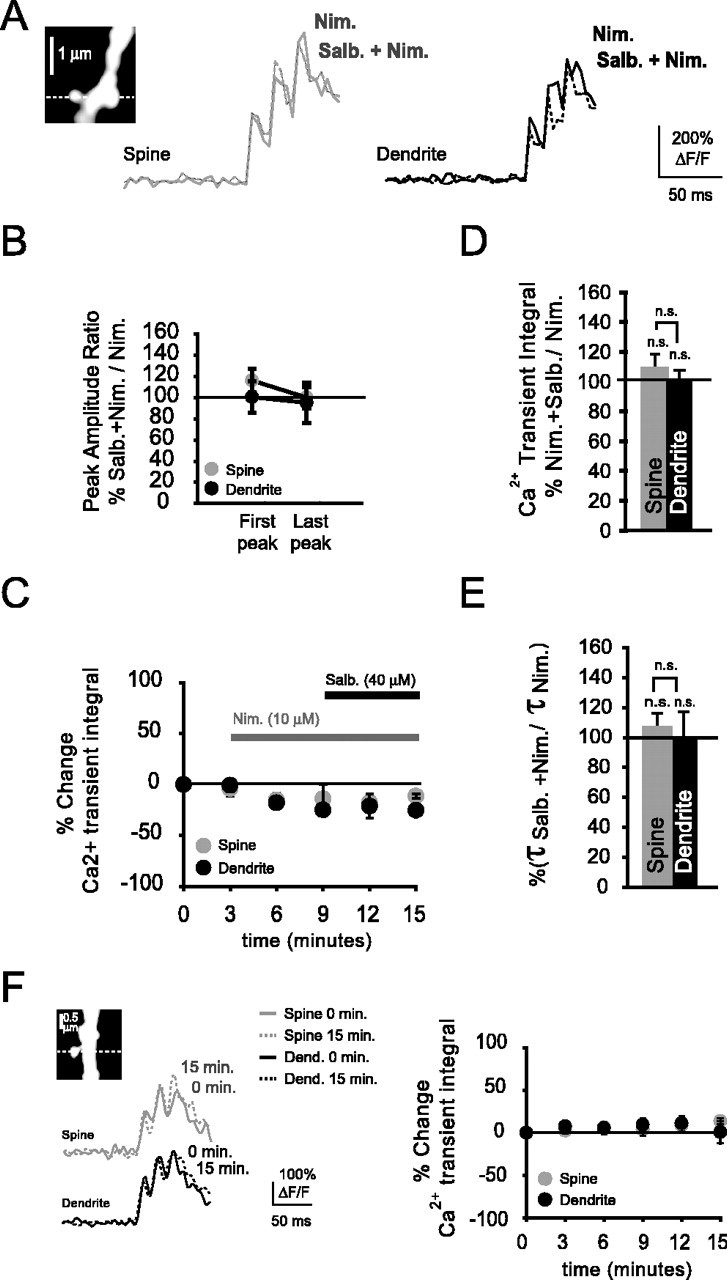
Blockade of L-type Ca2+ channels abolishes the spine-localized Ca2+ transient enhancement during β2-AR activation. A, Shown are bAP-evoked Ca2+ transients recorded from a spine and parent dendrite in the presence of nimodipine (Nim; 10 μm) and during bath application of salbutamol (Salb.; 40 μm). B, With L-type VDCCs blocked with nimodipine and β2-ARs activated with salbutamol, Ca2+ transient peak amplitudes were not significantly changed in spines and their parent dendrites during the first bAP (spines, n = 4, NS, p = 0.08; dendrites, n = 4, NS, p = 0.34) or last (spines, NS, p = 0.38; dendrites, NS, p = 0.17) bAP of a burst. C, Time course displaying the percentage change of the Ca2+ transient integral during application of the L-type Ca2+ channel blocker nimodipine and the β2-AR agonist salbutamol. D, Percentage change of the Ca2+ transient integrals in the presence of salbutamol and nimodipine with respect to the integrals in the presence of nimodipine alone. With L-type VDCCs blocked and β2-ARs activated, Ca2+ transient integrals did not differ significantly in spines or parent dendrites when β2-ARs were activated (spines, n = 4, NS, p = 0.38; dendrites, n = 4, NS, p = 0.13). E, Ca2+ transient decay times were nonsignificantly changed during activation of β2-ARs with salbutamol in the presence of nimodipine relative to nimodipine by itself, in both spines and their parent dendrites (spines, n = 4, NS, p = 0.14; dendrites, n = 4, NS, p = 0.34). F, Left, Ca2+ transients (single trials) in a basal spine and its parent dendrite (see inset) in the absence of any drugs at the start of recordings and 15 min later. Right, Time course displaying the Ca2+ transient integrals of spines and parent dendrites (n = 4) under no drug conditions. There were no reductions of the Ca2+ transient integrals during our recording time, showing that negligible rundown of Ca2+ channels occurred in spines and parent dendrites.
Altogether, these data suggest that the salbutamol-induced changes observed in the spines were mediated by L-type Ca2+ channels. To rule out that the reduction of Ca2+ transients in spines and their parent dendrites during nimodipine wash in was attributable to Ca2+ channel rundown, we also monitored Ca2+ transient integrals in spines and their parent dendrites during more prolonged recordings in the absence of any drugs (Fig. 7F). Ca2+ transient integrals were not significantly different from control 15 min after recordings begun (spines, 109 ± 2%, NS, p = 0.06; dendrites, 101 ± 13%, NS, p = 0.48), suggesting that, during this period, channel rundown did not strongly affect our measurements.
Role of PKA in mediating spine Ca2+ transient increases during β2-AR activation
The coupling of β2 adrenergic receptors to L-type Ca2+ channels has been well characterized in cardiac myocytes (Xiao et al., 1995; Zhou et al., 1997; Zhang et al., 2001; Xiao, 2001). This functional coupling involves PKA-dependent activation of the L-type Ca2+ channel (Naguro et al. 2001; Scott et al., 2000; Hulme et al., 2003), although evidence also exists for PKA-independent mechanisms (Jo et al., 2002). We wanted to test whether the spine Ca2+ transient enhancements observed after β2-AR stimulation were mediated by PKA. This would corroborate previous findings, which showed that CaV1.2 coimmunoprecipitates with this kinase (Davare et al., 1999). Figure 8A shows the spine and dendritic Ca2+ transients that were obtained in the presence of any of the PKA inhibitors alone (3 μm KT5720, 100 μm Rp-cAMPS, or 100 μm PKI) or in the presence of both salbutamol (40 μm) and one of the PKA inhibitors. Ca2+ transient amplitudes were not significantly different in the presence of salbutamol and each of the PKA inhibitors used in this study when compared with amplitudes in the presence of any of the PKA inhibitors alone (Fig. 8B). In the presence of KT5720 (3 μm), both spine and dendritic Ca2+ transient amplitudes were nonsignificantly increased during the first (spines, 105 ± 14%, n = 4, NS, p = 0.39; dendrites, 82 ± 12%, n = 4, NS, p = 0.19) and last bAP (spines, 100 ± 7%, NS, p = 0.36; dendrites, 103 ± 5%, NS, p = 0.44). In addition, when Rp-cAMPS was included in the patch pipette and β2-ARs were activated with salbutamol, both spine and dendritic Ca2+ transient amplitudes did not change significantly during either the first bAP (spines, 79 ± 4%, n = 3, NS, p = 0.09; dendrites, 92 ± 19%, n = 3, NS, p = 0.49) or the last bAP (spines, 113 ± 8%, NS, p = 0.15; dendrites, 109 ± 17%, NS, p = 0.27). Inclusion of PKI (100 μm) in the patch pipette confirmed results with the other PKA inhibitors. Ca2+ transient amplitudes were nonsignificantly changed in spines and their parent dendrites during the first (spines, 87 ± 15%, n = 4, NS, p = 0.18; dendrites, 106 ± 24%, n = 4, NS, p = 0.38) and last (spines, 106 ± 14%, NS, p = 0.24; dendrites, 112 ± 17%, NS, p = 0.18) bAP. Sequential bath application of KT5720 and salbutamol did not result in any significant changes of the spine and dendritic Ca2+ transient integrals (Fig. 8C). Ca2+ transient integrals also did not change significantly when PKA was blocked with any of the other PKA inhibitors but β2-ARs were activated (Fig. 8D). In the presence of KT5720, Ca2+ transient integrals revealed nonsignificant decreases in spines and their parent dendrites (spines, 95 ± 9%, n = 4, NS, p = 0.4; dendrites, 97 ± 5%, n = 4, NS, p = 0.4). With Rp-cAMPS, Ca2+ transient integrals were nonsignificantly changed in spines and their parent dendrites (spines, 90 ± 10%, n = 3, p = 0.2; dendrites, 102 ± 14%, n = 3, p = 0.5). Furthermore, when PKA was blocked with PKI during β2-AR activation, we also found no significant changes of integrals in spines or their parent dendrites (spines, 89 ± 5%, n = 4, p = 0.1; dendrites, 99 ± 6%, n = 4, p = 0.4). We observed a slight increase of the Ca2+ transient decay times in the presence of PKA inhibitors, but no significant differences were observed between spines and their parent dendrites (Fig. 8E). In the presence of KT5720 and salbutamol, spine and dendritic decay times did not increase significantly relative to Ca2+ transient decays with KT5720 alone (spines, 118 ± 14%, n = 4, NS, p = 0.18; dendrites, 111 ± 7%, n = 4, NS, p = 0.11). When PKA was blocked with Rp-cAMPS and β2-ARs were activated, neither spine nor dendritic decay times were significantly enhanced relative to decay times in the presence of Rp-cAMPS by itself (spines, 108 ± 5%, n = 3, NS, p = 0.2; dendrites, 106 ± 6%, n = 3, NS, p = 0.3). Finally, when we blocked PKA with PKI and activated β2-ARs with salbutamol, spine decay times did not increase significantly in spines or parent dendrites relative to Ca2+ transient decays in the presence PKI alone (spines, 117 ± 7%, n = 4, NS, p = 0.10; dendrites, 113 ± 14%, n = 4, NS, p = 0.16).
Figure 8.
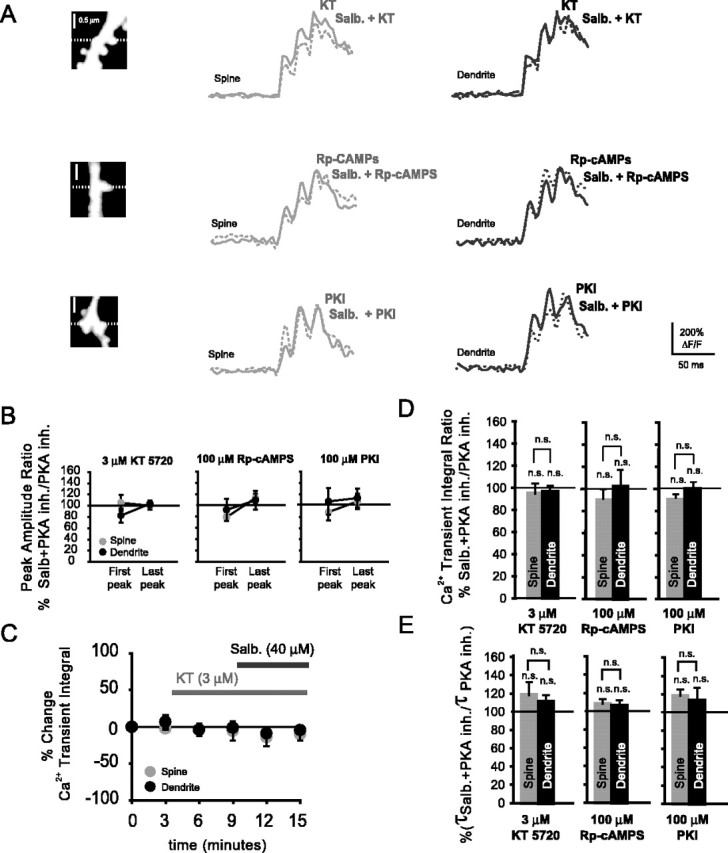
Blockade of protein kinase A abolishes β2-AR mediated enhancements of bAP-evoked spine Ca2+ transients. A, Ca2+ transients in spines and their parent dendrites in the presence of the PKA inhibitors KT5720 (KT; 3 μm), Rp-cAMPS and PKI (pipette concentrations of both 100 μm), and during additional activation of β2-ARs with salbutamol (40 μm). B, Percentage ratio of the Ca2+ transient amplitudes with salbutamol in the presence of a PKA inhibitor relative to Ca2+ transient amplitudes with PKA inhibitors alone. Ca2+ transient peak amplitudes did not change significantly during the first bAP (KT5720 spines, n = 4, NS, p = 0.39; KT5720 dendrites, n = 4, NS, p = 0.19; Rp-cAMPS spines, n = 3, NS, p = 0.09; Rp-cAMPS dendrites, n = 3, NS, p = 0.49; PKI spines, n = 4, NS, p = 0.18; PKI dendrites, n = 4, NS, p = 0.38) or the last bAP (KT5720 spines, NS, p = 0.36; KT5720 dendrites, NS, p = 0.44; Rp-cAMPS spines, NS, p = 0.15; Rp-cAMPS dendrites, NS, p = 0.27; PKI spines, NS, p = 0.24; PKI dendrites, NS, p = 0.18). B, Time course displaying the percentage change of the Ca2+ transient integrals with respect to control (no drug) in the presence of KT5720 (3 μm) and during additional activation of β2-ARs with salbutamol (40 μm). C, Percentage ratio of the Ca2+ transient integrals in the presence of KT5720, Rp-cAMPS, PKI, and salbutamol with respect to the integral in the presence of PKA inhibitors alone. Duringβ2-AR activation and with PKA blocked, Ca2+ transient integrals were not significantly different from integrals with PKA inhibitors alone, in both spines (KT5720, n = 4, NS, p = 0.4; Rp-cAMPS, n = 3, NS, p = 0.2; PKI, n = 4, NS, p = 0.1) and parent dendrites (KT5720, n = 4, NS, p = 0.4; Rp-cAMPS, n = 3, NS, p = 0.5; PKI, n = 4, NS, p = 0.4). E, Ratio of the decay times in the presence of salbutamol and PKA inhibitors versus decay times in the presence of PKA inhibitors alone. Both spine Ca2+ transient decay times (KT5720, n = 4, NS, p = 0.18; Rp-cAMPS, n = 3, NS, p = 0.2; PKI, n = 4, NS, p = 0.10) and dendritic decay times (KT5720, n = 4, NS, p = 0.11; Rp-cAMPS, n = 3, NS, p = 0.3; PKI, n = 4, NS, p = 0.16) did not differ significantly from control (only PKA inhibitors).
The above findings suggest that a highly compartmentalized signaling occurs from β2-ARs to spine-localized L-type Ca2+ channels and that this signaling requires PKA. Such coupling corroborates previous studies, which attribute increased Ca2+ channel activity to phosphorylation by PKA (Tsien et al., 1986; Johnson et al., 1994).
Discussion
Amplitudes and kinetics of bAP-evoked spine Ca2+ transients
We showed that bAP-evoked Ca2+ transient amplitudes in basal spines are similar to their parent dendrites. Previous studies in CA1 pyramidal neurons indicated that both basal and apical spine Ca2+ transients are significantly larger than those of their parent dendrites (Yuste et al., 1999; Majewska et al., 2000). The smaller diameter of basal dendrites (Megias et al., 2001) could account for a sharper rise of Ca2+ in these dendrites and thus cause similar increases as in the basal spines.
Decay times of the Ca2+ transients that we measured were well fit by single-exponential functions, conforming to neurons containing both slow- and fast-binding endogenous buffers (Lee et al., 2000). Spine Ca2+ transient decays were significantly faster than those of their parent dendrites. Interestingly, there were no significant differences in spine Ca2+ transient decays after single bAPs or a burst. Thus, even after a train of bAPs (three bAPs at 50Hz), spine Ca2+ transient decays remained fast, maintaining compartmentalization of Ca2+ during bursting activity. There may be a functional significance to this because postsynaptic bursting activity is important for synaptic potentiation (Pike et al., 1999; Paulsen and Sejnowski, 2000). Therefore, it will be of interest to study AP firing frequencies at which active Ca2+ extrusion mechanisms (smooth endoplasmic reticulum Ca2+-ATPase pumps and plasma membrane Ca2+-ATPase pumps) in spines become saturated.
Contribution of L-type VDCCs to bAP-evoked spine Ca2+ transients
In agreement with other studies (Sabatini and Svoboda, 2000; Yasuda et al., 2003), we found that R- and T-type VDCCs contribute primarily to bAP-evoked spine Ca2+ transients. The majority of spine Ca2+ influx is most likely mediated by R-type VDCCs because of their activation properties (Yasuda et al., 2003). However, T-type channel contribution cannot be ruled out completely and may contribute more strongly in spines of distal dendrites, in which bAPs are broadened (Randall and Tsien, 1997).
We hypothesized that bursts of bAPs prime L-type VDCCs as a result of their sensitivity to pre-depolarizations (Bourinet et al., 1994; Altier et al., 2001; Dzhura et al., 2000; Schjött and Plummer, 2000) and thus allow detection of their functional presence. Indeed, we found that, during brief bursts of bAPs, L-type VDCC blockers and activators accordingly modulate the resulting spine Ca2+ transients. The finding that L-type VDCCs are preferentially activated by EPSPs at room temperature (Mermelstein et al., 2000) has been challenged by others, who showed bAPs are equally effective in opening these channels at physiological temperatures (Liu et al., 2003). L-type VDCCs display three gating modes: mode 0, with channels not opening with depolarization; mode 1, characterized by multiple short openings; and mode 2, associated with long channel openings and high open probability (Hess et al., 1984). Gating mode 2 can be induced by L-type VDCC activators, stimulation of β-ARs (Yue et al., 1990), and through association of L-type channels with CaMKII (calcium/calmodulin-dependent protein kinase II) (Dzhura et al., 2000). Whereas L-type VDCC activators cause more Ca2+ influx as a result of a shift to mode 2 (Hess et al., 1984; Lauven et al., 1999), L-type channel blockers typically cause a stabilization of mode 0 (Hess et al., 1984).
In basal spines and their parent dendrites, Ca2+ transients were similarly reduced by L-type VDCC blockers. This was not the result of Ca2+ channel washout, because long-term recordings in the absence of these drugs did not cause a significant reduction of Ca2+ transient integrals. Interestingly, in basal spines, activators of L-type VDCCs caused a significantly larger enhancement of the Ca2+ transients than in their parent dendrites. One explanation for this preferential enhancement could be a higher density of L-type channels in the spines of basal dendrites. Other factors that affect channel kinetics and thereby the observed L-type channel contributions could include alternative splicing of channel subunits (α or β). Splice variants confer differences in DHP sensitivity (Soldatov et al., 1995; Welling et al., 1997). Furthermore, palmitoylation (Qin et al., 1998), phosphorylation state (Sculptoreanu et al., 1995), or truncation of the C terminus of the L-type channel α-subunit (Gao et al., 2001) could also differentially affect Ca2+ signaling through these channels in spines relative to dendrites. In addition, differences in voltage dependence have been described for class C, CaV1.2 and class D, CaV1.3 L-type channels (Xu and Lipscombe, 2001), which may be differentially targeted, although recent evidence suggests that CaV1.2 is found in both spines and dendrites (Obermair et al., 2004). Given the results, it is possible that L-type channels in basal dendrites are permissive for gating modes 0 and 1 but not mode 2. Although it cannot be ruled out completely, diffusion of Ca2+ from spines to parent dendrites did not appear to strongly affect the measured integrals, because we found that, during β2-AR activation, strong integral increases in spines did not result in significant enhancements of dendritic integrals or decay times.
Our data suggest that L-type VDCCs attribute a small amount to bAP-evoked basal spine Ca2+ increases. Modulation of L-type VDCCs, as seen with cardiac myocytes by β adrenergic receptors (Yue et al., 1990), could enhance the contribution of these channels similar to L-type Ca2+ channel activators and may control gene transcription. This effect on transcription can be relayed from synapses via translocation of calmodulin to the nucleus (Mermelstein et al., 2002). Calmodulin translocation requires Ca2+ influx through L-type Ca2+ channels and NMDA receptors (Deisseroth et al., 1998). Most interestingly, a recent study suggests that the activity of the central noradrenergic system during wakefulness could modulate neuronal gene transcription, thereby favoring synaptic potentiation (Cirelli and Tononi, 2004). Furthermore, L-type VDCCs appear to regulate transcription of the SNK (serum-inducible kinase) gene, the gene product of which selectively affects postsynaptic density and spine remodeling (Pak and Sheng, 2003), providing a homeostatic feedback to neurons experiencing intense synaptic activity and postsynaptic bursting.
Modulation of spine L-type VDCCs by the β2 adrenergic receptor
Modulation of VDCCs has been well documented in presynaptic terminals of the hippocampus. G-protein coupled receptors (GPCRs) modulating presynaptic VDCCs include GABAB, adenosine, neuropeptide Y, and muscarinic receptors (Wu and Saggau, 1994, 1995; Qian and Saggau, 1997; Qian et al., 1997). They each appear to preferentially target subsets of VDCCs. It is likely, therefore, that such membrane-delimited signaling complexes are present as well in dendritic spines and allow the precise fine-tuning of the Ca2+ sources that determine the polarity of synaptic plasticity. For example, it was shown that the neuromodulator GABA selectively affects spine VDCCs (Sabatini et al., 2000) through the GABAB receptor. Others have found that postsynaptic N-type Ca2+ channels can be selectively inhibited with low concentrations of the serotonin receptor agonist OH-DPAT [hydroxy-2(di-n-propylamino)tetralin] (Normann et al., 2000). We found a preferential enhancement of spine Ca2+ transients after activation of the β2-AR, also a GPCR. This enhancement was mediated most likely by an increased Ca2+ influx through spine-localized L-type VDCCs, because it was prevented by the L-type VDCC blocker nimodipine. Inhibition of PKA also prevented the β2-AR-induced enhancement of spine Ca2+ transients. Both stratum radiatum and stratum oriens of the hippocampal CA1 area receive noradrenergic inputs from the locus ceruleus (Loy et al., 1980). Noradrenergic terminals can synapse directly onto dendritic spines of cortical pyramidal neurons (Schroeter et al., 2000) and could thus modulate individual synapses.
Coimmunoprecipitation studies have revealed a macro-molecular complex that allows local modulation of L-type VDCCs. This complex consists of PKA, phosphatase 2A, protein kinase A anchoring proteins (AKAPs), and β2-ARs (Davare et al., 1999, 2000, 2001). Surprisingly, PKA alone appears to be insufficient to phosphorylate L-type VDCCs; rather, AKAPs are required (Gray et al., 1998, Johnson et al., 1994). AKAP79/150 coimmunoprecipitates with β2-AR, suggesting a compartmentalized signaling complex (Davare et al., 2001), and is found in high density in dendritic spines (Sik et al., 2000). AKAPs also regulate surface expression of L-type VDCCs (Altier et al., 2002). It is plausible, therefore, that preassembled signaling complexes consisting of both L-type VDCCs and β2-ARs are targeted to dendritic spines.
The role of the β2-ARs in synaptic plasticity remains unclear. Knock-outs of both receptor subtypes in mice demonstrated that β1-ARs, but not β2-ARs, could affect synaptic plasticity (Winder et al., 1999). However, the specific enhancement of spine L-type VDCCs after stimulation of β2-ARs might have an indirect effect on plasticity, because L-type VDCCs have been implicated in affecting both long-term depression and long-term potentiation in CA1 pyramidal neurons (Cavus and Teyler, 1996, 1998; Christie et al., 1997; Normann et al., 2000; Zakharenko et al., 2001; Evers et al., 2002). Interestingly, one study suggests that L-type spine VDCCs play an indirect role in tuning the susceptibility for long-term potentiation at apical synapses (Yasuda et al., 2003).
Our data suggest that only a small portion of bAP-evoked Ca2+ increases can be attributed to L-type VDCCs in basal dendritic spines of CA1 pyramidal neurons. We have shown, however, that Ca2+ influx through spine, but not dendritic, L-type VDCCs can be strongly modulated by activation of β2 adrenergic receptors and requires PKA. Our study thus adds to a growing body of evidence suggesting that VDCCs in spines can be locally modulated in much the same way as in presynaptic terminals that synapse onto CA1 pyramidal neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS33147 and EB001048 (P.S.). We thank Drs. G. Faas, A. Frick, and R. McQuiston for helpful discussions and B. Losavio and Dr. M. Mori for useful comments on this manuscript.
Correspondence should be addressed to Dr. P. Saggau, Department of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: psaggau@bcm.tmc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248416-12$15.00/0
References
- Altier C, Spaetgens RL, Nargeot J, Bourinet E, Zamponi GW (2001) Multiple structural elements contribute to voltage-dependent facilitation of neuronal alpha 1C (CaV1.2) L-type calcium channels. Neuropharmacology 40: 1050-1057. [DOI] [PubMed] [Google Scholar]
- Altier C, Dubel SJ, Barrere C, Jarvis SE, Stotz SC, Spaetgens RL, Scott JD, Cornet V, De Waard M, Zamponi GW, Nargeot J, Bourinet E (2002) Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J Biol Chem 277: 33598-33603. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Charnet P, Tomlinson WJ, Stea A, Snutch TP, Nargeot J (1994) Voltage-dependent facilitation of a neuronal alpha 1C L-type calcium channel. EMBO J 13: 5032-5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavus I, Teyler T (1996) Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. J Neurophysiol 76: 3038-3047. [DOI] [PubMed] [Google Scholar]
- Cavus I, Teyler TJ (1998) NMDA receptor-independent LTP in basal versus apical dendrites of CA1 pyramidal cells in rat hippocampal slice. Hippocampus 8: 373-379. [DOI] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D (1995) Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol 73: 2553-2557. [DOI] [PubMed] [Google Scholar]
- Christie BR, Schexnayder LK, Johnston D (1997) Contribution of voltagegated Ca2+ channels to homosynaptic long-term depression in the CA1 region in vitro. J Neurophysiol 77: 1651-1655. [DOI] [PubMed] [Google Scholar]
- Cirelli C, Tononi G (2004) Locus ceruleus control of state-dependent gene expression. J Neurosci 24: 5410-5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier RJ, Greenwood AC, Connor JA (2001) Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J Neurophysiol 85: 399-406. [DOI] [PubMed] [Google Scholar]
- Davare MA, Dong F, Rubin CS, Hell JW (1999) The A-kinase anchor protein MAP2B and cAMP-dependent protein kinase are associated with class C L-type calcium channels in neurons. J Biol Chem 274: 30280-30287. [DOI] [PubMed] [Google Scholar]
- Davare MA, Horne MC, Hell JW (2000) Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem 275: 39710-39717. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A beta2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98-101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198-202. [DOI] [PubMed] [Google Scholar]
- Denk W, Yuste R, Svoboda K, Tank DW (1996) Imaging calcium dynamics in dendritic spines. Curr Opin Neurobiol 6: 372-378. [DOI] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME (2000) Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol 2: 173-177. [DOI] [PubMed] [Google Scholar]
- Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bosl MR, Morellini F, Bartsch U, Dityatev A, Schachner M (2002) Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J Neurosci 22: 7177-7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Erick RT, Hosev MM (2001) C-terminal fragments of the alpha 1C (CaV1.2) subunit associate with and regulate L-type calcium channels containing terminal-truncated alpha 1C subunits. J Biol Chem 276: 21089-21097. [DOI] [PubMed] [Google Scholar]
- Graef IA, Mermelstein PG, Stankunas K, Neilson JR, Deisseroth K, Tsien RW, Crabtree GR (1999) L-type calcium channels and GSK-3 regulate the activity of NF-ATc4 in hippocampal neurons. Nature 401: 703-708. [DOI] [PubMed] [Google Scholar]
- Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates III JR, Scheuer T, Catterall WA, Murphy BJ (1998) Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron 20: 1017-1026. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK (1989) Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci 9: 2982-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci 12: 2685-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123: 949-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW (1984) Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311: 538-544. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Tsay D, Yuste R (2002) Calcium dynamics of spines depend on their dendritic location. Neuron 33: 425-437. [DOI] [PubMed] [Google Scholar]
- Hulme JT, Lin TW-C, Westenbroek RE, Scheuer T, Catterall WA (2003) β-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci USA 100: 13093-13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura Y, Kato N (1999) Action potential-induced dendritic calcium dynamics correlated with synaptic plasticity in developing hippocampal pyramidal cells. J Neurophysiol 82: 1993-1999. [DOI] [PubMed] [Google Scholar]
- Jo SH, Leblais V, Wang PH, Crow MT, Xiao RP (2002) Phosphatidylinositol 3-kinase functionally compartmentalizes the concurrent G(s) signaling during beta2-adrenergic stimulation. Circ Res 91: 46-53. [DOI] [PubMed] [Google Scholar]
- Johnson BD, Scheuer T, Catterall WA (1994) Voltage-dependent potentiation of L-type Ca2+ channels in skeletal muscle cells requires anchored cAMP-dependent protein kinase. Proc Natl Acad Sci USA 91: 11492-11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalchuk Y, Eilers J, Lisman J, Konnerth A (2000) NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci 20: 1791-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Caron MG (2001) Signal transduction. Bringing channels closer to the action! Science 293: 62-63. [DOI] [PubMed] [Google Scholar]
- Lauven M, Handrock R, Muller A, Hofmann F, Herzig S (1999) Interaction of three structurally distinct Ca2+ channel activators with single L-type Ca2+ channels. Naunyn Schmiedebergs Arch Pharmacol 360: 122-128. [DOI] [PubMed] [Google Scholar]
- Lee S-H, Schwaller B, Neher E (2000) Kinetics of Ca2+ binding to parvalbumin in bovine chromaffin cells: implications for [Ca2+] transients of neuronal dendrites. J Physiol (Lond) 525: 419-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Ren J, Murphy TH (2003) Decoding of synaptic voltage waveforms by specific classes of recombinant high-threshold Ca2+ channels. J Physiol (Lond) 553: 473-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy R, Koziell DA, Lindsey JD, Moore RY (1980) Noradrenergic innervation of the adult rat hippocampal formation. J Comp Neurol 189: 699-710. [DOI] [PubMed] [Google Scholar]
- Magee JC, Avery RB, Christie BR, Johnston D (1996) Dihydropyridine-sensitive, voltage-gated Ca2+ channels contribute to the resting intracellular Ca2+ concentration of hippocampal CA1 pyramidal neurons. J Neurophysiol 76: 3460-3470. [DOI] [PubMed] [Google Scholar]
- Majewska A, Brown E, Ross J, Yuste R (2000) Mechanisms of calcium decay kinetics in hippocampal spines, role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J Neurosci 20: 1722-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megias M, Emri Z, Freund TF, Gulyas AI (2001) Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102: 527-540. [DOI] [PubMed] [Google Scholar]
- Mermelstein PG, Bito H, Deisseroth K, Tsien RW (2000) Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J Neurosci 20: 266-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermelstein PG, Deisseroth K, Dagsupta N, Isaksen AL, Tsien RW (2002) Calmodulin priming: nuclear translocation of a calmodulin complex and the memory of prior neuronal activity. Proc Natl Acad Sci USA 99: 4132-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer Jr JR, Thompson LT, Black JP, Disterhoft JF (1992) Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol 68: 2100-2109. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM (1991) L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7: 625-635. [DOI] [PubMed] [Google Scholar]
- Naguro I, Nagao T, Adachi-Akahane S (2001) Ser(1901) of alpha(1C) subunit is required for the PKA-mediated enhancement of L-type Ca2+ channel currents but not for the negative shift of activation. FEBS Lett 489: 87-91. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K (2000) Calcium stores regulate the polarity and input specificity of synaptic modification. Nature 408: 584-588. [DOI] [PubMed] [Google Scholar]
- Normann C, Peckys D, Schulze CH, Walden J, Jonas P, Bischofberger J (2000) Associative long-term depression in the hippocampus is dependent on postsynaptic N-type Ca2+ channels. J Neurosci 15: 8290-8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE (2004) Differential targeting of the L-type Ca2+ channel alpha1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci 19: 2109-2122. [DOI] [PubMed] [Google Scholar]
- Pak DTS, Sheng M (2003) Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science 302: 1368-1373. [DOI] [PubMed] [Google Scholar]
- Paulsen O, Sejnowski TJ (2000) Natural patterns of activity and long-term synaptic plasticity. Curr Opin Neurobiol 10: 172-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AW, Paulsen O (1999) Rapid report: postsynaptic bursting is essential for “Hebbian” induction of associative long-term potentiation at excitatory synapses in rat hippocampus. J Physiol (Lond) 518: 571-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Saggau P (1997) Presynaptic inhibition of synaptic transmission in the rat hippocampus by activation of muscarinic receptors: involvement of presynaptic calcium influx. Br J Pharmacol 122: 511-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Colmers WF, Saggau P (1997) Inhibition of synaptic transmission by neuropeptide Y in rat hippocampal area CA1: modulation of presynaptic Ca2+ entry. J Neurosci 17: 8169-8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Costantin JL, Stefani E, Birnbaumer L (1998) Unique regulatory properties of the type 2a Ca2+ channel beta subunit caused by Palmitoylation. Proc Natl Acad Sci USA 95: 4690-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon y Cajal S (1891) Sur la structure de l'écorce cérébrale de quelques mammifères. Cellule 7: 124-176. [Google Scholar]
- Ramon y Cajal S (1894) The Croonian Lecture: la fine structure des centres nerveux. Proc R Soc Lond B Biol Sci 55: 444-468. [Google Scholar]
- Randall AD, Tsien RW (1997) Contrasting biophysical and pharmacological properties of T-type and R-type calcium channels. Neuropharmacology 7: 879-893. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Svoboda K (2000) Analysis of calcium channels in single spines using optical fluctuation analysis. Nature 408: 589-593. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K (2002) The life cycle of Ca2+ ions in dendritic spines. Neuron 33: 439-452. [DOI] [PubMed] [Google Scholar]
- Schjött JM, Plummer MR (2000) Sustained activation of hippocampal Lp-type voltage-gated calcium channels by tetanic stimulation. J Neurosci 20: 4786-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD (2000) Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol 420: 211-232. [PubMed] [Google Scholar]
- Scott JD, Dell'Acqua ML, Fraser ID, Tavalin SJ, Lester LB (2000) Coordination of cAMP signaling events through PKA anchoring. Adv Pharmacol 47: 175-207. [DOI] [PubMed] [Google Scholar]
- Sculptoreanu A, Figouroy A, De Groat WC (1995) Voltage-dependent potentiation of neuronal L-type calcium channels due to state-dependent phosphorylation. Am J Physiol 269: C725-C732. [DOI] [PubMed] [Google Scholar]
- Shankar S, Teyler TJ, Robbins N (1998) Aging differentially alters forms of long-term potentiation in rat hippocampal area CA1. J Neurophysiol 79: 334-341. [DOI] [PubMed] [Google Scholar]
- Sik A, Gulacsi A, Lai Y, Doyle WK, Pacia S, Mody I, Freund TF (2000) Localization of the A kinase anchoring protein AKAP79 in the human hippocampus. Eur J Neurosci 12: 1155-1164. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Bouron A, Reuter H (1995) Different voltage-dependent inhibition by dihyfropyridinies of human Ca2+ channel splice variants. J Biol Chem 270: 10540-10543. [DOI] [PubMed] [Google Scholar]
- Sorra KE, Harris KM (2000) Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 10: 501-511. [DOI] [PubMed] [Google Scholar]
- Spacek J, Harris KM (1997) Three-dimensional organization of smooth endoplasmic reticulum in hippocampal CA1 dendrites and dendritic spines of the immature and mature rat. J Neurosci 17: 190-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka O, Sakagami H, Kondo H (1995) Localization of mRNAs of voltage-dependent Ca2+ channels: four subtypes of alpha 1- and beta-subunits in developing and mature rat brain. Brain Res Mol Brain Res 30: 1-16. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420: 788-794. [DOI] [PubMed] [Google Scholar]
- Tsien RW, Bean BP, Hess P, Lansman JB, Nilius B, Nowycky MC (1986) Mechanisms of calcium channel modulation by beta-adrenergic agents and dihydropyridine calcium agonists. J Mol Cell Cardiol 18: 691-710. [DOI] [PubMed] [Google Scholar]
- Wang SS, Denk W, Hausser M (2000) Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci 3: 1266-1273. [DOI] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F (1997) Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ Res 81: 526-532. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER (1999) ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24: 715-726. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P (1994) Adenosine inhibits evoked synaptic transmission primarily by reducing presynaptic calcium influx in area CA1 of hippocampus. Neuron 12: 1139-1148. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P (1995) GABAB receptor-mediated presynaptic inhibition in guinea-pig hippocampus is caused by reduction of presynaptic Ca2+ influx. J Physiol (Lond) 485: 649-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP (2001) Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE 2001: RE15. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Ji X, Lakatta EG (1995) Functional coupling of the beta 2-adrenoceptor to a pertussis toxin-sensitive G protein in cardiac myocytes. Mol Pharmacol 47: 322-329. [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001) Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21: 5944-5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K (2003) Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948-955. [DOI] [PubMed] [Google Scholar]
- Yue DT, Herzig S, Marban E (1990) β-Adrenergic stimulation of calcium channels occurs by potentiation of high-activity gating modes. Proc Natl Acad Sci USA 87: 753-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Denk W (1995) Dendritic spines as basic functional units of neuronal integration. Nature 375: 682-684. [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Cash SS, Denk W (1999) Mechanisms of calcium influx into hippocampal spines, heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci 19: 1976-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4: 711-717. [DOI] [PubMed] [Google Scholar]
- Zhang ZS, Cheng HJ, Ukai T, Tachibana H, Cheng CP (2001) Enhanced cardiac L-type calcium current response to beta2-adrenergic stimulation in heart failure. J Pharmacol Exp Ther 298: 188-196. [PubMed] [Google Scholar]
- Zhou YY, Cheng H, Bogdanov KY, Hohl C, Altschuld R, Lakatta EG, Xiao RP (1997) Localized cAMP-dependent signaling mediates beta 2-adrenergic modulation of cardiac excitation-contraction coupling. Am J Physiol 273: H1611-H1618. [DOI] [PubMed] [Google Scholar]