

Abstract
Caenorhabditis elegans egg-laying behavior is inhibited by neurotransmitter signaling through the neural G-protein Gαo and serves as a model for analyzing Gαo signaling. Mutations that alter egg-laying frequency have identified genes encoding a number of signaling proteins that act with Gαo, but the receptors that activate Gαo remain mostly uncharacterized. To further analyze Gαo signaling, we cloned the egl-47 gene, which was identified by two dominant mutations that severely inhibit egg laying. egl-47 encodes two orphan G-protein-coupled receptor isoforms, which share all seven transmembrane domains but have different extracellular N termini. Both dominant mutations change the same alanine to valine in the sixth transmembrane domain, resulting in constitutively activated receptors. Deletion of the egl-47 gene caused no detectable egg-laying defects, suggesting that EGL-47 functions redundantly, or it inhibits egg laying under specific circumstances as yet unidentified. Using promoter::green fluorescent protein transgenes, we found that EGL-47 is expressed in a number of neurons, including the hermaphrodite-specific neurons (HSNs) that innervate the egg-laying muscles to stimulate contraction. Transgenic expression of constitutively active EGL-47 or constitutively active Gαo specifically in the HSNs was sufficient to inhibit egg-laying behavior. Our results suggest that EGL-47 regulates egg laying by activating Gαo in the HSN motor neurons to inhibit their activity. Because several neurotransmitters act through Gαo to inhibit HSN function, it appears that loss of any one receptor, such as EGL-47, causes only mild defects. Gαo apparently integrates signaling from multiple receptors in the HSNs, including EGL-47, to set the frequency of egg-laying behavior.
Keywords: EGL-47, egg-laying behavior, Caenorhabditis elegans, G-protein-coupled receptor, Gαo, HSN neuron
Introduction
Gαo is an abundant neural G-protein in humans and mediates signaling by many neurotransmitter receptors, but the mechanism and ultimate effects of Gαo signaling are not understood. Caenorhabditis elegans egg-laying behavior is inhibited by neuro-transmission through Gαo and serves as a model for studying Gαo signaling. Eggs are laid when the hermaphrodite-specific neurons (HSNs) release serotonin to stimulate contraction of egg-laying muscles (Desai et al., 1988). Serotonin may additionally feedback inhibit HSN activity by signaling through Gαo to terminate an episode of egg laying, although the serotonin autoreceptor(s) responsible for this effect have not been identified (Shyn et al., 2003). The ventral type C (VC) neurons, which synapse onto both the HSN processes and the egg-laying muscles, inhibit egg laying by releasing acetylcholine (White et al., 1986; Bany et al., 2003). This acetylcholine is hypothesized to signal through Gαo in the HSN presynaptic termini to inhibit neurotransmitter release, thus blocking egg laying. GAR-2, a G-protein-coupled receptor on the HSNs, mediates a portion of the effects of acetylcholine on egg laying; additional unidentified acetylcholine receptors must also inhibit HSN function (Bany et al., 2003). Both serotonin and acetylcholine are thus thought to act through Gαo to presynaptically inhibit HSNs. Gαo is also expressed in egg-laying muscles and may have additional functions in these cells (Mendel et al., 1995; Ségalat et al., 1995; Shyn et al., 2003).
The mechanism of neurotransmitter signaling in the egg-laying system has been investigated via genetic screens for mutants with altered rates of egg laying (Trent et al., 1983; Desai and Horvitz, 1989). Some of these mutants simply have developmental defects in the HSNs, VCs, or egg-laying muscle cells. Other mutants are anatomically normal but have defects in neurotransmitter signaling. These have been used to identify components of the Gαo signaling pathway. For example, mutations that result in hyperactive egg-laying behavior have identified the Gαo ortholog GOA-1 and the regulator of G-protein signaling (RGS) protein EAT-16 (Mendel et al., 1995; Ségalat et al., 1995; Hajdu-Cronin et al., 1999). Mutations that reduce egg-laying behavior cause the egg-laying-defective (Egl) phenotype and have identified the RGS protein EGL-10, the Gαq protein EGL-30, and the PLCβ protein EGL-8, a downstream effector of Gαq (Brundage et al., 1996; Koelle and Horvitz, 1996; Lackner et al., 1999). Studies of these mutants have demonstrated that Gαo signaling is antagonized by Gαq and that each G-protein is under RGS control.
Overall, genetic studies of egg laying suggest that Gαo directs presynaptic inhibition in the HSNs. However, this idea remains based primarily on indirect evidence because it has not been shown that Gαo activity in the HSNs is sufficient to inhibit HSN function, and the receptors that activate Gαo in the HSNs remain mostly unknown. We have addressed these issues by cloning and characterizing egl-47, a gene identified by mutations that block egg laying. Our analysis shows that EGL-47 is a G-protein-coupled receptor found in the HSNs, which can activate GOA-1 in the HSNs to inhibit neurotransmitter release.
Materials and Methods
Nematode culture. The wild-type strain was Bristol N2. Worms were cultured at 20°C under standard conditions, and double- and triple-mutant strains were generated using standard genetic techniques (Brenner, 1974).
Unlaid egg assay. Unlaid eggs were quantified as described (Koelle and Horvitz, 1996). The staged adults used in all assays were obtained by collecting late fourth larval stage (L4) animals and culturing at 20°C for 30 hr. In the case of the experiment shown in Figure 5, animals were aged for 40 hr after L4 to allow expression from the cell-specific promoters, which become active only at the L4-adult transition. For each strain analyzed, at least 30 staged adults were individually dissolved in 1.2% sodium hypochlorite, and their eggs, which survived because of their protective eggshells, were counted.
Figure 5.
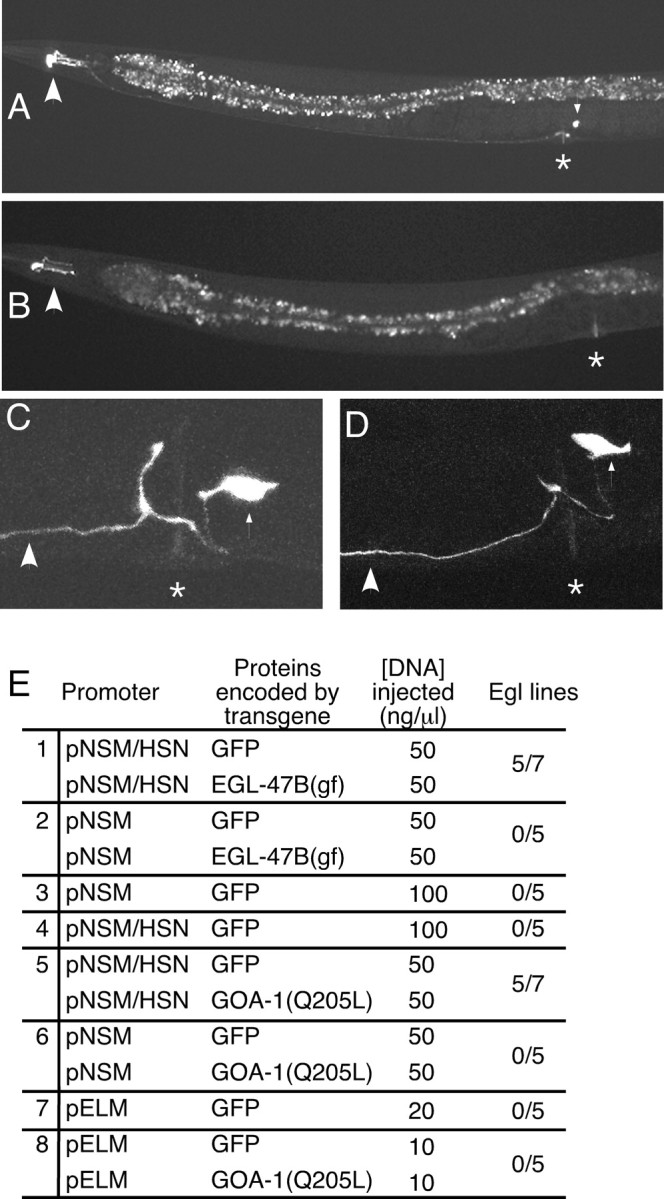
Effects of expression of EGL-47B(gf) or activated GOA-1 in the HSNs and egg-laying muscles. A, Adult with GFP expression driven from the pNSM/HSN promoter. The large arrowhead indicates the fluorescent NSMs; the small arrowhead indicates the fluorescent HSN cell body; the asterisk indicates the position of the vulva. The HSN process can be seen passing the vulva and extending to the head. The intestine, a large structure running the length of the body, shows prominent punctate autofluorescence but does not express GFP. B, Adult with GFP expression driven by the pNSM promoter. The large arrowhead indicates the fluorescent NSMs. No HSN expression is evident. C, D, HSN morphology in animals expressing EGL-47B(gf) (C) or activated GOA-1 (D) in the HSNs, revealed by GFP fluorescence. High-magnification micrographs of the vulval region are shown. The large arrowheads indicate HSN processes; small arrowheads indicate the HSN cell bodies; asterisks indicate the vulvas. HSN morphology appeared unaffected by expression of either EGL-47B(gf) or activated GOA-1 (compare with Fig. 1 D). E, Effects of expression of EGL-47B(gf) or activated GOA-1 in specific cells of the egg-laying system. Promoters pNSM or pNSM/HSN were used to drive expression of cDNAs for EGL-47B(gf) or activated GOA-1 (carrying the Q205L mutation), as indicated. In addition, promoter pELM was used to express GOA-1(Q205L) in egg-laying muscles. In every case, GFP was coexpressed to visualize cell morphology, as shown in A-D. As negative controls, GFP was expressed alone, i.e., without EGL-47(gf) or GOA-1(Q205L). At least five independent lines were assayed for each experiment. Lines were defined as Egl if >20% of adults contained at least 30 eggs. Activation of EGL-47 in the HSNs or activation of GOA-1 in the HSNs inhibited egg laying similarly, suggesting that EGL-47 activates GOA-1 in the HSNs to inhibit egg laying.
Pharmacological assays. Individual staged adult animals were placed in 50 μl of M9 buffer (Brenner, 1974), or M9 containing 7.5 mg/ml serotonin or 0.5 mg/ml fluoxetine. After 30 min, the number of released eggs was counted. N2 animals typically laid 0 eggs in M9 buffer, 3-8 eggs in serotonin, and 5-8 eggs in fluoxetine. egl-47(dm) animals and Egl transgenic lines typically laid 0 eggs in M9, >15 eggs in serotonin, and 0 eggs in fluoxetine. Egl animals release a large number of eggs in serotonin because they have many unlaid eggs at the start of the assay.
Mapping of egl-47. Triple-mutant unc-42(e270) egl-47(n1081dm) sqt-3(sc63ts) hermaphrodites were crossed with polymorphism-containing Hawaiian CB4856 males. Two hundred eighty F2 Sqt-non-Unc animals were isolated: these carried recombination events in the 1.75 map units between unc-42 and sqt-3. Recombinant chromosomes were homozygosed, and restriction digests of PCR products for nine polymorphic sites were used to determine whether Bristol or Hawaiian DNA was present, thereby determining the positions of recombination events. The presence or absence of the egl-47(n1081dm) mutation was determined by scoring for the Egl phenotype.
Transcript analysis. The cDNA clone yk1065f10, encoding EGL-47B (GenBank accession number NM_073148), was obtained from Dr. Yuji Kohara (National Institute of Genetics of Japan). RNA was prepared from mixed stage worms and analyzed by the 5′ rapid amplification of cDNA ends (RACE) technique (Frohman et al., 1988), identifying the transcript encoding EGL-47A (GenBank accession number AY532645).
egl-47(n1081dm) transformation. Genomic DNA was prepared was from n1081dm mutants and used for long-range PCR using the Gene-Amp XL PCR kit (PerkinElmer Life Sciences, Emeryville, CA). PCR products were purified using QIAEX II (Qiagen, Hilden, Germany) and injected into lin-15(765ts) animals at 1 ng/μl along with the lin-15-rescuing plasmid pL15EK at 50 ng/μl.
egl-47 gene knock-out. The egl-47(vs81) deletion mutation was identified by the method of Liu et al. (1999) using a PCR screen of DNA from a frozen C. elegans mutant library representing the progeny of 460,000 trimethylpsoralen-mutagenized animals. Deletion mutants were backcrossed four times to wild-type N2 animals to produce a clean genetic background. egl-47(vs81) is a 2501 bp deletion of sequences whose limits are TCATTTTTGTAGCGAGACAG... GCTCAACTGGTATGATGACT.
Egg-laying behavior of egl-47 knock-out. Egg-laying assays were performed according to the following references: inhibition by aldicarb (Bany et al., 2003), inhibition by octopamine (Tsalik et al., 2003), inhibition by dopamine or chronic serotonin (Schafer and Kenyon, 1995), response to starvation and refeeding (Dong et al., 2000), inhibition by vibration (Sawin, 1996), inhibition by liquid medium and stimulation by acute serotonin (Trent et al., 1983), and stimulation by fluoxetine (Weinshenker et al., 1995).
Wild-type and mutant egl-47 transgenes. Genomic DNA containing the coding sequences and 4851 bp 5′ of the first A-isoform exon and 733 bp of 3′ untranslated region were subcloned into pBluescript (Stratagene, La Jolla, CA) to make pJM5. Additional constructs contained one or two mutations introduced into pJM5. Single-nucleotide additions caused frameshifts in the second A-specific exon, first B-specific exon, or first common region exon. We also engineered the missense mutation found in n1081(dm). Transgenic strains were generated by injecting 10 ng/μl pJKL449.1 (myo-2-gfp coinjection marker; a gift from A. Fire, Carnegie Institute of Washington, Washington, DC), with either 50 ng/μl test construct and 20 ng/μl pBluescript or 2 ng/μl of test construct and 68 ng/μl pBluescript, into egl-47(vs81) deletion animals. We analyzed at least five independent transgenic lines for each injection and used only green fluorescent protein (GFP)-positive animals in the unlaid egg assay. Because extrachromosomal transgenes used are randomly lost during development to produce genetically mosaic animals, transgenes that induced the Egl phenotype did not always do so in 100% of animals within a line. An individual animal was defined as Egl if it contained at least 30 eggs. Any line with >20% Egl animals was defined as an Egl line. Transgenes that failed to produce Egl lines by this definition never produced >2% Egl animals.
GFP transgenes. A GFP reporter plasmid was constructed by inserting a fragment from cosmid C50H2 into pPD95.77 (a gift from A. Fire). This construct contained the promoter region and 5′ coding sequences of egl-47, such that the first common exon was fused in frame to the coding sequence for GFP. The construct contained the same promoter region as the egl-47 expression transgenes. Single-nucleotide additions to either the second A- or B-specific exons resulted in frameshift mutations to block expression of one isoform, thus generating isoform-specific expression patterns.
HSN and egg-laying muscle promoter transgenes. Based on the work of Sze et al. (2002), we generated two vectors to drive cell-specific expression. pNSM (pJM51A) contains a 377 bp fragment of the tph-1 promoter inserted into the HindIII--SphI site of pPD49.26 (a gift from A. Fire). When the GFP coding sequence was inserted between the KpnI and SacI sites of pJM51A, the resulting construct gave robust expression in the two NSM neurons: no other expression was detected. pNSM/HSN (pJM66A) contains a 3124 bp fragment of the tph-1 promoter inserted into the SphI site of pPD49.26. When the GFP coding sequence was inserted between the KpnI and SacI sites of pJM66A, the resulting plasmid gave robust GFP expression in the two NSMs and the two HSN neurons: no other expression was detected. To drive expression in egg-laying muscles, we used the pELM promoter, derived from pBH34.21 (Harfe and Fire, 1998), which contains a multimer of the NdeI-box enhancer in front of a minimal pes-10 promoter. pNSM and pNSM/HSN, and pELM were used to drive cDNAs encoding EGL-47B(gf) and GOA-1(Q205L). The EGL-47B cDNA clone yk1065f10 was modified to contain the gain-of-function mutation. pNSM::cDNA, pNSM/HSN::cDNA, and pELM::cDNA constructs were coinjected with the corresponding GFP constructs and the lin-15-rescuing plasmid pL15EK (50 ng/μl) into lin-15(n765ts) animals. Staged non-Muv adults were used in the unlaid egg assay.
Fluorescent visualization of HSN morphology. Transgenic worms containing pNSM/HSN (pJM60A) were paralyzed in a drop of 2 mm levami-sole atop a thin layer of 3% agarose on a glass slide. For each strain analyzed, the morphology of the HSNs in 10 worms was examined using a Bio-Rad (Hercules, CA) MRC 1024 confocal microscope.
Results
egl-47 dominant mutants have a defect in HSN neuron function
egl-47 was originally identified by two dominant mutations that cause a phenotype associated with a defect in HSN neuron activity (Desai and Horvitz, 1989). This phenotype has four characteristics: (1) animals accumulate unlaid eggs, suggesting that egg-laying behavior fails to occur; (2) animals can be stimulated to lay eggs by treatment with exogenous serotonin, demonstrating that the egg-laying muscles are functional and that the egg-laying defect is thus likely to result from lack of HSN function; (3) serotonin reuptake inhibitors, such as imipramine and fluoxetine, fail to stimulate egg laying, suggesting that the HSN neurons fail to release serotonin, whose action would otherwise be potentiated by these drugs; and (4) the HSN neurons appear morphologically normal, suggesting that they have functional rather than developmental defects.
We analyzed the egl-47 phenotype in detail. We found that the two dominant (dm) egl-47 mutants were phenotypically indistinguishable. egl-47(n1081dm) animals accumulated 44 ± 4 unlaid eggs, and egl-47(n1082dm) animals accumulated 44 ± 2 eggs, whereas the wild-type accumulated only 14 ± 1 eggs (Fig. 1A,B,F). The accumulation of eggs in egl-47(dm) animals was similar to that seen in egl-1(dm) mutant animals, which completely lack HSN neurons because of a developmental defect (Trent et al., 1983). Both egl-47 alleles were fully dominant because heterozygotes were indistinguishable from homozygotes (Fig. 1F). We verified that both mutants laid eggs in response to serotonin but not to fluoxetine (data not shown). Finally, we developed a GFP reporter that enabled us to analyze the fine details of HSN morphology in living animals. HSN cell bodies are normally located slightly posterior to the vulva and extend a single process that has a small branch near the vulva and continues anteriorly toward the nerve ring in the head (Fig. 1D). HSN morphology in egl-47(n1081dm) animals was within the variability seen for the wild type (Fig. 1D,E).
Figure 1.
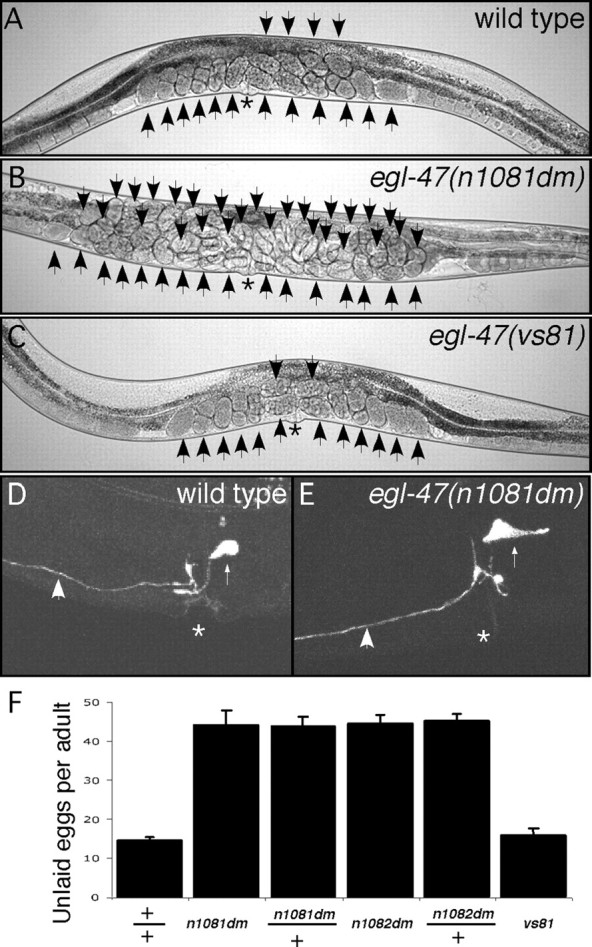
Characteristics of adult egl-47 mutants. A, Wild-type hermaphrodite. B, egl-47(n1081dm) gain-of-function mutant. C, egl-47(vs81) null mutant. Arrows indicate unlaid eggs; asterisks indicate the vulvas. n1081dm animals rarely lay eggs, causing an accumulation of unlaid eggs. D, E, HSN morphology in a wild-type and n1081dm hermaphrodite revealed by GFP fluorescence. High-magnification micrographs of the vulval region are shown. Large arrows indicate HSN processes; small arrows indicate the HSN cell bodies; asterisks indicate the vulvas. HSN morphology was not affected by egl-47(n1081dm). F, Number of unlaid eggs retained in wild-type or mutant strains. n ≥ 30 for all measurements. n1081dm and n1082dm mutations caused animals to fill with eggs and were fully dominant, as heterozygous and homozygous animals accumulated numbers of unlaid eggs that were not statistically different (p > 0.1, Student's t test). The vs81 null mutant was not statistically distinguishable from the wild type.
egl-47 mutants thus meet all four criteria outlined above, suggesting that they have severe defects in HSN function but normal HSN development. Similarly, null mutants for egl-10, the RGS protein that inactivates the G-protein GOA-1, also have the same four characteristics (Koelle and Horvitz, 1996). This similarity suggests that egl-47 dominant mutations, like egl-10 null mutations, might result in increased GOA-1 signaling. egl-10 mutants show a variety of behavioral defects, such as sluggish locomotion, that are attributable to increased GOA-1 signaling in cells outside the egg-laying system (Koelle and Horvitz, 1996). In contrast, we have not detected locomotion defects in egl-47 mutants, suggesting that EGL-47 function may be restricted to the egg-laying system.
egl-47 encodes two orphan G-protein-coupled receptors
We set out to molecularly identify the egl-47 gene. egl-47 had previously been mapped to a 403 kb interval between unc-42 and egl-3 on chromosome V (Desai and Horvitz, 1989). We identified single-nucleotide polymorphisms in this interval and used them for high-resolution mapping. egl-47 mapped to the 97 kb interval between polymorphisms vsP4 and vsP8. Seven recombinants within this interval did not separate egl-47 from two polymorphisms, vsP5 and vsP7, suggesting that egl-47 lies very close to these polymorphisms (Fig. 2A). We used transgenes to identify the gene in this region corresponding to egl-47. Because egl-47 mutations are dominant, we expected that transforming wild-type animals with a genomic clone containing the mutant egl-47 gene would induce the Egl phenotype. We used DNA from n1081dm animals as a template to generate long-range PCR products. Transgenic animals with the PCR product containing predicted open reading frame C50H2.2 phenocopied egl-47(dm) mutants: they were full of eggs, laid eggs in response to exogenous serotonin, and did not lay eggs in response to fluoxetine (data not shown). The C50H2.2 coding region was sequenced for both egl-47(dm) alleles. Both had the same mutation, changing an alanine to a valine (Fig. 2C). Thus, genetic mapping, transformation, and sequencing confirmed C50H2.2 as egl-47.
Figure 2.
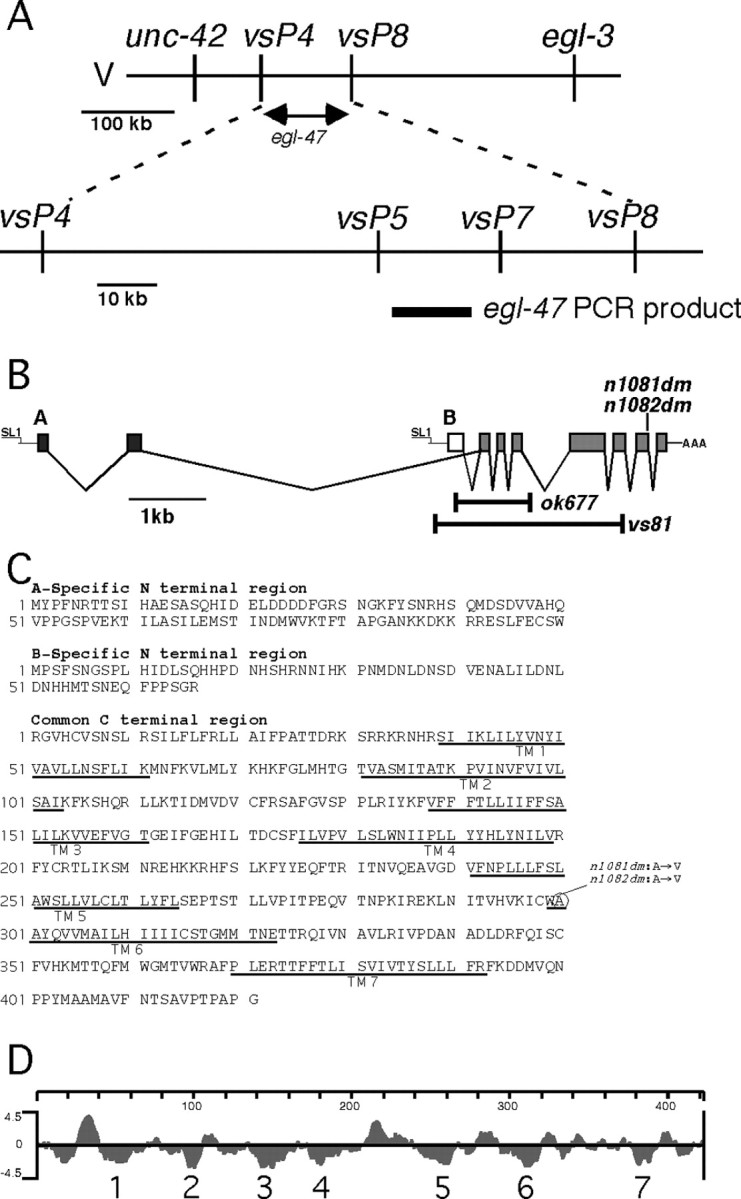
egl-47 mapping, exon structure, protein sequence, and hydrophilicity plot. A, Mapping and cloning strategy for egl-47. The top line schematizes the genetic positioning of egl-47 between single-nucleotide polymorphisms vsP4 and vsP8. The bottom line shows additional polymorphisms that were not separated from egl-47 by seven recombination events within this interval. The thick bar under the bottom line represents a PCR product that contains egl-47. When amplified from the n1081dm gain-of-function mutant and transformed into wild-type animals, this PCR product induced the egl-47 egg-laying defect. B, egl-47 gene structure and locations of mutations. Black boxes represent A-specific exons; the white box represents the B-specific exon; the gray boxes indicate exons common to both isoforms. The SL1 trans-splice leaders and poly(A) tail found at the ends of the transcripts are indicated. Bars indicate deleted regions of probable null alleles vs81 and ok677. C, Protein sequence of the A-specific region, B-specific region, and common region. Predicted transmembrane domains are underlined. The circle indicates the amino acid residue altered in dominant mutants n1081dm and n1082dm. D, Kyte-Doolittle hydrophilicity plot of the common region. Seven predicted transmembrane domains are indicated.
To analyze the egl-47 transcripts we obtained an egl-47 cDNA, which was full-length because it included an SL1 trans-spliced leader at its 5′ end and a poly(A) tract at its 3′ end. Using the RACE technique, we found that egl-47 produces two types of transcripts, one corresponding to the cDNA and the other containing different 5′ exons as well as an SL1 leader (Fig. 2B). EGL-47 therefore appears to use two different promoters to generate transcripts with two different 5′ ends. These transcripts encode two protein isoforms (EGL-47A and EGL-47B) with different N-terminal sequences and the same C-terminal sequence.
The deduced amino acid sequences contain seven predicted transmembrane domains encoded by the exons common to both isoforms (Fig. 2C,D), suggesting that EGL-47A and EGL-47B are G-protein-coupled receptors (GPCRs). The fact that an alanine to valine change in the sixth transmembrane domain is a dominant mutation in EGL-47 is consistent with the hypothesis that EGL-47 proteins are GPCRs because similar mutations cause constitutive activity in known GPCRs (Parnot et al., 2002). Ligand binding often occurs in the transmembrane domains of GPCRs, but the extracellular N termini can also contribute to ligand binding (Bockaert and Pin, 1999); thus, the different N termini of EGL-47A and EGL-47B could cause these isoforms to bind different ligands. GPCR sequences are highly diverse, but many can be assigned to subfamilies based on sequence similarity (Remm and Sonnhammer, 2000). Comparison of EGL-47 proteins to other GPCRs, however, shows that they cannot be clearly assigned to any subfamily. We performed a basic local alignment search tool (BLAST) search with the EGL-47 common region and identified the Drosophila orphan receptor Gr39a, a putative gustatory receptor (Clyne et al., 2000), as the most similar known sequence. No C. elegans receptors with high similarity to EGL-47 were identified. Thus, sequence analysis does not suggest which ligand(s) might activate EGL-47A and EGL-47B.
Our genetic and molecular analyses of egl-47 suggest a model in which the EGL-47 proteins function as receptors to activate the G-protein GOA-1. First, egl-47(dm) mutations block egg laying in a manner similar to that of mutations that increase GOA-1 signaling. Second, the EGL-47 proteins have the seven transmembrane domains characteristic of GPCRs, proteins that directly activate G-proteins. Third, the egl-47(dm) mutations are similar to mutations in other GPCRs that cause constitutive (ligand-independent) activation of G-proteins. Based on these lines of evidence, we hereafter refer to the EGL-47 proteins as orphan G-protein-coupled receptors. Definitive proof that EGL-47 proteins are receptors that directly activate GOA-1 would require expressing the EGL-47 proteins and showing that they stimulate GTP binding by GOA-1. However, we have so far been unable to achieve expression of EGL-47 protein in cultured cells for this purpose.
egl-47 null animals have no obvious phenotypic defects
We generated egl-47 null mutants to determine their phenotype. We screened a library of mutagenized animals to obtain deletion alleles of egl-47. We recovered one allele (vs81) from this library that had a 2501 bp deletion. The deletion removed the B-specific exon and five of the seven common exons (Fig. 2B). vs81 is thus a presumptive egl-47 null allele. egl-47(vs81) mutants appeared normal (Fig. 1C) and had wild-type levels of unlaid eggs (Fig. 1F). ok677, a deletion allele obtained from the C. elegans knock-out consortium, also appeared to be wild type. Wild-type animals alter their egg laying in response to multiple external factors, including the availability of food (Dong et al., 2000), the presence of vibration (Sawin, 1996), and immersion in liquid (Trent et al., 1983). In addition, treatment with several neurotransmitters or drugs can alter egg laying, including fluoxetine (Weinshenker et al., 1995), acute serotonin (Trent et al., 1983), chronic serotonin (Schafer and Kenyon, 1995), aldicarb (Bany et al., 2003), octopamine (Tsalik et al., 2003), and dopamine (Schafer and Kenyon, 1995). We tested the ability of vs81 animals to alter egg laying under all of these conditions and found vs81 animals were able to properly regulate egg laying in response to all of the conditions tested (Table 1). Although we failed to detect a defect in null mutants, EGL-47 may have subtle or redundant functions in egg laying. Another possibility is that EGL-47 is required for regulation of egg laying in response to yet another environmental condition that remains to be discovered.
Table 1.
Egg-laying analysis of egl-47 null animals
|
Egg-laying behavior assayeda |
Test conditions |
Wild-type measurement |
egl-47(vs81) measurement |
|---|---|---|---|
| Response to starvation and refeedingb | Starved/starved | 6.0 ± 4.0 | 6.0 ± 4.0 |
| Fed/fed | 64.5 ± 1.5 | 62.0 ± 4.0 | |
| Starved/fed | 50.0 ± 15.0 | 64.5 ± 15.5 | |
| Inhibition by vibrationc | No vibration | 33.2 ± 4.2 | 24.6 ± 4.5 |
| Vibration | 0.0 ± 0.0 | 0.0 ± 0.0 | |
| Inhibition in liquid mediumd | 0.0 ± 0.0 | 0.0 ± 0.0 | |
| Stimulation by fluoxetined | 0.5 mg/ml | 8.5 ± 0.9 | 9.3 ± 0.9 |
| Stimulation by acute serotonind | 7.5 mg/ml | 3.8 ± 1.2 | 3.7 ± 1.2 |
| Inhibition by chronic serotonine | 0 mg/ml | 0.0 ± 0.0 | 0.0 ± 0.0 |
| 3 mg/ml | 92.1 ± 3.7 | 87.5 ± 4.6 | |
| Inhibition by aldicarbf | 0 μM | 90.7 ± 9.0 | 94.0 ± 8.3 |
| 10 μM | 74.3 ± 7.7 | 81.4 ± 4.3 | |
| Inhibition by octopamineg | 0 mg/ml | 18.8 ± 1.3 | 20.2 ± 2.8 |
| 16 mg/ml | 2.0 ± 1.3 | 0.8 ± 0.4 | |
| Inhibition by dopamineh | 0 mg/ml | 55.8 ± 11.7 | 54.2 ± 7.5 |
|
|
6 mg/ml |
7.7 ± 4.2 |
7.6 ± 4.5 |
References for assays are given in Materials and Methods. Errors represent SEM. None of the differences between wild type and vs81 are statistically significant.
Ten animals were preconditioned for 2 hr on plates containing (to feed the animals) or not containing (to starve them) bacteria. Animals were then moved to assay plates with or without food, and the number of eggs laid in 30 min was counted. n = 2.
Eggs laid by five animals in 20 min on a plate with or without vibration. n = 5.
Eggs laid by one animal in 30 min in M9 liquid containing the indicated drug. n = 10.
Percentage of egg-laying-defective animals after 15 hr on a plate containing the indicated amounts of serotonin. n ≥ 50.
Eggs laid by 10 animals in 60 min on a plate containing the indicated amounts of aldicarb. n ≥ 3.
Eggs laid by two animals in 60 min on a plate containing the indicated amounts of octopamine. n = 5.
Eggs laid by 10 animals in 45 min on a plate containing the indicated amounts of dopamine. n = 6.
Overexpression of wild-type EGL-47 blocks egg laying
We used transgenes to overexpress EGL-47 in C. elegans to determine the effects of increasing EGL-47 signaling. In general, overexpression of GPCRs can lead to increased response to ligands as well as constitutive signaling by these receptors (Chen et al., 2000). We constructed a plasmid containing a fragment of genomic DNA including the entire wild-type egl-47 gene. To overexpress EGL-47, we injected a solution containing a high concentration (50 ng/μl) of this plasmid into worms to generate transgenic lines carrying high-copy tandem arrays of the plasmid. All five transgenic lines produced a significant number of Egl animals (Fig. 3B, row 1) that laid eggs in response to serotonin but not fluoxetine, phenocopying the genomic egl-47(dm) mutants. We modified the plasmid by inserting a stop codon into the first common exon (Fig. 3A, C stop) to prevent expression of either isoform of EGL-47. When injected at a high concentration, none of the five lines produced were Egl (Fig. 3B, row 2). Thus, EGL-47 protein expression is required to induce the Egl phenotype. To obtain lines with lower-copy transgenes and presumably lower EGL-47 expression, we injected a solution containing 2 ng/μl wild-type plasmid: this did not produce Egl lines (Fig. 3B, row 3). These experiments showed that an increase in EGL-47 signaling, induced by high-level expression of wild-type EGL-47, can cause an Egl phenotype. In addition, because egl-47(dm) mutations cause a similar Egl phenotype, these mutations appear to act by increasing the normal signaling function of EGL-47.
Figure 3.

Diagram and effects on egg laying of wild-type and mutant egl-47 transgenes. A, Diagram of egl-47 transgenes. Stop codons are shown that were included in certain transgenes so that one, both, or neither of the isoforms could be expressed. Also shown is the n1081dm gain-of-function (gf) mutation, included in some transgenes. The egl-47 coding exons are indicated by boxes; A-specific exons are black; the B-specific exon is white; common exons are gray. In addition to the coding regions shown, the transgenes contained 4.8 kb of 5′ promoter and 0.7 kb of 3′ untranslated region DNA. B, Effects of the transgenes. Indicated are the mutations included in individual transgenes, concentrations of DNA injected, and the number of Egl lines produced. Lines were defined as Egl if >20% of adults contained at least 30 eggs. When EGL-47 activity was increased, by overexpression or by mutation, egg laying was inhibited. Either isoform was capable of inhibiting egg laying.
Mutant EGL-47 is more active than wild-type EGL-47
To further analyze the effect of the egl-47(dm) mutation on protein function, we modified the egl-47 genomic clone to carry the n1081dm mutation (Fig. 3A). When injected at a low concentration, this modified clone conferred the Egl phenotype in four of six lines (Fig. 3B, row 4). Protein expression is required for this effect because insertion of a stop codon into the first common exon prevented the generation of any Egl lines (Fig. 3B, row 5). Because the wild-type clone did not induce the Egl phenotype at a low concentration, these results suggest that EGL-47 protein with the alanine to valine gain-of-function mutation is more potent than wild-type EGL-47 protein. This is consistent with n1081dm being an activating mutation that causes constitutive EGL-47 activity. Because the mutant proteins result in a gain of function, we hereafter refer to them as EGL-47(gf).
EGL-47A(gf) or EGL-47B(gf) can inhibit egg laying
To determine which isoform of EGL-47 is responsible for regulating egg laying, we constructed isoform-specific expression plasmids. We modified the egl-47(n1081dm)-containing construct to have a stop codon in either an A- or B-specific exon, so that only one isoform could be produced (Fig. 3A). When injected at the low concentration of 2 ng/μl, each of these constructs made four of five lines Egl (Fig. 3B, rows 6, 7). These experiments indicate that either EGL-47A(gf) or EGL-47B(gf) can induce the Egl phenotype.
egl-47 is expressed in the HSNs and other neurons
To examine the expression pattern of egl-47, we fused an egl-47 genomic fragment, containing the promoters for both egl-47 transcripts as well as the coding sequences up to the first common exon, to the coding sequences for the GFP. This construct was further modified to contain a stop codon in either an A- or B-specific exon. This allowed visualization of the expression pattern specific for each isoform. We examined transgenic animals carrying these constructs by confocal fluorescent microscopy. EGL-47A::GFP was expressed in a small subset of neurons in the head (Fig. 4A), the HSN neurons (Fig. 4B), and the PVQ interneurons of the tail (Fig. 4C). In the fourth larval stage, EGL-47A:: GFP was expressed in the vulval cells (Fig. 4D), but this was not detected in the adult. A similar pattern was observed for EGL-47B::GFP (Fig. 4E-H). The similar expression patterns of the two isoforms are consistent with those of the earlier transgenic experiments showing that either isoform can cause the Egl phenotype. The HSNs were the only cells in the adult egg-laying system to express EGL-47, suggesting that EGL-47 functions in the HSNs to inhibit egg laying. Expression outside of the egg-laying system was unexpected because the only obvious defect in egl-47 mutants is in egg-laying behavior.
Figure 4.
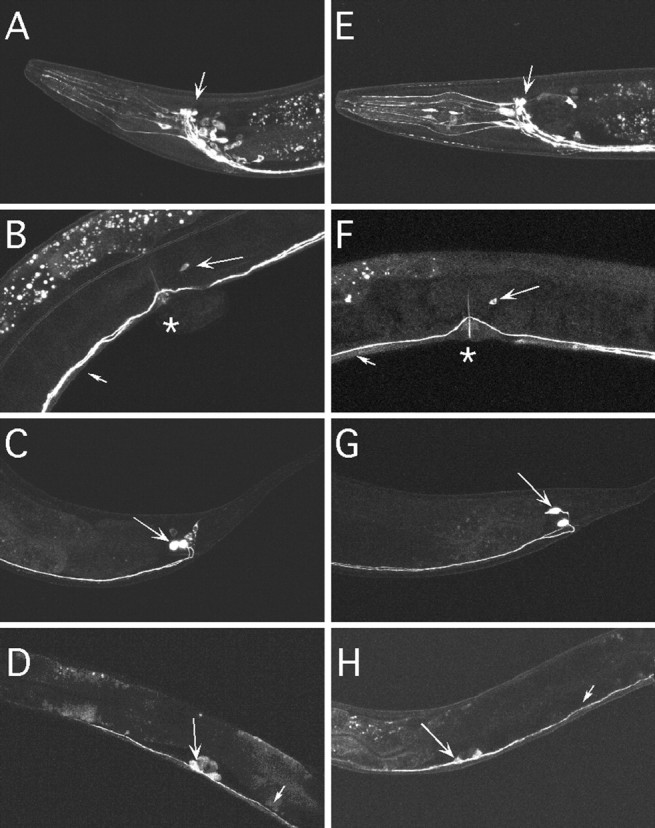
Expression patterns of egl-47::gfp reporter transgenes. A-D, GFP expression driven from the egl-47 isoform A promoter. E-H, GFP expression driven from the egl-47 isoform B promoter. A, E, Head region. Arrows point to fluorescent processes of the nerve ring flanked by a number of associated cell bodies. B, F, Midbody region. Small arrows indicate fluorescent neural processes of the ventral cord; large arrows indicate the HSN cell bodies; asterisks indicate the vulvas. C, G, Tail region. Arrows indicate the PVQ neural cell bodies, which send processes anteriorly along the ventral nerve cord. D, H, Midbody region of animals in the fourth larval stage. Large arrows indicate fluorescent vulval cells; small arrows indicate fluorescent neural processes of the ventral cord. EGL-47 isoforms A and B have similar expression patterns. In the adult, the HSNs are the only cells of the egg-laying system that express egl-47.
EGL-47(gf) expressed in the HSNs can induce the Egl phenotype
Because the egl-47(dm) phenotype suggested a functional defect in the HSNs, and the expression pattern described above demonstrates EGL-47 was expressed in the HSNs, we tested whether activation of EGL-47 specifically in the HSNs was sufficient to induce the Egl phenotype. For this purpose, we expressed a cDNA for EGL-47B(gf) in the HSNs and determined whether this conferred the Egl phenotype on wild-type animals. Because there was no completely HSN-specific promoter available, we drove cDNA expression using pNSM/HSN, a promoter that expresses only in the HSNs and the NSMs (Fig. 5A). The NSMs are a pair of neurons in the head with no known role in egg laying. To rule out effects attributable to NSM expression, we also used pNSM, a modified version of pNSM/HSN that lacks HSN expression but retains NSM expression (Fig. 5B). Expression of EGL-47B(gf) from pNSM/HSN caused the Egl phenotype in five of seven lines (Fig. 5E, row 1). Egl animals were similar to the egl-47(dm) mutants in that they laid eggs in response to serotonin but not fluoxetine. They were also similar to egl-47(dm) mutants in that they had normal HSN morphology, as visualized by coexpression of GFP in the HSNs (Fig. 5C). No Egl lines were produced when EGL-47B(gf) was expressed from pNSM (Fig. 5E, row 2), when only GFP was expressed from pNSM (Fig. 5E, row 3), or when GFP was expressed from pNSM/HSN (Fig. 5E, row 4). Thus, we conclude EGL-47 activity in the HSNs is sufficient to induce the Egl phenotype associated with egl-47(dm) mutants.
Activated GOA-1 expressed in the HSNs can induce the Egl phenotype
What G-protein is activated by EGL-47 to inhibit egg laying? The Gαo homolog GOA-1 is expressed in all neurons, including the HSNs. Null mutants for GOA-1 are hyperactive for egg laying, suggesting that this G-protein inhibits egg laying (Mendel et al., 1995; Ségalat et al., 1995). Deletion mutants for every Gα gene of C. elegans have been analyzed, and GOA-1 was the only Gα protein found to inhibit egg laying, although other G-proteins might also affect egg laying but could have escaped detection because of redundancy (Jansen et al., 1999). These observations suggest GOA-1 to be the G-protein activated by EGL-47 receptors in the HSNs to inhibit egg laying. As one test of this idea, we examined the phenotype of animals carrying a goa-1 null mutation and the egl-47(dm) mutation. We found goa-1; egl-47(dm) double mutants were similar to goa-1 single mutants: both strains laid eggs so frequently that they retained an average of fewer than three unlaid eggs per adult, compared with the 14 ± 1 unlaid eggs seen in the wild type or the 44 ± 4 unlaid eggs seen in egl-47(dm) single mutants. Thus, GOA-1 is required for the EGL-47(gf) protein to inhibit egg laying, consistent with EGL-47 signaling through GOA-1.
To determine whether GOA-1 activation in the HSNs is sufficient to inhibit egg laying, we expressed a constitutively active mutant of GOA-1 in the HSNs. We used the pNSM and pNSM/HSN promoters to express GOA-1(Q205L), a GTPase-deficient mutant in which the glutamine at residue 205 is replaced with a leucine (Mendel et al., 1995). GOA-1(Q205L) expressed in the NSMs and the HSNs caused five of seven lines to be Egl (Fig. 5E, row 5). In the negative control, GOA-1(Q205L) expressed in the NSMs did not generate any Egl lines (Fig. 5E, row 6). The Egl animals were stimulated to lay eggs in response to serotonin but not to fluoxetine (data not shown), phenocopying the egl-47(dm) mutants. They were also similar to egl-47(dm) mutants in that they had normal HSN morphology, as visualized by coexpression of GFP in the HSNs (Fig. 5D).
Previous work has shown that GOA-1 is expressed in the egg-laying muscles and may function in these cells to inhibit egg laying (Mendel et al., 1995; Ségalat et al., 1995, Shyn et al., 2003). Thus, we also tested whether expression of GOA-1(Q205L) in the egg-laying muscles affected egg laying. The pELM promoter (Harfe and Fire, 1998) was used to drive expression of GOA-1(Q205L) and/or GFP in all 16 egg-laying muscles. This resulted in very bright GFP fluorescence in the muscles, but did not cause animals to be Egl (Fig. 5, rows 7, 8). Although this negative result does not rule out a function for GOA-1 in the egg-laying muscles, it suggests that the predominant site at which GOA-1 inhibits egg laying may be in the HSN neurons, rather than in the egg-laying muscles.
In summary, activation of GOA-1 in the HSNs was sufficient to inhibit egg laying. Because expressing EGL-47B(gf) in the HSNs was similarly sufficient to inhibit egg laying, these results are consistent with the idea that the normal function of EGL-47 is to signal through GOA-1 in the HSNs to regulate egg laying.
Discussion
Activated EGL-47 prevents egg laying by activating GOA-1 to inhibit neurotransmitter release from the HSNs
We suggest a model in which activation of EGL-47 receptor in the HSN motor neurons activates the Gαo protein GOA-1 to inhibit neurotransmitter release, thereby inhibiting egg laying. This is supported by the following: (1) activating EGL-47 blocks egg laying; (2) serotonin release from the HSNs in egl-47(dm) animals is reduced because serotonin reuptake inhibitors have no effect on egg laying; (3) EGL-47 contains seven transmembrane domains and is thus a putative G-protein-coupled receptor; (4) the amino acid substitution found in egl-47(dm) mutants is consistent with causing constitutive receptor activation; (5) GFP reporters show EGL-47 is expressed in the HSNs; (6) expression of activated EGL-47 specifically in HSNs blocks egg laying, phenocopying egl-47(dm) mutants; (7) the effects of EGL-47 activation require the G-protein GOA-1; and (8) expression of activated GOA-1 specifically in HSNs blocks egg laying, phenocopying egl-47(dm) mutants.
Previous studies have suggested a mechanism by which GOA-1 can inhibit neurotransmitter release. GOA-1 signaling results in relocalization of the synaptic protein UNC-13 away from neurotransmitter release sites in certain ventral cord neurons (Nurrish et al., 1999). UNC-13 is required to prime synaptic vesicles for release and is recruited to membranes by binding diacylglycerol (Rosenmund et al., 2003). Genetic studies suggest that GOA-1 signaling reduced diacylglycerol levels, thereby releasing UNC-13 from presynaptic membranes and blocking neurotransmitter release (Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999).
What ligand activates EGL-47?
EGL-47A and EGL-47B, like the vast majority of GPCRs in both humans and model organisms, are orphan receptors whose activating ligand(s) remain unknown. Because each isoform has a different extracellular N-terminal domain, it is possible that each isoform binds a different ligand. BLAST analysis showed EGL-47 has weak similarity to a putative Drosophila gustatory receptor. A role for gustatory receptors in regulating egg laying might be expected because egg-laying behavior is affected by food (Dong et al., 2000). However, we found that EGL-47 was highly expressed in nonchemosensory neurons, such as PVQ interneurons and HSN motor neurons, suggesting that the EGL-47 ligand is more likely a neurotransmitter produced in the worm rather than an external chemosensory signal.
Several neurotransmitters affect egg laying when applied exogenously to C. elegans, including serotonin (Trent et al., 1983), acetylcholine (Weinshenker et al., 1995; Waggoner et al., 1998; Bany et al., 2003), dopamine (Schafer and Kenyon, 1995), and octopamine (Horvitz et al., 1982). However, it is unlikely that any of these neurotransmitters activate EGL-47 because EGL-47 shows little sequence similarity to known biogenic amine or acetylcholine receptors. Neuropeptides are also candidate EGL-47 ligands. There are >200 putative neuropeptides in C. elegans (Li et al., 1999: Nathoo et al., 2001), but mutants for only two neuropeptide genes have been described (Nelson et al., 1998; Rogers et al., 2003). One of these mutants, flp-1, has an abnormal temporal pattern of egg laying and also does not properly turn off egg laying when removed from food, demonstrating that the FLP-1 peptide can modulate egg laying (Waggoner et al., 2000). However, EGL-47 is not likely to be an FLP-1 receptor because we found that egl-47 null animals did properly turn off egg laying when removed from food.
GOA-1 integrates signals from multiple GPCRs in the HSNs to regulate egg laying
Multiple G-protein-coupled receptors on the HSNs appear to activate GOA-1 to inhibit neurotransmitter release. (Supplemental material, available at http://www.jneurosci.org, shows a model summarizing the signals thought to impinge on the HSNs.) Shyn et al. (2003) showed that an unidentified serotonin autoreceptor silences the HSNs, and this effect requires GOA-1. Thus, serotonin release from the HSNs into the neuromuscular junction causes contraction of the egg-laying muscles but also feeds back to inhibit HSN function. Bany et al. (2003) found acetylcholine released from the VC neurons also inhibits egg laying, and this effect is partially mediated by the G-protein-coupled acetylcholine receptor GAR-2, which is expressed on the HSNs (Bany et al., 2003). Our data, showing that activation of GOA-1 specifically in the HSNs is sufficient to inhibit egg laying, strengthens the models of Shyn et al. (2003) and Bany et al. (2003), in which serotonin and acetylcholine receptors inhibit egg laying by acting through GOA-1 to inhibit HSN activity. EGL-47A and EGL-47B are two additional receptors that use GOA-1 to regulate synaptic activity in the HSNs. EGL-47 may act as an HSN autoreceptor in a feedback mechanism similar to that suggested by Shyn et al. (2003) or to receive a signal from another neuron, as proposed for GAR-2 (Bany et al., 2003).
The multiple signals acting on the HSNs have all been demonstrated experimentally, but what biological purpose(s) do they serve? C. elegans regulates egg laying in multiple ways (Table 1), presumably to deposit its eggs in favorable circumstances. C. elegans also regulates egg laying so that clusters of eggs are laid approximately every 20 min (Waggoner et al., 1998), although any benefit of this clustering remains obscure. The egg-laying system thus requires a mechanism to integrate a number of factors to set a level of egg-laying activity. We hypothesize that GOA-1 activity in the HSN neurons serves as an integrator of multiple signals to set an appropriate level of HSN activity and thus egg-laying behavior. At the present time, we do not know which signals account for which aspect of egg-laying regulation. For example, the circumstances that lead to release of acetylcholine from the VC neurons or to the release of the EGL-47 ligand are not known. It has been speculated that feedback signaling by HSN-released serotonin could account for the clustering of egg-laying events (Shyn et al., 2003).
Why do EGL-47 null mutants, like null alleles of most GPCRs, have no obvious defects?
The C. elegans genome encodes up to 1000 G-protein-coupled receptors (Bargmann, 1998), making the GPCRs the largest family of proteins. However, genetic screens in C. elegans have identified only three GPCR genes: the odorant receptor ODR-10 (Sengupta et al., 1996), the neuropeptide receptor NPR-1 (de Bono and Bargmann, 1998), and EGL-47. Keating et al. (2003) used RNAi to knock down expression of 60 GPCRs predicted to bind neurotransmitters or neuropeptides and found only a small number with defects. Several mutagenesis screens have identified genes involved in G-protein control of egg laying (Trent et al., 1983; Desai and Horvitz 1989; Mendel et al., 1995; Robatzek and Thomas, 2000). These screens have yielded many mutations in G-proteins and other signaling proteins, but to date, EGL-47 is the only GPCR identified. Why are GPCR mutations so difficult to find? It was only possible to identify egl-47 with gain-of-function mutations, and these were exceedingly rare. In fact, both gain-of-function alleles resulted in the very same amino acid substitution, suggesting that there may only be one point mutation that can hyperactivate EGL-47. It appears that loss-of-function mutations in GPCRs generally do not have obvious phenotypic defects. Consistent with this idea, deletion alleles of egl-47 had no noticeable defects. The lack of defects in GPCR mutants could be attributed to redundancy. However, there is no other GPCR closely related to EGL-47, so we have no candidate to test for redundancy with EGL-47. Another hypothesis is that GPCRs like EGL-47 have subtle functions that are not obviously detected. For example, EGL-47 may inhibit egg laying in a special circumstance that we have not tested. As discussed above, EGL-47 is one of a number of GPCRs that all seem to act in the HSNs to signal through GOA-1 to inhibit egg laying. It appears that loss of any individual such receptor may cause only minor defects, whereas loss of GOA-1 or other shared downstream signaling components has dramatic effects.
EGL-47 is expressed in only a limited subset of neurons, just as that observed for other GPCRs (Troemel et al., 1995), and this is likely another factor contributing to the limited effects of GPCR mutations. We expect that the function of every neuron expressing EGL-47(gf) is inhibited as a result of overactivation of GOA-1. Even so, the only observed defect can be attributed to inactivation of just the HSN neurons. This is not entirely surprising. Many individual neurons in C. elegans can be ablated without causing obvious behavioral defects: such neurons in some cases have been shown to have subtle or redundant functions (Avery and Horvitz, 1989; Bargmann and Horvitz, 1991; McIntire et al., 1993). In the case of EGL-47, the neurons that most strongly expressed our egl-47::GFP reporter were the PVQ interneurons, and these are among the large set of C. elegans neurons for which no behavioral functions have been defined. We identified the EGL-47 receptor only because it acts in one neuron type (HSN) whose inactivation leads to a dramatic Egl defect and because powerful genetic screens for Egl mutants have allowed very rare gain-of-function mutations to be isolated.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS36918 and a Leukemia and Lymphoma Society Scholar Award (M.R.K.). We thank the Oklahoma group of the C. elegans Gene Knockout Consortium for the egl-47(ok677) mutant and the Caenorhabditis Genetics Center, which is supported by the NIH National Center for Research Resources, for additional strains.
Correspondence should be addressed to Dr. Michael Koelle, Department of Molecular Biophysics and Biochemistry, Yale University School of Medicine, 333 Cedar Street, Sterling Hall of Medicine, Room CE30, New Haven, CT 06520-8024. E-mail: michael.koelle@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248522-09$15.00/0
References
- Avery L, Horvitz HR (1989) Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans Neuron 4: 473-485. [DOI] [PubMed] [Google Scholar]
- Bany IA, Dong MQ, Koelle MR (2003) Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J Neurosci 22: 8060-8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI (1998) Neurobiology of the Caenorhabditis elegans genome. Science 282: 2028-2033. [DOI] [PubMed] [Google Scholar]
- Bargmann CI, Horvitz HR (1991) Control of larval development by chemosensory neurons in Caenorhabditis elegans Science 251: 1243-1246. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Pin JP (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J 18: 1723-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans Genetics 77: 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, Sternberg PW, Simon MI (1996) Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron 16: 999-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Way J, Armour S, Watson C, Queen K, Jayawickreme CK, Chen WJ, Kenakin T (2000) Use of constitutive G protein-coupled receptor activity for drug discovery. Mol Pharmacol 57: 125-134. [PubMed] [Google Scholar]
- Clyne PJ, Warr CG, Carlson JR (2000) Candidate taste receptors in Drosophila Science 287: 1830-1834. [DOI] [PubMed] [Google Scholar]
- de Bono M, Bargmann CI (1998) Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans Cell 94: 679-689. [DOI] [PubMed] [Google Scholar]
- Desai C, Horvitz HRH (1989) Caenorhabditis elegans mutants defective in the functioning of the motor neurons responsible for egg laying. Genetics 121: 703-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai C, Garriga G, McIntire S, Horvitz HR (1988) A genetic pathway for the development of the Caenorhabditis elegans HSN motor neurons. Nature 336: 638-646. [DOI] [PubMed] [Google Scholar]
- Dong MQ, Chase D, Patikoglou GA, Koelle MR (2000) Multiple RGS proteins alter neural G protein signaling to allow C. elegans to rapidly change behavior when fed. Genes Dev 14: 2003-2014. [PMC free article] [PubMed] [Google Scholar]
- Frohman MA, Dush MK, Martin GR (1988) Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA 85: 8998-9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajdu-Cronin YM, Chen WJ, Patikoglou G, Koelle MR, Sternberg PW (1999) Antagonism between Goα and Gqα in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Gqα activity. Genes Dev 13: 1780-1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harfe BD, Fire A (1998) Muscle and nerve-specific regulation of a novel NK-2 class homeodomain factor in Caenorhabditis elegans Development 125: 421-429. [DOI] [PubMed] [Google Scholar]
- Horvitz HR, Chalfie M, Sulston JE, Evans PD (1982) Serotonin and octopamine in the nematode C. elegans Science 216: 1012-1014. [DOI] [PubMed] [Google Scholar]
- Jansen G, Thijssen KL, Werner P, van der Horst M, Hazendonk E, Plasterk RH (1999) The complete family of genes encoding G proteins of Caenorhabditis elegans Nat Genet 21: 414-419. [DOI] [PubMed] [Google Scholar]
- Keating CD, Kriek N, Daniels M, Ashcroft NR, Hopper NA, Siney EJ, Holden-Dye L, Burke JF (2003) Whole-genome analysis of 60 G protein-coupled receptors in Caenorhabditis elegans by gene knockout with RNAi. Curr Biol 13: 1715-1720. [DOI] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR (1996) EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 84: 115-125. [DOI] [PubMed] [Google Scholar]
- Lackner MR, Nurrish SJ, Kaplan JM (1999) Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24: 335-346. [DOI] [PubMed] [Google Scholar]
- Li C, Kim K, Nelson LS (1999) FMRFamide-related neuropeptide gene family in Caenorhabditis elegans Brain Res 848: 26-34. [DOI] [PubMed] [Google Scholar]
- Liu LX, Spoerke JM, Mulligan EL, Chen J, Reardon B, Westlund B, Sun L, Abel K, Armstrong B, Hardiman G, King J, McCague L, Basson M, Clover R, Johnson CD (1999) High-throughput isolation of Caenorhabdtitis elegans deletion mutants. Genome Res 9: 859-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RHA, Sternberg PW (1995) Participation of the protein Go in multiple aspects of behavior in C. elegans Science 267: 1652-1655. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Kaplan J, Horvitz HR (1993) The GABAergic nervous system of Caenorhabditis elegans Nature 364: 337-341. [DOI] [PubMed] [Google Scholar]
- Miller KG, Emerson MD, Rand JB (1999) Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans Neuron 24: 323-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathoo AN, Moeller RA, Westlund BA, Hart AC (2001) Identification of neuropeptide-like protein gene families in Caenorhabditis elegans and other species. Proc Natl Acad Sci USA 98: 14000-14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LS, Rosoff ML, Li C (1998) Disruption of a neuropeptide gene, flp-1, causes multiple behavioral defects in Caenorhabditis elegans Science 281: 1686-1690. [DOI] [PubMed] [Google Scholar]
- Nurrish S, Ségalat L, Kaplan JM (1999) Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron 24: 231-242. [DOI] [PubMed] [Google Scholar]
- Parnot C, Miserey-Lenkei S, Bardin S, Corvol P, Clauser E (2002) Lessons from constitutively active mutants of G protein-coupled receptors. Trends Endocrinol Metab 13: 336-343. [DOI] [PubMed] [Google Scholar]
- Remm M, Sonnhammer E (2000) Classification of transmembrane protein families in the Caenorhabditis elegans genome and identification of human orthologs. Genome Res 10: 1679-1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robatzek M, Thomas JH (2000) Calcium/calmodulin-dependent protein kinase II regulates Caenorhabditis elegans locomotion in concert with a G(o)/G(q) signaling network. Genetics 156: 1069-1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers C, Reale V, Kim K, Chatwin H, Li C, Evans P, de Bono M (2003) Inhibition of Caenorhabditis elegans social feeding by FMRFamide-related peptide activation of NPR-1. Nat Neurosci 6: 1178-1185. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Rettig J, Brose N (2003) Molecular mechanisms of active zone function. Curr Opin Neurobiol 5: 509-519. [DOI] [PubMed] [Google Scholar]
- Sawin ER (1996) Genetic and cellular analysis of modulated behaviors in Caenorhabditis elegans PhD thesis, Massachusetts Institute of Technology.
- Schafer WR, Kenyon CJ (1995) A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans Nature 375: 73-78. [DOI] [PubMed] [Google Scholar]
- Ségalat L, Elkes DA, Kaplan JM (1995) Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans Science 267: 1648-1651. [DOI] [PubMed] [Google Scholar]
- Sengupta P, Chou JH, Bargmann CI (1996) odr-10 encodes a seven transmembrane domain olfactory receptor required for responses to the odorant diacetyl. Cell 84: 899-909. [DOI] [PubMed] [Google Scholar]
- Shyn SI, Kerr R, Schafer WR (2003) Serotonin and Go modulate functional states of neurons and muscles controlling C. elegans egg-laying behavior. Curr Biol 13: 1910-1915. [DOI] [PubMed] [Google Scholar]
- Sze JY, Zhang S, Li J, Ruvkun G (2002) The C. elegans POU-domain transcription factor UNC-86 regulates the tph-1 tryptophan hydroxylase gene and neurite outgrowth in specific serotonergic neurons. Development 129: 3901-3911. [DOI] [PubMed] [Google Scholar]
- Trent C, Tsung N, Horvitz HR (1983) Egg-laying defective mutants of the nematode Caenorhabditis elegans Genetics 104: 619-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, Bargmann CI (1995) Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans Cell 83: 207-218. [DOI] [PubMed] [Google Scholar]
- Tsalik EL, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O (2003) LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev Biol 263: 81-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner LE, Zhou GT, Schafer RW, Schafer WR (1998) Control of alternative behavioral states by serotonin in Caenorhabditis elegans Neuron 21: 203-214. [DOI] [PubMed] [Google Scholar]
- Waggoner LE, Hardaker LA, Golik S, Schafer WR (2000) Effect of a neuropeptide gene on behavioral states in Caenorhabditis elegans egg-laying. Genetics 154: 1181-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, Garriga G, Thomas JH (1995) Genetic and pharmacological analysis of neurotransmitters controlling egg laying in C. elegans J Neurosci 15: 6975-6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S (1986) The structure of the nervous system of the nematode Caenorhabditis elegans Philos Trans R Soc Lond B Biol Sci 275: 327-348. [DOI] [PubMed] [Google Scholar]