

Abstract
Alzheimer's disease (AD) is a progressive amnestic dementia that involves post-translational hyperphosphorylation, enzymatic cleavage, and conformational alterations of the microtubule-associated protein tau. The truncation state of tau influences many of its pathologic characteristics, including its ability to assume AD-related conformations and to assemble into filaments. Cleavage also appears to be an important marker in AD progression. Although C-terminal truncation of tau at D421 has recently been attributed to the apoptotic enzyme caspase-3, N-terminal processing of the protein remains mostly uncharacterized. Here, we report immunohistochemical staining in a cohort of 35 cases ranging from noncognitively impaired to early AD with a panel of three N-terminal anti-tau antibodies: Tau-12, 5A6, and 9G3-pY18. Of these three, the phosphorylation-independent epitope of 5A6 was the earliest to emerge in the pathological lesions of tau, followed by the appearance of the Tau-12 epitope. The unmasking of the Tau-12 epitope in more mature 5A6-positive tangles was not correlated with tau phosphorylation at tyrosine 18 (9G3-pY18). Still, later in the course of tangle evolution, the extreme N terminus of tau was lost, correlating temporally with the appearance of a C-terminal caspase-truncated epitope lacking residues 422-441. In addition, caspase-6 cleaved the N terminus of tau in vitro, preventing immunoreactivity with both Tau-12 and 5A6. Mass spectrometry confirmed that the in vitro caspase-6 truncation site is D13, a semicanonical and hitherto undescribed caspase cleavage site in tau. Collectively, these results suggest a role for caspase-6 and N-terminal truncation of tau during neurofibrillary tangle evolution and the progression of Alzheimer's disease.
Keywords: Alzheimer's disease, tau, neurofibrillary tangle, caspase, truncation, tyrosine phosphorylation
Introduction
A major feature of the molecular pathogenesis of Alzheimer's disease (AD) is the post-translational modification, misfolding, and aggregation of the microtubule-associated protein tau to form neurofibrillary tangles, dystrophic neurites, and neuritic plaques (Johnson and Jenkins, 1996; Duyckaerts et al., 1998; Johnson and Hartigan, 1998; Friedhoff et al., 2000; Uboga and Price, 2000; Mitchell et al., 2002). The evolution of tau structural changes in pathological specimens of AD brains can be inferred by comparing tangles in cases that vary in pathological severity. Such analyses have shown that through a complex series of phosphorylations, enzymatic cleavages, and conformational changes, tau transitions from its highly soluble and natively folded state to an insoluble accumulation of misfolded protein (Ghoshal et al., 2002; García-Sierra et al., 2003).
It is clear that enzymatic cleavage of tau plays an important role in the development of the neurofibrillary tangle as AD progresses. Cleavage at residue D421 is triggered by a caspase, most likely caspase-3 (Gamblin et al., 2003b; García-Sierra et al., 2003). The truncation state of tau influences many of its normal and pathologic characteristics, including its ability to bind to and stabilize microtubules (Fasulo et al., 1996; Canu et al., 1998), assume specific tangle-associated conformations (Carmel et al., 1996; Ghoshal et al., 2001), self-assemble into filaments (Perez et al., 1996; Abraha et al., 2000; Berry et al., 2003; Gamblin et al., 2003a), and induce apoptosis (Fasulo et al., 1998, 2000; Chung et al., 2001).
In contrast to C-terminal truncation of tau, little is known about its N-terminal processing. Tyrosine phosphorylation of tau at Y18 has recently been demonstrated in connection with activation of Fyn, a member of the src family of tyrosine protein kinases (Lee et al., 2004). Additionally, a number of studies have suggested that the N terminus of tau is lost as tangles become extracellular (Dickson et al., 1992; Endoh et al., 1993; Bondareff et al., 1994). At least one proteolytic cleavage has been postulated along a lengthy stretch of the molecule (between amino acids 9 and 210 of the epitopes of the Tau-12 and Tau-5 antibodies, respectively). The loss of N-terminal (Tau-12) immunoreactivity signifies evolution to more severe AD pathology (Ghoshal et al., 2002).
Here, we show that the phospho-independent antibody 5A6 is the first of three N-terminal antibodies to label early diffuse tangles. The Tau-12 epitope becomes unmasked as lesions assume a fibrillar morphology. This change appears to be unrelated to tyrosine phosphorylation at Y18 of tau, as detected by the 9G3-pY18 antibody. Subsequent to the unmasking of Tau-12, the extreme N-terminal epitopes of tau are lost from tangles of human AD brains, a process that correlates temporally with the appearance of the C-terminal caspase truncation-specific epitope at D421. Furthermore, we show that caspase-6 cleaves tau in vitro at amino acid D13, causing loss of immunoreactivity with both Tau-12 and 5A6 antibodies. These results suggest a role for caspase-6 in the N-terminal truncation of tau in tangle maturation and provide additional evidence for cleavage of tau by apoptosis-related proteases in Alzheimer's disease.
Materials and Methods
Epitope mapping of Tau-12, 5A6, and cross-reactive polyclonal. The antibodies used in this study are described in Table 1 and Figure 1 A. Epitopes for each antibody were defined by their reactivity or lack thereof with overlapping N-terminal internal deletion mutants of tau (Carmel et al., 1996). The expression and purification of full-length tau (hTau40, 2N4R) (Goedert et al., 1989) and tauΔ2-18 have been described previously (Carmel et al., 1996; Abraha et al., 2000; Gamblin et al., 2003a). Novel internal deletion mutations were generated using PCR on pT7c-ht40 with primers immediately flanking the desired deletion: tauΔ18-42:Tau18 F (5′-PO4-CGTCCCAGCGTGATCTTCCAATCAC) and Tau42 R (5′-PO4-CTGAAAGAATCTCCCCTGCAGACC); tauΔ18-30:Tau18 F (above) and Tau30 R (5′-PO4-ATGCACCAAGACCAAGAGGTGAC); tauΔ24-36:Tau24 F (5′-PO4-CCTGTCCCCCAACCCGTACGTC) and Tau36 R (5′-PO4-GGTGACACGGACGCTGGCCTGAAA). The amplified sequences were religated and expressed as described above. Proteins were analyzed by SDS-PAGE and Western blotting with Tau-12 (1:250,000), 5A6 (1:50,000), cross-reactive polyclonal (CR Polyclonal; 1:5000), and Tau-5 (1:50,000). Horseradish peroxidase (HRP)-conjugated secondary antibody (Vector Laboratories, Burlingame, CA) and enhanced chemiluminescence were used to visualize bands (Amersham Biosciences, Piscataway, NJ). The affinity of each antibody for purified recombinant hTau40 was also assessed by ELISA as described previously (Ghoshal et al., 2001).
Table 1.
Characteristics of the antibodies used in this study
|
Antibody |
Epitope |
Host/Class |
Comments |
References |
|---|---|---|---|---|
| Tau-12 | 9-18* | Mouse/IgG | Ghoshal et al., 2002 | |
| CR Polyclonal | 12-24* | Rabbit | Lee et al., 2004 | |
| 9G3-pY18 | 12-24, pY18 | Mouse/IgG | Phospho-Tyr18 specific | Lee et al., 2004 |
| 5A6 | 19-46* | Mouse/IgG | Johnson et al., 1997 | |
| Tau-5 | 210-230 | Mouse/IgG | Carmel et al., 1996 | |
| MN423 | Trunc-E391 | Mouse/IgG | Truncation specific at E391 | Wischik et al., 1988b; Novak et al., 1993 |
| Tau-C3 | Trunc-D421 | Mouse/IgG | Truncation specific at D421, associated with caspases | Gamblin et al., 2003b |
| Tau-46.1 |
428-441 |
Mouse/IgG |
Intact C terminus |
Kosik et al., 1988; Carmel et al., 1996 |
Asterisks denote epitopes that were remapped in this report.
Figure 1.
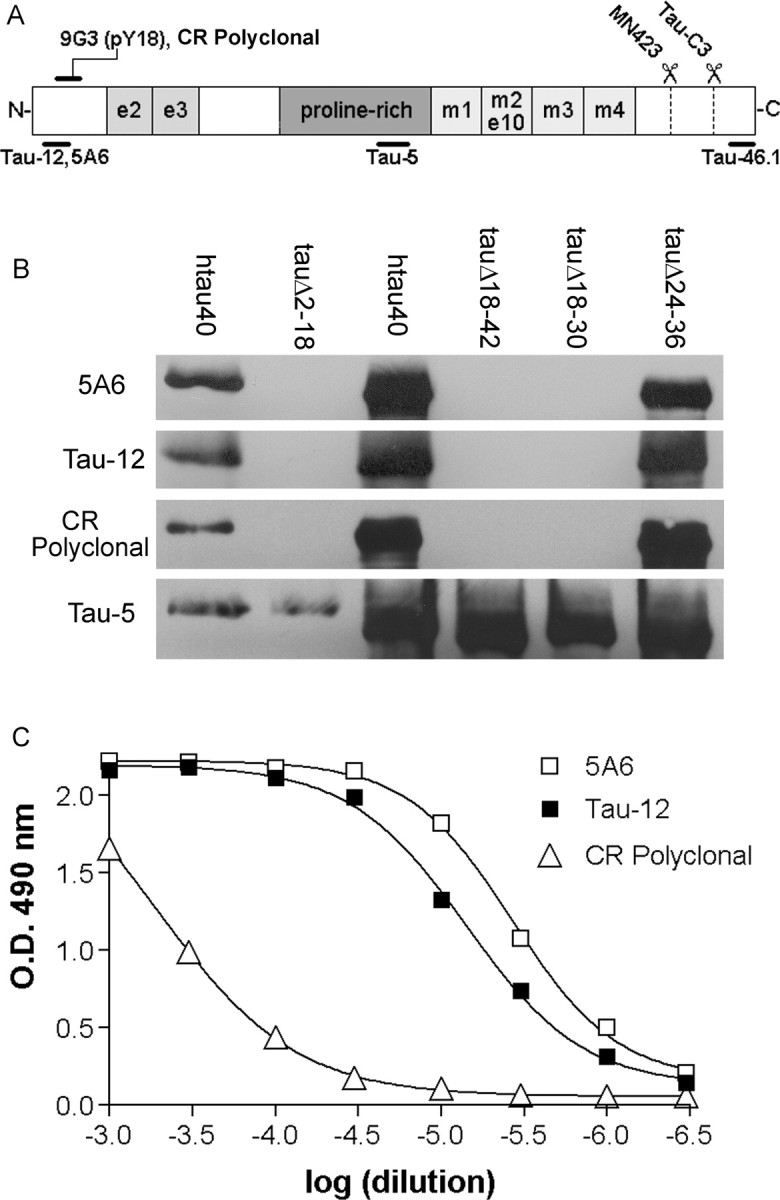
Epitope maps of the tau antibodies used. A, The structure of the longest human tau isoform found in brain (hTau40) with the approximate locations of the epitopes from the antibodies used in this study. B, The 5A6, Tau-12, and CR polyclonal phospho-independent epitopes were re-examined using internal deletion mutations of tau. Left two lanes, Purified recombinant protein; right four lanes, whole lysates of bacteria induced to express recombinant proteins. All three epitopes overlap (residues 2-23) and encompass tyrosine 18 of tau. Tau-5 shows total tau on the membrane. C, The relative affinities of the three antibodies for purified recombinant hTau40 were compared by ELISA. Tau-12 and 5A6 both show very high affinities.
Brain tissue. In experiments addressing the earliest changes in tau pathology, samples of middle temporal gyrus (MTG) were obtained from the Religious Orders Study (ROS) cohort at the Rush University Alzheimer's Disease Center (Chicago, IL). The 35 cases from the ROS have been characterized extensively, both clinically and pathologically (Mufson et al., 1999; Kordower et al., 2001; Bennett et al., 2002; Ghoshal et al., 2002; Wilson et al., 2002). To study more advanced N-terminal changes, additional tissue was obtained from five pathologically confirmed severe AD cases (Braak stage V-VI) (Braak and Braak, 1995) from the Northwestern University Cognitive Neurology and Alzheimer's Disease Center Brain Bank.
Immunohistochemistry. In single-label studies, immunohistochemistry was performed on adjacent 40 μm sections of middle temporal gyrus from the 35 ROS cases as described previously (Ghoshal et al., 2002) using the N-terminal monoclonal tau antibodies Tau-12 (1:500,000), 9G3-pY18 (1:5000), and 5A6 (1:10,000). In double-label experiments for brightfield microscopy, sections from the temporal lobes of severe AD cases were incubated with CR Polyclonal (1:5000) and either Tau-12 (1:250,000) or 5A6 (1:50,000). Appropriate secondary antibodies conjugated to either HRP or biotin and a Vectastain ABC-alkaline phosphatase (AP) solution were used, and Nova Red and AP substrate kit III were used as chromogens (Vector Laboratories).
Quantitative and statistical analysis of immunohistochemistry. Slides from the ROS cases were analyzed using StereoInvestigator software (MicroBrightField, Williston, VT) with the experimenter (P.M.H.) blind to all information about the slides (including case, diagnosis, and antibody stain). The seven cases with the most extensive pathology were analyzed using systematic random sampling (fractionator). In cases with less severe pathology, the gyrus was counted in its entirety. In all cases, only tangles containing a visible perinuclear region were counted. Because of the range in pathological severity of the cases studied, it was necessary to normalize the data to a fourth section labeled with all three primary antibodies to allow quantitative comparisons of tangle labeling across cases. A simple linear correlation analysis (Pearson r test) was performed on the normalized data using Prism 2.01 software (GraphPad Software, San Diego, CA). Normalized Tau-12, 9G3-pY18, and 5A6 staining across the cohort were compared by ANOVA and protected paired t tests.
Double-label immunofluorescence. Double-label immunofluorescence (IF) confocal microscopy was performed as described previously (García-Sierra et al., 2003) on temporal lobe tissue from severe AD cases. Tissue was incubated with two of the following primary antibodies: 5A6 (1:50,000), biotinylated Tau-12 (1:100), CR Polyclonal (1:2000), Tau-46.1 (1:4000), Tau-C3 (1:2500), and MN423 (1:500). Species- or biotin-specific secondary antibodies linked to Texas Red, FITC, or cyanine 5 (Cy5) were used (Jackson ImmunoResearch, West Grove, PA; Molecular Probes, Eugene, OR), and sections were viewed using the 60× objective on a 510-Zeiss (Thornwood, NY) laser scanning confocal microscope. Individual sections, two-dimensional projections, and three-dimensional rotations were analyzed with the software included with the microscope (Zeiss LSM Image Browser v.2.30.011). For quantification, projections of three fields (325 × 325 μm) encompassing cortical layers II-III of the superior temporal gyrus (STG) were analyzed per case (n = 3) per antibody pair.
Caspase-6 treatment of recombinant tau. A double-deletion mutant lacking both N and C termini (tauΔ2-18Δ422-441) was generated by restriction endonuclease digestion of both single-deletion mutants (Gamblin et al., 2003a,b) with XhoI and SacI (New England Biolabs, Beverly, MA). Digestion products were separated by agarose gel electrophoresis, and fragments containing the desired mutations were extracted from the gel, religated, and used to transform cells as described above. Control hTau40 and tau deletion mutants (100 ng/μl) were incubated with active caspase-6 (5 ng/μl) at 37°C for 24 hr (BioMol, Plymouth Meeting, PA). Products were analyzed by SDS-PAGE and Western blotting (described above). Representative data from three independent experiments are shown.
Fyn treatment of tauΔ422-441 and Y18F-tauΔ422-441. A tyrosine to phenylalanine mutant of hTau40 was generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The forward (F) and reverse (R) mutant primers used in the reaction were as follows: Y18F F, GATCACGCTGGGACGTTCGGGTTGGGGGACAGG; and Y18F R, CTGTCCCCCAACCCGAACGTCCCAGCGTG. This mutation was then excised with XhoI/SacI and ligated onto a tauΔ422-441 background as described above. All mutations were confirmed by DNA sequencing. Purified recombinant tauΔ422-441 and Y18F-tauΔ422-441 (100 ng/μl) were incubated for 1 hr at 37°C with or without 1 ng/μl active Fyn (Upstate Biotechnology, Lake Placid, NY). Active caspase-6 (5 ng/μl) was added directly to the phosphorylation reaction for 24 hr at 37°C, and the products were analyzed by Western blotting with 9G3-pY18, Tau-5, and Tau-12.
Mass spectrometry of untreated and caspase-6-treated tauΔ422-441. The intact molecular masses of untreated and caspase-6-treated tauΔ422-441 were determined in triplicate by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF MS) in linear mode on an Applied Biosystems (Foster City, CA) Voyager-DE STR. For peptide mass fingerprinting analyses, untreated and caspase-6-treated tauΔ422-441 were trypsinized, desalted by solid-phase ZipTip extraction (Millipore, Bedford, MA), and analyzed by MALDI-TOF MS in reflector mode.
Results
N-terminal tau epitopes differ in prevalence in tangles of early AD
The phospho-independent Tau-12 and 5A6 monoclonal antibodies and CR Polyclonal antibody (Fig. 1A, Table 1) were selected for investigation of early N-terminal changes because of their overlapping epitopes spanning the extreme N terminus of tau. The contribution of tyrosine phosphorylation of tau at Y18 was assessed with the phosphotyrosine-specific 9G3-pY18 antibody, raised against a phosphorylated peptide corresponding to amino acids 12-24 of tau (Lee et al., 2004). Tau-12 (previously attributed to residues 9-18) and 5A6 (residues 19-46) were both raised against purified recombinant human tau (Johnson et al., 1997; Ghoshal et al., 2002).
Because previous reports indicated that the epitopes of Tau-12, 5A6, and CR Polyclonal are in close proximity, they were remapped using novel, smaller, N-terminal internal deletion mutations of tau (Fig. 1B). These data show a great deal of similarity between the epitopes of all three antibodies and suggest that the epitope for 5A6 extends further N terminally than was previously thought. By virtue of binding to tauΔ24-36 but not tauΔ2-18 or tauΔ18-30, all three antibodies bind somewhere within a span of tau (residues 2-23) that includes the Fyn target Y18. Both 5A6 and Tau-12 show a strong affinity for purified recombinant human tau (Fig. 1C). However, because the affinity of an antibody for tau in fixed human tissue may differ from its affinity for recombinant tau in vitro, all antibodies were retitered on tissue before use.
To better understand the early tau changes that take place in the tangles of AD, tissue from a cohort of 35 cases ranging from no cognitive impairment to mild AD was immunolabeled with the three N-terminal monoclonal antibodies described above. MTG tissue was selected because of the wide range of pathological severities observed in this region of cortex (from a few isolated tangles or threads to widespread extensive tau lesions spanning cortical layers II-VI).
We found marked differences in the prevalence of the three N-terminal tau epitopes in neurofibrillary tangles. The number of tangles labeled with 5A6 did not significantly differ from the number of tangles stained with all three primary antibodies combined (p > 0.2), whereas Tau-12 and 9G3-pY18 labeled fewer tangles than 5A6 (Fig. 2A). Furthermore, in several of the pathologically mild cases, 5A6-labeled tangles were observed in the absence of Tau-12 labeling (five cases), whereas the reverse was never observed (Fig. 2B, left). In the case of Tau-12 and 5A6, these findings were unexpected, because our epitope mapping experiments suggested significant overlap of the two epitopes (see Discussion).
Figure 2.

N-terminal tau epitopes differ in prevalence in human brain tissue. A, Quantitative analysis in the ROS cohort shows that 5A6 labels far more tangles than either Tau-12 or 9G3-pY18 (89.0 ± 12.1, 13.1 ± 2.9, and 26.1 ± 3.0%, respectively; all 3 differ from each other at p < 0.001). B, Correlation analysis of normalized tangle densities shows a positive predictive relationship between the tangle labeling of Tau-12 and 5A6 (Pearson, r = 0.516; p = 0.0015), whereas 9G3-pY18 labeling is not significantly correlated with either of these antibodies (p = 0.40 and 0.20).
A correlation analysis was then performed on the data (Fig. 2B). Although labeling with the two phospho-independent antibodies showed a positive correlation, neither of these were significantly correlated with 9G3-pY18 labeling. These results suggest an in situ relationship between the Tau-12 and 5A6 epitopes despite the disparity in the number of tangles stained.
Morphologically, 5A6 labeled both early diffuse tangles and compact fibrillar tangles (Fig. 3A-C), whereas Tau-12 primarily labeled compact tangles (Fig. 3D,E), as did 9G3-pY18 (data not shown). The DAB staining of 5A6 also extended further into the apical dendritic arbor (Fig. 3C) than that of either Tau-12 (Fig. 3D,E) or 9G3-pY18 (data not shown). Interestingly, neuropil threads, thought to be the earliest manifestation of tau pathology (Ghoshal et al., 2002), were heavily labeled with 5A6 in many of the cases but were revealed only in more pathologically severe cases with Tau-12 and 9G3 (data not shown). The highlighting of neuritic plaques (Fig. 3F) followed a similar pattern. Thus, 5A6 labeled tau lesions earlier than either Tau-12 or 9G3-pY18.
Figure 3.

Tau pathology stained with N-terminal antibodies. A, A diffuse tangle in the MTG of a case from the ROS cohort is labeled with 5A6 and visualized with DAB. B, Diffuse (top) and fibrillar (bottom) tangles are labeled with 5A6. C, Transitional tangles exhibit both diffuse and fibrillar 5A6 labeling. Extensive labeling of the dendritic tree is often seen with 5A6. D, A representative Tau-12-positive tangle in the same case as A-C has a compact, fibrillar appearance and lacks extensive dendritic staining. E, Tau-12 stains dense neurofibrillary tangles in the inferior temporal gyrus of a severe AD case. F, 5A6 also readily labels other tau lesions, including neuritic plaques, in pathologically mild cases. G, Brightfield microscopy depicts three tangles in the hippocampus (CA1) of a severe AD case that are double labeled for 5A6 (red) and the polyclonal N-terminal antibody CR Polyclonal (blue). This antibody pair shows ∼100% cosegregation in tangles. H-J, Double-label laser scanning confocal microscopy demonstrates the colocalization of biotinylated Tau-12 (H) and 5A6 (I) in tangles of the STG of severe AD(merged image; J). Images A-G are at the same magnification as are H-J. Scale bars, 20 μm.
To test the hypothesis that the more mature and compact 5A6-positive tangles also label with Tau-12, sections from severe AD cases were double labeled with either 5A6 or Tau-12 and the N-terminal phospho-independent polyclonal peptide antibody CR Polyclonal (Fig. 3G). Both Tau-12 and 5A6 showed ∼100% double labeling with CR Polyclonal in these tangles, suggesting that they label the same population of tangles. In addition, confocal microscopy using 5A6 and biotinylated Tau-12 in severe AD brains demonstrated numerous double-labeled compact tangles (Fig. 3H-J). These data support the hypothesis that the Tau-12 epitope is unmasked in the more mature and fibrillar 5A6-positive tangles of more pathologically advanced AD cases.
Loss of N-terminal tau epitopes correlates temporally with the appearance of a C-terminal caspase cleavage epitope
Previous data suggested that tau is cleaved at a site between the Tau-12 and Tau-5 epitopes (amino acids 9-210) and that the loss of the Tau-12 epitope correlates with progression to more severe impairment on tests of episodic memory (Ghoshal et al., 2002). To determine the timing of this important change in individual tangles, a pathological timescale was used that makes use of a defined sequence of C-terminal truncations of tau (Kosik et al., 1988; Wischik et al., 1988a; Carmel et al., 1996; Ghoshal et al., 2002; Gamblin et al., 2003b; García-Sierra et al., 2003). Sections from severe AD cases were labeled with CR Polyclonal and three separate C-terminal antibodies (Fig. 4A): Tau-46.1 (intact C terminus), Tau-C3 (caspase-truncated C terminus at D421), and MN423 (late-stage C-terminal truncation at E391). As truncations of tau gradually remove epitopes external to the cleavage sites, this trio of antibodies provides a useful approach to characterizing in situ the timing of the changes tau undergoes in the tangles of AD brains.
Figure 4.
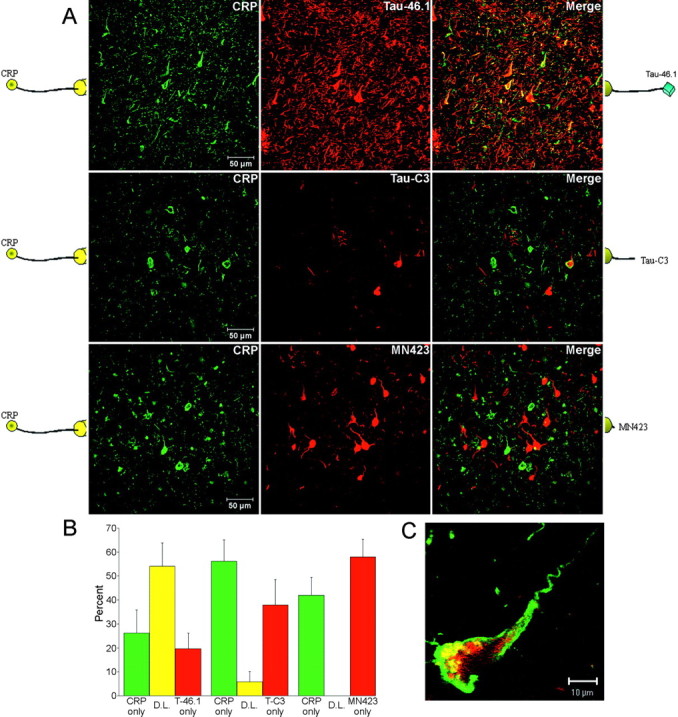
Loss of N-terminal tau epitopes correlates temporally with the appearance of Tau-C3. A, Tissue from severe AD brains is labeled with CR Polyclonal (green) and one of three C-terminal monoclonal antibodies (Tau-46.1, Tau-C3, and MN423 in red; epitopes shown to the right of the merged image). Images represent projections of several optical sections. B, CR Polyclonal labeling cosegregates well with Tau-46.1 [54.2 ± 9.7% double labeled (D.L.)] but is rarely seen in the same tangles as Tau-C3 (5.9 ± 4.3%) and is never observed with MN423. C, The tangles that double label with Tau-C3 and CR Polyclonal show a distinct pattern: Tau-C3 (red) appears mostly in the core of the tangle, whereas CR Polyclonal (green) primarily decorates the periphery of the somatodendritic compartment. This merged image represents a single optical section.
Quantitative analysis of these data showed that the extreme N-terminal marker CR Polyclonal cosegregates well with the intact C-terminal marker Tau-46.1 in tangles but is rarely seen in tangles with Tau-C3 and never with MN423 (Fig. 4B). In the rare cases of CR Polyclonal-Tau-C3 cosegregation, the tangles were usually chimeric (i.e., Tau-C3 staining occupied a central region of the tangle, and CR Polyclonal staining was restricted to the peripheral soma and neuronal processes) (Fig. 4C). These results indicate that the loss of the N terminus of tau correlates temporally with the appearance of a C-terminal truncation associated with caspase activation.
Caspase-6 cleaves tau in vitro at D13 causing loss of N-terminal epitopes
Examination of the N-terminal sequence of tau revealed a semicanonical caspase-6 site (MAEPRQEFEVMED*HA...) that would result in cleavage after aspartic acid 13 of tau, which is at the center of the Tau-12 and 5A6 epitopes. An in vitro caspase-6 assay was performed using recombinant tau with a deletion of the C terminus mimicking C-terminally caspase-truncated tau (tauΔ422-441). Subsequent Western blotting with Tau-5 showed that caspase-6 indeed cleaved tauΔ422-441 to generate a single lower molecular weight band (Fig. 5A, top). The truncated tau band was not immunoreactive with either Tau-12 or 5A6 antibodies (Fig. 5A, bottom), suggesting that the truncation was N terminal and not at another possible C-terminal caspase-6 site (D402). Tau harboring an additional N-terminal deletion mutation that removed the caspase site (tauΔ2-18Δ422-441) was not cleaved by caspase-6 (data not shown). Caspase-6 also cleaved full-length wild-type tau and tauΔ2-18 at D421, albeit with far lower efficiency than at the D13 site (data not shown). This is consistent with other reports showing low-efficiency caspase-6 cleavage at canonical DXXD caspase-3 sites (Talanian et al., 1997; Thornberry et al., 1997; Gamblin et al., 2003b).
Figure 5.

Caspase-6 cleaves tau N terminally in vitro at amino acid D13. A, Caspase-6 treatment of tauΔ422-441 causes the appearance of a single lower molecular weight band recognized by Tau-5 but not by Tau-12 or 5A6. B, Tyrosine phosphorylation at residue 18 of tau by Fyn (top) has little effect on the extent of caspase-6 cleavage as shown by Tau-5 Western blotting (bottom). C, MALDI-TOF mass spectra of in-solution tryptic digests of samples in A show a shift of the peaks at m/z 2053.9 and 2181.9 to 1046.5 and 1174.6 (arrows), respectively, as would be expected for cleavage at D13. All peaks labeled in the spectra correspond to predicted tryptic peptides of tauΔ422-441.
Although Y18 phosphorylation was not correlated with the 5A6 → Tau-12 progression of early tangles, we investigated its possible role in the caspase-6 cleavage of the N terminus. Compared with untreated tauΔ422-441, Fyn phosphorylation at Y18 did not greatly affect the extent of N-terminal cleavage (Fig. 5B). TauΔ422-441 Y18F, an unphosphorylatable mutant, was also studied to control for the possible direct effects of Fyn on caspase-6 activity. These data suggest that tyrosine phosphorylation of tau at Y18 would have little impact on the N-terminal processing of the protein by caspase-6 in AD.
To confirm the exact location of the in vitro N-terminal caspase-6 truncation, the control and caspase-6-treated samples were subjected to mass spectrometric analysis (Fig. 5C). In-solution digestion with trypsin yielded fragments that were identified monoisotopically using MALDI-TOF mass spectrometry. Trypsin cleaves intact tau to yield a doublet encompassing the predicted truncation site (Fig. 5C, top). In samples treated with caspase, the doublet is shifted to lower molecular mass, corresponding to the predicted cleavage of the peptide at D13 (Fig. 5C, bottom). These results were confirmed by determining the intact molecular masses of untreated and caspase-6-treated tauΔ422-441 (data not shown). These results suggest that N-terminal caspase cleavage at D13 of tau may contribute to the loss of N-terminal epitopes from tangles in AD brains.
Discussion
Early tau evolution in AD tangles
Characterizing the early changes that tau undergoes is critical to understanding the process of tangle formation and the role of tau evolution in the development of AD. In this study, we investigated the changes that take place in the extreme N terminus of tau, a relatively uncharted region of this molecule. Although the epitopes of Tau-12, 9G3-pY18, and 5A6 have been described separately (Johnson et al., 1997; Ghoshal et al., 2002; Lee et al., 2004), the appearance, timing, and relationships of the three extreme N-terminal tau antibodies have not been investigated previously together.
Here, we have shown that 5A6 is the first of the N-terminal antibodies to decorate tau pathology in AD tangle development. The 5A6 epitope predominates in tangles from the ROS cohort, a group that ranges clinically from no cognitive impairment to mild AD. In the cases with the mildest pathology, 5A6 labeling consistently appears in the absence or paucity of Tau-12 labeling. As pathological severity increases, the Tau-12 epitope is unmasked in fibrillar compact tangles, and Tau-12 is shown to colocalize with 5A6 in most tangles of severe cases. Although the trigger for this change remains unclear, it appears to be unrelated to Y18 phosphorylation and to occur before the activation of caspases.
Despite our inability to differentiate the Tau-12 and 5A6 epitopes at the molecular level using recombinant tau deletion mutants, the two antibodies clearly show distinct staining patterns in AD tissue. A number of explanations for this disparity can be proposed. Destruction of a portion of the epitope could reduce antibody affinity by removing important hydrophobic or charge-charge interactions between antibody and antigen. Also, removal of a nearby region of the molecule not directly involved in antibody binding could sufficiently disrupt the orientation or structure of surrounding regions to reduce antibody affinity. These possibilities may explain why 5A6 was unable to bind both tauΔ18-42 and tau cleaved at D13. Alternatively, conformational changes (see below) or as yet unidentified post-translational modifications at the N terminus of tau could influence the in situ presentation of the epitope. Such modifications could cause 5A6 alone to bind diffuse tangles while allowing both 5A6 and Tau-12 access to their epitopes when tau is arranged in a fibrillar morphology. Although 5A6 and Tau-12 show similarly high affinities for purified recombinant tau, changes in conformation and other post-translational modifications could cause them to have different affinities for tau in fixed AD tissue. A finer mapping of the epitopes of these two antibodies is warranted, as is investigation of the contributions of post-translational modifications to the affinities of these antibodies.
We also offer evidence that the subsequent loss of the N terminus of tau from AD tangles may be caused by caspase activation. First, the N-terminal marker CR Polyclonal cosegregated well with the intact C-terminal marker Tau-46.1 in tangles but poorly with Tau-C3, a marker of C-terminal caspase cleavage at D421. In addition, caspase-6 is shown to cleave tau specifically at the N terminus in vitro, causing loss of binding of N-terminal antibodies. Because caspases 3 and 6 are capable of activating each other (Cohen, 1997; Cryns and Yuan, 1998), tau may be the target of a coordinated onslaught of caspase activity.
D13 is novel and different from other caspase sites in tau
A number of studies have provided evidence that tau is an important substrate for caspases (Canu et al., 1998; Fasulo et al., 2000; Chung et al., 2001; Rohn et al., 2002; Gamblin et al., 2003b; Rissman et al., 2004). Although most of these reports focus on the C-terminal truncation of tau at D421, Rohn et al. (2002) describe another possible N-terminal caspase-3 cleavage site at D25 (DRKD*) by use of a polyclonal antibody to the putative cleavage site. However, in an in vitro study, this site was not cleaved by any known caspase (Gamblin et al., 2003b), and basic residues immediately N terminal to the cleavage site are known to inhibit cleavage by caspase-3 subfamily members (Talanian et al., 1997; Thornberry et al., 1997). Here, we provide direct evidence of in vitro N-terminal cleavage of tau at D13, a hitherto undescribed site. This site (VMED*) varies from the canonical caspase-6 site (VEID* in lamin A) (Cohen, 1997) in that the acidic and hydrophobic residues are reversed. The mass spectrometric data presented here do not rule out C-terminal cleavage at another putative caspase-6 site (D402); however, such a cleavage was not detected by Western blotting. Although we have described in vitro truncation of tau by caspase-6 specifically at D13, it is still unknown whether this truncation state of tau exists in AD brain. Furthermore, additional N-terminal truncations in more mature tangles may take place in vivo as well. Precedent for this type of processing of tau is found at its C terminus, where the Tau-C3 epitope is replaced by that of MN423 as the tangle evolves.
Timing and significance of N-terminal cleavage
Previous work has shown that loss of Tau-12 immunoreactivity in tangles indicates progression to more mature tau pathology (Ghoshal et al., 2002; García-Sierra et al., 2003). In this report, we show that the N terminus of tau is lost from tangles in AD brains at approximately the same time as caspase cleavage of the C terminus of tau (likely caused by caspase-3). The association of the bilateral cleavage of tau with activation of programmed cell death-associated proteases has grave implications with regard to neuronal viability and suggests an integral role for caspases in tangle evolution.
The N-terminal 15 amino acids of tau are required for the formation of the Alz-50 conformation of tau (Carmel et al., 1996; Jicha et al., 1997), a conformation known to be a marker for early tangles (Hyman et al., 1988; Ghoshal et al., 2002; García-Sierra et al., 2003). The Alz-50 conformation of tau is gradually replaced by another conformation in tangles, Tau-66 (Ghoshal et al., 2001; García-Sierra et al., 2003), which does not require an intact N terminus. Although removal of the N terminus of tau decreases the rate and extent of tau polymerization in vitro (Gamblin et al., 2003a), N-terminal truncation may stabilize existing filaments through a conformational shift from the Alz-50 to Tau-66 conformations. Thus, N-terminal cleavage of tau could play an integral part in tangle maturation in AD.
Role of caspases and tau in AD
Apoptosis has long been hypothesized as the mechanism of neuronal loss in AD (Smale et al., 1995; Cotman and Su, 1996; Desjardins and Ledoux, 1998). Altered levels of pro-apoptotic and anti-apoptotic proteins are found in transgenic models of AD (Hwang et al., 2002) and human AD tissue (Kitamura et al., 1998), including the p10 fragment of active caspase-6 (LeBlanc et al., 1999). The presence of active caspase-6, in particular, is necessary and sufficient to cause apoptosis in primary human neuronal cultures (Zhang et al., 2000). Interestingly, microinjection of these cultured neurons with levels of caspase-6 below a threshold increased the susceptibility of these cells to other insults without triggering immediate apoptosis. This suggests that although some tau may be truncated in AD neurons, indicating caspase activation, these cells may still be viable.
Amyloid β-induced neurotoxicity, which requires the presence of tau (Rapoport et al., 2002), is thought to involve apoptotic mechanisms, and caspase cleavage of tau and other substrates are associated with neurofibrillary tangle formation (Rohn et al., 2001; Gamblin et al., 2003b). Because tau cleavage products may be intrinsically neurotoxic (Canu et al., 1998; Fasulo et al., 2000; Chung et al., 2001), caspase cleavage of tau may represent part of a positive feedback loop in neurons of AD brains. This body of evidence also suggests that caspases may be a crucial link between amyloid β and tau lesions in AD (Canu et al., 1998; Rohn et al., 2002; Gamblin et al., 2003b). Overall, the data support the interpretation that caspase cleavage of tau is not merely a reflection or marker of enzyme activation in ailing neurons but is instead actively involved in the disease process.
Footnotes
This work was supported by National Institutes of Health Grants AG20418, AG20506, AG21661, AG09466, AG13854, AG10161, and AG15819. We thank Drs. Gloria Lee (University of Iowa, Iowa City, IA), Gail Johnson (University of Alabama at Birmingham, Birmingham, AL), Virginia Lee (University of Pennsylvania, Philadelphia, PA), and Michal Novak (Slovak Academia of Sciences, Bratislava, Slovakia) for the generous gifts of antibodies 9G3-pY18 and CR polyclonal, 5A6, Tau-46.1, and MN423, respectively. We continue to be indebted to the hundreds of nuns, priests, and brothers of the Religious Orders Study as well as those individuals who participated in the Northwestern University Cognitive Neurology and Alzheimer's Disease Center brain donation program. We also thank Julie Bach, Beth Howard, Dr. Eileen Bigio, the coordinators of the Religious Orders Study, the Rush Brain Bank, and Northwestern Brain Bank. We thank Dr. Nupur Ghoshal for her assistance in managing the ROS brain tissue and Drs. T. Chris Gamblin and Francisco García-Sierra for contributing to the artwork in several diagrams.
Correspondence should be addressed to Peleg M. Horowitz, Department of Cell and Molecular Biology, Northwestern University, 303 East Chicago Avenue, Chicago, IL 60611. E-mail: p-horowitz@md.northwestern.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247895-08$15.00/0
References
- Abraha A, Ghoshal N, Gamblin TC, Cryns V, Berry RW, Kuret J, Binder LI (2000) C-terminal inhibition of tau assembly in vitro and in Alzheimer's disease. J Cell Sci 113: 3737-3745. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J (2002) Natural history of mild cognitive impairment in older persons. Neurology 59: 198-205. [DOI] [PubMed] [Google Scholar]
- Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, Binder LI (2003) Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry 42: 8325-8331. [DOI] [PubMed] [Google Scholar]
- Bondareff W, Harrington C, Wischik CM, Hauser DL, Roth M (1994) Immunohistochemical staging of neurofibrillary degeneration in Alzheimer's disease. J Neuropathol Exp Neurol 53: 158-164. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1995) Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging 16: 271-278. [DOI] [PubMed] [Google Scholar]
- Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, Novak M, Cattaneo A, Bradbury A, Calissano P (1998) Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci 18: 7061-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmel G, Mager EM, Binder LI, Kuret J (1996) The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem 271: 32789-32795. [DOI] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK (2001) Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 8: 162-172. [DOI] [PubMed] [Google Scholar]
- Cohen GM (1997) Caspases: the executioners of apoptosis. Biochem J 326: 1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Su JH (1996) Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol 6: 493-506. [DOI] [PubMed] [Google Scholar]
- Cryns V, Yuan J (1998) Proteases to die for. Genes Dev 12: 1551-1570. [DOI] [PubMed] [Google Scholar]
- Desjardins P, Ledoux S (1998) Expression of ced-3 and ced-9 homologs in Alzheimer's disease cerebral cortex. Neurosci Lett 244: 69-72. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Ksiezak-Reding H, Liu WK, Davies P, Crowe A, Yen SH (1992) Immunocytochemistry of neurofibrillary tangles with antibodies to sub-regions of tau protein: identification of hidden and cleaved tau epitopes and a new phosphorylation site. Acta Neuropathol 84: 596-605. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Colle MA, Dessi F, Piette F, Hauw JJ (1998) Progression of Alzheimer histopathological changes. Acta Neurol Belg 98: 180-185. [PubMed] [Google Scholar]
- Endoh R, Ogawara M, Iwatsubo T, Nakano I, Mori H (1993) Lack of the carboxyl terminal sequence of tau in ghost tangles of Alzheimer's disease. Brain Res 601: 164-172. [DOI] [PubMed] [Google Scholar]
- Fasulo L, Ovecka M, Kabat J, Bradbury A, Novak M, Cattaneo A (1996) Overexpression of Alzheimer's PHF core tau fragments: implications for the tau truncation hypothesis. Alzheimer's Res 2: 195-200. [Google Scholar]
- Fasulo L, Visintin M, Novak M, Cattaneo A (1998) Tau truncation in Alzheimer's disease: expression of a fragment encompassing PHF core tau induces apoptosis in COS cells. Alzheimer's Reports 1: 25-32. [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A (2000) The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 75: 624-633. [DOI] [PubMed] [Google Scholar]
- Friedhoff P, von Bergen M, Mandelkow EM, Mandelkow E (2000) Structure of tau protein and assembly into paired helical filaments. Biochim Biophys Acta 1502: 122-132. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI (2003a) Tau polymerization: role of the amino terminus. Biochemistry 42: 2252-2257. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Abraha A, Miller R, Fu Y, Garcia-Sierra F, Lagalwar S, Berry RW, Binder LI, Cryns VL (2003b) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA 100: 10032-10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sierra F, Ghoshal N, Quinn B, Berry R, Binder LI (2003) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimer's Disease 5: 65-77. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, García-Sierra F, Fu Y, Beckett LA, Mufson EJ, Kuret J, Berry RW, Binder LI (2001) Tau-66: evidence for a novel tau conformation in Alzheimer's disease. J Neurochem 77: 1372-1385. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, Garcia-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI (2002) Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol 177: 475-493. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8: 393-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH, Cho JY, Hong JT, Song CW, Paik SG, Cho JS, Kim YK (2002) Alterations in behavior, amyloid beta-42, caspase-3, and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer's disease. FASEB J 16: 805-813. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Wolozin BL, Davies P, Kromer LJ, Damasio AR (1988) Alz-50 antibody recognizes Alzheimer-related neuronal changes. Ann Neurol 23: 371-379. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P (1997) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48: 128-132. [DOI] [PubMed] [Google Scholar]
- Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D (1997) The tau protein in human cerebrospinal fluid in Alzheimer's disease consists of proteolytically derived fragments. J Neurochem 68: 430-433. [DOI] [PubMed] [Google Scholar]
- Johnson GVW, Hartigan JA (1998) Tau protein in normal and Alzheimer's disease brain: an update. Alzheimer's Disease Rev 3: 125-141. [DOI] [PubMed] [Google Scholar]
- Johnson GVW, Jenkins SM (1996) Tau protein in normal and Alzheimer's disease brain. Alzheimer's Disease Rev 1: 38-54. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, Smith MA, Perry G, Whitehouse PJ, Taniguchi T (1998) Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res 780: 260-269. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Stebbins GT, DeKosky ST, Cochran EJ, Bennett D, Mufson EJ (2001) Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann Neurol 49: 202-213. [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM, Lee G (1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1: 817-825. [DOI] [PubMed] [Google Scholar]
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J (1999) Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem 274: 23426-23436. [DOI] [PubMed] [Google Scholar]
- Lee G, Thangavel R, Sharma VM, Litersky JM, Bhaskar K, Fang SM, Do LH, Andreadis A, Van Hoesen G, Ksiezak-Reding H (2004) Phosphorylation of tau by fyn: implications for Alzheimer's disease. J Neurosci 24: 2304-2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell TW, Mufson EJ, Schneider JA, Cochran EJ, Nissanov J, Han LY, Bienias JL, Lee VM, Trojanowski JQ, Bennett DA, Arnold SE (2002) Parahippocampal tau pathology in healthy aging, mild cognitive impairment, and early Alzheimer's disease. Ann Neurol 51: 182-189. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Chen EY, Cochran EJ, Beckett LA, Bennett DA, Kordower JH (1999) Entorhinal cortex beta-amyloid load in individuals with mild cognitive impairment. Exp Neurol 158: 469-490. [DOI] [PubMed] [Google Scholar]
- Novak M, Kabat J, Wischik CM (1993) Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J 12: 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Valpuesta JM, Medina M, Montejo de Garcini E, Avila J (1996) Polymerization of tau into filaments in the presence of heparin: the minimal sequence required for tau-tau interaction. J Neurochem 67: 1183-1190. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW (2004) Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest 114: 121-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Head E, Su JH, Anderson AJ, Bahr BA, Cotman CW, Cribbs DH (2001) Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer's disease. Am J Pathol 158: 189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E (2002) Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol Dis 11: 341-354. [DOI] [PubMed] [Google Scholar]
- Smale G, Nichols NR, Brady DR, Finch CE, Horton Jr WE (1995) Evidence for apoptotic cell death in Alzheimer's disease. Exp Neurol 133: 225-230. [DOI] [PubMed] [Google Scholar]
- Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW (1997) Substrate specificities of caspase family proteases. J Biol Chem 272: 9677-9682. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW (1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272: 17907-17911. [DOI] [PubMed] [Google Scholar]
- Uboga NV, Price JL (2000) Formation of diffuse and fibrillar tangles in aging and early Alzheimer's disease. Neurobiol Aging 21: 1-10. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Beckett LA, Barnes LL, Schneider JA, Back J, Evans DA, Bennett DA (2002) Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 17: 179-193. [PubMed] [Google Scholar]
- Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA (1988a) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA 85: 4884-4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A (1988b) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA 85: 4506-4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goodyer C, LeBlanc A (2000) Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7, and -8. J Neurosci 20: 8384-8389. [DOI] [PMC free article] [PubMed] [Google Scholar]