

Abstract
Mitochondria-produced reactive oxygen species (ROS) are thought to contribute to cell death caused by a multitude of pathological conditions. The molecular sites of mitochondrial ROS production are not well established but are generally thought to be located in complex I and complex III of the electron transport chain. We measured H2O2 production, respiration, and NADPH reduction level in rat brain mitochondria oxidizing a variety of respiratory substrates. Under conditions of maximum respiration induced with either ADP or carbonyl cyanide p-trifluoromethoxyphenylhydrazone,α-ketoglutarate supported the highest rate of H2O2 production. In the absence of ADP or in the presence of rotenone, H2O2 production rates correlated with the reduction level of mitochondrial NADPH with various substrates, with the exception of α-ketoglutarate. Isolated mitochondrial α-ketoglutarate dehydrogenase (KGDHC) and pyruvate dehydrogenase (PDHC) complexes produced superoxide and H2O2. NAD+ inhibited ROS production by the isolated enzymes and by permeabilized mitochondria. We also measured H2O2 production by brain mitochondria isolated from heterozygous knock-out mice deficient in dihydrolipoyl dehydrogenase (Dld). Although this enzyme is a part of both KGDHC and PDHC, there was greater impairment of KGDHC activity in Dld-deficient mitochondria. These mitochondria also produced significantly less H2O2 than mitochondria isolated from their littermate wild-type mice. The data strongly indicate that KGDHC is a primary site of ROS production in normally functioning mitochondria.
Keywords: mitochondria, reactive oxygen species, lipoamide dehydrogenase, ketoglutarate dehydrogenase, Parkinson, Alzheimer
Introduction
Reactive oxygen species (ROS) are thought to contribute to neuronal cell death caused by ischemia, excitotoxicity, and various acute and chronic neurological disorders (Dykens, 1994; Fiskum et al., 1999; Murphy et al., 1999; Fiskum, 2000; Nicholls and Budd, 2000). A compelling body of evidence indicates that mitochondria are the major source of ROS in normal tissues and under a variety of neurodegenerative conditions (Murphy et al., 1999). However, the mechanism and the sites of ROS production in mitochondria require additional research. The vast majority of studies on mitochondrial ROS generation have used heart mitochondria and respiratory chain inhibitors as tools to maximize ROS production and to identify potential sites of ROS generation. These studies revealed that inhibiting complexes I and III of the mitochondrial respiratory chain with specific mitochondrial toxins, such as rotenone and antimycin A, resulted in high rates of ROS production (Turrens, 1997; Murphy et al., 1999; Lenaz, 2001). Similar approaches have been used successfully to study ROS production by isolated (Kwong and Sohal, 1998) and in situ (Sipos et al., 2003) brain mitochondria, but no information is yet available regarding the specific sites or mechanisms of ROS generation in the absence of respiratory chain inhibitors (Sorgato et al., 1974; Patole et al., 1986; Cino and Del Maestro, 1989; Ramsay and Singer, 1992; Hensley et al., 1998; Kwong and Sohal, 1998; Sims et al., 1998; Tretter and Adam-Vizi, 2000; Sipos et al., 2003).
Previously, we suggested that mitochondrial matrix dehydrogenases other than complex I [e.g., α-ketoglutarate dehydrogenase enzyme complex (KGDHC)] can contribute to the observed ROS production in the absence of inhibitors of the mitochondrial respiratory chain (Starkov and Fiskum, 2002). It is known that reduced flavins (Massey, 1994) and flavoproteins (Chan and Bielski, 1974, 1980; Zhang et al., 1998; Bunik and Sievers, 2002) can generate superoxide in aqueous oxygenated solutions. Isolated lactate dehydrogenase (Chan and Bielski, 1974) and glyceraldehyde-3-phosphate dehydrogenase (Chan and Bielski, 1980) were shown to catalyze NADH-dependent superoxide production, whereas malate and isocitrate dehydrogenase did not produce superoxide (Chan and Bielski, 1974). The isolated dehydrogenase component of mitochondrial succinate dehydrogenase (SDH) complex was also capable of flavin-dependent superoxide production in the absence of an electron acceptor (Zhang et al., 1998). It has also been recently demonstrated that the flavin of the dihydrolipoamide dehydrogenase (Dld) component (EC 1.8.1.4) of isolated KGDHC can generate superoxide (Bunik and Sievers, 2002). The latter is of particular interest with regard to the mechanisms and sites of ROS production in mitochondria because the flavin of the Dld subunit is abundant in mitochondria (Kunz and Gellerich, 1993) and has a sufficiently negative redox potential (Em 7.4 = -283 mV) (Kunz and Kunz, 1985) to allow for superoxide production.
The data presented here demonstrate that KGDHC represents a significant source of ROS in brain mitochondria. The reduced Dld subunit of KGDHC is the most likely source of ROS in the mitochondrial matrix under conditions of an elevated NADPH/NADP+ ratio in the matrix of mitochondria. Preliminary results have been reported previously (Starkov and Fiskum, 2002).
Materials and Methods
Reagents. Oligomycin, antimycin A3, and rotenone (Sigma, St. Louis, MO) were dissolved in ethanol, and Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine; Molecular Probes, Eugene, OR) was dissolved in dimethylsulfoxide. All other reagents were purchased from Sigma. All reagents and ethanol were tested and exhibited no interference with the H2O2 assay at the concentrations used in our experiments.
Mouse and rat forebrain mitochondria were isolated as described previously (Rosenthal et al., 1987; Starkov and Fiskum, 2001, 2003), with modifications as follows. Sprague Dawley rats and knock-out Dld-deficient mice (Johnson et al., 1997) and their littermate controls were used. Animals were decapitated, and the brain was excised and placed into ice-cold isolation buffer containing 225 mm mannitol, 75 mm sucrose, 5 mm HEPES-KOH, pH 7.4, 1 mm EGTA, and 1 gm/l bovine serum albumin. Two mouse brains or a single rat brain were used per isolation. The cerebellum was removed, and the rest of the brain tissue was placed in a 15 ml Dounce homogenizer and homogenized manually with eight strokes of pestle A, followed by eight strokes of pestle B. The homogenate was diluted with 15 ml of isolation buffer, distributed into four centrifuge tubes, and centrifuged at 3000 × g for 4 min. The supernatant was separated and centrifuged again at 14,000 × g for 10 min. The pellet was resuspended in 15 ml of the ice-cold isolation buffer without BSA and kept on ice, and 30 μl of digitonin (10% stock solution in DMSO) was added. After a 4 min incubation with occasional stirring by slow inversion of tubes, the suspension was diluted with 15 ml of ice-cold isolation buffer containing BSA and centrifuged at 14,000 × g for 10 min. The pellet was resuspended in 8 ml of ice-cold isolation buffer containing neither BSA nor EGTA and centrifuged again at 14,000 × g for 10 min. The final pellet containing mitochondria of both synaptosomal and nonsynaptosomal origin was resuspended in isolation buffer without EGTA and BSA to a concentration of 25-30 mg of protein/ml, stored on ice, and used within 5 hr.
Alternatively, nonsynaptosomal rat and mouse forebrain mitochondria were isolated by the Percoll gradient separation method as described (Sims, 1990), and measurements of H2O2 production and respiration were performed. The Percoll gradient-isolated mitochondria generally exhibited higher H2O2 and respiration rates than digitonin-isolated mitochondria when expressed per milligram of mitochondrial protein, however the difference was not qualitative and would not justify repeating all the experiments reported here with mitochondria isolated by both methods. Therefore, only the data obtained with mitochondria isolated by the digitonin procedure are presented here, unless indicated otherwise.
Respiration of isolated mitochondria was measured at 37°C with a commercial Clark-type oxygen electrode (Hansatech, Norfolk, UK). The incubation medium composition and respiratory substrates are indicated in the legends to the figures.
The quality of the mitochondrial preparation was estimated by measuring the acceptor control ratio (ACR) defined as ADP-stimulated (state 3) respiration divided by resting (state 4) respiration. For these experiments, the incubation medium consisted of (in mm) 125 KCl, 20 HEPES, pH 7.0, 2 KH2PO4, 1 MgCl2, 5 glutamate, 5 malate, and 0.8 ADP. State 3 respiration was initiated by the addition of 0.5 mg/ml brain mitochondria to the incubation medium. State 3 respiration was terminated, and state 4 was initiated by the addition of 1 μm carboxyatractylate, an inhibitor of the ADP/ATP transporter. Only mitochondrial preparations that exhibited an ACR of >8 were used in this study.
Membrane potentials of isolated mitochondria were estimated using the fluorescence of safranine O (3 μm) with excitation and emission wavelengths of 495 and 586 nm, respectively (Votyakova and Reynolds, 2001; Starkov et al., 2002).
KGDHC activity in mouse mitochondria was measured fluorimetrically. The reaction medium was composed of 50 mm KCl, 10 mm HEPES, pH 7.4, 20 μg/ml alamethicin, 0.3 mm thiamine pyrophosphate (TPP), 10 μm CaCl2, 0.2 mm MgCl2, 5 mm α-ketoglutarate, 1 μm rotenone, and 0.2 mm NAD+. The reaction was started by adding 0.14 mm CoASH to permeabilized mitochondria (0.1-0.25 mg/ml). Reduction of NAD+ was followed at 460 nm emission after excitation at 346 nm. The scale was calibrated by adding known amounts of freshly prepared NADPH. KGDHC and pyruvate dehydrogenase complex (PDHC) activity in rat brain mitochondria were measured in the same way, except that reduction of NAD+ was followed by absorbance changes at 340 nm using the extinction coefficient, E340mm = 6.22 cm-1.
Succinate dehydrogenase activity was measured spectrophotometrically as described previously (Arrigoni and Singer, 1962). The reaction medium was composed of 50 mm KCl, 10 mm HEPES, pH 7.4, 20 μg/ml alamethicin, 10 mm succinate, 2 mm KCN, 1 μm rotenone, 50 μm 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone, 50 μm 2,6-dichlorophenol indophenol, and 20 μm EDTA; reaction was monitored at 600 nm, and the activity was calculated using E600mm = 21 cm-1 for 2,6-dichlorophenol indophenol.
Measurement of hydrogen peroxide was performed as follows. The incubation medium contained 125 mm KCl, 20 mm HEPES, pH 7.0, 2 mm KH2PO4, 4 mm ATP, 5 mm MgCl2, 1 μm Amplex Red, 5 U/ml horseradish peroxidase (HRP), and 20 U/ml Cu,ZnSOD and was maintained at 37°C. A change in the concentration of H2O2 in the medium was detected as an increase in Amplex Red fluorescence using excitation and emission wavelengths of 585 and 550 nm, respectively. The response of Amplex Red to H2O2 was calibrated either by sequential additions of known amounts of H2O2 or by continuous infusion of H2O2 at 100-1000 pmol/min. The concentration of commercial 30% H2O2 solution was calculated from light absorbance at 240 nm using E240mm = 43.6 cm-1; the stock solution was diluted to 100 μm with water and used for calibration immediately.
It is to be noted that HRP catalyzes the oxidation of NADH that might result in an underestimation of H2O2 in an HRP-dependent assay, such as a classical scopoletin-HRP assay or Amplex Red-HRP assay used in our study. However, it was found that although NADH can react with the oxidized form of HRP, it cannot effectively compete with scopoletin in an HRP-scopoletin H2O2 assay (Marquez and Dunford, 1995). Furthermore, rat brain mitochondria contain ∼5 nmol of total NAD/mg protein (C. Chinopoulos, unpublished observations) similar to the amount found in mitochondria from other tissues (3-7 nmol of total NAD/mg mitochondrial protein) (Tischler et al., 1977; Di Lisa et al., 2001). We have previously demonstrated that under the experimental conditions used in this study, rat brain mitochondria did not release more than ∼15% of their total NAD content (Chinopoulos et al., 2003). Similar amounts of NADH added exogenously did not affect the H2O2 rates detected by the Amplex Red-HRP assay (data not shown).
Superoxide production was followed spectrophotometrically with partially acetylated cytochrome c (Azzi et al., 1975) and calculated from cytochrome c absorbance at 550-540 nm using extinction coefficient E550-540mm = 19.2 cm-1.
The mitochondrial NADPH reduction state was measured fluorimetrically using an excitation wavelength of 346 nm and an emission wavelength of 460 nm. Maximal NADPH reduction was defined as the absorbance observed after the addition of the electron transport chain complex I inhibitor rotenone (1 μm), and maximal oxidation was defined as the absorbance obtained in the presence of the saturating amounts of respiratory uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (120-160 pmol/mg mitochondria).
Ubiquinones were extracted from frozen-thawed and sonicated mitochondrial samples into hexane using coenzyme Q6 as an internal standard. Samples were analyzed by HPLC using an MDA-50 (ESA, Chelmsford, MA) column and gradient separation. The detector was an eight-channel electrochemical array detector (CoulArray 5600; ESA) consisting of a series of five increasing oxidation potentials (ending with +800 mV) before a reducing channel of -800 mV and two additional oxidizing channels at +5 and +200 mV. Measurements were made on the final +200 mV channel (Gamache, 1999).
Mitochondrial protein was estimated by the Biuret method.
Statistical analysis. Data are expressed as mean ± SEM. Statistical analysis was performed using Student's t test.
Results
Previous attempts to understand the mechanisms of ROS production by mitochondria have been hampered by the low sensitivity of ROS detection methods and have therefore used inhibitors of either complex I or III of the respiratory chain to maximize ROS production. Recently, a novel fluorescent probe for H2O2, Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) (Zhou et al., 1997), has become available that is suitable for measurements with isolated mitochondria (Votyakova and Reynolds, 2001; Kushnareva et al., 2002). We have determined that it is sufficiently sensitive and its fluorescent response is linearly related to H2O2 even at levels that are generated by mitochondria in the absence of respiratory chain inhibitors (data not shown). With Amplex Red, it was possible to perform reliable measurements of H2O2 emission rates with the NAD+-dependent respiratory substrates malate, pyruvate, citrate, glutamate, and α-ketoglutarate and with succinate and compare them with rates of O2 consumption and the level of NADPH reduction of isolated rat and mouse brain mitochondria.
Figure 1 shows typical fluorescent recordings of H2O2 production by isolated rat brain mitochondria oxidizing NAD+-dependent substrates or succinate. H2O2 generation was dependent on the addition of a respiratory substrate (succinate) (Fig. 1, curve a) or α-ketoglutarate (Fig. 1, curves b, c) and was suppressed by mitochondrial uncoupler FCCP (Fig. 1, curves a, b).
Figure 1.

H2O2 production by rat brain mitochondria oxidizing α-ketoglutarate or succinate. Medium (37°C) contained 125 mm KCl, 2 mm MgCl2, 0.2 mm EGTA, 2 mm KH2PO4, 10 mm HEPES, pH 7.2, 5 U/ml HRP, 20 U/ml superoxide dismutase, and 1 μm Amplex Red. Curve a, Rat brain mitochondria oxidizing succinate (10 mm); curves b and c, rat brain mitochondria oxidizing α-ketoglutarate. Additions included 0.25 mg/ml rat brain mitochondria (Mito), 5 mm α-ketoglutarate (α-Keto), 0.5 μm rotenone (Rot), 1 mm NAD+, 120 pmol/mg FCCP, and 1 μm antimycin (Ant.A). Numbers near the tracings indicate the rates of H2O2 production in picomoles per minute per milligram of mitochondrial protein. Typical tracings are shown.
With NAD+-dependent substrates, H2O2 production was stimulated by rotenone, which inhibits NADH oxidation at complex I (Fig. 1, curves b, c). However, rotenone inhibited succinate-supported H2O2 production, indicating that it was fueled by reverse electron transfer from succinate to a site in complex I (Turrens, 1997). With both types of substrates, H2O2 production was stimulated by an inhibitor of complex III (antimycin A) that increases the coenzyme Q semiquinone level (Turrens, 1997). With both types of substrates, H2O2 production was unaffected by the addition of NAD+ (only the data with α-ketoglutarate is presented) (Fig. 1, curve c). It is well known that the inner mitochondrial membrane is impermeable to NAD+.
It is important to note that H2O2 production in the presence of a complex I inhibitor (rotenone) was two to three times higher with α-ketoglutarate than that with succinate (Fig. 1). Previously, we (Korshunov et al., 1997; Starkov et al., 2002; Starkov and Fiskum, 2003) and others (Hansford et al., 1997; Votyakova and Reynolds, 2001) demonstrated that regardless of the nature of the respiratory substrate, rates of H2O2 production were directly related to the magnitude of the membrane potential of mitochondria. However, the difference in H2O2 production demonstrated in Figure 1 cannot be explained by the difference in the membrane potential because the concentration of FCCP (120 pmol/mg mitochondria) used in these experiments was selected to completely uncouple mitochondria so that no membrane potential could be detected under these conditions by either the TPP+ electrode or safranine fluorescence method, and no additional increase in FCCP concentration would stimulate the respiration of brain mitochondria (data not presented). Previously, we demonstrated that at such low values the membrane potential no longer affects the H2O2 production supported by either succinate (Korshunov et al., 1997) or a NAD-dependent substrate (Starkov and Fiskum, 2003).
We (Starkov and Fiskum, 2003) and others (Kushnareva et al., 2002) also observed that with a NAD+-dependent substrate, the rate of H2O2 production was apparently modulated by the level of NADPH reduction in mitochondria. However, it is obvious that such a modulation could not explain the finding that rotenone inhibition of complex I induced much higher H2O2 production when α-ketoglutarate was the substrate (Fig. 1, traces b, c) compared with succinate-supported H2O2 production (Fig. 1, trace a). In these experiments, a sufficiently high concentration of rotenone was used (0.5 μm) that completely inhibited NADH oxidation (judging by the complete inhibition of respiration with pyruvate plus malate or glutamate plus malate; data not shown). Intuitively, one would expect essentially similar levels of the intramitochondrial NADPH reduction state when the oxidation of NADH is completely inhibited with rotenone, as in our experiments. If mitochondrial complex I is a sole source of H2O2 under such conditions, it seems difficult to understand why rotenone-inhibited complex I produces less H2O2 when its substrate NADH was reduced by succinate rather than by α-ketoglutarate.
It appeared that rates of H2O2 production varied significantly among different NAD+-linked substrates both in the absence (Fig. 2A) and in the presence (Fig. 2B) of the complex I inhibitor rotenone. The rates of H2O2 generation were not directly related to relative rates of state 4 (Fig. 2C) or state 3 (Fig. 2D) respiration. With most NAD+-dependent substrates, there was a reasonably good correlation between the level of NADPH reduction and the rate of H2O2 emission. However, α-ketoglutarate-supported H2O2 production was higher than expected under both conditions and did not correlate with the level of reduction of matrix pyridine nucleotides in either the presence or absence of rotenone (Fig. 2A,B). We therefore hypothesized that KGDHC itself could be a significant source of ROS.
Figure 2.

A typical relationship between H2O2 production rates by rat brain mitochondria oxidizing various substrates and the corresponding levels of reduction mitochondrial pyridine nucleotides and the rates of respiration. A, ΔH2O2 production by rat brain mitochondria in state 4. ΔNADPH was obtained by measuring the difference in NADPH fluorescence both in the absence and in the presence of 160 pmol/mg FCCP. ΔNADPH in the presence of succinate was taken as 100%. H2O2 production in the presence of succinate was 1123 ± 71 pmol/min/mg (data not shown). The H2O2 production rate in the presence of FCCP was subtracted from that in the absence of the uncoupler and presented as ΔH2O2. B, ΔH2O2 production rate plotted against ΔNADPH in the presence of 1 μm rotenone. C, H2O2 production rate plotted against the rate of respiration by mitochondria in state 4. D, H2O2 production rate plotted against the rate of respiration by mitochondria in state 3. Incubation medium (see Fig. 1) was maintained at 37°C. For D only, state 3 respiration was initiated by adding 0.4 mm ADP to mitochondrial suspension. Substrates were present at the following concentrations: malate and glutamate, 5 mm plus 5 mm; α-ketoglutarate, 7 mm; succinate, 5 mm; citrate, 5 mm; pyruvate, 10 mm; glutamate, 5 mm; malate, 5 mm. Mitochondria were added at 0.5 mg/ml.
In intact mitochondria, the activity of all dehydrogenases can affect each other through the common pool of pyridine nucleotides and through the inter-conversion of tricarboxylic acid (TCA) cycle metabolic intermediates. To test whether the rate of H2O2 production is substrate selective in the absence of this inter-conversion, we made mitochondria freely permeable to metabolites and other small molecules by the addition of the pore-forming peptide alamethicin (Gostimskaya et al., 2003). This treatment did not result in loss of malate dehydrogenase, pyruvate dehydrogenase, or KGDHC activities from the mitochondria (data not shown). Figure 3 shows that adding α-ketoglutarate stimulated H2O2 production in both alamethicin-permeabilized (Fig. 3, curves a, c) and intact (Fig. 3, curve b) rat brain mitochondria. Coenzyme A (CoA), a cofactor of KGDHC, stimulated H2O2 production only in alamethicin-treated mitochondria (Fig. 3, curves a, c) because it is impermeable to the inner membrane of intact mitochondria (Fig. 3, curve b); the addition of alamethicin stimulated H2O2 production in intact mitochondria by rendering their inner membrane permeable to CoA (Fig. 3, curve b). Catalase almost completely suppresses H2O2 production (Fig. 3, curve c). Under these conditions, the now-permeant NAD+ inhibited H2O2 production (Fig. 3, curves a-c), whereas rotenone (Fig. 3, curves a, b) stimulated production because its ability to inhibit complex I of the mitochondrial electron transport chain does not require an intact inner membrane. It causes net reduction of NAD+ to NADH and thereby overreduction of complex I and complex I-mediated ROS production in both intact and permeabilized mitochondria.
Figure 3.

H2O2 production by permeabilized rat brain mitochondria. Incubation medium was composed of 225 mm mannitol, 75 mm sucrose, 10 mm HEPES-KOH, pH 7.4, 2 mm KH2PO4, 1 mm MgCl2, 0.25 mm EGTA, 48 μm thiamine, 5 U/ml HRP, 20 U/ml superoxide dismutase, and 1 μm Amplex Red (t = 37°C). Curve a, Mitochondria (Mito; 1 mg) were incubated for 5 min with 20 μg/mg alamethicin, then centrifuged at 20,000 × g for 10 min and resuspended at 0.5 mg/ml for H2O2 measurement; curve b, intact rat brain mitochondria; curve c, mitochondria were pretreated as in curve a; 0.25 mg/ml catalase was included into the incubation medium. Additions included 10 mm α-ketoglutarate (α-Ktg), 0.2 mm NAD+, 0.12 mm CoASH(CoA), 1 μm rotenone (Rot), and 20 μg/mg alamethicin (Ala).
Results shown in Figure 4 indicate that both PDHC and KGDHC produce superoxide. The maximum superoxide production rate was obtained in the presence of all the same enzyme cofactors and substrates as needed for a normal enzymatic reaction catalyzed by these enzyme complexes. However, KGDHC produced approximately twice as much superoxide as PDHC (Fig. 4). As with permeabilized mitochondria (Fig. 3), NAD+ inhibited superoxide production (Fig. 4). Malate dehydrogenase in the presence of either malate or oxaloacetate did not produce detectable amounts of superoxide (data not shown).
Figure 4.

Cofactor and substrate dependence of superoxide production by isolated KGDHC and PDHC. Superoxide production was measured as described in Materials and Methods. Incubation medium contained 50 mm KH2PO4 buffer, pH 7.8, 50 μm acetylated cytochrome c, 10 μm CaCl2, and 0.2 mm MgCl2, maintained at t = 37°C. Where indicated, 0.12 mm CoASH, 0.3 mm TPP, 40 U/ml superoxide dismutase (SOD), 2 mm NAD+, and either 10 mm ketoglutarate (for KGDHC) or 7 mm pyruvate (for PDHC) were included into the incubation medium (substrate). Reaction was started by adding 0.9-3.6 U/ml PDHC or 0.6-2.4 U/ml KGDHC.
Both PDHC and KGDHC also produced H2O2. We explored the substrate and cofactor dependence for H2O2 production and for NAD+ reduction catalyzed by KGDHC. The H2O2 production rate by isolated KGDHC exhibited hyperbolic dose dependence to concentrations of CoA-SH (Fig. 5) and α-ketoglutarate (data not shown; more details can be found in an accompanying report by Tretter and Adam-Vizi, 2004). However, CoA-SH requirements for maximum H2O2 production appeared to be much lower than that for maximum NAD+ reduction; the apparent Km for CoASH was ∼0.03 μm for H2O2 production (Fig. 5), whereas the Km for NAD+ reduction was ∼30 μm (data not shown).
Figure 5.

CoA-SH dependence of H2O2 production by isolated KGDHC. H2O2 production was measured as described in Materials and Methods. Incubation medium contained 50 mm KH2PO4 buffer, pH 7.8, 10 μm CaCl2, 0.2 mm MgCl2, 0.3 mm TPP, 40 U/ml superoxide dismutase (SOD), 5 U/ml HRP, and 1 μm Amplex Red, maintained at t = 37°C. Medium was supplemented with 10 mm ketoglutarate.
We also measured enzymatic activities of PDHC and KGDHC in alamethicin-permeabilized rat brain mitochondria (see Materials and Methods). We found that PDHC activity was 0.245 U/mg mitochondria and that of KGDHC was 0.215 U/mg. Assuming that PDHC and KGDHC produce superoxide (Fig. 4) as their primary ROS, and that two superoxide molecules dismutate to produce one H2O2 molecule, we calculated that mitochondria produce a maximum of ∼560 pmol of H2O2/mg protein with α-ketoglutarate as substrate and ∼330 pmol of H2O2/mg protein with pyruvate, whereas minimum rates (in the presence of NAD+) are ∼70 and ∼90 pmol of H2O2/mg protein, respectively. These values are remarkably similar to the actual rates of H2O2 production observed with rat brain mitochondria, particularly for α-ketoglutarate (Figs. 1, 2, 3). Altogether, these results indicate that the Dld component of KGDHC, and to a lesser degree of PDHC, may be an important constitutive source of ROS in mitochondria.
Additional experiments used brain mitochondria isolated from knock-out heterozygous mice deficient in Dld (Dld+/-). Although Dld is shared between PDHC and KGDHC, a deficiency in Dld affected primarily mitochondrial KGDHC enzyme activity (Fig. 6A). As expected, maximum respiration rates were reduced only when α-ketoglutarate was oxidized; there was no significant difference in maximum respiration rates or respiratory control index when mitochondria oxidized pyruvate plus malate, glutamate plus malate, or succinate (Table 1). Therefore, Dld deficiency apparently did not indirectly affect other parts of the respiratory chain, such as complex I or complex III. Measurements of complex I and complex II activity in alamethicin-permeabilized mitochondria supported this conclusion (Fig. 6B). However, H2O2 production rates by Dld+/- mitochondria were significantly reduced with either succinate or α-ketoglutarate during state 4 respiration (Fig. 7A) and were similarly reduced in the presence of the respiratory chain inhibitors rotenone (Fig. 7B) and antimycin A (Fig. 7C).
Figure 6.

Activity of selected mitochondrial enzymes in brain mitochondria isolated from Dld-deficient mice compared with their littermate controls. A, Activity of KGDHC and PDHC. B, Activity of complex I (NADH:Q1 reductase) and SDH. KGDHC, PDHC, and SDH activities were measured as described in Materials and Methods. Mitochondrial complex I was measured with frozen-thawed mitochondrial samples fluorimetrically by following coenzyme Q1-induced rotenone-sensitive NADH oxidation at 346 nm excitation and 460 nm emission. Incubation medium was composed of 125 mm KCl, 2 mm MgCl2, 2 mm KH2PO4, 0.2 mg/ml BSA, 10 μm CaCl2, 2 mm KCN, 50 μm NADH, 20 μg/ml alamethicin, and 0.08-0.09 mg/ml mitochondria, at t = 37°C. Reaction was started by adding 40 μm coenzyme Q1 and terminated by adding 1 μm rotenone. Complex I activity was calculated as the difference between the NADH oxidation rate in the presence and in the absence of rotenone and presented in nanomole of NADH per minute per milligram. The scale was calibrated by adding known amounts of freshly prepared NADH.
Table 1.
Respiration of mouse brain mitochondria with different substrates
|
Substrate |
State 3 |
State 4 |
ACR |
|---|---|---|---|
| Pyruvate plus malate | |||
| Dld +/+ | 95.6 ± 9.7 | 6.2 ± 1.1 | 16.1 ± 1.3 |
| Dld +/− | 96.7 ± 5.7 | 6.7 ± 0.4 | 14.5 ± 1.0 |
| Glutamate plus malate | |||
| Dld +/+ | 98.4 ± 11.8 | 7.4 ± 0.9 | 13.4 ± 1.1 |
| Dld −/+ | 87.4 ± 8.3 | 6.6 ± 0.8 | 13.5 ± 1.0 |
| α-Ketoglutarate | |||
| Dld +/+ | 40.2 ± 6.7 | 8.4 ± 1.4 | 4.9 ± 0.4 |
| Dld −/+ | 26.8 ± 1.6 | 6.7 ± 0.7 | 4.1 ± 0.2 |
| Succinate | |||
| Dld +/+ | 38.7 ± 3.2 | 23.0 ± 2.6 | 1.7 ± 0.1 |
| Dld −/+ |
36.2 ± 4.7 |
21.8 ± 1.4 |
1.6 ± 0.1 |
Mitochondria were isolated from brains of Dld-deficient mice and their littermate wild-type mice. Incubation medium was as in Figure 7. Substrates were added at the following concentrations: pyruvate plus malate, 7 plus 1 mm; glutamate plus malate, 5 plus 5 mm; α-ketoglutarate, 5 mm; succinate (in the absence of rotenone), 5 mm. State 4 was induced by carboxyatractylate. Respiration was measured as described in Materials and Methods. The numbers represent respiration rates expressed in nanomoles of O2 per minute per milligram of mitochondria. ACR, Acceptor control index.
Figure 7.

Hydrogen peroxide production by brain mitochondria isolated from Dld-deficient mice compared with their littermate wild-type mice. Incubation medium was as in Figure 1, except that EGTA was omitted and 0.4 mm ADP and 0.2 mg/ml BSA were included. Where indicated, 5 mm succinate or 5 mm α-ketoglutarate A were included into the medium. The sequence of additions was as follows: mitochondria (0.125 mg/ml) were added into the incubation medium and incubated for 2 min, then phosphorylation was inhibited with 1.2 μm carboxyatractylate and H2O2 production was measured as described in Materials and Methods (state 4); then 1 μm rotenone and, finally, 1 μm antimycin A were added into the incubation medium. A, H2O2 production in state 4. B, H2O2 production induced by rotenone. C, H2O2 production induced by antimycin A. In B for α-ketoglutarate only and in C for both α-ketoglutarate and succinate, the presented rate of H2O2 production was obtained by subtracting the rate of H2O2 production in state 4 from the rate induced by rotenone (B) and the rate in the presence of rotenone from that was induced by antimycin A (C).
Generally, the rate of H2O2 generation depends on the magnitude of the mitochondrial membrane potential (Hansford et al., 1997; Korshunov et al., 1997; Votyakova and Reynolds, 2001; Starkov et al., 2002; Starkov and Fiskum, 2003), which in turn depends on the metabolic state and the quality (“coupling”) of mitochondria. Dld+/- mitochondria are deficient in KGDHC and therefore may possess lower membrane potential than Dld+/+ mitochondria. The Percoll isolation procedure yields very tightly coupled, mostly nonsynaptic mitochondria possessing high membrane potential. Therefore, we used Percoll-isolated (see Materials and Methods) mitochondria to measure the membrane potential and H2O2 production by mouse brain mitochondria. It appeared that the amplitude of the membrane potential was virtually identical in both Dld+/+ and Dld+/- mitochondria oxidizing either succinate (Fig. 8 A) or α-ketoglutarate (Fig. 8 B) either in the presence of ADP or in the resting state. Nevertheless, Dld+/- mitochondria produced significantly less H2O2 (Fig. 9) than Dld+/+ mitochondria under all metabolic conditions, except state 3 (Fig. 9A).
Figure 8.

The membrane potential of mouse brain mitochondria oxidizing succinate or α-ketoglutarate. Mouse brain mitochondria were isolated by the Percoll gradient procedure (see Materials and Methods). Incubation medium contained 125 mm KCl, 2 mm MgCl2, 2 mm KH2PO4, 10 mm HEPES, pH 7.2, 0.2 mg/ml BSA, 0.2 mm ADP, 5 mm succinate (A) or 5 mm α-ketoglurate (B), and 1 μm safranin O. Additions included 0.25 mg/ml mouse brain mitochondria (Mito), 1 μm rotenone (Rot), 1 μm carboxyatractylate (cAtr), and 1 μm antimycin A (Ant A). Typical tracings are shown. Solid lines, Dld+/+ mitochondria; dotted lines, Dld+/- mitochondria.
Figure 9.

Hydrogen peroxide production by brain mitochondria isolated by the Percoll procedure from Dld-deficient mice compared with their littermate wild-type mice. Mouse brain mitochondria were isolated by the Percoll gradient procedure (see Materials and Methods). Incubation medium was as in Figure 8, except that safranin O was omitted and 5 U/ml HRP, 20 U/ml superoxide dismutase, and 1 μm Amplex Red were included. Where indicated, 5 mm succinate or 5 mm α-ketoglutarate A were included into the medium. The sequence of additions was as follows: mitochondria (0.1-0.125 mg/ml) were added into the incubation medium and incubated for 2 min, then phosphorylation was inhibited with 1.2 μm carboxyatractylate, then 1 μm rotenone and, finally, 1 μm antimycin A were added into the incubation medium. A, H2O2 production in state 3. B, H2O2 production in state 4. C, H2O2 production induced by rotenone. D, H2O2 production induced by antimycin A. In C for α-ketoglutarate only and in D for both α-ketoglutarate and succinate, the presented rate of H2O2 production was obtained by subtracting the rate of H2O2 production in state 4 from the rate induced by rotenone (C) and the rate in the presence of rotenone from that induced by antimycin A (D).
We also measured the content and composition of coenzymes Q in mitochondrial membrane because there are several reports that the Q9/Q10 ratio and the total amount of coenzyme Q can affect ROS production (Boveris and Chance, 1973; Lass et al., 1997; Lass and Sohal, 1999, 2000). There was no difference in the Q9/Q10 ratio or coenzymes Q content in mitochondria from Dld+/- mice compared with their littermate controls (Fig. 10).
Figure 10.
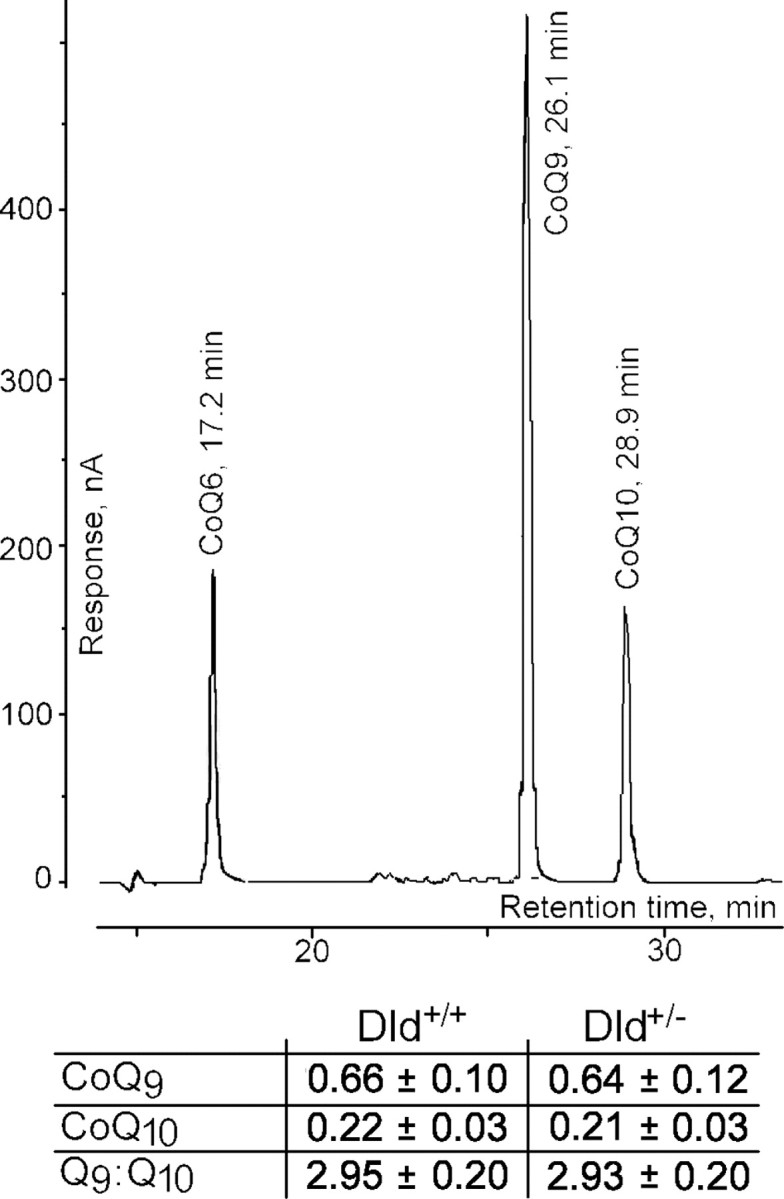
Coenzyme Q9 and coenzyme Q10 content in brain mitochondria isolated from Dld-deficient mice compared with their littermate wild-type mice. Coenzymes Q9 and Q10 were measured by HPLC as described in Materials and Methods. Data are presented in picomole of quinone per milligram of mitochondria.
Discussion
Our data suggest that complex I of the mitochondrial electron transport chain is not the exclusive source of H2O2 and that the Dld components of KGDHC and PDHC are substantial constitutive sources of free radicals in rat and mouse brain mitochondria under conditions of the elevated mitochondrial NADPH/NADP+ ratio.
Complex I of the mitochondrial electron transport chain has been viewed as a major site of mitochondrial ROS production (Barja, 1999; Herrero and Barja, 2000; Lenaz, 2001; Sipos et al., 2003). However, there are no data yet that demonstrate that complex I is the major site of ROS production in intact mitochondria, in the absence of respiratory chain inhibitors. There are three principal types of experiments that contributed to the concept that complex I is a major ROS-producing site: (1) experiments demonstrating that isolated complex I preparations generate ROS in the presence of NADH; (2) experiments with rotenone-inhibited mitochondria oxidizing NAD-dependent substrates; and (3) experiments with isolated mitochondria under conditions favoring reverse electron transfer from succinate to complex I. The latter reaction generates large amounts of ROS (Fig. 1, curve a) (Turrens, 1997; Lenaz, 2001; Votyakova and Reynolds, 2001). However, the possibility of reverse electron transfer under physiological conditions is not yet established. The interpretation of such experiments may be complicated because the source of ROS could be anything that is in a redox equilibrium with intramitochondrial NADPH. This difficulty also applies to experiments demonstrating the dependence of mitochondrial ROS production on the amplitude of the membrane potential (Hansford et al., 1997; Korshunov et al., 1997; Votyakova and Reynolds, 2001; Starkov and Fiskum, 2003) or intramitochondrial NADPH/NADP+ ratio (Kushnareva et al., 2002; Starkov and Fiskum, 2003).
Several research groups have demonstrated that isolated complex I preparations can generate ROS when reduced with NADH, although there is no consensus about the site of ROS production in complex I (Lenaz, 2001; Kushnareva et al., 2002; Liu et al., 2002).
However, some evidence argues against the concept that complex I in mitochondria, or in submitochondrial particles, can generate ROS in the absence or even in the presence of its inhibitors. The absence of a correlation between the inhibition of complex I activity by rotenone and other inhibitors and the production of ROS by submitochondrial particles was interpreted as an indication of the presence of a superoxide-producing rotenone-binding site other than complex I (Ramsay and Singer, 1992). The finding that H2O2 production is frequently reported as being almost absent in the presence of succinate and rotenone (Barja, 1999; Liu et al., 2002) is intriguing because the intramitochondrial NADPH/NADP+ ratio under such conditions is high. Stimulatory effects of ADP (Barja, 1999) and Ca2+ (Dykens, 1994; Kowaltowski et al., 1995, 1996, 1998a,b) on mitochondrial ROS production are also puzzling because both Ca2+ uptake/retention and ADP-induced oxidative phosphorylation dissipate energy and would be expected to decrease the level of reduction of complex I and hence the ROS production. It also appears that the stimulatory effect of the complex I inhibitor rotenone on ROS production that possibly originates from a site within complex I is species and tissue dependent; ROS stimulation by rotenone varies from ∼300% in guinea pig to 0% in horse heart submitochondrial particles (Herrero and Barja, 2000) and in whole intact rat heart mitochondria (Chen et al., 2003) to inhibition of ROS production in mouse kidney mitochondria (Kwong and Sohal, 1998).
Our data indicate that KGDHC may be a major ROS-producing site in mitochondria. Mammalian KGDHC is composed of multiple copies of three enzymes: α-ketoglutarate dehydrogenase (E1; EC 1.2.4.2), dihydrolipoamide succinyltransferase (E2; EC 2.3.1.12), and Dld (E3 or Dld; EC 1.6.4.3). E1 and E3 are noncovalently bound to a core formed by E2 (Wagenknecht et al., 1983; Sheu and Blass, 1999). Dld is also a part of other multienzyme complexes such as PDHC, branched chain ketoacid dehydrogenase complex, and glycine cleavage complex (Koike et al., 1974; Patel and Roche, 1990; Reed and Hackert, 1990). Dld is a flavoenzyme, the redox center of which is formed by a disulfide bridge coupled with a flavin ring. The catalyzed reaction proceeds via the formation of a charge transfer complex between those two groups (Matthews et al., 1977; Templeton et al., 1980). The catalytic mechanism of α-ketoacid dehydrogenase complexes was reviewed by Bunik (2003).
In the context of the enzymatic mechanism, results of experiments with isolated PDHC and KGDHC point to the flavin or the neighboring disulfide bridge in the catalytic center of the Dld component as an electron donor for superoxide formation. This explanation is in agreement with another recently published study (Bunik and Sievers, 2002) indicating that the flavin of the Dld component of KGDHC is involved in superoxide generation.
It is not clear yet why α-ketoglutarate-supported (and presumably, KGDHC-mediated) ROS production is much higher than pyruvate-supported production (Fig. 2) because both KGDHC and PDHC share the Dld component that generates ROS (Zhang et al., 1998). The reason for this discrepancy may be related to differing composition and molecular organization of these large enzyme complexes.
KGDHC activity is regulated by multiple mechanisms. The enzyme is inhibited by its own product, succinyl-CoA, by a high NADH/NAD+ ratio as well as by a high dihydrolipoate/lipoate ratio, thereby playing an important role in cellular redox regulation (Bunik, 2003). KGDHC is activated by low concentrations of Ca2+ and matrix ADP (Hamada et al., 1975; McMinn and Ottaway, 1977; LaNoue et al., 1983; Wan et al., 1989; Kiselevsky et al., 1990). Considering that KGDHC-mediated ROS production requires a fully active complex with all the cofactors and substrates (except NAD+) (Figs. 4, 5), the fact that the enzyme activity is stimulated by Ca2+ and ADP may perhaps account for previous findings that mitochondrial ROS production was increased by Ca2+ (Dykens, 1994; Kowaltowski et al., 1995, 1996, 1998a,b), Ca2+ in the presence of rotenone and succinate (Starkov et al., 2002), and by ADP (Barja, 1999). The results presented in the accompanying report by Tretter and Adam-Vizi (2004) demonstrate that Ca2+ activated ROS production by isolated KGDHC both in the presence and in the absence of pyridine nucleotides.
It is well known that the activity of KGDHC is severely reduced in a variety of neurodegenerative diseases associated with impaired mitochondrial functions, including Alzheimer's and Parkinson's diseases. However, the relationship between KGDHC activity and mitochondrial damage per se is much less clear. KGDHC is an integral mitochondrial enzyme tightly bound to the inner mitochondrial membrane on the matrix side (Maas and Bisswanger, 1990). It binds (specifically) to complex I of the mitochondrial respiratory chain (Sumegi and Srere, 1984) and may form a part of the TCA cycle enzyme supercomplex (Lyubarev and Kurganov, 1989). The mitochondrial TCA cycle enzymes aconitase, succinate dehydrogenase (SDH), and KGDHC itself are sensitive to oxidative inactivation both in vitro and in vivo (Tretter and Adam-Vizi, 2000; Gibson et al., 2002; Sadek et al., 2002). The close spatial and functional relationship of KGDHC to sensitive TCA cycle enzymes may result in specific targeting and damage to these enzymes by KGDHC-originated ROS. Mitochondria from different brain regions possess different amounts of KGDHC (Calingasan et al., 1994; Park et al., 2000), which may account for regional vulnerability. For instance, the cholinergic neurons of the nucleus basalis of Meynert have high levels of KGDHC, and these neurons are particularly vulnerable in Alzheimer's disease (Gibson et al., 1988). The conditions promoting KGDHC-mediated ROS production may be any that increase the intramitochondrial NADH/NAD+ ratio (e.g., inhibition of oxidative phosphorylation or inhibition of any segment of the mitochondrial electron transport chain). This hypothesis is strongly supported by the results presented in the accompanying report by Tretter and Adam-Vizi (2004) that demonstrated that ROS production by isolated KGDHC is strongly dependent on the NADH/NAD+ ratio. It also agrees well with the recent demonstration of a selective loss of KGDHC-enriched neurons in human brains with Alzheimer's disease (Ko et al., 2001). [Alternatively, as Dr. John Blass pointed out (personal communication), a regulatory mechanism might be in place decreasing the expression and/or assembly of a functional KGDH complex in response to the oxidative stress induced by either damage to mitochondria leading to an increase in their overall reduction potential, or by an oxidants external to mitochondria.]
In this respect, it is very interesting that the DLST gene encoding the E2 component of KGDHC also encodes truncated mRNA for another protein designated MIRTD that localizes to mitochondria, where it regulates the biogenesis of the mitochondrial respiratory chain (Kanamori et al., 2003).
In conclusion, we emphasize that although results presented here, and particularly those with Dld-deficient mice (Fig. 7), indicate that the Dld component of KGDHC may be a significant source of ROS in mitochondria, they do not rule out complex I (or other potential sites) as important sources of ROS. In fact, results of substrate-dependent ROS production experiments (Fig. 2B) indicate that some other dehydrogenases might also be involved in ROS production. The results presented here challenge the idea of a single “major” site of ROS production in mitochondria, whether it be complexes I, II, and III or the Dld component, and emphasize the necessity of additional systematic research on the mechanisms and regulation of mitochondrial ROS production.
Footnotes
This work was supported in part by National Institutes of Health Grants ES11838 and NS34152 (G.F.) and AG14930 (M.F.B.) and by the United States Army Medical Research and Materiel Command Neurotoxin Initiative, Mitochondrial Collaborative Group (DAMD17-99-1-9483 to G.F. and DAMD17-98-1-8620 to S.E.B.). We are grateful to Drs. John P. Blass, Gary E. Gibson, and Bill Nicklas for very helpful discussion of portions of this manuscript.
Correspondence should be addressed to Dr. M. Flint Beal, Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York Presbyterian Hospital, 525 East 68th Street, New York, NY 10021. E-mail: fbeal@med.cornell.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247779-10$15.00/0
References
- Arrigoni O, Singer TP (1962) Limitations of the phenazine methosulphate assay for succinic and related dehydrogenases. Nauchni Tr Vissh Med Inst Sofiia 193: 1256-1258. [DOI] [PubMed] [Google Scholar]
- Azzi A, Montecucco C, Richter C (1975) The use of acetylated ferricytochrome c for the detection of superoxide radicals produced in biological membranes. Biochem Biophys Res Commun 65: 597-603. [DOI] [PubMed] [Google Scholar]
- Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31: 347-366. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B (1973) The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunik VI (2003) 2-Oxo acid dehydrogenase complexes in redox regulation. Eur J Biochem 270: 1036-1042. [DOI] [PubMed] [Google Scholar]
- Bunik VI, Sievers C (2002) Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur J Biochem 269: 5004-5015. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Baker H, Sheu KF, Gibson GE (1994) Distribution of the alpha-ketoglutarate dehydrogenase complex in rat brain. J Comp Neurol 346: 461-479. [DOI] [PubMed] [Google Scholar]
- Chan PC, Bielski BH (1974) Enzyme-catalyzed free radical reactions with nicotinamide adenine nucleotides. II. Lactate dehydrogenase-catalyzed oxidation of reduced nicotinamide adenine dinucleotide by superoxide radicals generated by xanthine oxidase. J Biol Chem 249: 1317-1319. [PubMed] [Google Scholar]
- Chan PC, Bielski BH (1980) Glyceraldehyde-3-phosphate dehydrogenasecatalyzed chain oxidation of reduced nicotinamide adenine dinucleotide by perhydroxyl radicals. J Biol Chem 255: 874-876. [PubMed] [Google Scholar]
- Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 278: 36027-36031. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G (2003) Cyclosporin A-insensitive permeability transition in brain mitochondria: inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem 278: 27382-27389. [DOI] [PubMed] [Google Scholar]
- Cino M, Del Maestro RF (1989) Generation of hydrogen peroxide by brain mitochondria: the effect of reoxygenation following postdecapitative ischemia. Arch Biochem Biophys 269: 623-638. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P (2001) Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem 276: 2571-2575. [DOI] [PubMed] [Google Scholar]
- Dykens JA (1994) Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. J Neurochem 63: 584-591. [DOI] [PubMed] [Google Scholar]
- Fiskum G (2000) Mitochondrial participation in ischemic and traumatic neural cell death. J Neurotrauma 17: 843-855. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF (1999) Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab 19: 351-369. [DOI] [PubMed] [Google Scholar]
- Gamache PH, Freeto SM, Acworth IN (1999) Coulometric array HPLC analysis of lipid-soluble vitamins and antioxidants. Amer Clin Lab 18: 18-19. [Google Scholar]
- Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P (1988) Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer's disease. Arch Neurol 45: 836-840. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Zhang H, Xu H, Park LC, Jeitner TM (2002) Oxidative stress increases internal calcium stores and reduces a key mitochondrial enzyme. Biochim Biophys Acta 1586: 177-189. [DOI] [PubMed] [Google Scholar]
- Gostimskaya IS, Grivennikova VG, Zharova TV, Bakeeva LE, Vinogradov AD (2003) In situ assay of the intramitochondrial enzymes: use of alamethicin for permeabilization of mitochondria. Anal Biochem 313: 46-52. [DOI] [PubMed] [Google Scholar]
- Hamada M, Koike K, Nakaula Y, Hiraoka T, Koike M (1975) A kinetic study of the alpha-keto acid dehydrogenase complexes from pig heart mitochondria. J Biochem (Tokyo) 77: 1047-1056. [DOI] [PubMed] [Google Scholar]
- Hansford RG, Hogue BA, Mildaziene V (1997) Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr 29: 89-95. [DOI] [PubMed] [Google Scholar]
- Hensley K, Pye QN, Maidt ML, Stewart CA, Robinson KA, Jaffrey F, Floyd RA (1998) Interaction of alpha-phenyl-N-tert-butyl nitrone and alternative electron acceptors with complex I indicates a substrate reduction site upstream from the rotenone binding site. J Neurochem 71: 2549-2557. [DOI] [PubMed] [Google Scholar]
- Herrero A, Barja G (2000) Localization of the site of oxygen radical generation inside the complex I of heart and nonsynaptic brain mammalian mitochondria. J Bioenerg Biomembr 32: 609-615. [DOI] [PubMed] [Google Scholar]
- Johnson MT, Yang HS, Magnuson T, Patel MS (1997) Targeted disruption of the murine dihydrolipoamide dehydrogenase gene (Dld) results in perigastrulation lethality. Proc Natl Acad Sci USA 94: 14512-14517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori T, Nishimaki K, Asoh S, Ishibashi Y, Takata I, Kuwabara T, Taira K, Yamaguchi H, Sugihara S, Yamazaki T, Ihara Y, Nakano K, Matuda S, Ohta S (2003) Truncated product of the bifunctional DLST gene involved in biogenesis of the respiratory chain. EMBO J 22: 2913-2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselevsky YV, Ostrovtsova SA, Strumilo SA (1990) Kinetic characterization of the pyruvate and oxoglutarate dehydrogenase complexes from human heart. Acta Biochim Pol 37: 135-139. [PubMed] [Google Scholar]
- Ko LW, Sheu KF, Thaler HT, Markesbery WR, Blass JP (2001) Selective loss of KGDHC-enriched neurons in Alzheimer temporal cortex: does mitochondrial variation contribute to selective vulnerability? J Mol Neurosci 17: 361-369. [DOI] [PubMed] [Google Scholar]
- Koike K, Hamada M, Tanaka N, Otsuka KI, Ogasahara K, Koike M (1974) Properties and subunit composition of the pig heart 2-oxoglutarate dehydrogenase. J Biol Chem 249: 3836-3842. [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15-18. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE (1995) Ca(2+)-induced mitochondrial membrane permeabilization: role of coenzyme Q redox state. Am J Physiol 269: C141-C147. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Castilho RF, Vercesi AE (1996) Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett 378: 150-152. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Netto LE, Vercesi AE (1998a) The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanism. J Biol Chem 273: 12766-12769. [DOI] [PubMed] [Google Scholar]
- Kowaltowski AJ, Naia-da-Silva ES, Castilho RF, Vercesi AE (1998b) Ca2+-stimulated mitochondrial reactive oxygen species generation and permeability transition are inhibited by dibucaine or Mg2+. Arch Biochem Biophys 359: 77-81. [DOI] [PubMed] [Google Scholar]
- Kunz WS, Gellerich FN (1993) Quantification of the content of fluorescent flavoproteins in mitochondria from liver, kidney cortex, skeletal muscle, and brain. Biochem Med Metab Biol 50: 103-110. [DOI] [PubMed] [Google Scholar]
- Kunz WS, Kunz W (1985) Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim Biophys Acta 841: 237-246. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y, Murphy AN, Andreyev A (2002) Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J 368: 545-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LK, Sohal RS (1998) Substrate and site specificity of hydrogen peroxide generation in mouse mitochondria. Arch Biochem Biophys 350: 118-126. [DOI] [PubMed] [Google Scholar]
- LaNoue KF, Schoolwerth AC, Pease AJ (1983) Ammonia formation in isolated rat liver mitochondria. J Biol Chem 258: 1726-1734. [PubMed] [Google Scholar]
- Lass A, Sohal RS (1999) Comparisons of coenzyme Q bound to mitochondrial membrane proteins among different mammalian species. Free Radic Biol Med 27: 220-226. [DOI] [PubMed] [Google Scholar]
- Lass A, Sohal RS (2000) Effect of coenzyme Q(10) and alpha-tocopherol content of mitochondria on the production of superoxide anion radicals. FASEB J 14: 87-94. [DOI] [PubMed] [Google Scholar]
- Lass A, Agarwal S, Sohal RS (1997) Mitochondrial ubiquinone homologues, superoxide radical generation, and longevity in different mammalian species. J Biol Chem 272: 19199-19204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaz G (2001) The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52: 159-164. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fiskum G, Schubert D (2002) Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 80: 780-787. [DOI] [PubMed] [Google Scholar]
- Lyubarev AE, Kurganov BI (1989) Supramolecular organization of tricarboxylic acid cycle enzymes. Biosystems 22: 91-102. [DOI] [PubMed] [Google Scholar]
- Maas E, Bisswanger H (1990) Localization of the alpha-oxoacid dehydrogenase multienzyme complexes within the mitochondrion. FEBS Lett 277: 189-190. [DOI] [PubMed] [Google Scholar]
- Marquez LA, Dunford HB (1995) Transient and steady-state kinetics of the oxidation of scopoletin by horseradish peroxidase compounds I, II and III in the presence of NADH. Eur J Biochem 233: 364-371. [DOI] [PubMed] [Google Scholar]
- Massey V (1994) Activation of molecular oxygen by flavins and flavoproteins. J Biol Chem 269: 22459-22462. [PubMed] [Google Scholar]
- Matthews RG, Ballou DP, Thorpe C, Williams Jr CH (1977) Ion pair formation in pig heart lipoamide dehydrogenase: rationalization of pH profiles for reactivity of oxidized enzyme with dihydrolipoamide and 2-electron-reduced enzyme with lipoamide and iodoacetamide. J Biol Chem 252: 3199-3207. [PubMed] [Google Scholar]
- McMinn CL, Ottaway JH (1977) Studies on the mechanism and kinetics of the 2-oxoglutarate dehydrogenase system from pig heart. Biochem J 161: 569-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AN, Fiskum G, Beal MF (1999) Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab 19: 231-245. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315-360. [DOI] [PubMed] [Google Scholar]
- Park LC, Calingasan NY, Sheu KF, Gibson GE (2000) Quantitative alpha-ketoglutarate dehydrogenase activity staining in brain sections and in cultured cells. Anal Biochem 277: 86-93. [DOI] [PubMed] [Google Scholar]
- Patel MS, Roche TE (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J 4: 3224-3233. [DOI] [PubMed] [Google Scholar]
- Patole MS, Swaroop A, Ramasarma T (1986) Generation of H2O2 in brain mitochondria. J Neurochem 47: 1-8. [DOI] [PubMed] [Google Scholar]
- Ramsay RR, Singer TP (1992) Relation of superoxide generation and lipid peroxidation to the inhibition of NADH-Q oxidoreductase by rotenone, piericidin A, and MPP+. Biochem Biophys Res Commun 189: 47-52. [DOI] [PubMed] [Google Scholar]
- Reed LJ, Hackert ML (1990) Structure-function relationships in dihydrolipoamide acyltransferases. J Biol Chem 265: 8971-8974. [PubMed] [Google Scholar]
- Rosenthal RE, Hamud F, Fiskum G, Varghese PJ, Sharpe S (1987) Cerebral ischemia and reperfusion: prevention of brain mitochondrial injury by lidoflazine. J Cereb Blood Flow Metab 7: 752-758. [DOI] [PubMed] [Google Scholar]
- Sadek HA, Humphries KM, Szweda PA, Szweda LI (2002) Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch Biochem Biophys 406: 222-228. [DOI] [PubMed] [Google Scholar]
- Sheu KF, Blass JP (1999) The alpha-ketoglutarate dehydrogenase complex. Ann NY Acad Sci 893: 61-78. [DOI] [PubMed] [Google Scholar]
- Sims NR (1990) Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem 55: 698-707. [DOI] [PubMed] [Google Scholar]
- Sims NR, Williams VK, Zaidan E, Powell JA (1998) The antioxidant defences of brain mitochondria during short-term forebrain ischemia and recirculation in the rat. Brain Res Mol Brain Res 60: 141-149. [DOI] [PubMed] [Google Scholar]
- Sipos I, Tretter L, Adam-Vizi V (2003) Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J Neurochem 84: 112-118. [DOI] [PubMed] [Google Scholar]
- Sorgato MC, Sartorelli L, Loschen G, Azzi A (1974) Oxygen radicals and hydrogen peroxide in rat brain mitochondria. FEBS Lett 45: 92-95. [DOI] [PubMed] [Google Scholar]
- Starkov A, Fiskum G (2002) Generation of reactive oxygen species by brain mitochondria mediated by alpha-ketoglutarate dehydrogenase. Soc Neurosci Abstr 28: 194.17. [Google Scholar]
- Starkov AA, Fiskum G (2001) Myxothiazol induces H(2)O(2) production from mitochondrial respiratory chain. Biochem Biophys Res Commun 281: 645-650. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G (2003) Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86: 1101-1107. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Polster BM, Fiskum G (2002) Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem 83: 220-228. [DOI] [PubMed] [Google Scholar]
- Sumegi B, Srere PA (1984) Complex I binds several mitochondrial NAD-coupled dehydrogenases. J Biol Chem 259: 15040-15045. [PubMed] [Google Scholar]
- Templeton DM, Hollebone BR, Tsai CS (1980) Magnetic circular dichroism studies on the active-site flavin of lipoamide dehydrogenase. Biochemistry 19: 3868-3873. [DOI] [PubMed] [Google Scholar]
- Tischler ME, Hecht P, Williamson JR (1977) Effect of ammonia on mitochondrial and cytosolic NADH and NADPH systems in isolated rat liver cells. FEBS Lett 76: 99-104. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V (2000) Inhibition of Krebs cycle enzymes by hydrogen peroxide: a key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci 20: 8972-8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V (2004) Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J Neurosci 24: 7771-7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF (1997) Superoxide production by the mitochondrial respiratory chain. Biosci Rep 17: 3-8. [DOI] [PubMed] [Google Scholar]
- Votyakova TV, Reynolds IJ (2001) DeltaPsi(m)-dependent and -independent production of reactive oxygen species by rat brain mitochondria. J Neurochem 79: 266-277. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, Francis N, DeRosier DJ (1983) Alpha-ketoglutarate dehydrogenase complex may be heterogeneous in quaternary structure. J Mol Biol 165: 523-539. [DOI] [PubMed] [Google Scholar]
- Wan B, LaNoue KF, Cheung JY, Scaduto Jr RC (1989) Regulation of citric acid cycle by calcium. J Biol Chem 264: 13430-13439. [PubMed] [Google Scholar]
- Zhang L, Yu L, Yu CA (1998) Generation of superoxide anion by succinatecytochrome c reductase from bovine heart mitochondria. J Biol Chem 273: 33972-33976. [DOI] [PubMed] [Google Scholar]
- Zhou M, Diwu Z, Panchuk-Voloshina N, Haugland RP (1997) A stable nonfluorescent derivative of resorufin for the fluorometric determination of trace hydrogen peroxide: applications in detecting the activity of phagocyte NADPH oxidase and other oxidases. Anal Biochem 253: 162-168. [DOI] [PubMed] [Google Scholar]