

Abstract
The development of ethanol tolerance and dependence reflects neuroadaptive changes in response to continuous depression in synaptic activity. The present study used confocal imaging and electrophysiology procedures to assess the effects of prolonged ethanol exposure on NMDA receptor trafficking in cultures of hippocampal neurons. Neurons exposed to 50 mm ethanol for 4 d showed an increase in the colocalization of NMDA receptor type 1 (NR1) clusters with the presynaptic marker protein synapsin. This was accompanied by significant increases in the size and density of these synapsin-associated clusters with no change observed in nonsynapsin-associated NR1 clusters. Similar effects were observed with NR2B clustering after chronic ethanol exposure. The increase in synaptic NMDA receptor clustering was prevented by addition of a protein kinase A inhibitor or by coexposure to a low concentration of NMDA and was reversed when ethanol was removed from the cultures. No changes were observed in the synaptic content, cluster size, or density of AMPA receptors after ethanol exposure. Electrophysiological measurements on ethanol-treated neurons revealed a similar enhancement in synaptic NMDA currents with no change in AMPA-mediated events. After isolation of extrasynaptic NMDA receptors by MK801 (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (/) trapping, whole-cell responses to NMDA were not different between control and ethanol-treated neurons These observations demonstrate that neuroadaptive changes in NMDA receptors in response to prolonged ethanol exposure occur through activity-dependent processes that regulate their synaptic targeting and localization.
Keywords: NMDA receptors, ethanol, synaptic plasticity, extrasynaptic, protein kinase A, hippocampal cultures
Introduction
Neuroadaptive changes during chronic ethanol consumption are believed to play an important role in the development of tolerance and physical dependence to ethanol (Fadda and Rossetti, 1998). Although the cellular and molecular events that underlie these changes have yet to be clearly defined, alterations in glutamatergic neurotransmission have been observed after ethanol exposure. The critical role of glutamatergic neurotransmission in many forms of activity-dependent synaptic plasticity suggests that it may also play a role in long-lasting effects of ethanol on synaptic efficacy and connectivity. Adaptations within the glutamatergic system in response to prolonged ethanol exposure may, in some respects, engage physiological processes associated with experience-dependent synaptic plasticity (Chandler, 2003).
Numerous studies have demonstrated that movement of glutamate-activated AMPA receptors in and out of the synapse is an important mechanism for controlling synaptic efficacy (Lissin et al., 1998; O'Brien et al., 1998). Similar to AMPA receptors, NMDA receptors are also subject to activity-dependent trafficking and synaptic targeting (Carroll and Zukin, 2002; Wenthold et al., 2003). In addition to regulation of surface expression through insertion and internalization processes, the lateral movement of surface NMDA and AMPA receptors through the membrane between synaptic and extrasynaptic sites may be another important means for controlling synaptic efficacy (Choquet and Triller, 2003).
It is well established that intoxicating concentrations of ethanol inhibit NMDA receptor activity (Woodward, 2000). After chronic ethanol exposure, NMDA receptors appear to increase in number and/or function (Fadda and Rossetti, 1998). These increases presumably occur as an adaptive response to prolonged attenuation of receptor activity and may be generated through a process of homeostatic plasticity that regulates synaptic efficacy. Although ethanol-induced enhancement of NMDA receptor function has been widely reported, there is controversy concerning the cellular basis for this increase in function. Some studies have reported increases in NMDA receptor subunit protein and/or mRNA expression (Trevisan et al., 1994; Follesa and Ticku, 1996; Hu et al., 1996; Snell et al., 1996; Chen et al., 1997; Kumari, 2001), whereas others have observed increases in NMDA receptor function without changes in NMDA receptor levels (Chandler et al., 1997; Rudolph et al., 1997; Cebere et al., 1999; Winkler et al., 1999; Ferreira et al., 2001). These results suggest that although bulk increases in NMDA receptors can occur in response to prolonged ethanol exposure, additional processes that regulate NMDA receptor function are likely engaged (Chandler et al., 1998). Studies in hippocampal neuronal cultures demonstrate that prolonged attenuation of NMDA receptor activity leads to a compensatory increase in their synaptic localization (Rao and Craig, 1997). Because ethanol also inhibits NMDA receptor activity and can produce functional changes after prolonged exposure, the present study used immunohistochemistry with confocal imaging and electrophysiological approaches to examine the effects of prolonged ethanol exposure on the synaptic localization of NMDA and AMPA receptors. The results suggest that ethanol may induce homeostatic changes, at least in part, through activity-dependent alterations in the synaptic trafficking of NMDA receptors.
Materials and Methods
Primary hippocampal cultures. Primary hippocampal cultures were prepared as described previously (Sutton and Chandler, 2002) with modification for growing at low density. Brains from newborn rat pups (<24 hr of age) were removed and placed in isotonic saline containing 100 U of penicillin G, 100 μg of streptomycin, and 0.25 μg/ml of amphotericin B. Pooled hippocampi were digested in 2 ml of 0.25% trypsin (w/v) in isotonic salt solution, pH 7.4, treated with 80 μg of DNase I, and dissociated by trituration through a fire-polished Pasteur pipette. The dissociated cell suspension was then brought to a 50 ml volume with DMEM containing 10% platelet-poor horse serum (PPHS) (Sigma, St. Louis, MO). The cells were then pelleted by centrifugation at 600 × g for 10 min, resuspended in DMEM/PPHS, and plated at a density of 125,000 cells on 35 mm poly-l-lysine-coated plastic culture dishes (Nunc, Naperville, IL). Three hours after plating, the DMEM/PPHS media was replaced with Neurobasal media containing B-27 supplements, penicillin G/streptomycin, and 0.5 mm Glutamax. The Neurobasal media had been astroglia conditioned (without B-27) before addition to hippocampal cultures by incubation for 24 hr in astroglia-confluent flask (25 ml/150 mm flask). Three days after plating, 5 μm β-cytosine arabinoside (Sigma) was added to each dish. Cultures were maintained in this media with one-fourth media changes performed once per week. The glial flasks were prepared by plating 12 × 106 cells from hippocampal preps into poly-l-lysine-treated flasks and replacing the DMEM/PPHS media the following day with DMEM containing 10% FBS.
Chronic drug and ethanol exposure. Hippocampal cultures were chronically exposed to ethanol in vapor chambers as described previously (Chandler et al., 1993). In brief, a concentrated ethanol solution was prepared using culture growth media and added to 15- to 19-d-old cultures to produce 25, 50, and 100 mm ethanol in the culture media. Cultures were then placed in Tupperware containers along with a Nalgene (Naperville, IL) jar that contained 400 ml of an ethanol/water solution, 10% higher in concentration than the cultures. The water in the jar had been temperature and CO2 equilibrated just before the addition of ethanol. The vapor chamber was then filled with 8% balanced CO2 and returned to the CO2 incubators. Control dishes were treated in the same manner without ethanol. After addition of either 2.5 μm NMDA or 1 μm 9,12-epoxy-1H-diindolo(1,2,3-fg:3′,2′,1′-kl)pyrrolo(3,4-i)(1,6)benzodiazocine-10-carboxylic acid, 2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-, hexyl ester, (9α, 10β, 12α) (KT-5720) at the indicated time, dishes were returned to the vapor chambers with a fresh reservoir.
Ethanol measurements. The ethanol concentration in the culture media was routinely measured at the end of the chronic exposure period. Media samples were collected immediately after opening vapor chambers and stored at -20°C for subsequent analysis. Before determination of ethanol concentration on an Analox ethanol detection system (Analox Incorporated, Lunenburg, MA), samples were thawed at room temperature and centrifuged at 10,000 × g for 5 min. Analysis indicated that the ethanol concentration in the media was maintained at the appropriate concentration. For 50 mm ethanol treatments, the ethanol media concentration was found to be 48.0 ± 0.6 mm (n = 30).
Immunohistochemistry. Immunohistochemistry after chronic drug treatment was performed using 19- to 23-d-old cultures with the exception of ethanol withdrawal experiments, which were performed using 27- to 31-d-old cultures. To identify clustering of NMDA receptors and the presynaptic marker protein synapsin, neuronal cultures were rinsed twice in ice-cold PBS (in mm: 136 NaCl, 2.6 KCl, 1.8 KH2HPO4, 10 Na2HPO4, pH 7.4), fixed for 15 min in -20°C methanol, rinsed three times with PBS for 5 min each, and permeabilized in PBS containing 0.2% Triton X-100. After a 1 hr incubation with 1 ml of 5% normal donkey serum, dishes were rinsed with PBS and incubated at room temperature with mouse anti-NMDA receptor type 1 (NR1) (1:100; PharMingen, San Diego, CA) and rabbit anti-synapsin (1:500; Sigma) antibodies. To minimize the amount of antibody required, incubations were performed using CoverWell incubation chambers (Research Products International, Mt. Prospect, IL) that restricted the antibody solution to the center portion of the culture dish. After incubation for 2 hr at room temperature, the chambers were removed, and the dish was rinsed with PBS and then incubated with Alexa 488 anti-mouse (1:1000; Molecular Probes, Eugene, OR) and/or Alexa 594 anti-rabbit (1:4000; Molecular Probes) secondary antibodies in a volume of 1 ml. After 1 hr incubation, dishes were rinsed three times with PBS, dried and mounted with 1,4-diazabicyclo(2,2,2)octane containing mounting media. To identify NR2B containing NMDA receptors or AMPA receptors, the same procedure was performed using 4% paraformaldehyde/4% sucrose fixative with rabbit anti-NR2B (1:200; Chemicon, Temecula, CA) or rabbit anti-glutamate receptor 1 (GluR1) (1:500; Oncogene Sciences, Uniondale, NY) antibodies and mouse anti-synapsin (1:100; Chemicon). Synapsin and synaptophysin was visualized using 4% paraformaldehyde/4% sucrose with mouse anti-synapsin (1:100; Chemicon) and rabbit anti-synaptophysin (1:1000; Chemicon). For colabeling of actin and NR1, cultures were paraformaldehyde-fixed, and Alexa-conjugated phalloidin (1:40; Molecular Probes) was added for 20 min before incubation with anti-NR1 (1:1000; Chemicon) as described above.
Confocal imaging and data analysis. Multichannel imaging of proximal neuronal dendrites was performed with an Olympus (Tokyo, Japan) Fluoview FV 300 or a Zeiss (Thornwood, NY) LSM 510 confocal microscope using a 60× objective and a threefold zoom setting. Laser and detector settings were retained for all images collected within experimental groups. Control and experimental groups were always run in parallel within the same immunohistochemical procedure. MetaMorph 4.6 software was used to interactively define NR1 and synapsin clusters at twofold cytoplasmic background for evaluation of cluster density and size in single-channel analysis. Generating a dilated binary mask of synapsin clusters and overlaying regions of NR1 clusters from the same multichannel image determined cluster colocalization. All NR1 cluster regions with an average binary pixel value that was greater than zero were defined as colocalized with synapsin. Data collected in MetaMorph were exported to a Microsoft Excel (Microsoft, Seattle, WA) spreadsheet. Microsoft Excel data were transferred into Prism4 for analysis of statistical significance and graphing. Confocal images were prepared for display in Adobe Photoshop (Adobe Systems, San Jose, CA).
Whole-cell patch-clamp recordings. Whole-cell recordings of NMDA receptor currents were made using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA). Currents were digitized using an InstruTech (Port Washington, NY) ITC-16 interface and collected on a Macintosh (Apple Computers, Cupertino, CA) G4 computer running the pulse control module under the Igor Pro graphics platform (Wavemetrics, Lake Oswego, OR). Glass pipettes (3-7 mΩ) were filled with internal solution containing the following (in mm): 140 CsCl, 2 MgCl2, 2.5 EGTA, 2 tetraethylammonium, 4 K2ATP, 10 HEPES, pH 7.4, adjusted to 300-310 mOsm with sucrose. In some experiments, lidocaine N-ethyl bromide (5 mm) was added to the pipette filling solution to block voltage-dependent sodium channels. Cells were continuously perfused with an extracellular solution containing the following (in mm): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 5 HEPES, and 10 glucose, pH 7.4, adjusted to 320 mOsm with sucrose. Unless otherwise noted, glycine (10 μm) and tetrodotoxin (TTX; 1 μm) were added to the extracellular recording solution. Neurons selected for recording were held at -65 mV. Series resistance was routinely compensated by 60-80% and was monitored throughout each recording. For electrophysiological recording from neurons chronically exposed to ethanol, cells were patched immediately after the removal of the ethanol-containing culture media.
For recording glutamate-mediated miniature currents (minis), recordings were performed in the presence of bicuculline (25 μm) and strychnine (1 μm) to block GABAergic and glycinergic events, respectively. Although AMPA-mediated events are easily detected, the small size and slow kinetics of NMDA minis make it extremely difficult to distinguish them from background noise. Therefore, we used a technique that indirectly measures NMDA minis (Thomas et al., 2001). After recording in Mg2+-free buffer to capture miniature EPSCs (mEPSCs) that contained both AMPA and NMDA components, AMPA minis were isolated by perfusing the neuron with APV (10 μm) to block NMDA activity. Events were detected using the fast rise time of the AMPA component, and NMDA minis were calculated as the difference between currents recorded in the absence and presence of APV. GABA-mediated minis were measured directly in separate control and ethanol-treated cultures in the presence of AP-5 (10 μm) and 2,3-dihydroxy-6-nitro-7-sulfonylbenzo[f]quinoxaline (NBQX; 10 μm) to block glutamate-mediated events. Currents recorded for miniature synaptic events were filtered at 1-2 kHz and digitized at 5 kHz. Events (>100 per neuron) were detected off-line using the variable template detection routine in Axograph 4.6 software (Axon Instruments).
For measurement of agonist-induced currents, a multibarreled fast perfusion system (Warner Instruments, Hamden, CT) with a switching time of ∼8 msec was used. Currents were activated by jumping from the control barrel to one containing agonist and then back. To selectively block synaptic NMDA currents, neurons were first perfused with a solution containing 5 μm (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) plus 10 μm glycine for 5 min followed by a 10 min washout in glycine-free solution.
Results
Chronic ethanol enhances NMDA receptor clustering and synaptic targeting
To examine clustering and synaptic localization of NMDA receptors, low-density primary hippocampal neurons were maintained for 19-23 d in culture and subjected to immunohistochemical analysis of the NR1 subunit. A large majority of neurons exhibited typical pyramidal cell morphology, and only pyramidal-like neurons were selected for subsequent image analysis. Numerous punctate clusters containing NR1 immunoreactivity were observed along the dendritic arbors (Fig. 1). NR1 cluster analysis was restricted to the proximal portions of the dendrite within 150 μm of the soma. In preliminary control experiments, exposure (4 d) of neuronal cultures to the NMDA antagonist APV (100 μm) or block of synaptic activity by TTX (1 μm) enhanced NR1 clustering (data not shown) in a manner consistent with previous reports (Rao and Craig, 1997).
Figure 1.

Chronic ethanol exposure enhances the dendritic clustering of NMDA receptors. Immunohistochemistry of NR1 and confocal imaging was used to examine the effects of ethanol exposure on NMDA receptor clustering in hippocampal cultures. A-C, Confocal imaging of NR1 revealed punctate clustering of NMDA receptors in control cultures (A). A 4 d exposure of neuronal cultures to 25 mm ethanol (B) did not alter the cluster pattern, whereas 50 mm ethanol (C) exposure resulted in a striking increase in clustering. Scale bar, 40 μm. D, E, Quantification of the effects of ethanol on clustering revealed a significant increase in both NR1 cluster size (D) and density (E). The asterisk indicates a significant difference from control (p < 0.01, Student's t test; n = 90).
The next series of experiments examined the effects of prolonged ethanol exposure on NMDA receptor clustering. As shown in Figure 1, A and B, exposure of hippocampal cultures to 50 mm but not 25 mm ethanol for 4 d greatly increased the size and density of NR1 clusters. Increasing the duration of the 25 mm ethanol exposure to 8 d produced a significant increase in NR1 cluster size (control, 7.49 ± 0.30 vs ethanol-treated, 11.42 ± 0.41 pixels per cluster; p < 0.01; n = 30) and density (control, 6.87 ± 0.05 vs ethanol-treated, 12.05 ± 0.07 clusters per 10 μm;p < 0.01; n = 30), similar to that observed with 50 mm ethanol. Exposure of cultures to 100 mm ethanol for 2 d did not alter NR1 clustering, whereas 4 d of exposure produced an increase in NR1 cluster size (control, 7.59 ± 0.31 vs ethanol-treated, 11.54 ± 0.52 pixels per cluster; p < 0.01; n = 30) and density (control, 7.07 ± 0.49 vs ethanol-treated, 13.42 ± 0.72 clusters per 10 μm; p < 0.01; n = 30), similar to that observed with the 4 d 50 mm ethanol treatment. These observations indicate that ethanol exposure causes an increase in dendritic NMDA receptor clustering that is both time and dose dependent. Because 50 mm ethanol is well within the behaviorally relevant intoxicating dose range (equivalent to a blood ethanol concentration of ∼0.23 mg/dL), this concentration was used in all subsequent experiments.
To examine the subcellular localization of dendritic NR1 clustering, we performed dual immunohistochemistry of NR1 and the synaptic marker protein synapsin (Fig. 2). In control cultures, synapsin showed intense dendritic punctate staining with ∼50% colocalization with NR1 clusters (Fig. 2A,D). This suggests that approximately one-half of dendritic clustered NMDA receptors are localized in synapses, with the remaining clustered NMDA receptors being extrasynaptic. Exposure of neuronal cultures to ethanol (50 mm; 4 d) increased the synaptic localization of NR1 clusters to ∼80% (Fig. 2B,D). Additional analysis of the synaptic versus extrasynaptic NR1 clusters revealed that the ethanol-induced increase in NR1 clustering shown in Figure 1 occurred in the synaptic compartment (Fig. 2E,F). In contrast, ethanol had no effect on extrasynaptic NR1 cluster size or density. To determine whether the effect of ethanol on NR1 clustering was activity dependent, a low concentration of NMDA (2.5 μm) was included in the culture media during the final 2 d of ethanol exposure. As shown in Figure 2, addition of NMDA prevented the ethanol-induced increase in synaptic NR1 cluster size and density and reversed the enhanced colocalization. In contrast, NMDA had no effect on extrasynaptic NR1 clusters in ethanol-treated dishes. Together, these observations indicate that chronic ethanol exposure induces a selective, activity-dependent targeting of NMDA receptors to synapses. Chronic ethanol treatment also increased the size and density of synapsin clusters (Fig. 2G,H). This was an unexpected observation, because no changes in synaptophysin have been reported after prolonged exposure to APV or TTX (Rao and Craig, 1997; Crump et al., 2001; Fong et al., 2002). To determine whether the effect of ethanol was synapsin specific, we examined the effects of ethanol on clustering of synaptophysin. Dual immunohistochemistry of synapsin and synaptophysin revealed a high degree (>95%) of cluster colocalization. Again, ethanol (50 mm; 4 d) increased the degree of synaptophysin clustering similar to that observed for synapsin (Fig. 2I,J). Consistent with synaptic and extrasynaptic NMDA receptor populations, actin staining with phallotoxin to label dendritic spines revealed that some NR1 clusters were localized to spine heads and were presumably synaptic, whereas other NR1 clusters appeared to be localized on the main dendrite and were presumably extrasynaptic (Fig. 2K). Furthermore, some immature spines did not contain NR1 clusters.
Figure 2.
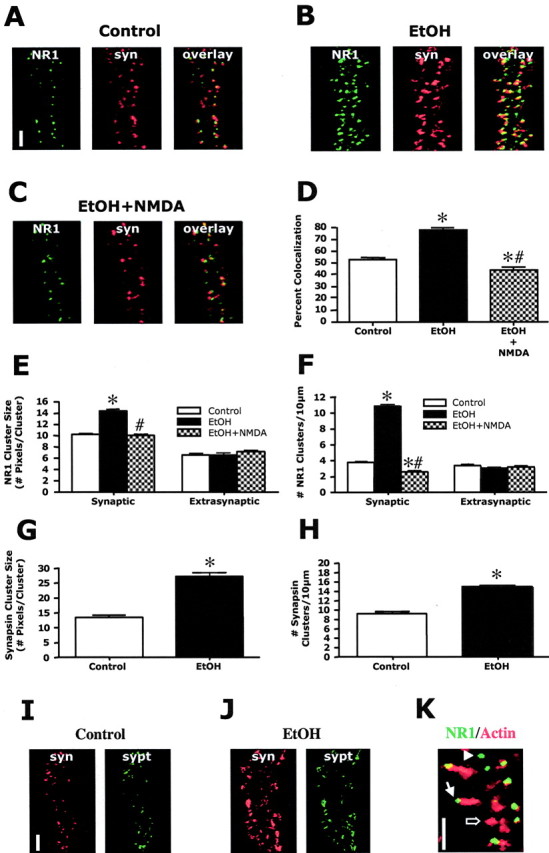
Chronic ethanol induces the synaptic targeting of NMDA receptors. A-F, Dual immunohistochemistry of NR1 (green) and synapsin (syn; red) revealed that ∼50% of the NR1 clusters colocalize with synapsin (A, D) under control conditions. Ethanol exposure (50 mm; 4 d) resulted in a significant increase in NR1/synapsin colocalization to ∼80% (B, D). The ethanol-induced increase in NR1 cluster size (E) and density (F) was limited to the synaptic compartment with no changes occurring in extrasynaptic NR1 clustering. The addition of NMDA (2.5 μm) completely prevented the ethanol-induced increases in NR1 synaptic colocalization (C, D) and increases in synaptic cluster size (E) and density (F). *, Significant difference from control; #, significant difference from ethanol treated (p < 0.01; ANOVA with Student Newman-Keuls; n = 90). Scale bar, 5 μm. G, H, Quantification of synapsin clusters revealed that ethanol also significantly increased cluster size (G) and density (H). The asterisk indicates significant difference from control (p < 0.01, Student's t test; n = 90). I, J, Dual immunohistochemistry of synapsin (red) and synaptophysin (sypt; green) revealed that ethanol (50 mm; 4 d) increased clustering of synaptophys in similar to that observed with synapsin. Scale bar, 5 μm. K, Actin staining with fluorescently labeled phallodin revealed a population of NR1 clusters that are localized to the heads of spines (filled arrow) and a population of NR1 clusters along the main dendrite that are not associated with spines (arrowhead). Some immature spines were without NR1 clusters (open arrow). Scale bar, 5 μm.
NMDA receptors in hippocampal neurons are composed of a requisite NR1 subunit and an NR2A and/or NR2B subunit. NR2 subunits impart distinct pharmacological and kinetic properties on the channel. For example, NR2B subunit-containing receptors are more sensitive to glutamate and exhibit slower channel deactivation times and therefore greater Ca2+ influx. To determine whether ethanol also induced changes in clustering of NR2 subunit-containing receptors, immunohistochemistry of the NR2B subunit was examined. As observed with the NR1 subunit (Fig. 1), ethanol (50 mm; 4 d) significantly increased dendritic NR2B cluster size and density (Fig. 3A-D). Dual immunohistochemistry revealed that under control conditions, ∼65% of NR2B clusters colocalized with synapsin, suggesting that they are synaptic (Fig. 3E-G). The degree of NR2B colocalization with synapsin increased to ∼80% after ethanol exposure. Furthermore, the increase in NR2B cluster size and density occurred primarily in the synaptic compartment (Fig. 3H,I). These observations indicate that synaptically targeted NMDA receptors contain the NR2B subunit. Attempts to examine clustering and targeting of NR2A subunits using several different commercially available NR2A specific antibodies were not successful.
Figure 3.
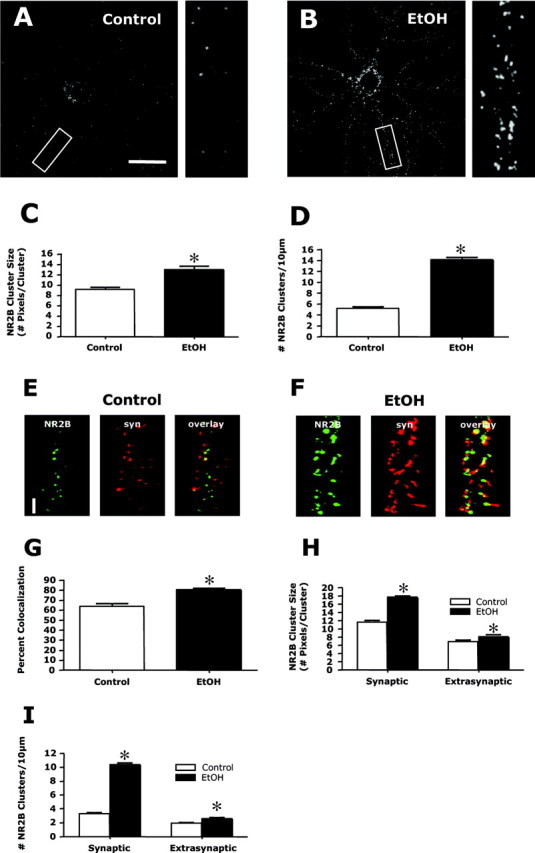
Chronic ethanol enhances the synaptic clustering of NR2B. A, B, Immunohistochemistry and confocal imaging of NR2B subunits revealed numerous small punctate clusters along dendrites similar to that observed with NR1. This clustering was greatly increased after exposure to ethanol (50 mm; 4 d) (compare A and B). Scale bar, 40 μm. C, D, Quantification of clustering indicated a significant increase in both cluster size (C) and density (D). The asterisk indicates a significant difference from control (p < 0.01, Student's t test; n = 90). E, G, Dual immunohistochemistry of NR2B (green) and synapsin (syn; red) revealed ∼65% colocalization of NR2B and synapsin clusters under control conditions (E, G). After ethanol exposure (50 mm; 4 d), this colocalization increased to ∼80% (F, G). The asterisk indicates significant difference from control (p < 0.01, Student's t test; n = 90). Scale bar, 5 μm. H, I, Analysis of clustering in the synaptic and extrasynaptic compartments revealed that ethanol treatment greatly increased the size (H) and density (I) of synaptic clusters, with only minor increases in extrasynaptic clusters. The asterisk indicates a significant difference from control (p < 0.01, Student's t test; n = 90).
Chronic ethanol does not alter GluR1 clustering
GluR1 immunoreactivity was examined to determine whether chronic ethanol exposure also altered the synaptic distribution of AMPA receptors. In control cultures, GluR1 showed intense dendritic punctate clustering similar to that for NR1 staining (Fig. 4). However, in contrast to NR1/synapsin colocalization, essentially all GluR1 clusters were colocalized with synapsin clusters under control conditions (control, 96.3 ± 1.1%). This indicates that in the older, more mature, hippocampal cultures used in this study, there was not a significant pool of clustered extrasynaptic GluR1 receptors and that synapses had already undergone significant “AMPAfication” (Zhu and Malinow, 2002). Ethanol (50 mm) for 4 d had no significant effect (Student's t test; n = 10) on GluR1/synapsin colocalization (ethanol, 95.3 ± 2.5%), GluR1 synaptic cluster size (control, 10.3 ± 0.7 pixels per cluster; ethanol, 12.1 ± 0.9 pixels per cluster), or density (control, 7.3 ± 0.7 cluster per 10 μm; ethanol, 7.4 ± 0.6 clusters per 10 μm).
Figure 4.

Chronic ethanol does not alter synaptic clustering of GluR1. Dual immunohistochemistry and confocal imaging of GluR1 and synapsin show almost complete colocalization of clusters in control cultures. Ethanol (50 mm; 4 d) had no effect on GluR1 clustering or colocalization with synapsin (n = 10). Scale bar, 20 μm.
Increased clustering of NMDA receptors is reversed after ethanol withdrawal and by inhibition of protein kinase A
If ethanol-induced synaptic targeting of NMDA receptors occurs in response to ethanol inhibition of synaptic activity, then NMDA receptor levels should revert to control levels after ethanol withdrawal. To test this hypothesis, cultures were removed from the ethanol vapor chambers after 4 d and returned to the incubator where the ethanol in the culture media was allowed to evaporate over time. Four days after removal of the dishes from the vapor chambers, the ethanol-induced increase in NR1 clustering was still present (data not shown). However, after 8 d of withdrawal, NR1 synaptic levels had completely reverted to that observed in age-matched controls (Fig. 5A,B) (control, 51.4 ± 1.7%; withdrawn, 52.2 ± 1.5%; n = 90). Similarly, synaptic NR1 cluster size (control, 11.8 ± 0.4 pixels per cluster; ethanol, 11.9 ± 0.5 pixels per cluster) and density (control, 5.1 ± 0.2 clusters per 10 μm; ethanol, 4.7 ± 0.2 clusters per 10 μm; n = 90) also returned to control levels. There was a slight decrease in the density (control, 5.0 ± 0.3 clusters per 10 μm; ethanol, 4.3 ± 0.2 clusters per 10 μm; p < 0.5; Student's t test) but not size (control, 6.6 ± 0.2 pixels per cluster; ethanol, 6.6 ± 0.2 pixels per cluster) of extrasynaptic NR1 clusters. These observations demonstrate that the ethanol-induced increase in synaptic NMDA receptors undergoes a time-dependent reversal after removal of ethanol.
Figure 5.

Ethanol-induced increase in synaptic clustering of NR1 is reversed after removal of ethanol. A, B, Culture dishes were removed from the ethanol vapor chambers, and ethanol was allowed to slowly evaporate from the culture media. NR1 (green) and synapsin (red) colocalization returned to control levels after 8 d. Scale bar, 20 μm.
A previous study with hippocampal cultures reported that protein kinase A (PKA) activity is required for activity-dependent targeting of NMDA receptors to synapses (Fong et al., 2002). In agreement with this finding, exposure of neuronal cultures to the PKA activator 8-Br-cAMP (10 μm) for 24 hr led to a large increase in synaptic NR1 clusters (data not shown). To test whether PKA activity was required for ethanol-induced synaptic trafficking of NMDA receptors, the effects of the PKA inhibitor KT-5720 on NR1 clustering were examined. Preliminary studies showed that treatment with KT-5720 (1 μm) for 2 d or longer induced significant toxicity, whereas a 1 d exposure had no noticeable effects on neuronal viability or morphology. Thus, KT-5720 (1 μm) was added only during the final day of the 4 d ethanol exposure period. Under this treatment protocol, KT-5720 slightly reduced NR1/synapsin colocalization in control cultures and completely reversed the ethanol-induced increase in NR1/synapsin colocalization (Fig. 6A,B). Analysis of NR1 clustering in synaptic versus extrasynaptic compartments revealed that KT-5720 also reversed the effect of ethanol on synaptic NR1 cluster size and density but did not alter extrasynaptic NR1 clusters (Fig. 6C,D). Together, these observations demonstrate that PKA activity is required for ethanol-induced synaptic targeting of NMDA receptors.
Figure 6.

The ethanol-induced increase in synaptic NR1 clustering is PKA dependent. Dual immunoblotting and confocal imaging were used to examine the effects of a PKA inhibitor on NR1 (green) and synapsin (red) colocalization. A, B, In control cultures, a 24 hr exposure to KT-5720 (KT; 1 μm) reduced NR1/synapsin colocalization. The addition of KT-5720 on the final day of ethanol exposure (50 mm; 4 d) completely prevented the ethanol-induced increase in clustering. *, Significant difference from control (p < 0.01); @, significant difference from control and ethanol alone (0.05); #, significant difference from ethanol alone (p < 0.01; ANOVA with Student Newman-Keuls). C, D, Examination of the effects of KT-5720 on NR1 clustering in the synaptic and extrasynaptic compartments revealed that the effect of KT-5720 on NR1 cluster size (C) and density (D) were specific for the synaptic receptor compartment. *, Significant difference from control (p < 0.01); @, significant difference from control and ethanol alone (p < 0.01); #, significant difference from ethanol alone (p < 0.01; ANOVA with Student Newman-Keuls). B-D, n = 60 for control, KT, and EtOH plus KT groups; n = 30 for EtOH group.
Chronic ethanol enhances synaptic NMDA currents but does not alter synaptic AMPA currents or extrasynaptic NMDA currents
The immunolabeling and confocal imaging studies clearly demonstrate that chronic exposure of hippocampal neurons to ethanol induces a preferential increase in NMDA receptors at synapses without changing AMPA receptors. To assess the functional relevance of these changes, several electrophysiological approaches were used to measure synaptic glutamatergic currents. In the first set of experiments, mEPSCs and mIPSCs were measured in control and ethanol-treated cultures. Chronic ethanol had no effect on the amplitude or frequency of AMPA minis (Fig. 7A) or the amplitude and frequency of GABAA minis (Fig. 7C). In contrast, ethanol significantly enhanced the NMDA/AMPA current ratio (Fig. 7B). The increase in the NMDA/AMPA ratio was attributable to an increase in NMDA mini amplitude (control, 5.22 ± 1.26 pA vs ethanol-treated, 8.81 ± 1.47 pA), because AMPA mini amplitude was not changed (control, 14.65 ± 2.05 pA vs ethanol-treated, 12.56 ± 1.29 pA). As observed with the immunohistochemical studies, the ethanol-induced increase in synaptic NMDA currents returned to control levels 8 d after the dishes were removed from the ethanol vapor chambers, and ethanol was allowed to evaporate from the culture dishes (Fig. 7B).
Figure 7.

Chronic ethanol exposure enhances synaptic but not extrasynaptic NMDA currents and does not affect synaptic AMPA currents. A, In the presence of APV (10 μm) to block NMDA receptors, ethanol (50 mm; 4 d) had no effect on either the amplitude or frequency of mEPSCsAMPA (Student's t test; n = 7). B, Ethanol treatment (50 mm; 4 d) increased the mEPSC NMDA/AMPA amplitude ratio. When culture dishes were removed from the ethanol vapor chambers and ethanol was allowed to evaporate from the culture media, the NMDA/AMPA current ratio returned to control levels. The asterisk indicates a significant difference from control (p < 0.05, Student's t test; n = 7). C, Ethanol (50 mm; 4 d) did not alter either the amplitude or frequency of mIPSCsGABAA (Student's t test; n = 3-4). D, Activation of synaptic NMDA receptors by the addition of the NMDA receptor coagonist glycine resulted in enhanced NMDA current amplitude in ethanol-treated cultures compared with untreated controls. The asterisk indicates a significant difference from control (p < 0.05, Student's t test; n = 5-8). E, F, Extrasynaptic NMDA receptor currents were isolated from synaptic currents by selectively blocking synaptic NMDA receptors using the MK-801 trapping technique. D, Stimulation of extrasynaptic NMDA receptors by application of NMDA revealed extrasynaptic currents constituted approximately two-thirds of the total current (compare the size of the before and after MK-801 trace). E, Ethanol treatment (50 mm; 4 d) did not alter the NMDA concentration-response curve for activation of extrasynaptic NMDA receptors (n = 5).
As an additional measure of the effects of ethanol on synaptic NMDA receptors, whole-cell currents were monitored before and during local application of glycine to selectively activate synaptic NMDA receptors (Lu et al., 2001). Cultures were bathed in magnesium-free and glycine-free media containing TTX, bicuculline, and strychnine. Brief application of 1 or 10 μm glycine to voltage-clamped neurons via a local perfusion pipette resulted in an increased whole-cell current (measured as total charge transfer normalized to cell capacitance) that arises from activation of synaptic NMDA receptors by glutamate released spontaneously from presynaptic terminals (Fig. 7D). The magnitude of this glycine-evoked synaptic NMDA receptor current was enhanced in neurons previously treated with ethanol.
To test whether ethanol also induced changes in extrasynaptic NMDA currents, the MK-801 trapping technique was used to eliminate synaptic NMDA receptors (Lu et al., 2001; Tovar and Westbrook, 2002). Blocking synaptic receptors with MK-801 reduced responses to NMDA by ∼33% (Fig. 7E). Using this technique, extrasynaptic NMDA dose responses were compared in control and ethanol-treated cultures. Under these conditions, chronic ethanol treatment had no effect on NMDA-evoked currents, consistent with the lack of effect of ethanol on extrasynaptic receptor clustering (Fig. 7F).
Because the immunohistochemical and electrophysiology studies showed that chronic ethanol exposure enhanced the synaptic content of NMDA receptors, we hypothesized that this would result in an increase in NMDA receptor driven network activity in the cultures after abrupt removal of ethanol. To test this, we first measured spontaneous activity under conditions where NMDA receptor function is maximized (zero magnesium and bicuculline in the bath). Neurons from control cultures showed spontaneous glutamatergic EPSCs containing AMPA (Fig. 8A) and NMDA (Fig. 8B) components. Both the amplitude and frequency of these events were dramatically enhanced in neurons undergoing withdrawal from chronic ethanol exposure. However, the omission of Mg2+ from the extracellular buffer creates an abnormal condition for activation of synaptic NMDA receptors. Therefore, to determine whether withdrawal from chronic ethanol exposure would also enhance excitability under more physiological conditions, we performed current-clamp recordings in media containing 1 mm Mg2+ with no added bicuculline. As shown in Figure 8C, the frequency of spiking in control neurons was ∼1.2 Hz. This increased to ∼2.1 Hz in chronic ethanol-treated cultures after washout of the ethanol-containing culture media. Bath application of the NMDA antagonist APV resulted in the near complete collapse of network activity and spontaneous firing in these low-density cultures (data not shown). These observations demonstrate that chronic ethanol exposure results in enhanced glutamatergic activity that is unmasked after ethanol removal and supports the suggestion that NMDA receptor hyperactivity may contribute to CNS hyperexcitability during ethanol withdrawal.
Figure 8.

Chronic ethanol exposure enhances spontaneous EPSCs and network activity. A, B, In the absence of extracellular Mg2+, the addition of APV to isolate EPSCsAMPA (A) or NBQX to isolate EPSCsNMDA (B) revealed a significant increase in the amplitude and frequency of both AMPA and NMDA components of the EPSC when measured immediately after the removal of the ethanol-containing culture media. Symbols indicate significant difference from control (*p < 0.01; @ p < 0.05; Student's t test; n = 3). C, In the presence of extracellular Mg2+, chronic ethanol treatment resulted in an increase in network driven spontaneous spike activity. The asterisk indicates a significant difference from control (p < 0.05, Student's t test; n = 5-6).
Discussion
There is considerable evidence indicating that glutamatergic neurotransmission plays an important role in the behavioral actions of both acute and chronic ethanol (Fadda and Rossetti, 1998;Krystal et al., 2003). The results of the present study demonstrate that enhanced glutamatergic activity after prolonged ethanol exposure is associated with a selective increase in synaptic NMDA receptors without changes in extrasynaptic receptors. The ability of ethanol to differentially alter the expression and trafficking of NMDA receptors between spatially resolved and functionally distinct membrane domains provides an elegant and dynamic means for adapting to ethanol-induced attenuation of synaptic signaling. These observations provide additional support for the emerging concept that normal experience-dependent plastic processes in the brain are adversely co-opted by ethanol and other drugs of abuse.
Chronic blockade of synaptic activity with TTX or APV induces the clustering of NMDA receptors at synapses (Rao and Craig, 1997). In the present study, prolonged exposure to ethanol increased the size and density of both NR1 and NR2B subunit clusters. Although these immunohistochemical studies did not distinguish between surface and internalized receptors, they did differentiate synaptic from extrasynaptic NMDA receptor clusters on the basis of their colocalization with the presynaptic marker protein synapsin. Analysis of NMDA/synapsin cluster colocalization revealed that the increase in NMDA receptor cluster size and density was restricted to the synaptic pool, because no changes were observed in the extrasynaptic domain. Prolonged ethanol exposure has been reported to enhance NMDA receptor synthesis (Chen et al., 1999). Although these newly synthesized receptors could be inserted directly into the synaptic membrane at the postsynaptic density, this is considered unlikely, because receptor endocytosis-exocytosis is thought to occur extrasynaptically (Roche et al., 2001; Blanpied et al., 2002). Rather, it appears that lateral movement of receptors, including NMDA, through the membrane may be a more important process in the delivery of receptors to the synapse (Choquet and Triller, 2003).
In the present study, ethanol-induced changes in the synaptic localization of NR1 subunit immunoreactivity were mirrored by nearly identical changes in synaptic immunoreactivity of the NR2B subunit. Studies have suggested that NR2B-containing receptors may contribute to enhanced plasticity of the young brain compared with the adult brain (Okabe et al., 1998). In addition to conferring distinct gating and pharmacological properties to heteromeric NMDA receptors, NR2A- and NR2B-containing receptors couple to distinct and often opposing intracellular signaling pathways and appear to differentially traffic to the synapse (Sheng and Pak, 2000; Barria and Malinow, 2002; Krapivinsky et al., 2003; Liu et al., 2004). Thus, the presence of extrasynaptic NR2B-containing receptors likely has important functional significance for activity-dependent synaptic plasticity. Unfortunately, attempts to analyze NR2A subunits were unsuccessful, because the commercial NR2A antibodies tested were not satisfactory for immunohistochemical analysis of clustering. Regardless, given the likely importance of NR2B containing receptors in experience-dependent plasticity, the ability of prolonged ethanol to increase the synaptic localization of NR2B subunits suggests a mechanism for ethanol-induced changes in synaptic plasticity similar to learning and memory processes. Interestingly, the NR2B subunit has been implicated in neuroadaptive changes in the central amygdala (Roberto et al., 2004) and hippocampus (Miyakawa et al., 1997; Yaka et al., 2003b) in response to ethanol exposure.
Calcium influx through NMDA receptors stimulates the rapid insertion of AMPA receptors in the synapse via a calcium/calmodulin-dependent protein kinase II-dependent process (Shi et al., 1999). Thus, prolonged inhibition of NMDA receptor activity by ethanol would be expected to reduce the clustering of AMPA receptors in synapses. However, chronic ethanol exposure did not affect the clustering or synaptic localization of the AMPA subunit GluR1. GluR1 clusters in control cultures showed a remarkably high degree of colocalization with synapsin, suggesting that these neurons were fully mature at the time they were examined (19-23 d in culture). This maturation may account for the apparent lack of AMPA receptor-based plasticity in response to ethanol. Such an age-dependent difference between AMPA and NMDA receptor plasticity would not be a unique observation, because studies in adult rat hippocampal slices have reported enhanced numbers of synaptic NMDA receptors and not AMPA receptors after long-term potentiation (LTP) (Grosshans et al., 2002). In addition, the adaptive increase in NMDA receptor activity after ethanol exposure may have negated (or reversed) changes in AMPA receptors in these cultures.
The immunohistochemical studies were confirmed by functional studies showing that chronic ethanol increased synaptic NMDA receptors. This was reflected by the increase in NMDA mEPSC amplitude and the enhanced current response to the local application of the NMDA coagonist glycine. The ethanol-induced change in synaptic NMDA currents was reversed after evaporation of ethanol contained in the media. In addition, ethanol treatment did not alter AMPA mEPSC amplitude. The selective effect of ethanol on synaptic NMDA currents was verified by using an MK-801/glycine exposure protocol and showed that currents produced by extrasynaptic receptors were similar in control and ethanol-treated neurons. The increase in the density and function of synaptic NMDA receptors after chronic ethanol exposure would be expected to significantly enhance neuronal excitability under conditions where action potential firing is intact. This was examined directly by recording glutamatergic EPSCs evoked by spontaneous network activity. Ethanol-treated neurons showed a significant increase in the number and amplitude of spontaneous NMDA and AMPA-mediated EPSCs. The ethanol-induced increase in synaptic NMDA receptors thus results in an enhanced release of glutamate and larger postsynaptic glutamatergic currents. However, these recordings were performed in the absence of magnesium and in the presence of blockers of inhibitory transmission (bicuculline and strychnine) to provide maximal excitatory drive. Under more physiological current-clamp recording conditions (extracellular buffer containing 1 mm Mg2+ and no bicuculline), chronic ethanol exposure of neuronal cultures again resulted in an increase in network driven neuronal firing activity. Together, these results support the suggestion that behavioral hyperexcitability and enhanced seizure activity observed during ethanol withdrawal may result from enhanced drive of synaptic NMDA receptors (Fadda and Rossetti, 1998).
The signaling mechanism(s) underlying ethanol-induced synaptic targeting of NMDA receptors is not clear. We observed that exposure of control cultures to the PKA inhibitor KT-5720 led to a selective reduction in the synaptic clustering of NMDA receptors, confirming previous findings (Crump et al., 2001). Inhibition of PKA also completely reversed the ethanol-induced increase in NMDA receptor clustering. Again, this effect was selective for synaptically localized receptors. Ethanol has been reported to activate PKA signaling, and studies have linked changes in cAMP/PKA signaling with chronic ethanol exposure (Diamond and Gordon, 1997) and have implicated PKA in the development of ethanol tolerance. For example, mice with genetic disruptions in cAMP/PKA signaling exhibit increased ethanol sensitivity and do not develop tolerance to its sedative effects (Thiele et al., 2000; Wand et al., 2001). It has also been reported that in the nucleus accumbens, acute tolerance to ethanol involves alterations in serine phosphorylation of the NR1 subunit through a PKA-dependent process (Maldve et al., 2002). Although our findings with a PKA inhibitor do not unequivocally prove that chronic ethanol induces clustering through activation of PKA, they are consistent with such a suggestion. Activity controls NMDA receptor synaptic delivery by regulating forward trafficking at the endoplasmic reticulum (ER) (Mu et al., 2003). Note that the coordinated phosphorylation of the NR1 subunit by PKA and protein kinase C (PKC) mediates release of the receptor from the ER before enhanced surface expression and trafficking to the synapse (Scott et al., 2003). Thus, it is reasonable to speculate that chronic ethanol enhances activity-dependent export of NMDA receptors from the ER via changes in PKA and PKC phosphorylation of the NR1 subunit. Alternatively, it has been reported that alterations in tyrosine phosphorylation of NR2 subunits by Fyn tyrosine kinase may be important in the development of ethanol tolerance (Miyakawa et al., 1997; Yaka et al., 2003a, b). Together with the results of the present study, the above observations suggest that rapid changes in NMDA receptors after acute ethanol involve phosphorylation-dependent alterations in channel conductance and/or surface expression, and slower changes during chronic ethanol exposure require alterations in their synaptic localization.
Finally, Hebbian mechanisms of plasticity are thought to play an important role in modifying neuronal circuitry in response to changes in activity. However, if considered an isolated phenomenon, Hebbian-based plasticity would be particularly destabilizing over time, because increases or decreases in activity would ultimately drive neuronal activity toward runaway excitation or depression, respectively. Thus, homeostatic plasticity is a higher-order form of plasticity that is thought to impart stability to neuronal circuits (Turrigiano and Nelson, 2004). By dynamically scaling synaptic strength, homeostatic processes may prevent neuronal circuits from becoming hyperactive or hypoactive in the face of an ongoing destabilizing force such as ethanol. Although acute ethanol may disrupt Hebbian-based plastic events such as LTP and neuronal survival during development, continued ethanol exposure must engage homeostatic processes to return stability to the neuronal network. Our data suggests that dynamic regulation of trafficking of NMDA receptors to the synapse is an important mechanism underlying ethanol-induced neuroadaptive plasticity.
Footnotes
This work was supported by National Institutes of Health Grants AA010983 and RR15776 to L.J.C., AA09986 and AA00238 to J.J.W., AA010761 to L.J.C. and J.J.W., and a grant from the Alcoholic Beverage Medical Research Foundation to L.J.C. E.C-.H. was supported by National Research Service Award AA13355.
Correspondence should be addressed to L. Judson Chandler, Department of Physiology and Neuroscience, Medical University of South Carolina, 67 President Street, Charleston, SC 29425. E-mail: chandj@musc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247859-10$15.00/0
References
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345-353. [DOI] [PubMed] [Google Scholar]
- Blanpied TA, Scott DB, Ehlers MD (2002) Dynamics and regulation of clathrin coats at specialized endocytic zones of dendrites and spines. Neuron 24: 435-449. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS (2002) NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci 25: 571-577. [DOI] [PubMed] [Google Scholar]
- Cebere A, Cebers G, Liljequist S (1999) Enhancement of NMDA-induced functional responses without concomitant NMDA receptor changes following chronic ethanol exposure in cerebellar granule cells. Naunyn Schmiedebergs Arch Pharmacol 360: 623-632. [DOI] [PubMed] [Google Scholar]
- Chandler LJ (2003) Ethanol and brain plasticity: receptors and molecular networks of the postsynaptic density as targets of ethanol. Pharmacol Ther 99: 311-326. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sumners C, Crews FT (1993) Ethanol inhibits NMDA receptor-mediated excitotoxicity in rat primary neuronal cultures. Alcohol Clin Exp Res 17: 54-60. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Norwood D, Sumners C, Crews FT (1997) Chronic ethanol increases N-methyl-d-aspartate stimulated nitric oxide formation but not receptor density in cultured cortical neurons. Mol Pharmacol 51: 733-740. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Harris RA, Crews FT (1998) Ethanol tolerance and synaptic plasticity. Trends Pharmacol Sci 19: 491-495. [DOI] [PubMed] [Google Scholar]
- Chen X, Michaelis ML, Michaelis EK (1997) Effects of chronic ethanol treatment on the expression of calcium transport carriers and NMDA/glutamate receptor proteins in brain synaptic membranes. J Neurochem 69: 1559-1569. [DOI] [PubMed] [Google Scholar]
- Chen X, Moore-Nichols D, Nguyen H, Michaelis EK (1999) Calcium influx through NMDA receptors, chronic receptor inhibition by ethanol and 2-amino-5-phosponopentanoic acid, and receptor protein expression. J Neurochem 72: 1969-1980. [DOI] [PubMed] [Google Scholar]
- Choquet D, Triller A (2003) The role of receptor diffusion in the organization of the postsynaptic membrane. Nat Rev Neurosci 4: 251-265. [DOI] [PubMed] [Google Scholar]
- Crump FT, Dillman KS, Craig AM (2001) cAMP-dependent protein kinase mediates activity-regulated synaptic targeting of NMDA receptors. J Neurosci 21: 5079-5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond I, Gordon AS (1997) Cellular and molecular neuroscience of alcoholism. Physiol Rev 77: 1-20. [DOI] [PubMed] [Google Scholar]
- Fadda F, Rossetti ZL (1998) Chronic ethanol consumption: from neuroadaptation to neurodegeneration. Prog Neurobiol 56: 385-431. [DOI] [PubMed] [Google Scholar]
- Ferreira VM, Frausto S, Browning MD, Savage DD, Morato GS, Valenzuela CF (2001) Ionotropic glutamate receptor subunit expression in the rat hippocampus: lack of an effect of a long-term ethanol exposure paradigm. Alcohol Clin Exp Res 25: 1536-1541. [PubMed] [Google Scholar]
- Follesa P, Ticku MK (1996) Chronic ethanol-mediated up-regulation of the N-methyl-d-aspartate receptor polypeptide subunits in mouse cortical neurons in culture. J Biol Chem 271: 13297-13299. [DOI] [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM (2002) Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J Neurosci 22: 2153-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans D, Clayton DA, Coultrap SJ, Browning MD (2002) LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci 5: 27-33. [DOI] [PubMed] [Google Scholar]
- Hu XJ, Follesa P, Ticku MK (1996) Chronic ethanol treatment produces a selective upregulation of the NMDA receptor subunit gene expression in mammalian cultured cortical neurons. Brain Res Mol Brain Res 36: 211-218. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and Ras-GRF1. Neuron 40: 775-784. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC (2003) N-methyl-d-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther 99: 79-94. [DOI] [PubMed] [Google Scholar]
- Kumari M (2001) Differential effects of chronic ethanol treatment on N-methyl-d-aspartate R1 splice variants in fetal cortical neurons. J Biol Chem 276: 29764-29771. [DOI] [PubMed] [Google Scholar]
- Lissin DV, Gomperts SN, Carroll RC, Christine CW, Kalman D, Kitamura M, Hardy S, Nicoll RA, Malenka RC, von Zastrow M (1998) Activity differentially regulates the surface expression of synaptic AMPA and NMDA glutamate receptors. Proc Natl Acad Sci USA 95: 7097-7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304: 1021-1024. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29: 243-254. [DOI] [PubMed] [Google Scholar]
- Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, Fienberg AA, Leslie SW, Gonzales RA, Morrisett RA (2002) DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nat Neurosci 5: 641-648. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H (1997) Fyn-kinase as a determinant of ethanol sensitivity: relation to NMDA-receptor function. Science 278: 698-701. [DOI] [PubMed] [Google Scholar]
- Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD (2003) Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron 40: 581-594. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067-1078. [DOI] [PubMed] [Google Scholar]
- Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M, McKay RD (1998) Hippocampal synaptic plasticity in mice overexpressing an embryonic subunit of the NMDA receptor. J Neurosci 18: 4177-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Craig AM (1997) Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron 19: 801-812. [DOI] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR (2004) Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J Neurosci 24: 1594-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ (2001) Molecular determinants of NMDA receptor internalization. Nat Neurosci 4: 794-802. [DOI] [PubMed] [Google Scholar]
- Rudolph JG, Walker DW, Iimuro Y, Thurman RG, Crews FT (1997) NMDA receptor binding in adult rat brain after several chronic ethanol treatment protocols. Alcohol Clin Exp Res 21: 1508-1519. [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Ehlers MD (2003) Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulated surface delivery of NMDA receptors. Neuropharmacol 45: 755-767. [DOI] [PubMed] [Google Scholar]
- Sheng M, Pak DTS (2000) Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol 62: 755-778. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R (1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811-1816. [DOI] [PubMed] [Google Scholar]
- Snell LD, Nunley KR, Lickteig RL, Browning MD, Tabakoff B, Hoffman PL (1996) Regional and subunit specific changes in NMDA receptor mRNA and immunoreactivity in mouse brain following chronic ethanol ingestion. Brain Res Mol Brain Res 40: 71-78. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ (2002) Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem 82: 1097-1105. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Willis B, Stadler J, Reynolds JG, Bernstein IL, McKnight GS (2000) High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20: RC75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC (2001) Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci 4: 1217-1223. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34: 255-264. [DOI] [PubMed] [Google Scholar]
- Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, Nestler EJ (1994) Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem 62: 1635-1638. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97-107. [DOI] [PubMed] [Google Scholar]
- Wand G, Levinem M, Zweifel L, Schwindinger W, Abel T (2001) The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci 21: 5297-5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS (2003) Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol 43: 335-358. [DOI] [PubMed] [Google Scholar]
- Winkler A, Mahal B, Kiianmaa K, Zieglgansberger W, Spanagel R (1999) Effects of chronic alcohol consumption on the expression of different NR1 splice variants in the brain of AA and ANA lines of rats. Brain Res Mol Brain Res 72: 166-175. [DOI] [PubMed] [Google Scholar]
- Woodward JJ (2000) Ethanol and NMDA receptor signaling. Crit Rev Neurobiol 14: 69-89. [DOI] [PubMed] [Google Scholar]
- Yaka R, Phamluong K, Ron D (2003a) Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci 23: 3623-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Tang KC, Camarini R, Janak PH, Ron D (2003b) Fyn kinase and NR2B-containing NMDA receptors regulate acute ethanol sensitivity but not ethanol intake or conditioned reward. Alcohol Clin Exp Res 27: 1736-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Malinow R (2002) Acute versus chronic NMDA receptor blockade and synaptic AMPA receptor delivery. Nat Neurosci 5: 513-514. [DOI] [PubMed] [Google Scholar]