

Abstract
It is widely believed that long-term depression (LTD) and its counterpart, long-term potentiation (LTP), involve mechanisms that are crucial for learning and memory. However, LTD is difficult to induce in adult cortex for reasons that are not known. Here we show that LTD can be readily induced in adult cortex by the activation of NMDA receptors (NMDARs), after inhibition of glutamate uptake. Interestingly there is no need to activate synaptic NMDARs to induce this LTD, suggesting that LTD is triggered primarily by extrasynaptic NMDA receptors. We also find that de novo LTD requires the activation of NR2B-containing NMDAR, whereas LTP requires activation of NR2A-containing NMDARs. Surprisingly another form of LTD, depotentiation, requires activation of NR2A-containing NMDARs. Therefore, NMDARs with different synaptic locations and subunit compositions are involved in various forms of synaptic plasticity in adult cortex.
Keywords: LTD, LTP, NR2B, NR2A, NMDARs, depotentiation
Introduction
NMDA receptor (NMDAR)-dependent long-term potentiation (LTP) and long-term depression (LTD) are forms of synaptic plasticity that may be critically involved in learning and memory (Bliss and Collingridge, 1993; Bear and Abraham, 1996; Kemp and Bashir, 2001). However, NMDAR-dependent LTD is not readily induced in adult animals, for reasons that are not known (Bear and Abraham, 1996; Kemp and Bashir, 2001). However, possibilities that may account for this include the subunit composition and location of NMDARs in adult tissue.
NMDARs are heteromers (for review, see Dingledine et al., 1999; Cull-Candy et al., 2001) composed of two essential NR1 subunits (Behe et al., 1995) and two or three NR2 subunits (Premkumar and Auerbach, 1997; Laube et al., 1998). There are four NR2 subunits (A-D), but the NR2A and NR2B subunits predominate in the fore-brain. Each of the NR2 subunits imparts different characteristics on functional NMDARs (Kutsuwada et al., 1992; Loftis and Janowsky, 2003); for example NR2A-containing NMDARs have rapid kinetics compared with those of NR2B-containing NMDARs (Monyer et al., 1994). During postnatal development there are changes in the expression of NR2A compared with NR2B subunits (Monyer et al., 1994; Sheng et al., 1994; Loftis and Janowsky, 2003). These changes in NR2A-NR2B ratio may explain the effects on the decay kinetics of NMDAR-mediated synaptic responses that occur during development (Hestrin, 1992; Monyer et al., 1994; Flint et al., 1997). Recent work suggests that these changes in NMDAR subunit composition may also have consequences for activity-dependent cortical development (Quinlan et al., 1999; Barth and Malenka, 2001) and coupling to intracellular cascades (Sala et al., 2000; Hardingham et al., 2002). In addition to the changes in subunit composition, another potentially important factor regulating NMDAR-dependent synaptic function is that in adult cortex NR2A-containing NMDARs may be preferentially targeted to synaptic sites, whereas NR2B-containing NMDARs may be targeted to extrasynaptic sites (Stocca and Vicini, 1998; Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999). However, the consequences for synaptic plasticity (LTD, LTP, and depotentiation) of the potential differential localization of NR2A- and NR2B-containing NMDARs are not known.
In this study we report that LTD is induced in adult perirhinal cortex under conditions that result in stimulation of extrasynaptic NMDARs. This form of LTD is blocked by antagonists of NR2B-containing NMDARs but is not prevented by an antagonist of NR2A-containing NMDARs. In contrast, LTP and depotentiation show the opposite sensitivity to subtype-selective NMDA receptor antagonists. Thus, surprisingly, different forms of LTD, de novo LTD and depotentiation, require the activation of different NR2 subunits. These results suggest that both subunit composition and postsynaptic location of NMDARs are critical determinants of their roles in synaptic plasticity.
Materials and Methods
Materials. Slices of perirhinal cortex were prepared from 7- to 12-week-old DA rats. Animals were anesthetized with a halothane-oxygen mixture, decapitated, and the brain was rapidly removed and placed in ice-cold artificial CSF (aCSF; bubbled with 95% O2 and 5% CO2) which comprised (in mm): NaCl, 124; KCl, 3; NaHCO3, 26; NaH2PO4, 1.25; CaCl2, 2; MgSO4, 1; and d-glucose, 10. A mid-sagittal section was made, the rostral and caudal parts of the brain were removed by single scalpel cuts made at ∼45° to the dorsoventral axis, and each hemisphere was glued by its caudal end to a vibroslice stage (Campden Instruments, Sileby, UK). Slices (400 μm) that included perirhinal, entorhinal, and temporal cortices were stored submerged in aCSF (20-25°C) for 1-2 hr before transferring to the recording chamber. A single slice was placed in a submerged recording chamber (28-30°C, flow rate, ∼2 ml/min) when required.
The following drugs were bath-applied after dilution into the aCSF from concentrated stock solutions: NMDA, d-AP-5, l-trans-2,4-PDC, dl-threo-β-benzyloxyaspartate (dl-TBOA, (+)-MK-801, LY431495, and ifenprodil were obtained from Tocris Cookson (Bristol, UK). R-(R*,S*)-α-(4-hydroxyphenyl)-β-methyl-4-(phenylmethyl)-1-piperidine propranol (Ro 25-6981) hydrochloride was obtained from Sigma-Aldrich (Poole, UK). NVP-AAM077 was a generous gift from Novartis Pharma (AG, Switzerland).
Recording. Standard extracellular recording techniques were used to monitor evoked field responses from layers II and III. Stimuli (constant voltage) were delivered alternately to the two stimulating electrodes (each electrode 0.033 Hz). The amplitude of the evoked field EPSPs was measured and expressed relative to the pre-conditioning baseline. Baseline responses were set to ∼70% of the maximal response. Effects of low-frequency stimulation (LFS) (900 stimuli, 1 Hz) or high-frequency stimulation (HFS) (four 1 sec trains of 100 Hz stimulation with a 10 sec interval between each train) were measured at appropriate time points (averaged over a 5 min period) after delivering LFS or HFS. To obtain NMDAR-mediated field EPSPs, picrotoxin (50 μm) and NBQX (10 μm) were bath-applied, and the stimulus intensity was increased. Whole-cell recordings were made from layer II and III neurons held at -70 mV. Pipette (4-7 MΩ) solutions (280 mOsm, pH 7.2) comprised of (in mm): CsMeSO3, 130; NaCl, 8; Mg-ATP, 4; Na-GTP, 0.3; EGTA, 0.5; HEPES 10; and QX-314, 6. Series resistance (Rs) for whole-cell experiments was 26 ± 5 MΩ. Only experiments in which there was <20% change in Rs were included in analysis. When required, MK-801 (3 mm) was added to the whole-cell filling solution. In these experiments depolarization (to -10 mV for 10 sec, once a min for 10 min) was applied to facilitate the MK-801 block (Woodhall et al., 2001). To ensure that the MK-801 block was still effective at the end of the experiment, neurons were depolarized (to -40 mV) in the presence of NBQX, and the stimulus intensity was increased to ensure that there was no residual NMDA EPSC.
Data analysis. Data were only analyzed from one slice per rat (n = number of rats). Data were recorded using an Axopatch 200B (for whole-cell) or Axoclamp 2B (for extracellular) amplifier (Axon Instruments, Foster City, CA), monitored, and analyzed on-line and re-analyzed off-line (Anderson and Collingridge, 2001). Data pooled across slices are expressed as the mean ± SEM, and effects of conditioning stimulation were measured after induction of LTP or LTD, as indicated in the results. Significance (p < 0.05) from baseline was tested using two-tailed t tests.
Results
LFS (1 Hz, 900 stimuli) is a widely used protocol for the study of NMDAR-dependent LTD. However, as in many other regions of the adult CNS (Kemp and Bashir, 2001), this stimulation protocol does not result in the induction of LTD in adult perirhinal cortex under normal conditions (5 ± 4% depression; n = 7; p > 0.05) (Fig. 1A). However, as in the hippocampus (Lee et al., 1998), bath application of NMDA (20 μm, 5 min) to adult perirhinal cortex slices results in robust synaptic depression (31 ± 4% depression; n = 9; p < 0.05; data not shown).
Figure 1.
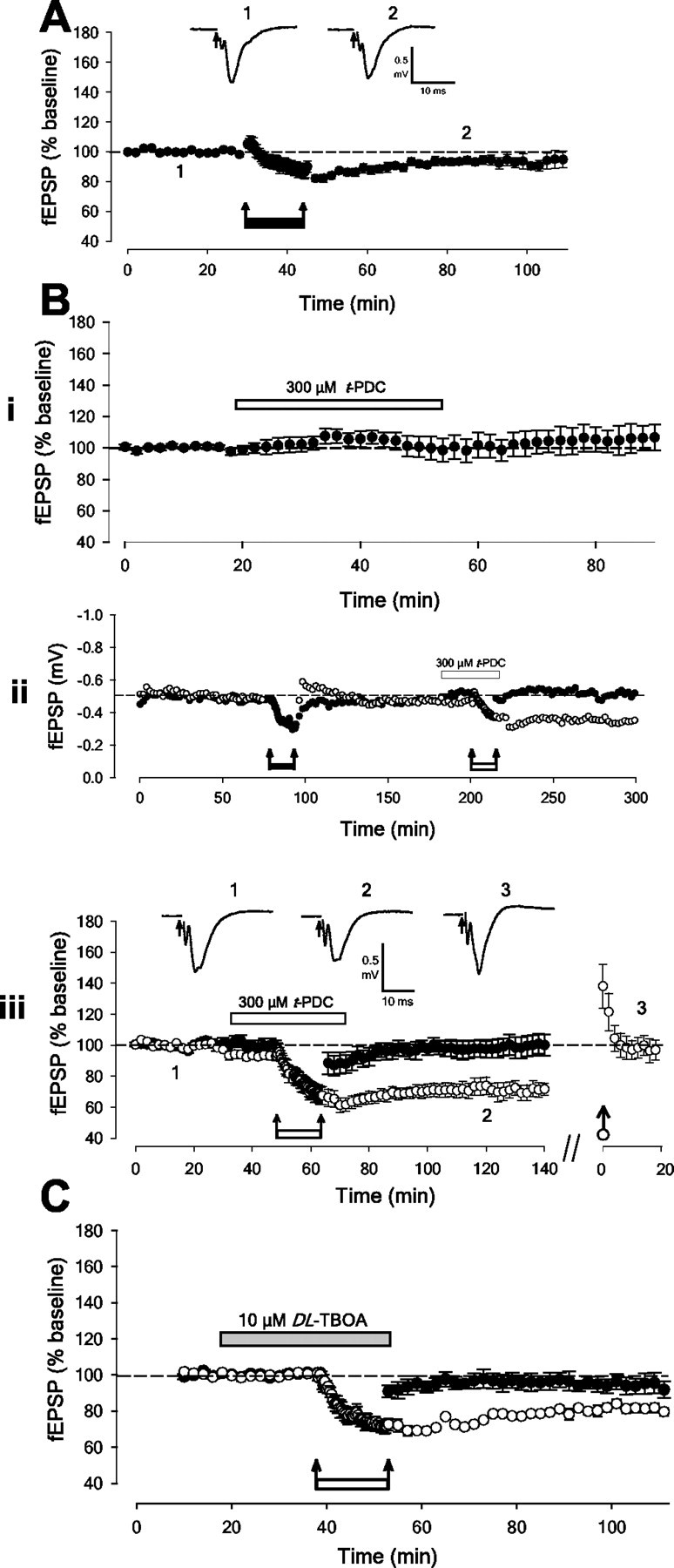
Activation of extrasynaptic NMDARs results in the induction of LTD. A, Pooled data (n = 7) to show that 1 Hz stimulation (900 stimuli) does not result in significant LTD in adult perirhinal slices. The magnitude of the plastic change is measured 60 min after conditioning stimulation, unless otherwise stated. In all experiments two independent inputs were stimulated, although in some cases only one input is illustrated for simplicity. B, t-PDC enables LFS to induce LTD. i, Pooled data (n = 4) shows that t-PDC alone has no effect on basal synaptic transmission. ii, Single example showing that LFS does not result in LTD (filled circles) unless delivered in the presence of t-PDC (open circles). iii, Pooled data (n = 8) for t-PDC. Note that subsequent HFS reverses LTD and that there is no LTD in the heterosynaptic input (2 ± 6% depression; p > 0.05; n = 7). C, Pooled data (n = 5) showing that LFS in the presence of dl-TBOA also results in the induction of homosynaptic LTD.
Spillover of glutamate allows LFS-induced LTD
One possible explanation for this effect is that NMDA, but not LFS, induces LTD because it also activates extrasynaptic NMDARs. If this is the case, the activation of extrasynaptic NMDARs (either alone or in addition to synaptic NMDARs) may be crucial for the induction of LTD. Therefore, we sought an approach whereby synaptically released glutamate could activate extrasynaptic NMDARs. A method for achieving this is to block glutamate transporters, because the spread of synaptically released glutamate under normal conditions is tightly controlled by active uptake mechanisms. Thus, LFS was delivered in the presence of the glutamate uptake inhibitor l-trans-pyrrolidine-2,4-dicarboxylate (t-PDC; 300 μm). The data in Figure 1B shows that t-PDC did not produce any lasting change in synaptic transmission per se (105 ± 9% 30 min after t-PDC; n = 4). However, in contrast to control conditions, robust LTD was induced by LFS in the presence of t-PDC (29 ± 6% depression; p < 0.05; n = 8). Furthermore, subsequent HFS reversed this form of LTD (to 96 ± 7% of initial baseline; n = 7) (Fig. 1B). Similarly, another glutamate uptake inhibitor, dl-TBOA (10 μm; Shimamoto et al., 1998), which does not reverse glutamate transporters or increase ambient glutamate levels at the concentration used in the present study (Diamond, 2001), had a similar effect. Thus, dl-TBOA had no effect on synaptic transmission per se, but in the presence of dl-TBOA, LFS resulted in the induction of LTD (20 ± 3% depression; p < 0.05; n = 5) (Fig. 1C). Together these data suggest that blocking glutamate uptake results in the induction of LTD by allowing glutamate released by LFS to activate receptors not normally activated during LFS.
LTD relies on activation of NMDARs
LTD in the CNS can be induced by the activation of NMDARs, mGlu receptors, or both, depending on the brain region and experimental conditions (Bear and Abraham, 1996; Kemp and Bashir, 2001). Therefore, to determine which receptors are required during the activity-dependent spill-over of glutamate we applied selective antagonists. First, LFS was delivered in the presence of t-PDC and the mGlu receptor antagonist LY341495 (100 μm) at a concentration that blocks all known presynaptic and postsynaptic mGluRs (Fitzjohn et al., 1998). This treatment did not block the induction of LTD (22 ± 4% depression; n = 4) (Fig. 2A). Second, LFS was delivered in the presence of the NMDAR antagonist AP-5 (100 μm). Under these conditions LTD was completely blocked (104 ± 3% relative to baseline; n = 7; p > 0.05) (Fig. 2B).
Figure 2.

LTD in adult cortex is induced by NMDARs. A, Pooled data (n = 4) to show that LTD induced in the presence of t-PDC is not blocked by the broad-spectrum mGlu receptor antagonist LY341495. B, LTD induced in the presence of t-PDC is blocked by the NMDAR antagonist d-AP5 (n = 7).
LTD relies on NR2B-containing NMDARs
Because there is good electrophysiological evidence that extrasynaptic NMDARs are predominantly composed of NR2B-containing heteromers (Stocca and Vicini, 1998; Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999), we first tested whether blockade of glutamate uptake allows the activation of NR2B-containing NMDARs and secondly tested whether this NMDAR subtype is specifically involved in LTD.
The application of t-PDC caused a significant prolongation (control decay constant: 100 ± 15 msec; in t-PDC: 133 ± 19 msec; p < 0.05; n = 4) (Fig. 3A) of the decay of NMDAR-mediated EPSCs. This is in agreement with other studies (Arth-Jensen et al., 2002; Clark and Cull-Candy, 2002). This prolongation was reversed (89 ± 8 msec) (Fig. 3A) by the selective NR2B antagonist Ro 25-6981 (Fischer et al., 1997). When t-PDC was applied in the presence of Ro 25-6981 there was no change in the time course of NMDAR-mediated EPSCs (control decay constant: 87 ± 3 msec; in Ro 25-6981: 86 ± 8 msec; in Ro 25-6981 and t-PDC: 76 ± 7 msec; n = 5). Together these results suggest that NR2B-containing NMDARs are preferentially activated during block of glutamate uptake.
Figure 3.
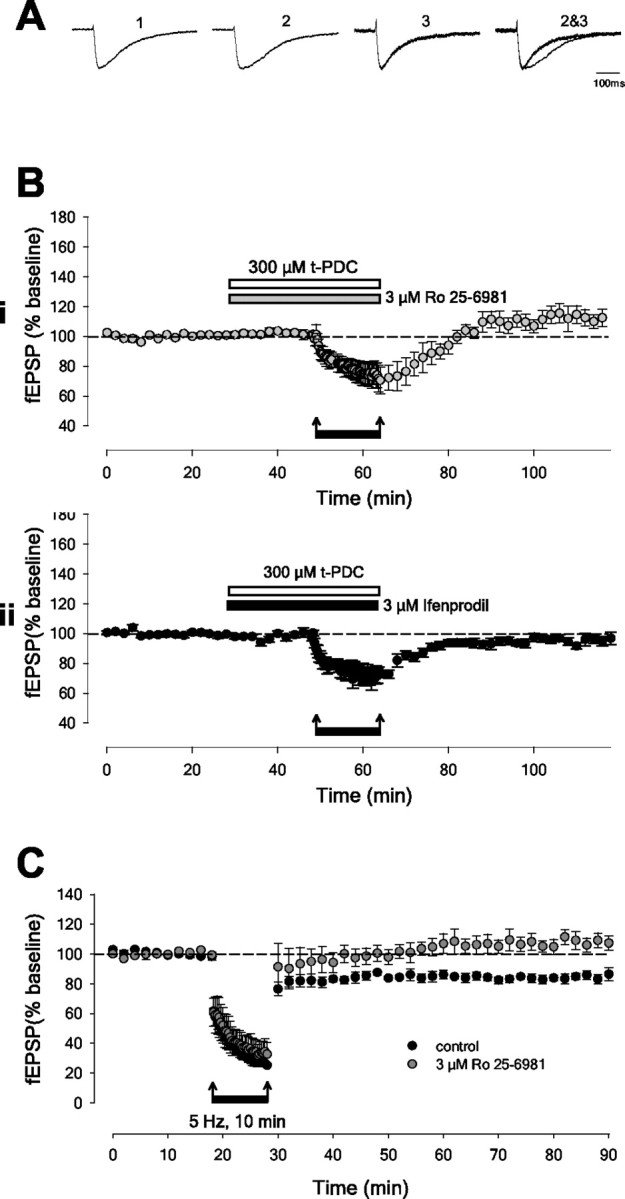
LTD is induced by NR2B-containing NMDARs. A, NMDA EPSCs (1) are prolonged by the application of t-PDC (2). This prolongation is reversed by the NR2B antagonist Ro 25-6981 (3). To illustrate the effect on the decay and thus highlight the NR2B component of the NMDA EPSC, traces are scaled to peak amplitude and superimposed (2, 3). B, NR2B-specific antagonists block the induction of LTD in the presence of t-PDC. i shows the effects of Ro 25-6981 (n = 5), and ii shows the effects of ifenprodil (n = 5). C, LTD induced by 5 Hz stimulation, in the absence of a glutamate uptake inhibitor, is blocked by Ro 25-6981. The graph plots pooled data from controls (n = 5; black circles) and Ro 25-6981-treated (n = 5; gray circles) slices.
Importantly, the induction of LTD by LFS, in the presence of t-PDC, was completely prevented by either of the NR2B antagonists Ro 25-6981 (3 μm; 112 ± 6% relative to baseline; n = 5; p > 0.05) (Fig. 3B) or ifenprodil (3 μm; 97 ± 4% relative to baseline; n = 5; p > 0.05) (Fig. 3B). Ro 25-6981 also blocked LTD induced in the presence of dl-TBOA (data not shown). These results show that LTD in adult perirhinal cortex critically relies on the activation of NR2B-containing NMDARs.
We were interested to know whether NR2B-containing receptors could be activated to induce LTD without the addition of a glutamate uptake blocker. We found that a higher frequency train (5 Hz, 10 min) could induce LTD (17 ± 3% depression; p < 0.05; n = 5) that was fully blocked by Ro 25-6981 (109 ± 5% relative to baseline; n = 5; p > 0.05) (Fig. 3C). This raises the possibility that under appropriate conditions these receptors can be physiologically activated by synaptic activity to induce LTD.
LTP and depotentiation rely on NR2A-containing NMDARs
In contrast to LTD, neither NMDAR-dependent LTP nor NMDAR-dependent depotentiation (Ziakopoulos et al., 1999) was blocked by the NR2B antagonist Ro 25-6981 (control LTP: 123 ± 4%, n = 5; LTP in Ro 25-6981: 134 ± 3%, n = 5; control depotentiation: 102 ± 3%, relative to pre-LTP baseline; depotentiation in Ro: 106 ± 7%) (Fig. 4A,B).
Figure 4.
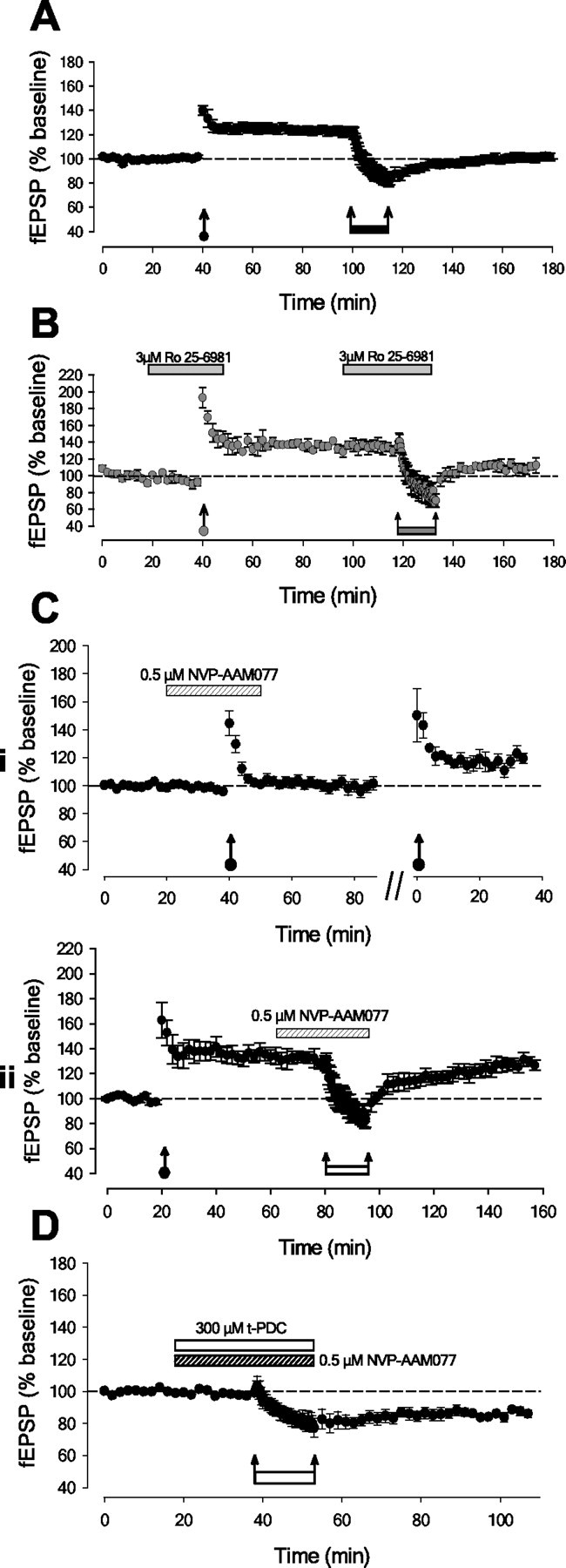
LTP and depotentiation require NR2A-containing NMDARs but not NR2B-containing NMDARs. A, Control experiments (n = 5) showing induction of LTP and depotentiation for interleaved experiments in Figure 4, B and C. B, LTP and depotentiation are not blocked by NR2B antagonist Ro 25-6981 (n = 5). C, LTP and depotentiation are blocked by a selective NR2A antagonist. i, interleaved experiments showing block of LTP using NVP-AAM077 (n = 5). ii, interleaved experiments showing block of depotentiation using NVP-AAM077 (n = 5). D, Selective inhibition of NR2A-containing NMDARs does not prevent LTD in the presence of t-PDC (n = 6).
Interestingly, the novel NR2A-selective antagonist NVP-AAM077 (the active diastereomer of (1RS,1′S)-PEAQX, 0.5 μm; Auberson et al., 2002) completely blocked both LTP and depotentiation (LTP: 102 ± 3%, n = 5, p > 0.05; depotentiation: 131 ± 4%, n = 5, p > 0.05 compared with pre-LTP baseline) (Fig. 4C). In contrast, LTD was induced in the presence of NVP-AAM077 (17 ± 4%; n = 6; p < 0.05) (Fig. 4D). These results suggest that although NR2A-containing NMDARs may contribute to the magnitude of LTD, unlike NR2B-containing NMDARs, they are not essential for its induction.
Activation of extrasynaptic NMDARs alone results in LTD
We next investigated whether the stimulation of extrasynaptic NMDARs alone is sufficient for the induction of LTD. To examine this question, MK-801 (10 μm) was bath-applied, and LFS was delivered (in the absence of glutamate uptake inhibitors) to selectively block synaptically activated NMDARs (Fig. 5A). To subsequently activate extrasynaptic NMDARs, MK-801 was washed out for 60 min, and then LFS was delivered in the presence of the glutamate uptake inhibitor t-PDC. Under these conditions, LFS still resulted in the induction of significant LTD (10 ± 1% depression; n = 4; p < 0.05) (Fig. 5A), although this was smaller than under normal conditions.
Figure 5.

Postsynaptic, extrasynaptic NMDARs mediate LTD. A, Synaptic activation of extracellular NMDARs is sufficient to induce LTD. A schematic diagram (i), a single example (ii), and data from four experiments (iii) illustrate the method for selective activation of extrasynaptic NMDARs. 1, MK-801 irreversibly blocks synaptic NMDARs activated by glutamate during LFS. 2, After washout of MK-801 for 60 min, LFS was delivered in the presence of t-PDC to activate only extrasynaptic NMDARs. iv, NMDAR-mediated synaptic transmission is still completely blocked 60 min after washout of MK-801. Traces illustrate example NMDAR field EPSPs before and 60 min after MK-801 showing a complete block of NMDA EPSPs by MK-801 both at washout and 60 min later. B, LTD induced in the presence of t- PDC is blocked by MK-801 applied postsynaptically via the patch pipette (n = 5; filled circles). Control LTD obtained during interleaved whole-cell recordings is shown superimposed (n = 5; open symbols). The traces are averages of four responses obtained 5 min into baseline and 25 min after LTD protocol at the times indicated.
We considered alternative possibilities that could explain the induction of LTD after block of synaptic NMDARs with MK-801. This could result from MK-801 dissociating from synaptic NMDAR channels over the course of 60 min. Alternatively this could be caused by unblocked extrasynaptic NMDAR channels migrating to synaptic sites (Tovar and Westbrook, 2002) and therefore subsequently being involved in the induction of LTD. However, the data in Figure 5A shows NMDAR-mediated synaptic transmission is still completely blocked 60 min after MK-801 washout, which suggests that neither of these possibilities is likely to account for the induction of LTD in the presence of MK-801 and t-PDC. Instead these results suggest that the activation of extrasynaptic NMDARs alone can trigger the induction of LTD in intact adult cortex slices.
LTD requires the activation of postsynaptic NMDARs
It has been reported that NR2B-containing NMDARs are located presynaptically (Woodhall et al., 2001) and that presynaptic NMDARs may be important in the induction of LTD in cerebellum (Casado et al., 2002) and neocortex (Sjostrom et al., 2003). To determine the localization of the NMDARs that are important for synaptically induced LTD in perirhinal cortex, we performed whole-cell recording experiments. Under control conditions LTD was induced by LFS in the presence of t-PDC (44 ± 11% depression; n = 5; p < 0.05) (Fig. 5B). In contrast, LTD was not induced (8 ± 4% depression; n = 5; p > 0.05) (Fig. 5B) in cells in which postsynaptic NMDARs were selectively blocked by the use-dependent channel blocker MK-801 contained in the whole-cell filling solution (Woodhall et al., 2001). These results show that the NMDARs involved in the induction of LTD are postsynaptically located.
Discussion
In this study we demonstrate, first, that LTD can be readily induced in adult cortex under conditions that enable activation of extrasynaptic NMDARs. Second, we show that LTD is blocked by antagonism of NR2B-containing NMDARs. Therefore the most likely explanation for these results is that activation of extrasynaptic NR2B-containing NMDARs is critically required for the induction of LTD in adult cortex.
Third, we show that this form of LTD is induced postsynaptically, but surprisingly does not require the activation of synaptic NMDARs. Fourth, our results demonstrate a differential role for NR2A- and NR2B-containing NMDARs in different forms of synaptic plasticity in adult cortex. Thus, we find that NR2A containing NMDARs are required for LTP, whereas NR2B-containing NMDARs are required for LTD. Moreover, we show that the direction of synaptic change is not simply related to NR2 subunit composition, because de novo LTD requires NR2B-containing receptors, whereas depotentiation requires NR2A-containing receptors.
Extrasynaptic NMDARs mediate LTD in adult cortex
To test whether LTD could be induced by activation of extrasynaptic NMDARs by synaptically released glutamate, we blocked glutamate uptake (Chen and Diamond, 2002; Clark and Cull-Candy, 2002). We used two different inhibitors of glutamate transport. In the presence of t-PDC, LTD was induced by LFS. However, t-PDC has been reported to not only block uptake but to reverse glutamate transport (Griffiths et al., 1994; Volterra et al., 1996) and increase ambient glutamate levels. However, in our studies, the application of t-PDC had no effect on synaptic transmission in the absence of LFS. This suggests that any increase in ambient glutamate per se does not have a detectable role in LTD but rather it is the spillover of synaptically released glutamate that is required for LTD. As a further control we also used the uptake blocker dl-TBOA, which does not reverse glutamate transporters (Shimamoto et al., 1998). At the concentration used in the present study (10 μm) dl-TBOA has been shown not to affect ambient glutamate levels (Diamond, 2001), although much higher concentrations can (Jabaudon et al., 1999). In the presence of dl-TBOA, LTD was also induced by LFS. Thus, it is unlikely that the induction of LTD can be explained by increases in ambient glutamate levels. Rather these results indicate that spillover of glutamate released by LFS results in the induction of LTD. Of course, the distinction between synaptic and extrasynaptic NMDARs is not easily definable: in the present study however we refer to extrasynaptic receptors as those that are not activated by LFS at 1 Hz, unless a glutamate uptake blocker is applied (Chen and Diamond, 2002; Clark and Cull-Candy, 2002).
Postsynaptic and presynaptic mGlu receptors, which are known to be activated after block of glutamate uptake, are not likely to play a role in this form of LTD because antagonism of all known mGluRs by LY341495 did not block LTD. However, LTD was blocked by AP-5, and it therefore seems likely that LTD was caused by activation of extrasynaptic NMDARs. Although in this study we have blocked glutamate uptake to activate extrasynaptic receptors, it has been suggested that neuronal depolarization can reduce the effectiveness of glutamate transporters (Wadiche et al., 1995; Diamond, 2001). Thus, an intriguing possibility is that LTD in adult cortex may be induced by mechanisms that rely on a physiological suppression of glutamate uptake. Furthermore, we could induce NMDAR-dependent LTD in the absence of glutamate uptake blockers by altering the frequency of stimulation during LFS. Further studies will be required to establish the physiological conditions that enable NMDAR-dependent LTD to be induced and its role in vivo.
The localization of NMDARs involved in LTD
Although classically LTD is induced by NMDARs located postsynaptically, there is also recent evidence that LTD may involve presynaptic NMDARs in certain brain regions (Casado et al., 2002; Sjostrom et al., 2003). However, our finding that LTD was blocked by MK-801 applied via the patch-pipette demonstrates the postsynaptic locus of LTD in the adult perirhinal cortex. By the use of bath application of MK-801, we were also able to provide evidence that synaptic NMDARs are not required to induce LTD. Thus, we found that although synaptic NMDARs were completely blocked by MK-801 under control conditions, it was still possible to induce LTD in the presence of a glutamate uptake blocker. Thus, we conclude that NMDARs located postsynaptically in the extrasynaptic membrane are involved in LTD. Interestingly it has recently been shown that NMDA treatment causes internalization of extrasynaptic AMPARs in cultured hippocampal neurons (Ashby et al., 2004). This raises the possibility that internalization of AMPARs occurs close to sites where extrasynaptic NMDARs are activated.
The role of NMDAR subtype in LTD and other forms of synaptic plasticity
The finding that two selective inhibitors of NR2B-containing NMDARs prevented the induction of LTD demonstrates the involvement of this subtype in this form of synaptic plasticity. Surprisingly, selective inhibition of NR2A-containing NMDARs did not prevent the induction of LTD. It is not yet known whether the NR2A antagonist NVP-AAM077 blocks NR1/2A/2B triheteromers. Therefore we cannot exclude the possibility that such heteromers are involved in LTD. Interestingly, Hrabetova et al. (2000) also previously suggested a role for NR2A/B-containing NMDARs in LTP, but not in LTD, in juvenile hippocampus. Our data extends these observations by showing that it is the NR2A-containing, rather than the NR2B-containing NMDARs that are important for LTP. The finding that either NR2B antagonists or an NR2D antagonist (Hrabetova et al., 2000) can block LTD raises the possibility that NR1/2B/2D triheteromers, sensitive to both classes of antagonists, may be involved in LTD.
Surprisingly, our results show a dissociation between the two forms of synaptic depression, because LTD from a basal state (de novo LTD) required activation of NR2B-containing NMDARs, whereas depotentiation required activation of NR2A-containing NMDARs. Furthermore, LTP required activation of NR2A-containing NMDARs. An interesting question that arises from these studies is how different NMDAR subunits can produce different forms of plasticity. Although this remains a matter of speculation, one possible explanation is that NR2A-containing and NR2B-containing NMDAR EPSCs by virtue of their different kinetics (Cull-Candy et al., 2001) provide the different levels of intracellular calcium required for LTP and LTD (Lisman, 1989, Cho et al., 2001). An alternative possibility arises from data suggesting that LTD and depotentiation are induced by different signaling cascades and by dephosphorylation at different sites on GluR1 (Lee et al., 2000). Thus, different NMDAR subtypes may trigger the separate signaling cascades required for different forms of synaptic plasticity. Whereas the precise signaling cascades remain to be determined, data suggesting that NR2A and NR2B subunits couple variously to different intracellular substrates (Bayer et al., 2002; Li et al., 2002) may provide clues as to how extrasynaptic NR2B receptors may produce one form of plasticity and synaptic NR2A receptors may produce two different forms of plasticity.
Since submission of this manuscript, a paper has been published (Liu et al., 2004) suggesting a role for NR2A-containing receptors in LTP and NR2B-containing receptors in LTD in juvenile hippocampus. These results fully agree with our present findings and our preliminary report (Massey et al., 2003). Based on these studies it can be concluded that differential roles of NR2A- and NR2B-containing NMDA receptors in synaptic plasticity are not specific to either a single brain region or a particular stage of development and may therefore represent a general property of the CNS. Our findings extend the observations of Liu et al. (2004) by showing that two forms of synaptic depression, LTD and depotentiation, require the activation of different NR2-containing NMDA receptors. The former requires NR2B-containing, whereas the latter requires NR2A-containing NMDA receptors. Importantly, we provide a possible explanation for why NR2B-containing NMDARs are required for LTD in adult cortex. First, the induction of LTD is promoted by blocking glutamate uptake, which enables greater activation of the extrasynaptically located NR2B-containing NMDARs. Second, the NR2B receptors required for LTD are most likely postsynaptically located in the extrasynaptic membrane. This raises the possibility that a reason for the difficulty in inducing LTD in adult tissue may be attributable to a more efficient glutamate uptake mechanism.
Footnotes
This work was supported by the Biotechnology and Biological Sciences Research Council and Medical Research Council. We are grateful to Novartis Pharma AG, Switzerland for the gift of NVP-AAM077.
Correspondence should be addressed to Zafar I. Bashir at the above address. E-mail: z.i.bashir@bris.ac.uk.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247821-08$15.00/0
References
- Anderson WW, Collingridge GL (2001) The LTP programme: a data acquisition programme for online analysis of long-term potentiation and other synaptic events. J Neurosci Methods 108: 71-83. [DOI] [PubMed] [Google Scholar]
- Arth-Jensen A, Jaboudon D, Scanziani M (2002) Cooperation between independent hippocampal synapses in controlled by glutamate uptake. Nat Neurosci 5: 325-331. [DOI] [PubMed] [Google Scholar]
- Ashby MC, De La Rue SA, Ralph GS, Uney J, Collingridge GL, Henley JM (2004) Removal of AMPA receptors (AMPARs) from synapses is preceded by transient endocytosis of extrasynaptic AMPARs. J Neurosci 24: 5172-5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auberson YP, Allgeier H, Bischoff S, Lingenhoel K, Moretti R, Schmutz M (2002) 5-Phosphonomethylquinoxalinediones as competitive NMDA receptor antagonists with a preference for the human 1A/2A, rather than 1A/2B receptor composition. Bioorg Med Chem Lett 12: 1099-1102. [DOI] [PubMed] [Google Scholar]
- Barth AL, Malenka RC (2001) NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci 4: 235-236. [DOI] [PubMed] [Google Scholar]
- Bayer K-U, De Konink P, Leonard AS, Hell JW, Schulman H (2002) Interaction with the NMDA receptor locks CaMKII in an active confirmation. Nature 411: 801-805. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC (1996) Long-term depression in hippocampus. Annu Rev Neurosci 19: 437-462. [DOI] [PubMed] [Google Scholar]
- Behe P, Stern P, Wyllie DJ, Nassar M, Schoepfer R, Colquhoun D (1995) Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc R Soc Lond B Biol Sci 262: 205-213. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Casado M, Isope P, Ascher P (2002) Involvement of presynaptic N-methyl-d-aspartate receptors in cerebellar long-term depression. Neuron 33: 123-130. [DOI] [PubMed] [Google Scholar]
- Chen S, Diamond JS (2002) Synaptically released glutamate activates extrasynaptic NMDA receptors on cells in the ganglion cell layer of rat retina. J Neurosci 22: 2165-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI (2001) An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol (Lond) 532: 459-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG (2002) Activity dependent recruitment of extrasynaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci 22: 4428-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy S, Brickely S, Farrant M (2001) NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol 11: 327-335. [DOI] [PubMed] [Google Scholar]
- Diamond JS (2001) Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci 21: 8328-8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynellis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Fischer G, Mutel V, Trube G, Malherbe P, Kew JNC, Mohacsi E, Heitz MP, Kemp JA (1997) Ro- 25-6981, a highly potent and selective blocker of N-methyl-d-aspartate receptors containing the NR2B subunit. Characterisation in vitro. J Pharmacol Exp Ther 283: 1285-1292. [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL (1998) The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology 37: 1445-1458. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstien AR, Monyer H (1997) NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci 17: 2469-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R, Dunlop J, Gorman A, Senior J, Grieve A (1994) l-trans-Pyrrolidine-2,4-dicarboxylate and cis-1-aminocyclobutane-1,3-dicarboxylate behave as transportable, competitive inhibitors of the high affinity glutamate transporters. Biochem Pharmacol 47: 267-274. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nature Neuroscience 5: 405-414. [DOI] [PubMed] [Google Scholar]
- Hestrin S (1992) Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature 357: 686-689. [DOI] [PubMed] [Google Scholar]
- Hrabetova S, Serrano P, Blace N, Tse HW, Skifter DA, Jane DE, Monaghan DT, Sactor TC (2000) J Neurosci 20: RC81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA 96: 8733-8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI (2001) Long-term depression: a cascade of induction and expression mechanisms. Prog Neurobiol 65: 339-365. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Ksiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M (1992) Molecular diversity of the NMDA receptor channel. Nature 358: 36-41. [DOI] [PubMed] [Google Scholar]
- Laube B, Kuhse J, Betz H (1998) Evidence for a tetrameric structure of recombinant NMDA receptors. J Neurosci 18: 2954-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in the hippocampus. Neuron 21: 1151-1162. [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405: 955-959. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA (2002) Differential regulation of synaptic and extrasynaptic NMDA receptors. Nat Neurosci 5: 833-834. [DOI] [PubMed] [Google Scholar]
- Lisman JA (1989) A mechanism for the Hebb and the anti-Hebb process underlying learning and memory. Proc Natl Acad Sci USA 86: 9574-9578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson Y, Wang YT (2004) Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304: 1021-1024. [DOI] [PubMed] [Google Scholar]
- Loftis JM, Janowsky A (2003) The N-methyl-d-aspartate receptor subunit NR2B: localisation, functional properties, regulation and clinical implications. Pharmacol Ther 97: 55-85. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Brown MW, Molnar E, Bashir ZI (2003) Extrasynaptic NR2B-containing NMDA receptors are required for LTD in the adult perirhinal cortex in vitro. Soc Neurosci Abstr 29: 169.5. [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12: 529-540. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Auerbach A (1997) Stochiometry of recombinant N-methyl-d-aspartate receptor channels inferred from single channel current patterns. J Gen Physiol 110: 485-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999) Rapid, experience dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2: 352-357. [DOI] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S (1999) Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci 19: 10603-10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M (2000) Developmentally regulated NMDA receptor dependent dephosphorylation of cAMP response element binding protein (CREB) in hippocampal neurons. J Neurosci 20: 3529-3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY (1994) Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368: 144-147. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998) dl-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195-201. [DOI] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB (2003) Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron 39: 641-654. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S (1998) Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol (Lond) 507: 13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19: 4180-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34: 255-264. [DOI] [PubMed] [Google Scholar]
- Volterra A, Bezi P, Rizzini BL, Trotti D, Ullensvang K, Danbolt NC, Racagni G (1996) The competitive transport inhibitor l-trans-pyrrolidine-2,4-dicarboxylate triggers excitotoxicity in rat cortical neuron-astrocyte co-cultures via glutamate release rather than uptake inhibition. Eur J Neurosci 8: 2019-2028. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Amara SG, Kavanaugh MP (1995) Ion fluxes associated with excitatory amino acid transport. Neuron 15: 721-728. [DOI] [PubMed] [Google Scholar]
- Woodhall G, Evand DI, Cunningham MO, Jones RSG (2001) NR2B-containing NMDA autoreceptors at synapses on entorhinal cortical neurones. J Neurophysiol 86: 1644-1651. [DOI] [PubMed] [Google Scholar]
- Ziakopoulos Z, Tillet CW, Brown MW, Bashir ZI (1999) Input- and layer-dependent synaptic plasticity in the rat perirhinal cortex in vitro. Neuroscience 92: 459-472. [DOI] [PubMed] [Google Scholar]