

Abstract
Although the basic molecular components that promote regulated neurotransmitter release are well established, the contribution of these proteins as regulators of the plasticity of neurotransmission and refinement of synaptic connectivity during development is elaborated less fully. For example, during the period of synaptic growth and maturation in brain, the expression of synaptosomal protein 25 kDa (SNAP-25), a neuronal t-SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) essential for action potential-dependent neuroexocytosis, is altered through alternative splicing of pre-mRNA transcripts. We addressed the role of the two splice-variant isoforms of SNAP-25 with a targeted mouse mutation that impairs the shift from SNAP-25a to SNAP-25b. Most of these mutant mice die between 3 and 5 weeks of age, which coincides with the time when SNAP-25b expression normally reaches mature levels in brain and synapse formation is essentially completed. The altered expression of these SNAP-25 isoforms influences short-term synaptic function by affecting facilitation but not the initial probability of release. This suggests that mechanisms controlling alternative splicing between SNAP-25 isoforms contribute to a molecular switch important for survival that helps to guide the transition from immature to mature synaptic connections, as well as synapse regrowth and remodeling after neural injury.
Keywords: SNARE, neuroexocytosis, synaptic development, neurotransmission, alternative splicing, plasticity
Introduction
Synaptic transmission occurs by fusion of vesicles with the presynaptic plasma membrane. The core of the protein complex responsible for regulated transmitter release is composed of vesicle-associated membrane protein (VAMP)-2/synaptobrevin, plasma membrane-associated syntaxin 1A, and synaptosomal protein 25 kDa (SNAP-25) (Sollner et al., 1993; Chen and Scheller, 2001). These are prototypical members of the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein family, distinguished by conserved α-helical domains, that promotes membrane fusion for constitutive and regulated membrane trafficking events in eukaryotic cells (Weimbs et al., 1997; Bock et al., 2001). The diversity and subcellular localization of SNARE family members suggests that different SNARE protein complexes could direct membrane traffic between organelles and for extracellular secretion (McNew et al., 2000). Although an attractive proposal, promiscuous associations found between cognate SNAREs (Fasshauer et al., 1999; Yang et al., 1999) indicate that spatial and temporal patterns of expression must play a significant role(s) in SNARE-mediated targeting of membrane fusion and traffic.
The t-SNARE SNAP-25 is regulated during brain growth and synaptogenesis at the level of expression and by alternative splicing between tandem arranged exons (Bark et al., 1995). This results in a developmental switch between 1 and 3 weeks of age from expression of predominately SNAP-25a to SNAP-25b transcripts that ultimately constitute >80% of SNAP-25 mRNA in adult mouse brain (Bark et al., 1995; Boschert et al., 1996; Jacobsson et al., 1999). In contrast, SNAP-25a remains the predominant isoform in adult neuroendocrine cells (Bark et al., 1995; Gonelle-Gispert et al., 1999; Grant et al., 1999). The isoforms differ by only 9 of 39 residues encoded by the alternatively spliced exons (Bark and Wilson, 1994; Bark et al., 1995), including nonconservative changes within the N-terminal helical SNARE domain (Fasshauer et al., 1998) and clustered cysteine residues that provide sites for palmitoylation (Hess et al., 1992; Veit et al., 1996) involved in membrane association (Lane and Liu, 1997; Gonzalo and Linder, 1998; Gonzalo et al., 1999; Koticha et al., 1999) and disassembly of the SNARE complex after exocytosis (Washbourne et al., 2001).
We considered that SNAP-25 isoforms might play a role in “tuning” the exocytotic machinery toward different modes of transmitter release. Few physiological differences, however, between SNAP-25 isoforms have been observed; for example, both isoforms support Ca2+-stimulated insulin secretion (Gonelle-Gispert et al., 1999). Nevertheless, although either SNAP-25 isoform directs Ca2+-evoked catecholamine release in SNAP-25-deficient chromaffin cells, their ability to stabilize the pool of primed vesicles for secretion differs in these cells (Sørensen et al., 2003). To explore the function of SNAP-25 isoforms in central synapses, we generated a mutation that perturbs alternative splicing and limits expression of SNAP-25b in mice. Most of the mutants overexpressing SNAP-25a die between postnatal day 25 (PN25) and PN35 when this switch normally occurs. Facilitation of synaptic transmission at hippocampal synapses of mutant mice is increased, although the probability of release is unaltered. This supports the idea that assembly of different SNARE complexes affects properties of membrane fusion and that the developmental switch between SNAP-25 isoforms alters the efficacy of synaptic transmission that may contribute to the solidification of developing neural circuitry.
Materials and Methods
Generation of targeting vector constructs for homologous recombination in the Snap25 locus. The Snap25Tkneo mutation was generated by targeted gene replacement in embryo stems cells using standard methods with positive selection for G418 resistance (Mansour et al., 1988). The gene targeting construct was derived from mouse Snap25 genomic λ phage clone p4.2 (Washbourne et al., 2002), and restriction maps were obtained for selected subclones spanning exons 5a/5b, 6, and 7. Multiplex PCR was used to generate a recombinant fragment in which the mouse exon 5b was replaced with chicken exon 5a sequences using two genomic templates: a PvuI-KpnI fragment derived from the chicken gene encoding SNAP-25 (Bark, 1993) that spans most of the 5a exon, and a mouse genomic KpnI-StyI DNA fragment containing most of the intervening sequence between exons 5a and 5b and the 5′ part of the mouse exon 5b. The resulting PCR product was cloned as a KpnI-HindIII fragment into the vector pBluescript (Stratagene, San Diego CA). Sequencing confirmed that this fragment contained the mouse intron between exon 5a and 5b downstream from the internal KpnI site, a chicken exon 5a replacing mouse exon 5b, and downstream sequences to the HindIII site located immediately 3′ of the original exon 5b in the mouse gene. This resulted in the substitution of 31 of the 118 nucleotides in the mouse 5b exon coding sequence to encode the exon 5a protein domain without changing any nucleotides in noncoding sequences and presumably leaving intact all control sequences for the regulation of alternative splicing. A series of constructs in the pBluescript KS II+ vector (Stratagene) were then developed to reconstitute part of the mouse gene spanning upstream from the original exon 5a to the SacI site located downstream of exon 6 (see Fig. 1 A). Finally, a SalI-BglII fragment from the Tkneo gene surrounded by loxP sites (O'Gorman et al., 1997) was introduced in the intron upstream of exon 6 in the targeting vector. The resulting construct, termed B52/loxP, had the neo gene in the same transcriptional orientation as the endogenous Snap25 gene (see Fig. 1 A). The embryonic stem (ES) cell transfection and initial chimeric mice were made at The Scripps Research Institute (TSRI) Transgenic Animal Facility. For the studies reported here, mice were obtained from heterozygote matings of mice backcrossed to a C57BL/6J background for six to seven generations. Studies were done in accordance with guidelines of TSRI and University of New Mexico Health Sciences Center Laboratory Animal Care and Use Committees, and the National Institutes of Health.
Figure 1.
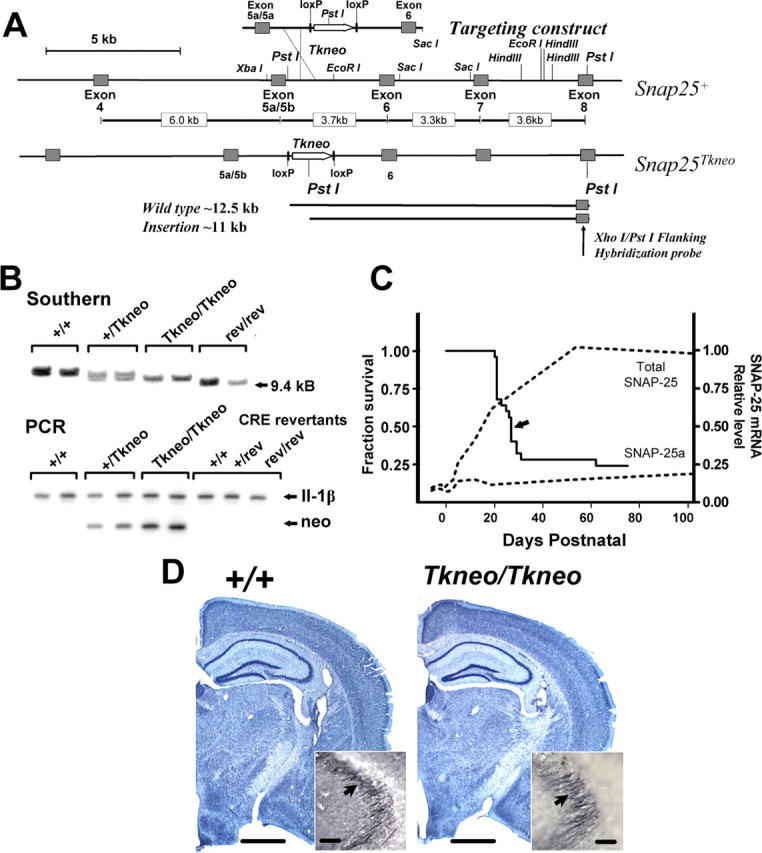
Targeted insertion of Tkneo selection transgene downstream of exon 5a and 5b in the Snap25 gene results in postnatal lethality. A, Schematic diagram demonstrating the generation and structure of the mutation Snap25Tkneo. The floxed Tkneo selection gene was inserted at the EcoRI site between the tandemly arranged exons 5a and 5b and exon 6 in a targeting construct generated from the wild-type gene (Snap25+) described in Washbourne et al. (2002). In the targeting construct, a PvuI-HindIII fragment containing exon 5a and 5b sequences was exchanged for an exon 5a-5a sequence produced by multiplex PCR (C. Bark, unpublished data). Sequence analysis of genomic PCR products obtained from DNA of homozygous mutant mice demonstrated that exon 5a and 5b in fact were intact in Snap25Tkneo mutant mice (see Material and Methods), indicating that the 5′ site of homologous recombination was downstream of exon 5. The bottom of the panel depicts the 3′ flanking XhoI-PstI fragment used to screen ES cell and genomic DNAs for homologous recombination by Southern blotting and the expected PstI fragments detected from wild-type (∼12.5 kb) and the targeted mutant (∼11 kb) alleles. A more representative diagram of the genomic structure of the Snap25 gene locus with the lengths of the respective intron sequences is shown between the maps of the endogenous Snap25+ and mutant Snap25Tkneo alleles. B, Representative genotyping by Southern blot of PstI restriction fragment length and by a genomic PCR assay that compares products of semiquantitative amplification of the inserted TKneo gene (neo) and of IL-1β, a flanking control gene on mouse chromosome 2. C, Survival curve demonstrating the postnatal lethality of homozygous Snap25Tkneo mutants (n = 25). Seven of 25 mice died before weaning, which is represented by the sharp drop at P21, and 6 of 25 survived >75 d; the median age of survival of P27 is indicated by an arrow. No remarkable postnatal lethality was observed for heterozygote or revertant mice, and their survival appears comparable with wild type. For comparison, the expression profiles of SNAP-25a and total SNAP-25 mRNAs, modified from Bark et al. (1995), are shown (dotted lines). D, Nissl staining reveals grossly normal morphology of mutant adult (PN78) brains (right, Tkneo/Tkneo) compared with wild-type (left, +/+) littermates; note the well structured development of the hippocampal formation in the mutant. Inset, Immunohistochemical staining for SNAP-25 of mossy fiber presynaptic terminals adjacent to the CA3 region of hippocampus (arrows) shows the characteristic accumulation of SNAP-25 in mutant mice, comparable with wild type. Scale bars: D, 1 mm; inset in D, 50 μm.
Genotyping. Genotyping was performed on tail DNA either by Southern blotting using a XhoI-PstI flanking probe (see Fig. 1) or by estimating gene dosage using a semiquantitative 32P-labeled PCR assay designed to compare amplification of the Tkneo gene insertion with that of the region spanning exons 2 and 3 of the IL-1β gene (proximal to Snap25 on mouse chromosome 2) as an internal control. PCR reactions (25 μl) contained 0.1 mm NTPs, 1 μCi of 32P-dCTP (3000 Ci/mmol; PerkinElmer, Boston, MA) 4 U of AmpliTaq Stoffel Fragment (Roche, Indianapolis, IN), and each of the following primers (0.5 μm): NeoForward, 5′-GCACGCAGGTTCTCCGGCCGC-3′; NeoReverse, 5′-TCAAGCGTATGCAGCCGCCGC-3′; IL-β1 exon 2, 5′-CCTGAACTCAACTGTGAAATGCCAC-3′; IL-β1 exon 3, 5′-GTCCGTCAACTTCAAAGAACAGTTC-3′. Amplification was limited to 22 cycles with annealing temperature of 68°C, which was determined empirically to be within the linear range of signal amplification. PCR products were run on 8% polyacrylamide gels in Tris-borate EDTA buffer, pH 8.0, and visualized on a Storm PhosphoImager and quantified using the ImageQuant program (Molecular Dynamics, Sunnyvale, CA). Genotypes were assigned based on the ratio of neo (360 bp) to IL-1β (603 bp) products in each DNA amplification and by comparison with control DNAs of wild-type, heterozygote, and homozygous mutant DNAs. In these PCR assays, homozygous mutant DNAs were distinguished by a ratio of neo to IL-1β PCR products that was twice that obtained from heterozygotes, and wild-type littermates lacked detectable neo amplification products. When necessary, genotypes were confirmed by Southern blotting analysis; however, in the vast majority of cases the PCR assay proved to correctly assign the Snap25 genotype.
Histological, RNA, and protein analysis. For Nissl and immunohistochemical staining, mice were anesthetized with pentobarbital (50 μg/gm) and perfused with 0.1 m phosphate buffer (PB) followed by 4% paraformaldehyde in PB. After cryoprotection in 30% sucrose, 40 μm sections were cut on a sliding microtome, and alternative sections were processed either for Nissl staining using 0.5% cresyl violet solution or immunostaining using polyclonal rabbit sera made to the SNAP-25 C terminus (Oyler et al., 1989), followed by horseradish peroxidase-conjugated secondary anti-rabbit and detection using the Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA). For RT-PCR, total cellular RNA was extracted from brain regions dissected on ice by standard methods using guanidinium thiocyanate and phenol-chloroform extraction. Reactions (25 μl) were performed on 1 μg of RNA using the Titan RT-PCR one-tube system (Roche) according to the manufacturer's protocol using primers to the start and stop codons of SNAP-25 coding sequence (primers also contained BamHI and EcoR1 cloning sites, respectively): forward, 5′-CGGGATCCCCCACCACTACCATGGCCGAGGAC-3′; reverse, 5′-CGGAATTCTTAACCACTTCCCAGCATCTTTGT-3′. After amplification with 34 cycles, DNA products were ethanol precipitated and 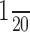 of each reaction was digested overnight with 7.5 U each of StyI and PvuI. The cleaved products were run on 8% polyacrylamide gels in Tris-borate EDTA buffer, pH 8.0; gels were dried, and the fragments were visualized and quantified by phosphoimaging as described above.
of each reaction was digested overnight with 7.5 U each of StyI and PvuI. The cleaved products were run on 8% polyacrylamide gels in Tris-borate EDTA buffer, pH 8.0; gels were dried, and the fragments were visualized and quantified by phosphoimaging as described above.
For protein analysis, brains were immersed in ice-cold PBS, and regions of interest were dissected over ice before freezing in liquid nitrogen. Extracts were prepared by homogenization and analyzed by SDS-electrophoresis and immunoblotting as described previously (Washbourne et al., 2002). Monoclonal antibodies to SNAP-25 and syntaxin 1 were obtained from Sternberger Monoclonals (Lutherville, MD) and Sigma (St. Louis, MO), respectively, and SNAP-23 rabbit polyclonal was from Affinity BioReagents (Golden, CO). Westerns were developed with species-appropriate HRP-conjugated secondary IgGs followed by chemiluminescence assay (ECL-Plus, Amersham Biosciences, Little Chalfont, UK) and quantified on the Storm PhosphorImager.
Electrophysiology. PN21-25 mice were anesthetized with ketamine (100 μg/gm) and killed by decapitation. Coronal slices (400 μm) of whole brain with hippocampi were cut with a vibratome at 0°C and incubated for 1 hr at 34°C in low-Ca2+ high-Mg2+ artificial CSF (ACSF) bubbled with 95% O2/5% CO2 containing (in mm): 124 NaCl, 5 KCl, 1.24 NaH2PO4, 0.5 CaCl2, 10 MgSO4, 26 NaHCO3, 10 d-glucose. For intracellular and extracellular recordings, slices were then placed in regular ACSF (as above but with 2.4 mm CaCl2 and 1.3 mm MgSO4) at 25°C for at least 1 hr before being transferred to the recording chamber (Scientific Systems Design, Mercerville, NJ) with a bath volume of 3 ml and a per-fusion rate of 2-5 ml/min with oxygenated ACSF at 34°C. Intracellular recordings from CA1 pyramidal neurons were made using 80-120 MΩ glass microelectrodes filled with 4 m KAc attached to an Axoclamp 2A amplifier and digitized with a Digidata 1200 analog-to-digital converter under control of pCLAMP 8.2 (Axon Instruments, Foster City, CA). Tip potentials were compensated, and no noticeable DC drift was observed. Only neurons producing action potentials to depolarizing pulses were used. Concentric bipolar stimulating electrodes (200 μm tip diameter) were placed in the stratum radiatum between the CA3 subfield and the recording electrode to deliver pulses of 0.15 msec duration at an intensity set to obtain EPSPs of approximately one-half threshold amplitude. EPSPs were recorded in response to pulse pairs at interpulse intervals (IPIs) between 50 and 300 msec. No summation of PSPs was observed at these interpulse intervals. Extracellular field EPSPs (fEPSPs) were recorded using borosilicate glass electrodes (2-3 μm tip diameter) containing 3 m NaCl and digitized and amplified as above. fEPSPs were elicited with stimuli of 0.1 msec duration to the CA1 subfield of the stratum radiatum at a frequency of 0.05 Hz. Five potential recordings were averaged on-line and stored using pCLAMP 8.2 software (Axon Instruments). For patch-clamp recording, individual slices were transferred to a submerged-type recording chamber (Warner Instruments, Hamden, CT) and perfused continuously at 34°C at a flow rate of 2 ml/min with oxygenated ACSF or ACSF to which drugs or vehicle had been added. Patch pipettes were filled with intracellular solution containing (in mm): 135 K-gluconate, 10 MgCl2, 0.1 CaCl2, 1 EGTA, 10 HEPES, 2 Na2ATP, pH 7.25 (adjusted with KOH), 270-280 mOsm. Whole-cell currents were amplified with an Axopatch 200B amplifier (Axon Instruments) and a Digidata 1322A interface, and pCLAMP 9.0 software (Axon Instruments) was used for data acquisition and analysis. Experiments measuring the time course of decay of the NMDA component of the EPSC were performed as described previously (Hessler et al., 1993). Briefly, stable NMDA EPSCs in CA1 pyramidal neurons were pharmacologically isolated with 20 μm bicucul-line and 10 μm CNQX in 0 Mg2+ ACSF and elicited every 10 sec. Cells were held at -65 mV. Stimulation was then stopped, and 50 μm MK-801 was added for a minimum of three bath exchanges. Stimulation was resumed (t = 0), and the exponential time course of block of NMDA EPSCs was measured by plotting peak EPSC amplitude as a function of time.
Statistical analysis was done using one-way and two-way factorial ANOVA and Bonferroni post hoc test (GraphPad Prism 4.0, San Diego, CA); for post hoc comparisons, p values <0.05 were considered significant.
Results
Generation of mice with a targeted insertion of Tkneo in the Snap25 gene locus
A mouse mutant overexpressing SNAP-25a was generated by homologous recombination in ES cells using a targeting vector construct of genomic DNA spanning exons 5 and 6 containing a Tkneo selection gene flanked by loxP recombination sites (floxed Tkneo) (Fig. 1A) (see Materials and Methods). G418-resistant transfected ES cells were screened by Southern blots of PstI-digested genomic DNAs using a unique flanking probe situated downstream of exon 7 as described previously (Washbourne et al., 2002). Homologous recombinants were identified by decreased PstI fragment length, from ∼11.5 to 10 kb, caused by insertion of a novel PstI site within the Tkneo selection gene. One of the selected ES cell lines gave rise to germline transmission, which was confirmed by Southern blot and genotyping subsequently by semiquantitative PCR amplification of the inserted Tkneo gene (Fig. 1B). Analysis of the targeted allele by Southern blotting and by genomic PCR and DNA sequencing of the amplified product demonstrated that the 5′ site of homologous recombination occurred between the exon 5a/5a sequence and downstream Tkneo selection gene of the targeting DNA construct (data not shown), which resulted in the insertion of the floxed Tkneo gene between exons 5 and 6 of the Snap25 gene. Nevertheless, as described below, insertion of the constitutively active Tkneo gene, positioned in the same transcriptional orientation as the Snap gene, was sufficient to disrupt the developmental regulation of alternative splicing between exons 5a and 5b. We designated this mutant allele Snap25Tkneo.
Homozygous Snap25Tkneo mutants were readily distinguishable from heterozygote and homozygous wild-type littermates. At 3-4 weeks after birth, the homozygous mutants were significantly smaller, with 72-82% of the body weight of their heterozygous and wild-type littermates (data not shown). Strikingly, homozygous mutants exhibited postnatal lethality (Fig. 1C). The majority of Snap25Tkneo/Tkneo mutants (17 of 25) died before PN60, with a median survival of 27 d after birth, and approximately one-third (8 of 25) died between the earliest point detected at PN14 and weaning at PN21. In most cases, the cause of death was uncertain, but when observed, death was preceded by indications of forebrain seizures, including vocalization before displaying straub tail, splayed hindlimbs, and facial clonus (S. L. Peterson and M. C. Wilson, unpublished observations). Those mutants that survived beyond PN50-60 (∼30%) were clearly distinguished by a disheveled appearance and lack of spontaneous activity compared with heterozygote or wild-type littermates. After several attempts to breed them with wild-type littermates, both males and females appeared to be infertile. Heterozygote mice, in contrast, appeared normally fecund and showed no evidence of postnatal lethality (data not shown). Gross histological examination of brains from homozygous mutant adult (Fig. 1D) and PN22-24 (data not shown) mice revealed no apparent neuroanatomical abnormalities, and particularly no morphological deficits in hippocampal formation were apparent. Furthermore, immunohistological staining demonstrated comparable accumulation of SNAP-25 in presynaptic mossy fiber terminals, a characteristic of SNAP-25 expression (Oyler et al., 1989; Geddes et al., 1990), again suggesting that hippocampal development and morphogenesis are not greatly impaired.
SNAP-25 isoform expression is altered in Snap25Tkneo mutants
To provide a quantitative assay for the relative expression of SNAP-25 isoform mRNAs, we took advantage of sequence-specific differences in the alternatively spliced exons that generate PvuI and StyI sites in the PCR products amplified from SNAP-25a and SNAP-25b mRNAs, respectively (Fig. 2A). Analysis of cortex, hippocampus, striatum, and cerebellum of newly weaned mice at PN23-31 and of adults (>100 d old) showed that SNAP-25a isoform mRNA was highly overexpressed compared with SNAP-25b (Fig. 2B-D) (striatum data not shown). In particular, the relative level of SNAP-25a mRNA in cortex of both homozygous mutant weanlings and adults was more than fourfold greater than wild type (PN23-31: Snap25Tkneo/Tkneo, 0.57 ± 0.02 vs Snap25+/+, 0.13 ± 0.01, p > 0.001; adult: Snap25Tkneo/Tkneo, 0.47 ± 0.01 vs Snap25+/+, 0.10 ± 0.01, p > 0.001). In hippocampus, SNAP-25a mRNA expression was also significantly elevated in young and mature surviving mutant mice (PN23-31: Snap25Tkneo/Tkneo, 0.48 ± 0.06 vs Snap25+/+, 0.20 ± 0.3, p > 0.001; adult: Snap25Tkneo/Tkneo, 0.49 ± 0.04 vs Snap25+/+, 0.10 ± 0.02, p > 0.001). Consistent with the delayed maturation of the hippocampal formation and increased expression of SNAP-25b during postnatal development (Bark et al., 1995), the relative level of SNAP-25a mRNA in hippocampus of PN23-30 wild-type mice was greater than that of either cortex of comparably aged mice or adult hippocampus of wild-type mice. In wild-type weanling and adult mice, SNAP-25a mRNA expression was considerably lower in cerebellum than the levels found in telencephalic structures such as cerebral cortex, hippocampus, or striatum (Fig. 2D). Nevertheless, despite a low level of expression, SNAP-25a transcripts in cerebellum of homozygous mutants were increased 2.5- to 3.5-fold both at PN23-31 and in adult compared with wild type (PN23-31: Snap25Tkneo/Tkneo, 0.055 ± 0.014 vs Snap25+/+, 0.014 ± 0.001, p > 0.05; adult: Snap25Tkneo/Tkneo, 0.053 ± 0.010 vs Snap25+/+, 0.020 ± 0.003, p > 0.05). These data demonstrate that increased levels of SNAP-25a persist in adult mutant mice and indicate that those mice that survive the initial period of postnatal lethality do not escape this critical period by reestablishing a normal pattern of alternative splicing.
Figure 2.

Increased levels of SNAP-25a mRNA are expressed in Snap25Tkneo mutant mice. A, Representative RT-PCR assay for SNAP-25 isoform mRNAs isolated from cortex of wild-type (+/+), heterozygote (+/Tkneo), homozygote mutant (Tkneo/Tkneo) and Cre-revertant (rev/rev) mice. PvuI and StyI restriction fragments that correspond to the 25a and 25b isoforms are indicated on the right. Below, a diagram depicts the open reading frame of the isoform mRNAs with the relative positions of PvuI and StyI sites within exon 5 (filled) that are diagnostic for spliced exon 5a or 5b; AUG and TAA indicate translational start and stop codons, respectively. Arrowheads indicate the position of the primers for RT-PCR. B-D, Quantification of SNAP-25a mRNA expression, relative to total SNAP-25 mRNA, in cortex, hippocampus, and cerebellum in wild-type, mutant, and revertant control mice. Data are expressed as the mean (±SEM) relative proportion of SNAP-25a mRNA in the total cleaved PCR product from preparations of individual mice at the ages shown (n = 3-11). Significantly increased SNAP-25a mRNA expression was seen in cortex and hippocampus of weaned (PN24-30) and adult (PN > 100) homozygous mutants (**p < 0.001; *p < 0.05; one-way ANOVA), but not in PN24-30 revertant mice, whereas in cerebellum an increased level of SNAP-25a mRNA was detected transiently in PN24-31 but not in PN35-45 mutant mice. Note that SNAP-25a mRNA expression in general is considerably lower in cerebellum, but particularly in weanlings (both in wild type and in mutants) compared with cortex and hippocampus, which may reflect either postnatal development or synaptic plasticity of this brain region.
Western blot analysis demonstrated that SNAP-25 protein expression in cortex and cerebellum of homozygous Snap25Tkneo mutants was reduced by ∼50% compared with wild type (Fig. 3A). Similar results were found for striatum (data not shown). In contrast, there was no effect on expression of the partner t-SNARE syntaxin 1A, as previously found for Snap25 knock-out mutants (Washbourne et al., 2002). The level of expression of SNAP-23, the constitutively expressed homolog of SNAP-25 (Ravichandran et al., 1996), was unchanged in cortex, hippocampus, and cerebellum (Fig. 3B). Although SNAP-23 can participate in the formation of neural SNARE complexes when overexpressed in chromaffin cells, it does not support Ca+2-triggered exocytotic bursts in chromaffin cells (Sørensen et al., 2003). This suggests that that there is little or no compensatory regulation in the expression of SNAREs or other components of the exocytotic machinery to affect evoked release at central synapses.
Figure 3.

Decreased levels of SNAP-25 protein in the Snap25Tkneo mutants but not Cre-revertant mice. Western blots of total protein isolated from indicated brain regions of 24- to 40-d-old mice of the Tkneo mutants and Cre revertants were probed for SNAP-25 [monoclonal antibody (mAb) SMI 81] and syntaxin 1A (mAb HPC-1) (A, C), and SNAP-25 (mAb SMI 81) and SNAP-23 [polyclonal antibody (pAb) PA1-783] (B). A, Semiquantitative Western blot analysis of 31- to 37-d-old Tkneo mutants shows ∼50% reduction of SNAP-25 protein in mutant cortex and cerebellum, but no similar decrease of syntaxin 1A expression in these brain regions. The ratios of the intensity of SNAP-25 and syntaxin 1A for each genotype were normalized to the mean value obtained for wild type (+/+). B, In 24-d-old Tkneo mutants, SNAP-25 also showed a ∼50% reduction in cortex, hippocampus, and cerebellum without a comparable alteration in SNAP-23 levels. Data are represented as the relative levels of SNAP-23 and SNAP-25 for each genotype normalized to the mean values obtained for wild type (+/+). C, SNAP-25 protein levels were not reduced in cortex or cerebellum of 35- to 40-d-old Cre-revertant mice. The relative levels of SNAP-25 compared with syntaxin in the revertants were calculated as in A. Representative images of the immunoreactive protein bands are shown above the histograms derived by PhosphorImager quantification of band intensities. Where indicated, the data represent the mean ± SEM obtained from duplicate or triplicate determinations.
Normal phenotype is restored in Cre-mediated revertants
The contribution of the Tkneo gene in producing altered levels of SNAP-25 isoforms was tested by excision of the inserted gene by Cre-mediated homologous recombination at the flanking pLox sites. Heterozygous mutants were cross-bred with Prm1Cre mice that express Cre recombinase and effectively direct homologous recombination between loxP sites in testis (O'Gorman et al., 1997). Excision of the floxed Tkneo gene sequence was demonstrated by the change in size of the PstI genomic fragment detected in the unique downstream flanking probe and loss of the neo sequence as determined by PCR amplification (Fig. 1). The decreased size of the genomic fragment was consistent with excision of an ∼1400 bp floxed Tkneo transgene, suggesting that either insertion or excision of neo resulted in the generation of a PstI site 5′ of the floxed Tkneo. Mice homozygous for the excised Tkneo Snap25 locus, termed Snap25rev, did not exhibit the postnatal lethality of Snap25Tkneo/Tkneo mice and were fertile. Importantly, both expression of the mRNAs encoding the two isoforms and the abundance of SNAP-25 protein in the revertant mice were restored to normal levels (Figs. 2, 3C).
Overexpression of SNAP-25a enhances facilitated but not unfacilitated release
To assess the consequences of the continued expression of the early-expressed SNAP-25a isoform, we used intracellular (sharp electrode), field potential, and whole-cell patch-clamp recordings from CA1 pyramidal neurons in hippocampal slices. In these studies, we used PN21-25 mice to avoid potential concern that mice that escape the apparent critical period from PN18 to PN30 could have developed compensatory mechanisms that might diminish the effects of overexpression of SNAP-25a. Figure 4A shows a significant genotype effect on paired-pulse facilitation (PPF), with increased facilitation for Snap25Tkneo/Tkneo mutants that decreases with increasing interpulse intervals. PPF is accepted to be a measure of presynaptic function that represents a facilitatory process (probably residual [Ca2+]i) after an initial EPSP that is greater than any transient depression of release that may follow the initial EPSP (Katz and Miledi, 1968; Regehr and Stevens, 2001). Thus, the increase in PPF seen in Figure 4A could result from an increase in the effectiveness of the facilitatory process or a decrease in the depression of release after the initial EPSP in the mutants. In an effort to distinguish between these two possibilities, we performed the experiments in which we assessed the amount of PPF as a function of the size of the initial EPSP over the range from threshold to saturation. In addition to comparing Snap25Tkneo/Tkneo mutants with wild-type littermates, heterozygote Snap25+/- mice that express comparably (50%) reduced levels of SNAP-25 protein expression (Washbourne et al., 2002) and the Cre-revertant Snap25rev/rev mice were analyzed at the same ages. For extracellular recordings, PPF was measured at 50 msec interpulse intervals between paired stimuli at stimulus strengths determined to produce EPSP values at threshold (0) and 25, 50, and 75% of maximum (0.25, 0.5, and 0.75, respectively), or at maximal EPSP response (1.0). As shown in Figure 4B, in wild-type (Snap25+/+), heterozygote (Snap25+/-), and the Cre-revertant (Snap25rev/rev) animals, there was a gradual increase in the amount of PPF between stimuli producing threshold and half-maximum EPSPs, and a decrease in the amount of PPF between stimuli producing half-maximum and saturated EPSPs. Input-output curves measured under these conditions showed no difference between mutant and control genotypes (Fig. 4C), consistent with the change in PPF representing presynaptic plasticity. The most parsimonious explanation for this observation is that paired pulses activate predominantly facilitatory processes at lower amplitude EPSPs (below half-maximum) while producing a transient depression at larger amplitude EPSPs (above half-maximum). Interestingly, the Snap25Tkneo/Tkneo mutants showed a significant increase in PPF for lower-amplitude EPSPs, suggesting that an important component of the effect of this mutation is an enhancement of the facilitatory process.
Figure 4.

Presynaptic short-term plasticity at CA1 hippocampal synapses is enhanced in SNAP-25a-overexpressing mutants. A, PPF assayed by intracellular microelectrode recordings. %PPF = 100 * (EPSP2 - EPSP1)/EPSP1 determined from EPSPs in response to approximately half-maximum stimulation of Schaffer collateral at IPIs between 50 and 300 msec. **Significant difference in facilitation at 50 msec between mutant and wild type (p < 0.01; unpaired t test). Representative traces are presented on the right. B, PPF was measured with extracellular field recordings of paired stimuli 50 msec apart at stimulus strengths determined to give threshold: 25, 50, 75, or 100% maximum field responses as indicated. Two-way ANOVA showed significant effect of interaction between genotype and relative stimulus strength (F(12,90) = 2.05; p < 0.05). Post hoc tests showed significantly increased facilitation in Snap25Tkneo/Tkneo animals compared with wild-type, heterozygote null, and revertant controls at threshold (**p > 0.01) and from heterozygote and revertant controls at 0.25 maximal stimulus (*p < 0.5). Note that increased facilitation was observed using both intracellular and extracellular recording methods, varying stimulus interval or stimulus response, respectively. C, Input-output curve calculated from extracellular recordings shown in B. A Two-way ANOVA with repeated measures showed significant interaction between genotype and %fEPSP (F(12,72) = 2.358; p = 0.0128) but no effect of genotype (F(3,72) = 1.654; p > 0.2) or difference between genotypes at any value for %fEPSP (Bonferroni post-tests; p > 0.5), indicating no substantial difference between mutant and control synaptic responses under these conditions. Representative traces of extracellular paired-pulse recordings from wild-type (+/+) and Snap25Tkneo/Tkneo mutants (Tkneo/Tkneo) at threshold (TH), 25 and 50% of maximal EPSC response used to generate the PPF and input-output curves are shown on the right. Error bars represent SEM.
The short-term facilitation observed in PPF has been attributed to a transient increase in probability of release attributable to residual [Ca2+]i after the initial stimulus (Katz and Miledi, 1968; Regehr and Stevens, 2001; Zucker and Regehr, 2002). To determine whether the enhanced facilitation observed in Snap25Tkneo/Tkneo mutant synapses was reflected by a change in the baseline probability of release, we used the use-dependent NMDA receptor (NMDAR) blocker MK-801 (Hessler et al., 1993; Rosenmund et al., 1993) to assess the probability of glutamate release at CA1 synapses independently of short-term presynaptic plasticity. As shown in Figure 5, the overall decay rate (τ) of EPSPs from Snap25Tkneo/Tkneo mutants measured during repeated stimulation at 0.1 Hz in the presence of 50 μm MK-801 was not significantly different from that of wild-type or heterozygote mice. This suggests that the basal probability of release measured at threshold or above is not substantially increased at SNAP-25a-overexpressing synapses, despite an apparent increase in facilitation during minimal stimulation. Although variable between experiments, consistent differences were not found in the peak amplitude of NMDA currents obtained from mutant and control slices before application of MK-801 (Snap25Tkneo/Tkneo, 78.9 ± 15.8 pA; Snap25+/Tkneo, 93.7 ± 21.7 pA; Snap25+/+, 88.5 ± 17.5 pA; one-way ANOVA; F(2,18) = 0.1786; p > 0.8), suggesting that increased SNAP-25a expression also did not alter synaptic connectivity or postsynaptic responsivity at these CA1 hippocampal synapses.
Figure 5.

Enhanced short-term facilitation is not caused by increased probability of release. The time course of MK-801 block of NMDAR EPSCs in response to Schaffer collateral stimulation was used to measure basal probability of glutamate release. No difference was seen between genotypes in the time course of the EPSC decay, and a one-way ANOVA showed no significant effects on the averaged individual decay curves (left inset, τ) between genotypes (Tkneo/Tkneo, 73.2 ± 8.3 sec, n = 9; heterozygote +/-, 68.6 ± 13.8 sec, n = 6; wild type +/+ (74.2 ± 7.6 sec, n = 7; F(2,20) = 0.2276; p > 0.5). Representative traces of NMDA currents recorded before MK-801 application are shown in the right insets. Error bars indicate SEM.
Quantal release at synapses is thought to be determined by the product of the size of the pool of readily releasable vesicles (N) and the probability of release (P) of individual quanta from that pool of vesicles (Katz, 1969). Under certain conditions, however, a change in release probability has been related to changes in the size of the readily releasable pool (RRP), in particular during synaptic depression caused by rapid stimulation and vesicle depletion (Dobrunz and Stevens, 1997). In contrast, our MK-801 data suggest that the two SNAP-25 isoforms do not differentially affect recruitment of vesicles into the readily releasable pool of neuronal synapses and yet still affect short-term facilitation. To further evaluate whether the size of the readily releasable pool needs to be altered to produce the difference in the short-term plasticity observed in hippocampal synapses of the SNAP-25a overexpressing mutant, we examined what affect altering the pool size and probability of release had on facilitation and depression in a mathematical model of short-term synaptic plasticity (see Appendix). According to this simplified model, the relative change in probability of release after the initial and second stimuli, together with the proportion of the initial vesicle pool affected by the change in basal probability of release, is sufficient to drive either enhanced (facilitation) or decreased (depression) release after short interpulse intervals. Moreover, consistent with our experimental data, facilitation is independent of the initial size of the readily releasable pool.
Discussion
Our results suggest that the developmental shift in SNAP-25 isoform expression, mediated by alternative splicing, contributes to synaptic transmission by affecting the facilitatory enhancement of neurotransmitter release. These findings help to define the role of proteins that mediate neuroexocytosis in presynaptic mechanisms of plasticity both in development and in response to nervous system injury.
Early studies based on the expression profile of SNAP-25 isoforms (Bark et al., 1995), together with antisense studies (Osen-Sand et al., 1993) and botulinum neurotoxin inhibition studies (Osen-Sand et al., 1996; Morihara et al., 1999) led to the view that SNAP-25a could serve as a t-SNARE involved in exocytotic processes required for membrane trafficking and addition during axonal outgrowth. Our recent analysis of SNAP-25 null mutant mice, however, demonstrated that early steps of neural development occur in the absence of SNAP-25 and consequently without evoked synaptic transmission supported by SNAP-25 (Molnar et al., 2002; Washbourne et al., 2002). These steps include cell migration, differentiation including axonal elongation, targeting, and the development of action potential-independent synaptic transmission. Membrane trafficking events responsible for neurite outgrowth are instead likely to be mediated by other t-SNARE homologs. Such SNARE complexes might include, for example, the constitutively expressed SNAP-25 homolog, SNAP-23, which is common to excitable and nonexcitable cells (Ravichandran et al., 1996; Wang et al., 1997), in conjunction with other more specialized SNARE components, such as tetanus neurotoxin-insensitive VAMP (TI-VAMP) (Martinez-Arca et al., 2001). The role of the early-expressed SNAP-25a, therefore, must lie downstream of initial synaptic contact, consistent with the suggestion that the shift between these isoforms may be involved in maturation of synaptic communication through shaping evoked neurotransmitter release. Although two Snap25 exon sequences have been described in vertebrates, human (Bark and Wilson, 1994), rat (Jacobsson et al., 1996), mouse (Bark et al., 1995), chicken (Bark, 1993), and zebrafish (Risinger et al., 1998), only one isoform is present in Drosophila (Risinger et al., 1997). This suggests that tandem duplication of exon 5 occurred during the evolutionary development of the vertebrate brain and contributes to increased functional plasticity and organization of these higher-ordered neural systems. Interestingly, we find that impeding the developmental shift toward SNAP-25b has a striking affect on survival, underscoring the importance of this transition in brain development.
The differential expression of SNAP-25 isoforms during development (Bark et al., 1995; Boschert et al., 1996) and in response to CNS injury (Boschert et al., 1996; Patanow et al., 1997) is consistent with the dynamic regulation of alternative splicing between the tandemly arranged, divergent copies of exon 5. Analysis of the genomic structure at the site of the Snap25Tkneo mutation showed that it was limited to insertion of the floxed Tkneo selection gene downstream of exon 5b. That Cre-mediated excision of the floxed Tkneo selection cassette from the Snap25Tkneo mutation completely restored a normal phenotype at the level of mRNA and protein expression is evidence that the insertion of the Tkneo mini-transgene is responsible for the altered phenotype of the mutant and does so by dysregulating the processes that select between alternative splice sites. The effect of an actively expressed selection marker on gene expression is not unprecedented; interference by the Pgkneo selection marker gene construct has been shown to result in compromised expression levels and altered splicing (Nagy, 2000; Lewandoski, 2001). Moreover, such hypomorphic mutations are stable and have proven useful, for example, in elucidating the function of FGF receptors (Meyers and Martin, 1999; Trumpp et al., 1999; Lewandoski et al., 2000; Sun et al., 2002). In the case of the Snap25Tkneo mutation, this promotes an expression profile of roughly equal levels of SNAP-25a and SNAP-25b transcripts in major telencephalic regions of adult brain that corresponds to the relative levels found in neonatal (approximately PN5-7) forebrain (Bark et al., 1995).
In addition to prolonging expression of SNAP-25a, the mutants exhibit ∼50% reduced levels of total SNAP-25 protein with a comparable decrease in both forebrain and cerebellum. Quantitative RNA analysis from microarray hybridizations has also shown a similar 50% reduction of SNAP-25 mRNA in hippocampus of PN13 and PN20 homozygous mutants (J. R. Matthews and Wilson, unpublished observations). Together, these results indicate that decreased SNAP-25 expression in the Snap25Tkneo/Tkneo mutant brain is not caused by instability of the overexpressed SNAP-25a isoform, but likely results from decreased transcription or RNA transcript stability. Nevertheless, several observations suggest that the phenotypic deficits of the Snap25Tkneo/Tkneo mouse cannot be simply attributed to reduced SNAP-25 protein expression. First, there appears to be little or no compensatory change in expression of presynaptic proteins in response to decreased SNAP-25 levels as evidenced by levels of syntaxin 1A or SNAP-23 in Snap25Tkneo/Tkneo mutant brain (Fig. 3), or after complete ablation of SNAP-25 expression in knock-out null mutants (Washbourne et al., 2002). Moreover, mice heterozygous for a Snap25 null mutation, which have a similar reduced level of SNAP-25 expression without altering the relative expression of the two isoforms (data not shown), exhibit neither the early lethality nor the increased PPF seen in the Snap25Tkneo/Tkneo mutant (Fig. 4). Although an interaction between reduced overall expression and altered splicing cannot be ruled out entirely, our data argue strongly that the dysregulation of splicing underlies the abnormal phenotype seen in these mutants.
Our present results extend the recent demonstration of functional differences between SNAP-25 isoforms in promoting neurosecretion (Sørensen et al., 2003). In these studies, expression of exogenous SNAP-25b in Snap25 null mutant fetal chromaffin cells was shown to be more effective than SNAP-25a in mediating Ca2+-triggered secretion. The difference was attributed to the ability of SNAP-25b to support a larger pool of primed vesicles, suggesting that SNAP-25 isoforms affect the rate of reversible priming necessary to sustain pools of releasable secretory vesicles in these neurosecretory cells. Although SNAP-25b is more efficient in this system, SNAP-25a is the predominant isoform expressed in adult rodent chromaffin cells (Bark et al., 1995; Grant et al., 1999), suggesting that SNAP-25a may be matched more physiologically to secretion from large dense-core vesicles.
In Snap25Tkneo mutant neurons, the sustained expression of SNAP-25a resulted in increased facilitation of glutaminergic transmission for 50-100 msec after an initial stimulus in CA1 hippocampal synapses. The enhanced facilitation seen at mutant synapses was not observed in heterozygote null mutants, despite a similar decreased level of total SNAP-25 protein, nor was it observed in Cre-mediated revertants. This alteration in synaptic transmission can be attributed to overexpression of the SNAP-25a isoform and not to an overall decrease in expression or disruption of the Snap25 gene. Moreover, analysis of the probability of release, measured by progressive blockade of open NMDAR channels with MK-801, showed that this facilitation was not accompanied by an increase in basal probability of release, an observation reflected in our simple model of short-term plasticity. This is consistent with SNAP-25a enhancing short-term plasticity of synaptic transmission in neurons by facilitating release after an initial conditioning stimulus, rather than by regulating the size of releasable vesicle pools. One attractive mechanism for this effect on plasticity is a multiple-site hypothesis in which SNAP-25a-containing SNARE complexes may preferentially recruit high-affinity or moderately high-affinity Ca2+ binding proteins to the exocytotic machinery that would sense residual [Ca2+]i from an initial stimulus to enhance vesicular fusion after subsequent stimuli (for review, see Zucker and Regehr, 2002). Given that SNAP-25a expression coincides with the period of synaptogenesis in neonatal brain (Bark et al., 1995), this facilitation could contribute to short-term plasticity involved in strengthening selective synaptic contacts during activity-dependent synapse elimination (Lichtman and Colman, 2000). Synaptic transmission in mutants deficient in Rab3A, the small GTP-binding protein associated with synaptic vesicle fusion, also shows increased PPF without an overall change in probability of release (Geppert et al., 1997). This suggests that, as the switch between SNAP-25 isoforms, the recruitment of Rab3a into synaptic vesicle exocytotic machinery may contribute to the refinement of synaptic transmission in mature rather than immature synaptic contacts.
The increased expression of SNAP-25a observed after hippocampal injury in adult rats (Boschert et al., 1996; Patanow et al., 1997) is consistent with the reenactment of developmental patterns of expression as damaged brain regions attempt to reestablish connections. Certain brain and spinal cord nuclei in adult rodents continue to express predominantly SNAP-25a into adulthood (Bark et al., 1995; Boschert et al., 1996; Jacobsson et al., 1999), although its selective function remains unclear. Nevertheless, the behavioral affect of seizures and premature death seen in these mutant mice suggests that generalized maintenance of SNAP-25a expression might disrupt normal brain function. It is interesting to consider that continued expression of this isoform may maintain the plasticity characteristic of early stages of synaptic development and thereby impair synaptic maturation and consolidation of neural circuits in these mutant mice. Alterations in the expression of various components of the presynaptic protein machinery for transmitter release also appear to contribute to the genetic underpinnings of certain neuropsychiatric deficits, such as schizophrenia (Mirnics et al., 2000). It will be important to examine whether deficiencies in SNAP-25 expression, as revealed in heterozygote null and isoform mutants, can act in concert with other genetic and potential environmental influences to increase the vulnerability to complex behavioral and neuropsychiatric disorders.
Appendix
A model for short-term synaptic plasticity
We have constructed a simple mathematical model for paired-pulse plasticity that accounts for both PPF and paired-pulse depression (PPD). This model is based on the following assumptions.
Paired-pulse plasticity on a time scale of <300 msec results from alterations of release from an RRP of vesicles.
The RRP has two interchangable states, a basal (State1) and a facilitated (State2), in which, for instance, the amount of [Ca2+]residual determines the fraction of the RRP in each state (Zucker and Regehr, 2002). There are N1 vesicles in State1 released with a probability of P1 and N2 vesicles in State2 released with a probability of P2.
State2 has a higher probability of release than State1.
During PPF or PPD, the response to the first stimulus depends entirely on release from State1, whereas the response to the second stimulus depends on a mixture of release from State1 and State2.
Replenishment of the RRP takes place on a slower time course than the transition between State1 and State2 and is thus not a factor in PPF or PPD.
The fraction of vesicles moving from State1 to State2 is independent of the release history of a synapse (Stevens and Wang, 1995). Because the transition between State1 and State2 does not depend on whether there is release at an individual synaptic release site, all release sites can be grouped together and the sum of their RRPs treated as a single RRP.
In the case of a pair of stimuli, A followed by B
Before stimulus A, all vesicles are in State1 and the number of vesicles released in response to stimulus A is NA = P1N1.
Immediately after stimulus A, a fraction of those that were not released move to State2 as follows: N2 = a(N1 - NA), where a is the fraction of vesicles affected by [Ca2+]residual that depends on the size of pulse A and the length of time after this pulse. For example, in a typical paired-pulse stimulus paradigm, 0 < a ≤ 1 at t = 0 and a = 0 at t = 300 msec.
The number of vesicles released in response to stimulus B comes from State1 and State2 and is as follows: NB = P1(N1 - NA - N2) + P2N2. (The first term is related to the amount of paired-pulse depression and the second term to the amount of paired-pulse facilitation.)
Expanding the parentheses and substituting the equations for N2 and NA results in the following: NB = P1N1 - P1NA - P1N2 + P2N2 = P1N1 - P1P1N1 - P1aN1 + P1aP1N1 + P2aN1 - P2aP1N1.
Thus, the paired pulse ratio (PPR) is as follows: PPR (= NB/NA) = 1 - P1 - a + aP1 +aP2/P1 - aP2 = 1 - a + P1(a - 1) + aP2(1/P1 - 1).
Because when PPR = 1, there is no short-term plasticity; whether PPR reflects depression or facilitation depends only on the relative values of P1, P2, and a. Consequently, this model predicts that PPF and PPD are not affected by the initial size of the RRP. Thus, PPF reflects the percentage of vesicles going to State2 with an enhanced probability of release (a), and depression reflects a diminution of the vesicles immediately available for release.

For example, based on this relationship, with P1 = 0.3 [the average probability of release observed for hippocampal synapses(Rosenmund et al., 1993)] and varying P2 and the fraction of vesicles affected by [Ca2+]residual (a), the following plot can be drawn predicting values for PPR where PPF (red) and PPD (blue) occur.

Alternatively, the transition between PPF and PPD can be viewed by plotting a transformation of this relationship, where a = P1/(-1 + P1 + (P2/P1) - P2) and varying P1, P2. In this case PPD occurs at combinations of P1, P2, and a below the surface and PPF for combinations above the surface. The model thus predicts that facilitation is greater for smaller values of initial probability of release (P1) (Dobrunz and Stevens, 1997).

Footnotes
This work was supported by National Institutes of Health Grant MH48989, in part by the Dedicated Health Research Funds of the University of New Mexico, School of Medicine (M.C.W.), and by the Swedish Natural Science Research Council, Magnus Bergvalls Foundation, and travel funds from the Karolinska Institutet (C.B.). We thank Steve Peterson for advice and comments on seizure phenotype, Maria Picchi and Andrea Allan for help in the survival curve and statistical analyses, and Lee Anna Cunningham for help with histology. We also thank C. William Shuttle-worth, Adrian Schiess, and C. Fernando Valenzuela for valuable discussions on the model and comments on this manuscript. We acknowledge the excellent technical support by Sayuri Nixon in genotyping and Ed Padilla at the University of New Mexico Health Sciences Center Animal Resource Facility for maintaining the mouse colony.
Correspondence should be addressed to Michael C. Wilson, Department of Neurosciences, University of New Mexico Health Sciences Center, Albuquerque, NM 87131. E-mail: mwilson@salud.unm.edu.
F. P. Bellinger's present address: Pacific Biomedical Research Center, University of Hawaii, Honolulu, HI 96822.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248796-10$15.00/0
References
- Bark IC (1993) Structure of the chicken gene for SNAP-25 reveals duplicated exon encoding distinct isoforms of the protein. J Mol Biol 233: 67-76. [DOI] [PubMed] [Google Scholar]
- Bark IC, Wilson MC (1994) Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene 139: 291-292. [DOI] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC (1995) Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci USA 92: 1510-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JB, Matern HT, Peden AA, Scheller RH (2001) A genomic perspective on membrane compartment organization. Nature 409: 839-841. [DOI] [PubMed] [Google Scholar]
- Boschert U, O'Shaughnessy C, Dickinson R, Tessari M, Bendotti C, Catsicas S, Pich EM (1996) Developmental and plasticity-related differential expression of two SNAP-25 isoforms in the rat brain. J Comp Neurol 367: 177-193. [DOI] [PubMed] [Google Scholar]
- Chen YA, Scheller RH (2001) SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol 2: 98-106. [DOI] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF (1997) Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18: 995-1008. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998) Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci USA 95: 15781-15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasshauer D, Antonin W, Margittai M, Pabst S, Jahn R (1999) Mixed and non-cognate SNARE complexes. Characterization of assembly and biophysical properties. J Biol Chem 274: 15440-15446. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Hess EJ, Hart RA, Kesslak JP, Cotman CW, Wilson MC (1990) Lesions of hippocampal circuitry define synaptosomal-associated protein-25 (SNAP-25) as a novel presynaptic marker. Neuroscience 38: 515-525. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Stevens CF, Sudhof TC (1997) The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature 387: 810-814. [DOI] [PubMed] [Google Scholar]
- Gonelle-Gispert C, Halban PA, Niemann H, Palmer M, Catsicas S, Sadoul K (1999) SNAP-25a and -25b isoforms are both expressed in insulin-secreting cells and can function in insulin secretion. Biochem J 339: 159-165. [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Linder ME (1998) SNAP-25 palmitoylation and plasma membrane targeting require a functional secretory pathway. Mol Biol Cell 9: 585-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo S, Greentree WK, Linder ME (1999) SNAP-25 is targeted to the plasma membrane through a novel membrane-binding domain. J Biol Chem 274: 21313-21318. [DOI] [PubMed] [Google Scholar]
- Grant NJ, Hepp R, Krause W, Aunis D, Oehme P, Langley K (1999) Differential expression of SNAP-25 isoforms and SNAP-23 in the adrenal gland. J Neurochem 72: 363-372. [DOI] [PubMed] [Google Scholar]
- Hess DT, Slater TM, Wilson MC, Skene JH (1992) The 25 kDa synaptosomal-associated protein SNAP-25 is the major methionine-rich polypeptide in rapid axonal transport and a major substrate for palmitoylation in adult CNS. J Neurosci 12: 4634-4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366: 569-572. [DOI] [PubMed] [Google Scholar]
- Jacobsson G, Piehl F, Bark IC, Zhang X, Meister B (1996) Differential subcellular localization of SNAP-25a and SNAP-25b RNA transcripts in spinal motoneurons and plasticity in expression after nerve injury. Brain Res Mol Brain Res 37: 49-62. [DOI] [PubMed] [Google Scholar]
- Jacobsson G, Bark C, Meister B (1999) Differential expression of SNAP-25a and SNAP-25b RNA transcripts in cranial nerve nuclei. J Comp Neurol 411: 591-600. [DOI] [PubMed] [Google Scholar]
- Katz B (1969) The release of neural transmitter substances. Liverpool, UK: Liverpool UP.
- Katz B, Miledi R (1968) The role of calcium in the neuromuscular junction of the frog. J Physiol (Lond) 195: 481-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koticha DK, Huddleston SJ, Witkin JW, Baldini G (1999) Role of the cysteine-rich domain of the t-SNARE component, SYNDET, in membrane binding and subcellular localization. J Biol Chem 274: 9053-9060. [DOI] [PubMed] [Google Scholar]
- Lane SR, Liu Y (1997) Characterization of the palmitoylation domain of SNAP-25. J Neurochem 69: 1864-1869. [DOI] [PubMed] [Google Scholar]
- Lewandoski M (2001) Conditional control of gene expression in the mouse. Nat Rev Genet 2: 743-755. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Sun X, Martin GR (2000) Fgf8 signaling from the AER is essential for normal limb development. Nat Genet 26: 460-463. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Colman H (2000) Synapse elimination and indelible memory. Neuron 25: 269-278. [DOI] [PubMed] [Google Scholar]
- Mansour SL, Thomas KR, Capecchi MR (1988) Disruption of the protooncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336: 348-352. [DOI] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouille P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, Galli T (2001) A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci 21: 3830-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNew JA, Parlati F, Fukuda R, Johnston RJ, Paz K, Paumet F, Sollner TH, Rothman JE (2000) Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature 407: 153-159. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Martin GR (1999) Differences in left-right axis pathways in mouse and chick: functions of FGF8 and SHH. Science 285: 403-406. [DOI] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P (2000) Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53-67. [DOI] [PubMed] [Google Scholar]
- Molnar Z, Lopez-Bendito G, Small J, Partridge LD, Blakemore C, Wilson MC (2002) Normal development of embryonic thalamocortical connectivity in the absence of evoked synaptic activity. J Neurosci 22: 10313-10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara T, Mizoguchi A, Takahashi M, Kozaki S, Tsujihara T, Kawano S, Shirasu M, Ohmukai T, Kitada M, Kimura K, Okajima S, Tamai K, Hirasawa Y, Ide C (1999) Distribution of synaptosomal-associated protein 25 in nerve growth cones and reduction of neurite outgrowth by botulinum neurotoxin A without altering growth cone morphology in dorsal root ganglion neurons and PC-12 cells. Neuroscience 91: 695-706. [DOI] [PubMed] [Google Scholar]
- Nagy A (2000) Cre recombinase: the universal reagent for genome tailoring. Genesis 26: 99-109. [PubMed] [Google Scholar]
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y (1997) Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci USA 94: 14602-14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osen-Sand A, Catsicas M, Staple JK, Jones KA, Ayala G, Knowles J, Grenningloh G, Catsicas S (1993) Inhibition of axonal growth by SNAP-25 antisense oligonucleotides in vitro and in vivo. Nature 364: 445-448. [DOI] [PubMed] [Google Scholar]
- Osen-Sand A, Staple JK, Naldi E, Schiavo G, Rossetto O, Petitpierre S, Malgaroli A, Montecucco C, Catsicas S (1996) Common and distinct fusion proteins in axonal growth and transmitter release. J Comp Neurol 367: 222-234. [DOI] [PubMed] [Google Scholar]
- Oyler GA, Higgins GA, Hart RA, Battenberg E, Billingsley M, Bloom FE, Wilson MC (1989) The identification of a novel synaptosomal-associated protein, SNAP-25, differentially expressed by neuronal sub-populations. J Cell Biol 109: 3039-3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patanow CM, Day JR, Billingsley ML (1997) Alterations in hippocampal expression of SNAP-25, GAP-43, stannin and glial fibrillary acidic protein following mechanical and trimethyltin-induced injury in the rat. Neuroscience 76: 187-202. [DOI] [PubMed] [Google Scholar]
- Ravichandran V, Chawla A, Roche PA (1996) Identification of a novel syntaxin- and synaptobrevin/VAMP-binding protein, SNAP-23, expressed in non-neuronal tissues. J Biol Chem 271: 13300-13303. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Stevens CF (2001) Physiology of synaptic transmission and short-term plasticity. In: Synapses (Cowan WM, Sudhof TC, Stevens CF, eds), pp 135-175. Baltimore: Johns Hopkins UP.
- Risinger C, Deitcher DL, Lundell I, Schwarz TL, Larhammar D (1997) Complex gene organization of synaptic protein SNAP-25 in Drosophila melanogaster Gene 194: 169-177. [DOI] [PubMed] [Google Scholar]
- Risinger C, Salaneck E, Soderberg C, Gates M, Postlethwait JH, Larhammar D (1998) Cloning of two loci for synapse protein Snap25 in zebrafish: comparison of paralogous linkage groups suggests loss of one locus in the mammalian lineage. J Neurosci Res 54: 563-573. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262: 754-757. [DOI] [PubMed] [Google Scholar]
- Sollner TH, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE (1993) SNAP receptors implicated in vesicle targeting and fusion. Nature 362: 318-324. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E (2003) Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell 114: 75-86. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Wang Y (1995) Facilitation and depression at single central synapses. Neuron 14: 795-802. [DOI] [PubMed] [Google Scholar]
- Sun X, Mariani FV, Martin GR (2002) Functions of FGF signaling from the apical ectodermal ridge in limb development. Nature 418: 501-508. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR (1999) Cremediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first brachial arch. Genes Dev 13: 3136-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit M, Sollner TH, Rothman JE (1996) Multiple palmitoylation of synaptotagmin and the t-SNARE SNAP-25. FEBS Lett 385: 119-123. [DOI] [PubMed] [Google Scholar]
- Wang G, Witkin JW, Hao G, Bankaitis VA, Scherer PE, Baldini G (1997) Syndet is a novel SNAP-25 related protein expressed in many tissues. J Cell Sci 110: 505-513. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Cansino V, Mathews JR, Graham M, Burgoyne RD, Wilson MC (2001) Cysteine residues of SNAP-25 are required for SNARE disassembly and exocytosis, but not for membrane targeting. Biochem J 357: 625-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19-26. [DOI] [PubMed] [Google Scholar]
- Weimbs T, Low SH, Chapin SJ, Mostov KE, Bucher P, Hofmann K (1997) A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc Natl Acad Sci USA 94: 3046-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Gonzalez LJ, Prekeris R, Steegmaier M, Advani RJ, Scheller RH (1999) SNARE interactions are not selective. Implications for membrane fusion specificity. J Biol Chem 274: 5649-5653. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]