

Abstract
The c-Jun N-terminal kinase (JNK) signaling pathway plays a critical role in mediating apoptosis in the nervous system; however, the mechanisms by which JNK triggers neuronal apoptosis remain incompletely understood. Recent studies suggest that in addition to inducing transcription of pro-apoptotic genes, JNK also directly activates the cell death machinery. Here, we report that JNK catalyzed the phosphorylation of the BH3-only protein Bcl-2 interacting mediator of cell death (BimEL) at serine 65, both in vitro and in vivo. The JNK-induced phosphorylation of BimEL at serine 65 promoted the apoptotic effect of BimEL in primary cerebellar granule neurons. We also characterized the role of the JNK-BimEL signaling pathway in apoptosis that was triggered by overexpression of the p75 neurotrophin receptor (p75NTR). We found that activation of p75NTR induced the JNK-dependent phosphorylation of endogenous BimEL at serine 65 in cells. The genetic knockdown of BimEL by RNA interference or the expression of a dominant interfering form of BimEL significantly impaired the ability of activated p75NTR to induce apoptosis. Together, these results suggest that JNK-induced phosphorylation of BimEL at serine 65 mediates p75NTR-induced apoptosis. Our findings define a novel mechanism by which a death-receptor pathway directly activates the mitochondrial apoptotic machinery.
Keywords: apoptosis, survival, neuron, BH3-only, signal transduction, p75 neurotrophin receptor, protein kinase
Introduction
Programmed cell death of neurons is an indispensable part of the development of the nervous system and contributes to neuronal loss in neurologic injury and disease. Two canonical cell death pathways have been characterized: an apoptotic pathway that is triggered by the activation of cell-surface death receptors and leads to direct activation of the caspase cascade, and a cell-intrinsic apoptotic pathway that requires the release of mitochondrial proteins for caspase activation (Strasser et al., 2000; Kaufmann and Hengartner, 2001).
The pro-apoptotic BH3-only proteins play an important role in the activation of the cell-intrinsic cell death program by acting as sentinels of cellular damage (Huang and Strasser, 2000). Once activated, some BH3-only proteins including Bid are thought to directly activate the pro-apoptotic multidomain Bcl-2 family members, whereas others including Bad interact with and inhibit the prosurvival Bcl-2 family members including Bcl-2 and Bcl-XL (Gross et al., 1999; Huang and Strasser, 2000).
Among the BH3-only proteins, Bcl-2 interacting mediator of cell death (Bim) is of particular interest to studies of apoptosis in the nervous system. Developmentally regulated neuronal apoptosis is delayed in mice in which the bim gene is disrupted (Putcha et al., 2001; Whitfield et al., 2001), suggesting a critical role for Bim in neuronal cell death. An important question raised by these studies is how Bim-induced death is regulated in neurons. Previously, it has been shown that the apoptotic stimuli of growth factor and neuronal activity withdrawal induce bim transcription in sympathetic and cerebellar granule neurons, in part through the c-Jun N-terminal kinase (JNK)-mediated phosphorylation and activation of the transcription factor c-Jun (Harris and Johnson, 2001; Putcha et al., 2001; Whitfield et al., 2001).
Bim exists in three major isoforms that are generated by alternative splicing: BimEL, BimL, and BimS. Among the distinct Bim isoforms, BimL and BimEL are the predominant isoforms in neurons (O'Reilly et al., 2000). Interestingly, BimEL is constitutively expressed in many neuronal cell types, suggesting that BimEL function might be regulated by posttranslational modifications in neuronal cells after exposure to an apoptotic stimulus (O'Reilly et al., 2000). Recent studies suggest that Bim is phosphorylated in response to several apoptotic stimuli, including growth factor withdrawal in primary neurons (Lei and Davis, 2003; Putcha et al., 2003).
The stress-activated protein kinases (SAPKs), including JNK and p38 mitogen-activated protein kinase (p38MAPK), are commonly activated in neurons in response to apoptotic stimuli (Mielke and Herdegen, 2000; Harper and LoGrasso, 2001). In addition to mediating trophic factor withdrawal-induced apoptosis, the JNK signaling pathway is a critical mediator of neuronal cell death after activation of the p75 neurotrophin receptor (p75NTR) (Friedman, 2000; Harrington et al., 2002; Roux and Barker, 2002). A widely held view is that activation of p75NTR in the absence of stimulation of the tyrosine kinase family of neurotrophin receptors constitutes an important trigger of apoptosis (Frade et al., 1996; Kaplan and Miller, 2000). p75NTR is thought to play a critical role during development and after pathogenic stimuli in the adult brain (Yaar et al., 1997; Bamji et al., 1998; Roux et al., 1999; Dechant and Barde, 2002).
The downstream effector pathways of p75NTR-activated and JNK-mediated neuronal apoptosis remain to be characterized. In contrast to trophic factor withdrawal-induced apoptosis, in which JNK-mediated c-Jun-dependent transcription plays a critical role, in p75NTR-induced cell death JNK activation is required, yet activation of c-Jun appears to be dispensable (Harrington et al., 2002; Palmada et al., 2002; Troy et al., 2002). These findings raise the interesting possibility that p75NTR-activated JNK might directly phosphorylate components of the mitochondrial cell death machinery and thereby induce neuronal apoptosis.
In this study, we have delineated a novel mechanism by which JNK mediates p75NTR-induced neuronal apoptosis. We found that JNK catalyzes the phosphorylation of the BH3-only protein BimEL in vitro and in vivo at the distinct site of serine 65. The phosphorylation of BimEL at serine 65 promotes the proapoptotic activity of BimEL in primary neurons. Overexpression of p75NTR was found to induce JNK-mediated phosphorylation of endogenous BimEL at serine 65. In addition, we demonstrated that BimEL and JNK-induced phosphorylation of BimEL at serine 65 are required for p75NTR-induced apoptosis. Together, these findings define the JNK-BimEL signaling pathway as a novel link between death receptor activation and the cell death machinery.
Materials and Methods
Plasmids and reagents. Full-length BimEL cDNA was cloned by RT-PCR from RNA isolated from rat cerebellar granule neurons. BimEL cDNA was then subcloned into the polylinker of the bacterial glutathione S-transferase (GST) gene fusion vector pGEX-4T-3 (Amersham Biosciences, Piscataway, NJ) and into the polylinker of the mammalian expression vector pCDNA3 (Invitrogen, Carlsbad, CA) with an NH2-terminal hemagglutinin (HA)-tag. Point mutations of Ser-55, Ser-65, Ser-73, Ser-100, and Thr-112 were made using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Similarly, the splicing donor site for BimL (T126 to G, without amino acid change) and the BH3-domain of BimEL (Leu-150 to Ala and Asp-155 to Ala) were mutated. Mutations were verified by sequencing. Activated MAPK kinase 3 (MKK3) was purchased from Stratagene. The activated MAPK kinase kinase (MEKK1) plasmid has been described previously (Lee et al., 1997). The dominant-negative JNK plasmid was kindly provided by Dr. Yang Shi (Harvard Medical School, Boston, MA).
The U6/BS-Bim RNA interference vector was designed as described (Gaudilliere et al., 2002; Sui et al., 2002). The sense strand of the encoded hairpin RNA was designed to specifically target the 20-nucleotide region GGTATTTCTCTTTTGACACAG in Bim RNA. The targeted region in Bim RNA showed no significant homology with any other gene by BLAST (basic local alignment search tool).
Preparation of recombinant adenovirus expressing green fluorescent protein (AdGFP), β-galactosidase (AdLacZ), full-length p75NTR (Adp75NTR), and the Flag-tagged JNK-binding domain (JBD) of JNK interacting protein (JIP) (AdJBD) have been described previously (Roux et al., 2002). All adenoviruses were amplified in human embryonic kidney (HEK) 293A cells and purified on a sucrose gradient as described (Roux et al., 2002). Titers are expressed as plaque-forming units.
To generate the phospho65-Bim antibody, a phosphopeptide of the sequence CLAPPApSPGPFATR (Tufts Synthesis Facility, Tufts Medical School, Boston, MA), was synthesized and coupled to keyhole limpet hemocyanin using the Imject Maleimide Activated mcKLH Kit (Pierce, Rockford, IL). The antigen was injected into New Zealand White rabbits (Covance Research Products, Denver, PA), from which serum was collected approximately every 3 weeks. Serum was affinity purified by subsequently passing it on an Immunopure immobilized protein A column (Pierce) and an agarose-iodoacetyl column (Pierce) to which a synthetic peptide of the sequence CLAPPASPGPFATR (Tufts Synthesis Facility) was coupled. The final eluate was desalted and concentrated using Amicon Ultra centrifugal filter devices (Millipore, Bedford, MA). The phospho65 antibody is available from Bioscience (Camarillo, CA).
Other antibodies used in this study include polyclonal anti-Bim antibody (Stressgen, Victoria, British Columbia, Canada; catalog #AAP-330), polyclonal anti-Bim antibody (Calbiochem, La Jolla, CA; catalog #202000), monoclonal anti-GST antibody (Santa Cruz Biotechnology, Santa Cruz, CA; catalog #s.c.-138), polyclonal anti-HA antibody (Santa Cruz Biotechnology; catalog #s.c.-805), polyclonal phospho-JNK antibody (Cell Signaling, Beverly, MA; catalog #9251), polyclonal JNK antibody (Upstate Biotechnology, Lake Placid, NY; catalog #06-748), polyclonal phospho-p38MAPK antibody (Cell Signaling; catalog #9211), polyclonal phospho-c-Jun antibody (Cell Signaling; catalog #9261), monoclonal 14-3-3 β antibody (Santa Cruz Biotechnology; catalog #s.c.-1657), monoclonal p42 MAP kinase antibody (Cell Signaling; catalog #9107), monoclonal anti-β-galactosidase antibody (Promega, Madison, WI; catalog #Z3781), monoclonal anti-actin antibody (Santa Cruz Biotechnology; catalog #s.c.-8432), and a polyclonal antibody against cleaved caspase-3 (Cell Signaling; catalog #9661). The p75NTR antibody has been described previously (Roux et al., 1999). Secondary antibodies conjugated to horseradish peroxidase or cyanine dye were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
The JNK inhibitor II (SP600125) and the p38MAPK inhibitor (SB203580) were purchased from EMD Biosciences Inc. (San Diego, CA).
In vitro kinase assays. In vitro kinase assays were done as described in the Upstate Biotechnology procedure for the JNK1α1 protein kinase assay. Briefly, JNK1α1 (Upstate Biotechnology), 5 μg of recombinant GST-Bim, 100 μm ATP, and 5 μCi of [γ-32P]ATP in 2 mm MOPS, pH 7.2, 2.5 mm β-glycerol phosphate, 0.5 mm EGTA, 0.1 mm sodium orthovanadate, 0.1 mm DTT, and 15 mm magnesium chloride were incubated at 30°C for 1 hr. The reactions were stopped by adding SDS sample buffer and boiling. Samples were analyzed by 15% SDS-PAGE, Coomassie staining, and autoradiography. In the experiments depicted in Figure 2 B, 200 μm unlabeled ATP was used, and the samples were analyzed by SDS-PAGE and immunoblotting.
Figure 2.
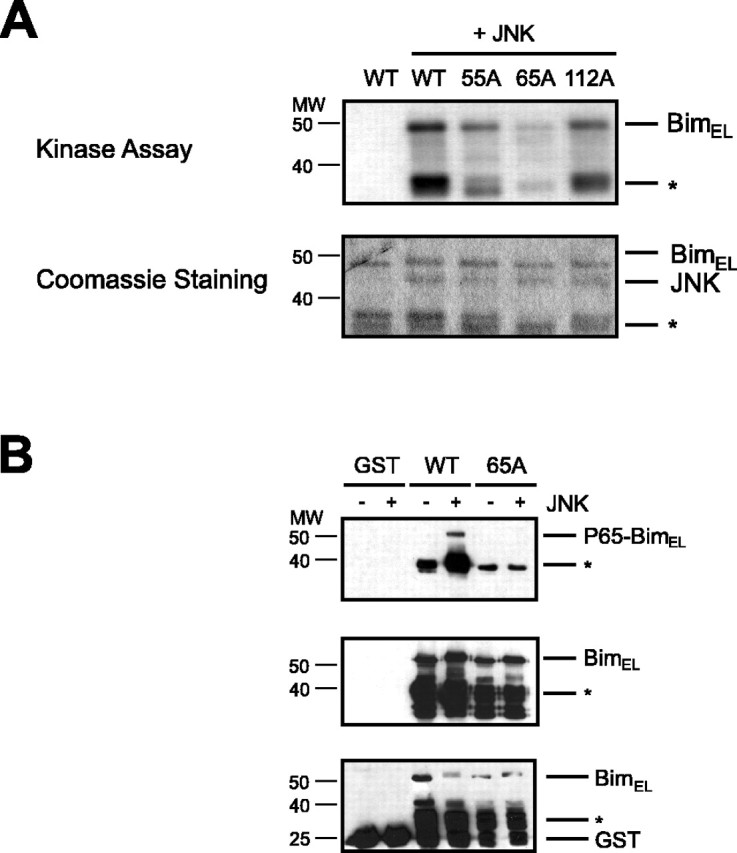
JNK phosphorylates BimEL at the distinct site of serine 65 in vitro. A, Recombinant GST-BimEL (WT) and recombinant BimEL phosphorylation mutants GST-BimELS55A (55A), GST-BimELS65A (65A), and GST-BimELT112A (112A) were incubated with no kinase or recombinant JNK1α1 and subjected to an in vitro kinase assay using [γ-32P]ATP. Phosphorylated substrates were separated by SDS-PAGE, stained with Coomassie blue (bottom panel), and analyzed by autoradiography (top panel). Degradation products of bacterially expressed GST-BimEL are indicated with an asterisk. B, Recombinant GST, GST-BimEL (WT), and GST-BimELS65A (65A) were subjected to an in vitro kinase assay with recombinant JNK1α1. Phosphorylated substrates were separated by SDS-PAGE and immunoblotted with the phospho65-Bim, Bim, or GST antibody. Degradation products of bacterially expressed proteins are marked with an asterisk.
Mass spectrometry. After SDS-PAGE and Coomassie staining, the JNK-phosphorylated BimEL band was cut from the gel and digested with trypsin in-gel. Peptides were separated by nanoscale microcapillary HPLC as described (Stemmann et al., 2001). Eluting peptides were ionized by electrospray ionization and analyzed by an LCQ-DECA ion trap mass spectrometer as described (Stemmann et al., 2001).
Cell culture, transfection, and infection of HEK293T and PC12 cells. HEK293T cells (American Type Culture Collection, Manassas, VA) were grown in DMEM (Invitrogen) supplemented with 10% fetal calf serum (Invitrogen), 2 mm l-glutamine, and 100 μg/ml penicillin-streptomycin. Rat pheochromocytoma PC12rtta cells were maintained at 37°C in 10% CO2 in air in DMEM (Hyclone, Logan, UT) supplemented with 6% horse serum (BioWhittaker, East Rutherford, NJ), 6% bovine calf serum (Bio-Whittaker), 1% glutamine, and 1% penicillin-streptomycin.
Cell lines were plated 18-24 hr before transfection and harvested 24 hr after transfection. HEK293T transfections were performed by a standard calcium phosphate transfection method. PC12rtta cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Forty-eight hours after transfection, PC12rtta cells were infected with 100 multiplicity of infection (MOI) of adenovirus per well unless indicated otherwise.
Western analysis. Proteins from cell extracts were separated by PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were blocked for 1 hr in a Tris-buffered saline solution (TBS) containing 0.02-0.05% Tween (TBST) and 5% (w/v) dried skim milk powder. All primary antibody incubations were performed in TBST supplemented with 5% bovine serum albumin (USB) for 1 hr at room temperature, except for incubations with the phospho-BimS65 antibody, which were done in blocking solution overnight at 4°C. Antibody binding was detected by enhanced chemiluminescence (ECL, PerkinElmer Life Sciences, Boston, MA) using a secondary antibody conjugated to horseradish peroxidase (dilution: 1:20,000).
Survival assays in cerebellar granule neurons. Cerebellar granule cells were prepared as described (D'Mello et al., 1993; Shalizi et al., 2003) and transfected using a calcium phosphate transfection method. For the experiments depicted in Figure 4, cultures from post-natal day 6 that were in culture for 8 d were transfected with Bim wild-type or mutant DNA (0.1 μg per well) or the control vector pCDNA3 (0.1 μg per well), together with a plasmid encoding β-galactosidase (0.25 μg per well) and a carrier plasmid (2 μg per well pECE) in a 24-well plate. The calcium phosphate precipitate was placed on granule neurons that had been starved for 1 hr in DMEM for 20 min, and cultures were subsequently returned to conditioned medium [basal medium Eagle (BME) (Invitrogen) supplemented with 10% calf serum (Hyclone) and 30 mm KCl]. After overnight incubation with conditioned medium, cultures were deprived of conditioned medium and left in BME in the presence or absence of insulin (10 μg/ml) for 8 hr. Cultures were fixed with 4% paraformaldehyde (PFA) in PBS and subjected to indirect immunofluorescence using an antibody to β-galactosidase (dilution, 1:500). Cell survival and death were assessed in β-galactosidase-expressing neurons based on the integrity of neurites and the morphology of the nucleus as determined using the DNA dye bis-Benzimide Hoechst 33258 trihydrochloride (Sigma, St. Louis, MO). Cell counts were done in a blinded manner (n = 100 cells per condition) and analyzed for statistical significance by ANOVA (Fisher's protected least significance difference).
Figure 4.
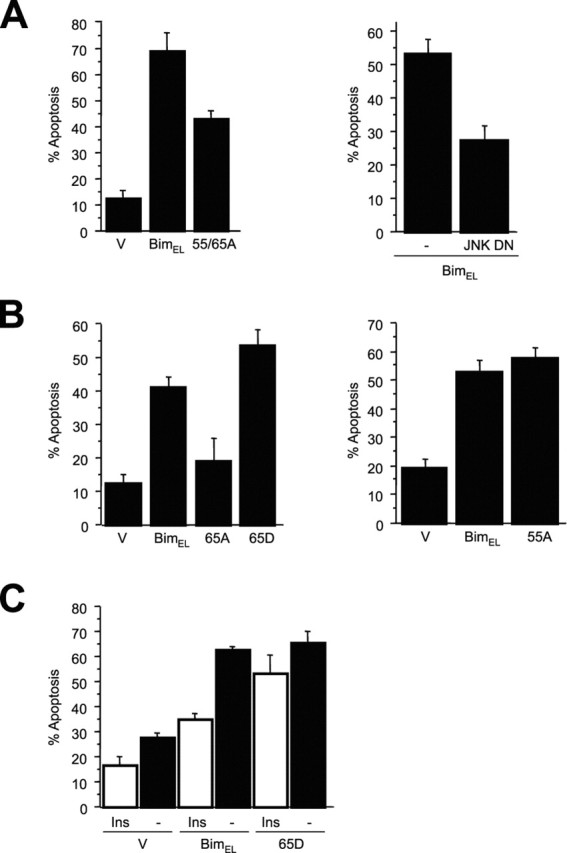
Phosphorylation of BimEL at serine 65 promotes BimEL-mediated apoptosis. A, Cerebellar granule neurons were transfected with control vector (V), BimEL, or BimELS55/65A (55/65A) and β-galactosidase (left panel) or with control vector (V) or JNK DN together with BimEL and β-galactosidase (right panel). One day after transfection, cultures were switched from full medium to basal medium in the absence of survival factors. Cells were fixed after 8 hr and subjected to indirect immunofluorescence. Percentage of apoptosis is represented as mean ± SE. The expression of BimELS55/65A induced significantly less apoptosis than BimEL (n = 3; ANOVA; p < 0.0005). Expression of JNK DN significantly reduced BimEL-induced apoptosis (n = 3; ANOVA; p < 0.005). B, Cerebellar granule neurons were transfected with plasmids encoding nonspliceable cDNAs of BimEL, BimELS55A (55A), BimELS65A (65A), BimELS65D (65D), or control vector (V) and β-galactosidase. Cultures were treated as described above and analyzed by indirect immunofluorescence. The expression of BimELS65A induced significantly less apoptosis than BimEL and BimELS65D (mean ± SE; n = 4; ANOVA; p < 0.01 and p < 0.0005). The expression of BimELS55A did not reduce the apoptotic effect compared with wild-type BimEL (mean ± SE; n = 6; ANOVA; p = 0.4). C, Cerebellar granule neurons were transfected with plasmids encoding nonspliceable cDNAs of wild-type BimEL, BimELS65D (65D), or control vector (V) and β-galactosidase. One day after transfection, cultures were switched from full medium to basal medium in the presence (Ins) or absence (-) of insulin (10 μg/ml) to activate the IGF-I receptor. Cells were fixed after 8 hr and subjected to indirect immunofluorescence. Activation of the IGF-I receptor reduced Bim-mediated apoptosis (mean ± SE; n = 4; ANOVA; p < 0.0001) but had little effect on BimELS65D-induced apoptosis (p > 0.04).
Caspase-3 activation assay in PC12 cells. PC12rtta cells were transfected with the indicated plasmids encoding GFP, pU6/BS control vector, pU6/BS-Bim RNA interference plasmid, pU6/BS-cyclin-dependent kinase 2(Cdk2) RNA interference plasmid, pCDNA3 control vector, Bim wild-type BH3-domain mutant plasmid, or Bim S65A BH3-domain mutant plasmid. Forty-eight hours after transfection, cells were infected with either AdLacZ or Adp75NTR and fixed 24 hr later with 4% PFA in PBS. GFP-positive cells were scored for the presence of cleaved caspase-3 by indirect immunocytochemistry using an antibody against cleaved caspase-3 (dilution, 1:1000). Transfected cells were scored by a blind observer (n = 150 cells per condition), and the composite data were analyzed for statistical significance by ANOVA (Tukey's honestly significant difference).
Results
JNK phosphorylates BimEL at serine 65 in vitro
We first investigated whether JNK might directly regulate the BH3-only protein BimEL. BimEL contains six putative proline-directed sites for JNK-mediated phosphorylation (Fig. 1A). Using an in vitro kinase assay, we found that recombinant JNK robustly catalyzed the phosphorylation of a recombinant GST-BimEL fusion protein (Fig. 1B). To determine which putative phosphorylation sites are targeted by JNK, we subjected recombinant GST-BimEL that was phosphorylated by JNK to tandem mass spectrometry. Tryptic peptides were identified that matched with the entire BimEL sequence. Phosphate-containing peptides were identified by a differential mass of +80 Da relative to the theoretical mass of nonphosphorylated peptides. Analysis of the peptides revealed that JNK phosphorylated BimEL at all six putative phosphorylation sites. As an example, the MS/MS spectrum of a phosphopeptide containing phospho-serine 55 and phospho-serine 65 is shown (Fig. 1C).
Figure 1.
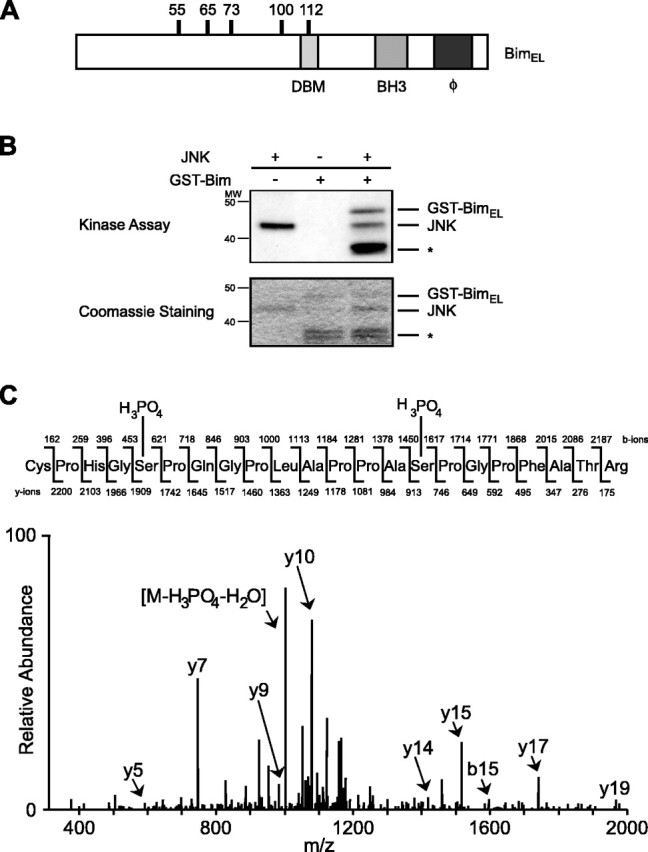
JNK phosphorylates BimEL in vitro. A, The structure of BimEL is shown schematically. The location of the six putative proline-directed phosphorylation sites, the dynein binding motif (DBM), the BH3-only domain, and the hydrophobic domain (ϕ) are indicated. B, Recombinant GST-BimEL was incubated with recombinant JNK1α1 and subjected to an in vitro kinase assay using [γ-32P]ATP. Phosphorylated GST-BimEL was separated by SDS-PAGE, stained with Coomassie blue (bottom panel), and analyzed by autoradiography (top panel). Degradation products of bacterially expressed GST-BimEL are indicated with an asterisk. C, Representative tandem mass spectrum of one of the phosphopeptides (amino acids 51-71) derived from JNK-phosphorylated GST-BimEL. Fragment ions in the spectrum were sequenced from both the N and C termini (b- and y-type ions). Both serine 55 and serine 65 show additional mass from a phosphate residue.
To identify the major sites of JNK-induced phosphorylation among the six sites that were identified by mass spectrometry, we generated BimEL mutants in which the phosphorylation sites at serine 55, serine 65, and threonine 112 were replaced with alanine and subjected the recombinant BimEL mutant proteins to an in vitro kinase reaction. We found that although all three BimEL mutants were phosphorylated by JNK, the phosphorylation was dramatically diminished in the BimELS65A mutant and to a lesser degree in the other mutants (Fig. 2A), suggesting that serine 65 is the predominant site that is phosphorylated by JNK in vitro.
To establish that JNK phosphorylates BimEL at serine 65, we raised a phosphospecific antibody to recognize BimEL only when it is phosphorylated at serine 65 (phospho65-Bim antibody). Recombinant BimEL was phosphorylated in vitro by JNK and then immunoblotted with the phospho65-Bim antibody or an antibody that recognizes Bim regardless of its phosphorylation state. We found that JNK robustly phosphorylated BimEL at serine 65 (Fig. 2B). The phospho65-Bim reactivity was specific, because it failed to recognize the BimELS65A mutant that was incubated with JNK (Fig. 2B). Together, our results indicate that JNK phosphorylates Bim specifically at serine 65 in vitro.
Stress-activated protein kinases phosphorylate Bim at serine 65 in vivo
We next determined whether activation of the JNK signaling pathway induces the phosphorylation of BimEL at serine 65 within cells. We expressed in HEK293T cells either wild-type BimEL or BimEL mutants in which the in vitro sites of JNK-induced phosphorylation were replaced with alanine in the presence or absence of an expression plasmid encoding a truncated and constitutive active form of the kinase MEKK1 (MEKK1Δ). MEKK1 is the prototypical MAPK that activates the JNK signaling pathway. Extracts from transfected HEK293T cells were prepared and subjected to immunoblotting using an anti-Bim antibody.
The expression of activated MEKK1 together with BimEL in HEK293T cells resulted in the retardation of the electrophoretic mobility of BimEL (Fig. 3A). The electrophoretic mobility shift of BimEL reflected BimEL phosphorylation, because the shift completely collapsed after treatment of the cell lysates with alkaline phosphatase (data not shown). Compared with wild-type BimEL, the phosphorylation mutants BimELS55A and BimELS65A displayed reduced electrophoretic mobility shifts, and the shift was completely absent in the BimELS55/65A mutant. In contrast to the S55/65A mutation, mutation of each of the other three in vitro sites of JNK-induced phosphorylation S73, S100, or T112 modestly reduced the electrophoretic mobility shift of BimEL (Fig. 3A). Immunoblotting of lysates of HEK293T cells in which BimEL was expressed together with MEKK1Δ using the phospho65-Bim antibody revealed that activated MEKK1 robustly induced the phosphorylation of BimEL at serine 65 (Fig. 3B). Together, these results indicate that activation of the MEKK1-induced SAPK signaling pathways in vivo induces the phosphorylation of BimEL at serine 65.
Figure 3.
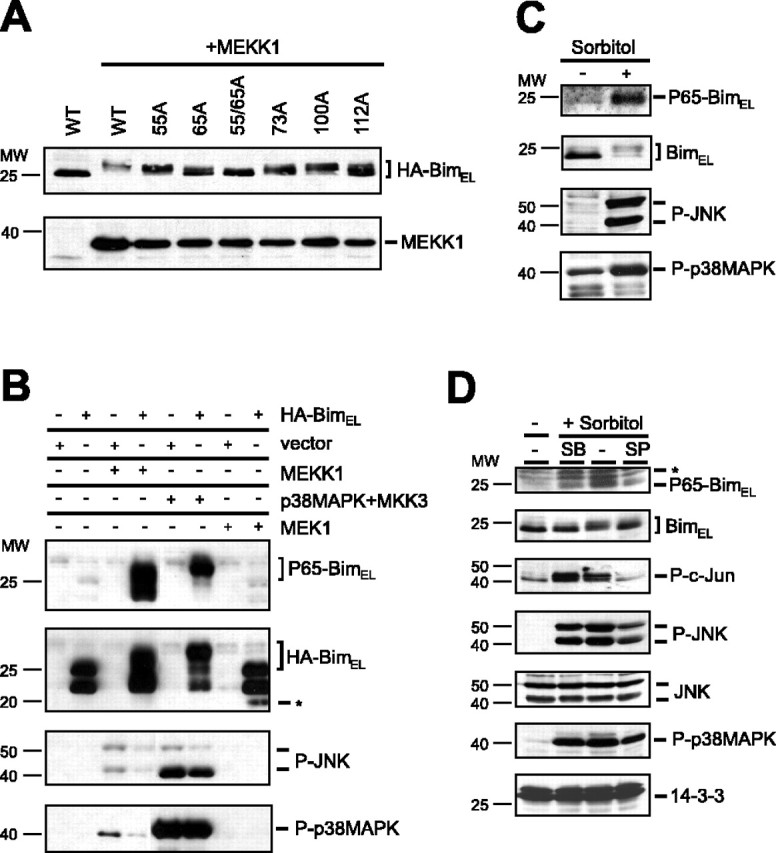
JNK phosphorylates BimEL in vivo. A, HEK293T cells were transfected with 1 μg of wild-type BimEL (WT) or the BimEL phosphorylation mutants BimELS55A (55A), BimELS65A (65A), BimELS73A (73A), BimELS100A (100A), and BimELT112A (112A) alone or together with 0.5 μg of activated MEKK1. The electrophoretic mobility of the BimEL proteins was assessed by SDS-PAGE and immunoblotting with the HA antibody. B, HEK293T cells were transfected with 1 μg of wild-type BimEL or vector control alone or together with 0.5 μg of activated MEKK1, p38MAPK plus MKK3, or activated MEK1. Lysates of transfected cells were immunoblotted with the phospho65-Bim, Bim, phospho-JNK (P-JNK), or phospho-p38MAPK (P-p38MAPK) antibody. Untagged BimEL is indicated with an asterisk. C, HEK293T cells were treated with sorbitol (500 mm) for 3 hr. Lysates of cells were immunoblotted with the phospho65-Bim, Bim, phospho-JNK (P-JNK), or phospho-p38MAPK (P-p38MAPK) antibody. D, HEK293T cells were treated with sorbitol (500 mm) for 3 hr in the presence of vehicle or the p38MAPK and JNK inhibitors SB203580 (5 μm) and SP600125 (10 μm), respectively. Lysates of cells were immunoblotted with the phospho65-Bim, Bim, phospho-c-Jun (P-c-Jun), phospho-JNK (P-JNK), JNK, phospho-p38MAPK (P-p38MAPK), or 14-3-3 antibody. Aspecific bands recognized by the phospho65-Bim antibody are indicated with an asterisk.
MEKK1 has been shown previously to induce protein kinase cascades that culminate in the activation of the stress-activated MAPKs JNK and p38MAPK (Harper and LoGrasso, 2001). In HEK293T cells, activated MEKK1 did induce the phosphorylation of both JNK and p38MAPK at sites that reflect their activation as determined by immunoblotting of HEK293T extracts using antibodies that recognize specifically the phosphorylated forms of JNK and p38MAPK (Fig. 3B). The expression of a constitutive active form of JNK also stimulated the phosphorylation of BimEL at serine 65, although to a lesser degree than MEKK1 (data not shown), suggesting that both JNK and p38MAPK might phosphorylate BimEL downstream of MEKK1. In other experiments, the expression of a constitutive active form of the p38MAPK activator MKK3 together with p38MAPK and BimEL in HEK293T cells strongly induced the phosphorylation of BimEL at serine 65 (Fig. 3B). Interestingly, the expression of MKK3 activated both p38MAPK and JNK in HEK293T cells (Fig. 3B), consistent with the conclusion that both of these kinases might contribute to the phosphorylation of BimEL at serine 65. In agreement with this interpretation, we found that the pharmacological inhibitors SB203580 (5 μm) and SP600125 (10 μm), which inhibit p38MAPK and JNK, respectively, each partially reduced the MEKK1-mediated phosphorylation of BimEL at serine 65 (data not shown).
Recently, BimEL has been suggested to be the target of the extracellular signal-related kinase (ERK) 1/2 signaling pathway (Ley et al., 2003); however, the expression of a constitutive active form of the ERK1/2 activator MEK1 together with BimEL had little effect on the phosphorylation of BimEL at serine 65 (Fig. 3B). Together, these data suggest that among the MAPKs, the SAPKs JNK and p38MAPK may selectively induce the phosphorylation of BimEL at serine 65.
To determine whether activation of the SAPK signaling pathways induces the phosphorylation of endogenous BimEL in vivo, we tested whether exposure of cells to hyperosmotic stress, a stimulus known to activate SAPK signaling pathways, triggers the phosphorylation of BimEL at serine 65. We found that exposure of HEK293T cells to high concentrations of the hyperosmotic stimulus sorbitol induced the phosphorylation of endogenous BimEL at serine 65 (Fig. 3C). Inhibition of JNK and p38MAPK activity by the inhibitors SP600125 and SB203580 partially reduced the sorbitol-mediated phosphorylation of BimEL at serine 65 (Fig. 3D). The activity of the JNK inhibitor was confirmed by the observation that it effectively reduced sorbitol-induced phosphorylation of the transcription factor c-Jun (Fig. 3D). Together, these results suggest that endogenous SAPKs mediate the phosphorylation of endogenous BimEL at serine 65 in vivo after osmotic stress.
JNK-induced phosphorylation of BimEL at serine 65 activates the apoptotic function of BimEL
To determine the biological effect of the JNK-induced phosphorylation of BimEL on the function of Bim, we expressed in primary cerebellar granule neurons BimEL or BimEL mutants in which the JNK sites of phosphorylation were mutated. After transfection, granule neurons were deprived of extracellular survival factors to provide a stimulus that activates the JNK signaling pathway in neurons (Coffey et al., 2002). The expression of BimEL robustly induced apoptosis of survival factor-deprived cerebellar granule neurons (Fig. 4A). We found that a dominant interfering form of JNK (JNK DN), when coexpressed with BimEL, significantly reduced BimEL-induced apoptosis in survival factor-deprived granule neurons, suggesting that the JNK signaling pathway stimulates the apoptotic function of BimEL in these neurons. Because the in vitro (Fig. 2A) and in vivo (Fig. 3A) evidence suggested that serines 55 and 65 might represent key sites of JNK-mediated phosphorylation, we first tested the effect of the double S55/65A mutation on BimEL-induced apoptosis. We found that expression of the BimELS55/65A mutant induced significantly less neuronal apoptosis than expression of BimEL (Fig. 4A), suggesting that JNK-induced phosphorylation of BimEL at these sites might promote BimEL-dependent apoptosis.
We next set out to determine the relative contribution of each of the two phosphorylation sites, serines 55 and 65, in BimEL-mediated neuronal apoptosis. In our initial experiments, the effect of these phosphorylation site mutants on neuronal apoptosis was somewhat variable. Remarkably, the BimEL cDNA contains internal splicing donor and acceptor sites such that the BimEL cDNA may lead to the generation of both BimEL and BimL mRNA (Shinjyo et al., 2001). According to Shinjyo et al. (2001), cells transfected with BimEL cDNA may produce both BimEL and BimL proteins (Shinjyo et al., 2001). BimL, a potent inducer of apoptosis, does not contain the JNK phosphorylation sites serines 55 and 65 and is not subject to the posttranslational regulation at these sites. We therefore reasoned that in the survival assays in granule neurons, the potential expression of BimL encoded by BimEL cDNA might mask the regulatory effect of serines 55 and 65 on BimEL-induced neuronal cell death. Therefore, before further testing of the functional consequences of BimEL phosphorylation, we first addressed the concern that the unusual splicing of BimEL cDNA might interfere with the interpretation of the survival assays. To circumvent the potential expression of BimL encoded by BimEL cDNA, we mutated the splice donor site in the BimEL expression plasmids and used these cDNAs for all subsequent survival assays.
The expression of BimEL encoded by the nonspliceable BimEL cDNA strongly induced apoptosis in survival factor-deprived granule neurons (Fig. 4B,C). Using nonspliceable BimEL cDNAs, we found that the BimEL mutant in which serine 55 was replaced with an alanine (BimELS55A) induced apoptosis in neurons as effectively as BimEL (Fig. 4B). In contrast, the nonspliceable BimEL S65A mutant (BimELS65A) strikingly reduced neuronal apoptosis to levels that were similar to those in control vector-transfected neurons (Fig. 4B). In other experiments we found that replacement of all five potential JNK phosphorylation sites by alanine reduced the apoptotic effect of BimEL but not more effectively than the single S65A mutation (Fig. 4B) (data not shown). Together, our findings suggest that JNK-induced phosphorylation of BimEL at serine 65 promotes the pro-apoptotic activity of BimEL in primary cerebellar granule neurons.
To begin to characterize how the phosphorylation at serine 65 promotes BimEL-dependent apoptosis, we expressed a BimEL mutant in which serine 65 was replaced with aspartate to mimic phosphorylation at this site (BimELS65D). We found that BimELS65D robustly induced apoptosis in survival factor-deprived neurons (Fig. 4B). Under these conditions, the apoptotic effect of BimELS65D was slightly greater than that of BimEL, but the difference did not reach statistical significance. Although BimEL-mediated apoptosis was inhibited by the expression of JNK DN (Fig. 4A), we found that coexpression of JNK DN had little effect on BimELS65D-induced cell death (data not shown). These results are consistent with the interpretation that the phosphorylation of BimEL at serine 65 acts downstream of JNK in promoting neuronal apoptosis.
We next asked whether phosphorylation of BimEL at serine 65 might have an effect on the apoptotic effect of BimEL under survival conditions. We found that activation of the IGF-I receptor and its downstream survival pathways by high doses of insulin (Dudek et al., 1997) significantly reduced BimEL-induced apoptosis (Fig. 4C); however, insulin failed to inhibit the apoptotic effect of BimELS65D (Fig. 4C). These results raise the possibility that the phosphorylation at serine 65 might promote the apoptotic function of BimEL by opposing survival factor inhibition of BimEL-mediated apoptosis.
The JNK-BimEL signaling pathway in p75NTR-induced apoptosis
Having identified the JNK-induced phosphorylation of BimEL at serine 65 as a mechanism for JNK to directly activate the cell death machinery, we next investigated whether the JNK-BimEL signaling pathway is a critical mediator of specific stimuli known to induce apoptosis in the nervous system. Recent studies suggest that JNK triggers neuronal apoptosis after activation of p75NTR via a c-Jun-independent mechanism (Palmada et al., 2002; Bhakar et al., 2003). These observations suggest that p75NTR might directly activate the cell death machinery via the JNK-BimEL signaling pathway.
To characterize the potential role of the JNK-BimEL signaling pathway in p75NTR-mediated apoptosis, we used a well established paradigm of overexpression of full-length p75NTR to trigger p75NTR-dependent cell death (Roux et al., 2001; Wang et al., 2001; Bhakar et al., 2003). We infected PC12 cells with adenovirus encoding full-length p75NTR or control LacZ protein and immunoblotted the PC12 cell extracts with the phospho65-Bim antibody or an anti-Bim antibody that recognizes BimEL regardless of its phosphorylation state. Expression of p75NTR induced the phosphorylation of BimEL at serine 65 (Fig. 5A). To determine whether p75NTR-induced phosphorylation of BimEL at serine 65 is dependent on JNK activation, we coexpressed with p75 the JBD of the scaffold protein JIP that functions as a dominant-negative inhibitor of JNK signaling (Harding et al., 2001). The expression of JBD blocked p75NTR-induction of JNK activity and cell death (Bhakar et al., 2003) (data not shown). We found that JBD expression also blocked the p75NTR-induced phosphorylation of BimEL at serine 65 (Fig. 5A). Together, these results suggest that JNK mediates p75NTR-induced phosphorylation of BimEL at serine 65.
Figure 5.

p75NTR-induced apoptosis activates the JNK-dependent phosphorylation of BimEL and is dependent on BimEL. A, PC12 cells were infected with p75NTR or control adenovirus (LacZ) together with either 5 or 15 MOI of JBD-JIP (J) or control (L) adenovirus. Lysates were immunoblotted with phospho65-Bim or Bim antibody. Aspecific bands recognized by the phospho65-Bim antibody are indicated with an asterisk. B, Lysates of PC12 cells transfected with increasing amounts of control vector (U6), BimEL RNA interference plasmid (Bim RNAi), or control RNAi plasmid (Cdk2 RNAi) were immunoblotted with the Bim (top panel), p42 MAP kinase (ERK1/2; middle panel), or actin antibody (bottom panel). C, PC12rtta cells were transfected with the indicated plasmids and subsequently infected with p75NTR (p75) or control adenovirus (LacZ). Twenty-four hours after infection, cells were fixed and immunostained for cleaved caspase-3. Double asterisk indicates a difference of p < 0.001 between p75NTR-infected, pCDNA3-transfected cells (bar 8) and mock or LacZ-infected cells (bars 1-7), and p75NTR-infected, U6-transfected cells (bar 11) and Bim RNAi-transfected cells (bar 12). Single asterisk indicates a difference of p < 0.05 between p75NTR-infected, pCDNA3-transfected cells (bar 8) and BimELS65A-transfected cells (bar 10), as determined by ANOVA.
To investigate whether BimEL mediates p75NTR-induced apoptosis, we used a DNA template-based method of RNA interference (RNAi) to reduce the levels of endogenous BimEL in PC12 cells. Expression of Bim hairpin RNA but not the control RNAi plasmid or the expression of hairpin RNA to the unrelated protein Cdk2 significantly reduced endogenous BimEL protein levels as determined in transfection experiments in PC12 cells (Fig. 5B). To determine the effect of BimEL knockdown on p75NTR-induced apoptosis, we transfected PC12 cells with GFP alone or GFP together with the distinct RNAi plasmids and then infected these cells with the p75NTR or control adenovirus. We found that the genetic knockdown of BimEL significantly reduced the p75NTR-induced apoptosis of PC12 cells as assayed by measuring the cleavage of caspase-3 (Fig. 5C). Together, these data suggest that BimEL plays a crucial role in p75NTR-induced cell death.
To test directly whether the phosphorylation of BimEL at serine 65 mediates p75NTR-induced cell death, we tested whether a mutant allele of BimEL in which serine 65 was replaced by an alanine acts in a dominant-negative manner to inhibit the p75NTR-mediated apoptotic response. Because BimEL is a potent inducer of cell death, we replaced serine 65 with alanine in the context of a full-length BimEL in which the BH3 domain was also mutated. Overexpression of BimEL harboring two BH3 domain mutations but that was otherwise wild-type, including an intact serine 65, failed to alter the ability of p75NTR to induce cell death (Fig. 5C). In contrast, the expression of BimEL containing both the BH3-domain mutations and the S65A mutation remarkably inhibited p75NTR-induced cell death (Fig. 5C). These results suggest that the nonphosphorylatable BimEL protein acts in a dominant-negative manner to inhibit p75NTR-induced cell death. Together with the BimEL knockdown results, our findings support the conclusion that JNK-induced phosphorylation of BimEL at serine 65 mediates p75NTR-induced cell death.
Discussion
In this study, we have defined a novel mechanism by which activation of JNK by several distinct stimuli, including activated p75NTR, mediates the phosphorylation and activation of the BH3-only protein BimEL. The JNK-induced phosphorylation of BimEL at the distinct site of serine 65 promoted the apoptotic function of BimEL in primary neurons. The genetic knockdown of BimEL by RNA interference or the expression of a dominant interfering form of BimEL in which serine 65 was replaced by an alanine significantly inhibited apoptosis induced by p75NTR overexpression. Together, our findings suggest that JNK-induced phosphorylation and activation of BimEL mediates p75NTR-induced cell death.
Activation of JNK has long been implicated in neuronal apoptosis (Mielke and Herdegen, 2000; Harper and LoGrasso, 2001). JNK is thought to induce neuronal apoptosis via transcription-dependent and -independent mechanisms. JNK-induced phosphorylation and activation of the transcription factor c-Jun that in turn mediates the expression of proapoptotic proteins has been characterized extensively (Ham et al., 2000). Accumulating evidence suggests that in addition to transcription-dependent mechanisms, JNK also directly activates the cell death machinery (Tournier et al., 2000; Lei et al., 2002); however, the connections between JNK and the cell death machinery remain to be elucidated. Our results in this study suggest that JNK phosphorylation of BimEL at serine 65 promotes BimEL-mediated apoptosis. We have shown previously that JNK phosphorylates and thereby activates the BH3-only protein Bad (Donovan et al., 2002). Together, our findings suggest that BH3-only proteins are critical targets of JNK, the phosphorylation of which directly activates apoptotic machinery.
The mechanism by which JNK-mediated phosphorylation leads to the activation of BimEL remains to be characterized. In non-neuronal cells, Bim has been shown to be bound to dynein light chain (DLC1) and thereby sequestered to the cytoskeleton (Puthalakath et al., 1999). Apoptotic stimuli thus are believed to cause the disruption of the Bim-DLC1 complex, thereby allowing Bim to function at the mitochondria. UV-activated JNK catalyzes the phosphorylation of exogenous BimL at threonine 56, which lies within the DLC1-binding motif, thereby releasing Bim from DLC1 (Lei and Davis, 2003); however, a role for DLC1 in the regulation of BimEL function in neuronal cells remains unclear. The primary site of BimEL localization in neurons appears to be the mitochondria, and the JNK-induced phosphorylation of BimEL has not been shown to alter BimEL localization (Putcha et al., 2003) (E. B. E. Becker and A. Bonni, unpublished data). Together, these data suggest that JNK activates BimEL in neuronal cells by a mechanism other than the release of BimEL from the cytoskeleton. Current investigations are underway to characterize the mechanism by which the phosphorylation of BimEL at serine 65 promotes the apoptotic function of BimEL.
In our studies of regulation of Bad, we found that JNK-induced phosphorylation of Bad at serine 128 opposes Akt-inhibition of Bad-mediated apoptosis (Donovan et al., 2002; Konishi et al., 2002). In an analogous manner, our results obtained in survival assays performed in primary neurons support the hypothesis that JNK-induced phosphorylation of BimEL at serine 65 promotes the apoptotic function of BimEL in neurons in part by opposing Akt-promoted cell survival. Insulin, a survival signal that robustly induces Akt (Dudek et al., 1997), blocked wild-type BimEL but not BimELS65D-mediated cell death in granule neurons (Fig. 4B), suggesting that JNK-induced phosphorylation of BimEL at serine 65 counteracts Akt-mediated neuronal survival. The mechanisms underlying the antagonism of Akt and the JNK-BimEL signaling pathway remain to be characterized. Akt has been reported to inhibit the activation of JNK in neurons by interacting with JIP1 (Kim et al., 2002), raising the possibility that Akt might inhibit JNK-induced phosphorylation of BimEL at serine 65. An alternative interpretation of our results is that the serine 65 phosphorylation might counteract the ability of an insulin-induced survival signal to directly inhibit BimEL-induced apoptosis.
Although we focus in this study on the phosphorylation of BimEL by JNK, the p38MAPK signaling pathway might also play a role in phosphorylating BimEL at serine 65. Similar to JNK, p38MAPK has been reported to be involved in a number of neuronal cell death paradigms, including growth factor withdrawal and exposure to glutamate (Mielke and Herdegen, 2000; Harper and LoGrasso, 2001). It will be interesting to determine whether the JNK and p38MAPK signaling pathways have overlapping functions in phosphorylating BimEL at serine 65 under the same apoptotic conditions or whether distinct stimuli activate these signaling pathways to differentially induce the phosphorylation of BimEL.
One cell death trigger that is specifically associated with the activation of JNK is activation of the neurotrophin receptor p75NTR (Harrington et al., 2002). In contrast to other extrinsic cell-death receptor pathways, p75NTR activation does not induce caspase-8 and subsequent cleavage of Bid, but p75NTR has been suggested to directly activate the cell-intrinsic apoptotic machinery at the mitochondria (Roux and Barker, 2002). In this study, we show that overexpression of p75NTR induces the JNK-mediated phosphorylation of BimEL at serine 65 and thereby triggers apoptosis. We have found recently that the p75NTR-JNK pathway also stimulates the phosphorylation of Bad at serine 128 and thus activates Bad-mediated apoptosis (Bhakar et al., 2003). Therefore, we propose that one mechanism by which activation of p75NTR leads to apoptosis is the JNK-mediated direct phosphorylation of BH3-only proteins. Consistent with our results, Putcha et al. (2003) found that BimEL is phosphorylated downstream of the JNK signaling pathway in neurons; however, whereas Putcha et al. (2003) focused on the phosphorylation of BimEL after trophic factor withdrawal, we here provide evidence that BimEL is phosphorylated after activation of p75NTR, thereby linking this cell-death receptor pathway directly to the cell-intrinsic apoptotic machinery.
p75NTR has been implicated in several neuropathological conditions, including focal ischemia, stroke, axotomy, and neurodegenerative disorders (Dechant and Barde, 2002). It will be interesting to determine whether the JNK-induced phosphorylation of BimEL at serine 65 contributes to neuronal cell loss under these neuropathological conditions.
Footnotes
This work was supported by an American Federation for Aging Research (AFAR)/Pfizer grant (A.B.) and by National Institutes of Health Grant R01-NS41021-01 (A.B.). A.B. is the recipient of a career development award from the Burroughs Wellcome Fund, a fellowship from the Alfred P. Sloan Foundation, a Robert H. Ebert Clinical Scholar Award from the Esther A. and Joseph Klingenstein Fund, and a Sidney Kimmel Foundation Award. E.B.E.B. is supported by the Boehringer Ingelheim Fonds and an Albert J. Ryan Foundation award. We thank R. Tomaino and S. Gygi for analysis of JNK-phosphorylated BimEL by mass spectrometry, S. Vasquez for technical assistance, and A. Shalizi for critical reading of this manuscript.
Correspondence should be addressed to Azad Bonni, Department of Pathology, Harvard Medical School, 77 Avenue Louis Pasteur, Boston, MA 02115. E-mail: azad_bonni@hms.harvard.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248762-09$15.00/0
References
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Howell JL, Paul CE, Salehi AH, Becker EB, Said F, Bonni A, Barker PA (2003) Apoptosis induced by p75NTR overexpression requires Jun kinase-dependent phosphorylation of BAD. J Neurosci 23: 11373-11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, Courtney MJ (2002) c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J Neurosci 22: 4335-4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant G, Barde YA (2002) The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 5: 1131-1136. [DOI] [PubMed] [Google Scholar]
- D'Mello SR, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 90: 10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan N, Becker EB, Konishi Y, Bonni A (2002) JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J Biol Chem 277: 40944-40949. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665. [DOI] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166-168. [DOI] [PubMed] [Google Scholar]
- Friedman WJ (2000) Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci 20: 6340-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A (2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277: 46442-46446. [DOI] [PubMed] [Google Scholar]
- Gross A, McDonnell JM, Korsmeyer SJ (1999) BCL-2 family members and the mitochondria in apoptosis. Genes Dev 13: 1899-1911. [DOI] [PubMed] [Google Scholar]
- Ham J, Eilers A, Whitfield J, Neame SJ, Shah B (2000) c-Jun and the transcriptional control of neuronal apoptosis. Biochem Pharmacol 60: 1015-1021. [DOI] [PubMed] [Google Scholar]
- Harding TC, Xue L, Bienemann A, Haywood D, Dickens M, Tolkovsky AM, Uney JB (2001) Inhibition of JNK by overexpression of the JNL binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem 276: 4531-4534. [DOI] [PubMed] [Google Scholar]
- Harper SJ, LoGrasso P (2001) Signaling for survival and death in neurones: the role of stress-activated kinases, JNK and p38. Cell Signal 13: 299-310. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO (2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22: 156-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CA, Johnson EM Jr (2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276: 37754-37760. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A (2000) BH3-only proteins-essential initiators of apoptotic cell death. Cell 103: 839-842. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Hengartner MO (2001) Programmed cell death: alive and well in the new millennium. Trends Cell Biol 11: 526-534. [DOI] [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV (2002) Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 35: 697-709. [DOI] [PubMed] [Google Scholar]
- Konishi Y, Lehtinen M, Donovan N, Bonni A (2002) Cdc2 phosphorylation of BAD links the cell cycle to the cell death machinery. Mol Cell 9: 1005-1016. [DOI] [PubMed] [Google Scholar]
- Lee FS, Hagler J, Chen ZJ, Maniatis T (1997) Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell 88: 213-222. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, BarSagi D, Davis RJ (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol Cell Biol 22: 4929-4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ (2003) Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem 278: 18811-18816. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T (2000) JNK and p38 stress kinases-degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol 61: 45-60. [DOI] [PubMed] [Google Scholar]
- O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML, Huang DC, Strasser A (2000) The proapoptotic BH3-only protein BIM is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol 157: 449-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, Carter BD (2002) c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol 158: 453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615-628. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM (2003) JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 38: 899-914. [DOI] [PubMed] [Google Scholar]
- Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A (1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 3: 287-296. [DOI] [PubMed] [Google Scholar]
- Roux PP, Barker PA (2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67: 203-233. [DOI] [PubMed] [Google Scholar]
- Roux PP, Colicos MA, Barker PA, Kennedy TE (1999) p75 neurotrophin receptor expression is induced in apoptotic neurons after seizure. J Neurosci 19: 6887-6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Bhakar AL, Kennedy TE, Barker PA (2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 276: 23097-23104. [DOI] [PubMed] [Google Scholar]
- Roux PP, Dorval G, Boudreau M, Angers-Loustau A, Morris SJ, Makkerh J, Barker PA (2002) K252a and CEP1347 are neuroprotective compounds that inhibit mixed-lineage kinase-3 and induce activation of Akt and ERK. J Biol Chem 277: 49473-49480. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A (2003) Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci 23: 7326-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinjyo T, Kuribara R, Inukai T, Hosoi H, Kinoshita T, Miyajima A, Houghton PJ, Look AT, Ozawa K, Inaba T (2001) Downregulation of Bim, a proapoptotic relative of Bcl-2, is a pivotal step in cytokine-initiated survival signaling in murine hematopoietic progenitors. Mol Cell Biol 21: 854-864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmann O, Zou H, Gerber SA, Gygi SP, Kirschner MW (2001) Dual inhibition of sister chromatid separation at metaphase. Cell 107: 715-726. [DOI] [PubMed] [Google Scholar]
- Strasser A, O'Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69: 217-245. [DOI] [PubMed] [Google Scholar]
- Sui G, Soohoo C, Affar el B, Gay F, Shi Y, Forrester WC (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 99: 5515-5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870-874. [DOI] [PubMed] [Google Scholar]
- Troy CM, Friedman JE, Friedman WJ (2002) Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J Biol Chem 277: 34295-34302. [DOI] [PubMed] [Google Scholar]
- Wang X, Bauer JH, Li Y, Shao Z, Zetoune FS, Cattaneo E, Vincenz C (2001) Characterization of a p75(NTR) apoptotic signaling pathway using a novel cellular model. J Biol Chem 276: 33812-33820. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629-643. [DOI] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA (1997) Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J Clin Invest 100: 2333-2340. [DOI] [PMC free article] [PubMed] [Google Scholar]