

Abstract
We examined the role of programmed cell death (PCD) pathways in retinal degeneration caused by a mutation in the norpA gene. norpA degeneration shows morphological hallmarks of programmed cell death, specifically cytoplasmic condensation and engulfment of the dying photoreceptor cells by neighboring retinal pigment cells. However, genetic mosaic analysis of adult photoreceptors lacking rpr, hid, and grim show that these PCD inducers are not required for norpA degeneration. We showed previously that ectopic expression of either rpr or hid triggers rapid PCD in adult photoreceptors, and this is completely suppressed by the coexpression of the baculoviral P35 caspase inhibitor. In contrast, expression of P35 does not suppress norpA retinal degeneration, although a small delay in the rate of degeneration is observed in low light-low temperature conditions. P35 does not alter the morphological characteristics of norpA cell death. Overexpression of the Drosophila inhibitor of apoptosis Diap1 or a dominant-negative form of the Dronc caspase, even when coexpressed with P35, does not dramatically alter the time course of norpA degeneration. These results establish that the pathways responsible for PCD in development do not play a major role in adult retinal degeneration caused by norpA.
Keywords: retinal degeneration, rpr, hid, diap1, Dronc, baculovirus P35, norpA, programmed cell death, Drosophila vision
Introduction
Programmed cell death (PCD) occurs as a normal process in the development and homeostasis in both vertebrates and invertebrates (Song and Steller, 1999). All cells appear capable of executing PCD, and the decision is controlled by the interplay of PCD activators and suppressors. In Drosophila melanogaster, PCD activator genes include reaper (rpr), head involution defective (hid), and grim. These genes play a role in developmental PCD decisions and also PCD triggered by chromosomal damage (White et al., 1994; Grether et al., 1995; Chen et al., 1996b). Rpr- and Hid-induced PCD is blocked by caspase inhibitors, such as the baculoviral P35 gene product (Grether et al., 1995; White et al., 1996), and Diap1, a Drosophila inhibitor of apoptosis (Hay et al., 1995). The dominant-negative mutant of the Dronc caspase, DroncDN, also known as DroncC318S, also suppresses Rpr- and Hid-induced cell death (Meier et al., 2000).
In addition to its role during development, PCD is implicated in the death process of differentiated cells. In the case of retinal diseases, mutations in a large number of different photoreceptor genes trigger retinal degeneration in both vertebrates (Clarke et al., 2000) and Drosophila (Ranganathan et al., 1995). Experimental results from several retinal disease models show that degeneration occurs by PCD processes (Chang et al., 1993; Lolley et al., 1994; Portera-Cailliau et al., 1994; Smith et al., 1995; Davidson and Steller, 1998), and there is evidence that multiple cell death pathways are involved in light-triggered retinal degeneration (Hao et al., 2002). The potential use of anti-apoptotic therapeutic agents, such as P35 and Bcl-2, to slow or eliminate the PCD process has been validated in some investigations (Chen et al., 1996a; Davidson and Steller, 1998) but not in others (Joseph and Li, 1996). It is clear that additional information is needed on the molecular mechanisms underlying PCD processes in diseased photoreceptors to allow further evaluation of these therapeutic strategies.
Drosophila norpA encodes the photoreceptor-specific phospholipase C that is activated by light and essential for the photoresponse (Bloomquist et al., 1988). norpA mutants show rapid light-induced retinal degeneration (Stark and Sapp, 1989; Zinkl et al., 1990). In the work reported here, we performed mosaic analysis to determine whether the H99 region, containing the rpr, hid, and grim genes, was required for norpA cell death. We also overexpressed the baculoviral P35 protein Diap1 and DroncDN in adult Drosophila photoreceptors to determine whether they could alter the degeneration processes seen in norpA mutants. Our results indicate that Rpr, Hid, Grim, Diap1, and Dronc activities are not required for norpA-induced cell death. Under certain conditions, P35 and DroncDN delay the retinal degeneration process in norpA mutants but cannot prevent cell death. These results show that norpA triggers retinal degeneration through cell death processes that are significantly different than those responsible for developmental PCD.
Materials and Methods
Drosophila strains. All transgenic strains used in the study were described previously: P[ninaEhid] (Hsu et al., 2002), P[GMRp35] and P[GMRhid] (Hay et al., 1994), P[GMRdiap1] (Hay et al., 1995), and P[GMRDroncDN] (Hawkins et al., 2000). We verified that the diap1 and DroncDN transgenes were active by showing they partially protect from GMRhid-driven cell death using the rescue of the small eye phenotype as described previously (Hay et al., 1995; Hawkins et al., 2000). However, unlike P35 (Hsu et al., 2002), they were not effective in the rescue of ninaEhid (data not shown).
Creation of Df(H99) mosaics. Genetic mosaics were created to analyze the effect of homozygous Df(H99) on norpAP24 photoreceptor cells. The mosaics were generated using the flippase recombinase/flippase recombinase target (FLP/FRT) system using the w+ FRT 80B chromosome designated 80-w+ (Xu and Rubin, 1993) and eyFLP (Newsome et al., 2000). Standard genetic crosses were used to generate the Df(3L)H99 FRT 80B chromosome. To facilitate construction of this chromosome, the P{lacW}Mi-2j3D4 w+ P element present within the 76C salivary chromosome region was recombined onto the FRT 80B chromosome. Because Df(H99) is located in the 75C salivary chromosome region, loss of this w+ P element marker identified likely Df(3L)H99 FRT 80B recombinant chromosomes from Df(3L)H99/w+ FRT 80B females. A chromosome identified in this manner was checked for lethality in the Df(3L)H99 FRT 80B/df(3L)H99 genotype. The presence of Df(3L)H99 on this chromosome was confirmed by our observation that 25% of the embryos from Df(3L)H99 FRT 80B/TM2 parents lack acridine orange staining, because homozygous Df(3L)H99 FRT embryos lack nearly all embryonic cell death and hence do not stain (White et al., 1994). To generate norpA/Y; df(H99) FRT 80B/w+ FRT 80B (mosaic) males, subsequently referred to as norpA; ΔH99 mosaics, w norpA eyFLP; w+ FRT 80B/TM3 females were crossed to w/Y; df(3L)H99 FRT 80B/TM2 males. The norpA+/norpA; ΔH99 mosaic female progeny from this cross provided the norpA+; ΔH99 mosaic controls.
Analysis of Drosophila mutant retina. Retinal degeneration was triggered by the amorphic mutant allele norpAP24 (Pearn et al., 1996) in most experiments. ninaED1 (Kurada and O'Tousa, 1995) and rdgB1 (Vihtelic et al., 1991) were used in experiments shown in Figure 5. Relevant genotypes were generated by standard genetic crosses; typically, a cross scheme was designed such that the progeny contained both experimental and control animals to minimize genetic background and environmental rearing differences. Unless specified in the text, flies were raised at room temperature (22°C) in a 12 hr light/dark cycle with a light intensity of ∼2900 lux. The deep pseudopupil (DPP) was visualized in red-eyed flies as a bright trapezoid structure when a bright white light illuminates the retina from the back of the head (Franceschini, 1975). During degeneration, the DPP becomes increasingly diffused before being completely lost. The DPP was scored as negative as soon as it was judged to be more diffused than would ever be observed in a wild-type retina. In all DPP experiments, the displayed data were compiled from three to five independent experimental trials that contained 30-50 flies of each genotype.
Figure 5.
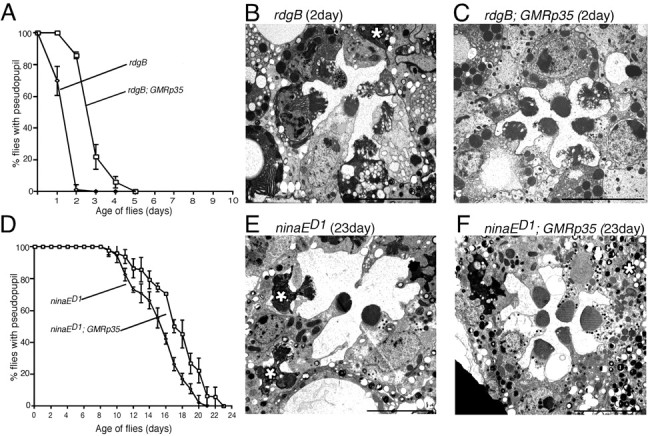
The influence of P35 in the control of photoreceptor cell death in other retinal degeneration mutants. Flies were reared under constant light at 22°C. A, Retinal degeneration assessed by deep pseudopupil analysis in rdgB and rdgB; P35. B, C, Electron micrographs of 2-d-old rdgB (B) and rdgB; P35 (C) photoreceptors. The degenerated photoreceptor (marked by * in B) appears in the process of being phagocytosed. D, Retinal degeneration assessed by deep pseudopupil analysis in ninaED1 and ninaED1; P35. E, F, Electron micrographs of 23-d-old ninaED1 retina (E) and ninaED1; P35 (F). The degenerated photoreceptors (marked by * in E) are being phogocytosed. Scale bars, 5 μm.
Microscopic analysis. Eyes were prepared for light and electron microscopy using procedures described by Washburn and O'Tousa (1992). Light microscopy sections ∼1 μm thick were stained with equal parts 1% methylene blue in 1% borax and 1% azure II. Electron microscopy sections were ∼80-100 nm thick, stained first in 5% uranyl acetate in 50% EtOH and then in Reynold's lead citrate. The micrographs shown in all figures are taken from ommatidia cross-sectioned at the depth of the R1-R6 photoreceptor nuclei to present similar views of the various genotypes.
GMR promoter expression analysis. Standard genetic crosses created wild-type, norpA, rdgB, and ninaED1 flies expressing green fluorescent protein (GFP) (P{UAS-EGFP}34 strain; Bloomington Stock Center, Bloomington, IN) under the control of the glass multimer receptor (GMR) promoter (GMR-GAL4) (Ellis et al., 1993). Flies were collected within 1 d of eclosion, before degeneration, and at later time points, to determine whether GFP expression from GMR was altered in any of the mutant backgrounds. Two heads of each genotype were homogenized in 30 μl of lysis buffer, and 15 μl was used for SDS-PAGE. The gel was blotted overnight at 30 V, blocked in 5% nonfat dry milk in TTBS (TBS and Tween 20) for 30 min, and incubated in mouse anti-GFP antibody (Clontech, Palo Alto, CA) diluted 1:1000 in TTBS. Secondary antibody (anti-mouse HRP; Pierce, Rockford, IL) was diluted 1:10,000 in TTBS, and chemiluminescent detection was performed with Pierce Supersignal reagents.
Results
norpA photoreceptors show morphological features of programmed cell death
In cross-sectioned wild-type cells (Fig. 1A), the photoreceptor rhabdomere (R) appears as an organized microvillar structure extending into the intraommatidial space. Pigment cells located on the basal side of the photoreceptor opposite the rhabdomere surround and insulate the photoreceptors of a single ommatidium. The pigment cells, even in white-eyed flies lacking the large pigment granules, are distinguished from photoreceptors by the homogeneous and granular appearance of their cytoplasm. The rhabdomeres of 1-d-old norpA photoreceptors are at early stages of degeneration (Fig. 1B), appearing smaller and less organized. The photoreceptor cell bodies are reduced in size, and pigment cells swell to occupy this space. At later stages of degeneration (Fig. 1C,D), rhabdomeres are further deteriorated, and cell bodies are shrunken with many aberrant vesicles and electron-dense material. Similar morphological changes are seen in adult photoreceptors destined to die because of either the Hid or Rpr proteins (Hsu et al., 2002).
Figure 1.
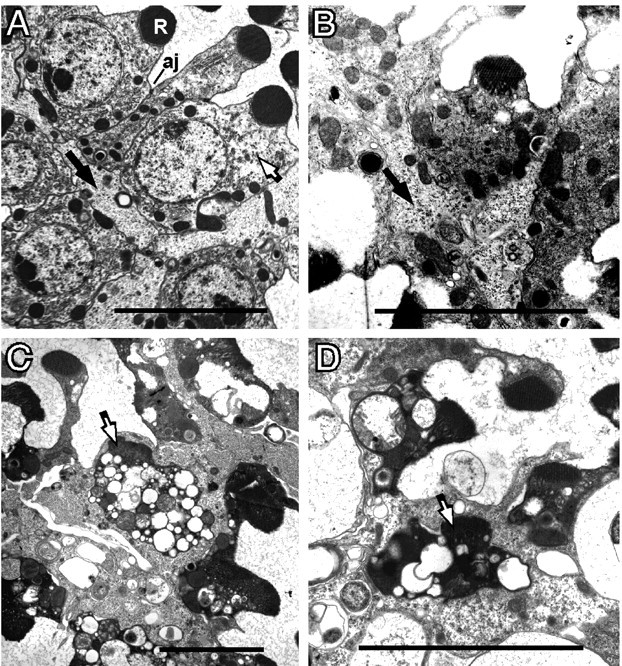
Morphology of degenerating norpA cells. Flies were reared at room temperature under constant light. A, The norpA+ retina shows the normal position of a photoreceptor (white arrow). The photoreceptor contains a well organized rhabdomere (R) and has adherens junctions (aj) connecting neighboring photoreceptor cells. The pigment cells (black arrows) possess a homogeneous granular cytoplasm and are located on the basal side of the photoreceptor cell opposite the rhabdomere. B, The 1-d-old norpA photoreceptors, at initial stages of degeneration, possess smaller, less organized, rhabdomeres and condensed cytoplasm. The pigment cell (black arrow) has expanded and begins to surround the photoreceptor as it loses contact with neighboring phototoreceptors. C, D, At 2 d, many photoreceptors (white arrows) are completely engulfed by an enlarged pigment cell. The photoreceptors become filled with vesicles and vacuoles, and the rhabdomeres are further disintegrated. Scale bars, ∼5 μm.
Figure 1, C and D, documents the engulfment of the dying norpA photoreceptors. The photoreceptor in the middle of each micrograph has lost contact with neighboring cells. The pigment cell is the phagocytic cell responsible for engulfing the photoreceptor. Additional evidence that the pigment cell is the engulfing cell is gained from similar temporal studies using w+ norpA (red-eyed) flies. In these studies, the engulfing cell always contains numerous large pigment granules found only in pigment cells (data not shown).
norpA cells degenerate in absence of Rpr, Hid, and Grim activity
The rpr, hid, and grim genes are clustered in a small chromosome region referred to as the H99 region (White et al., 1994). A homozygous deletion of the H99 region, referred to here as ΔH99, prevents PCD that normally occurs during embryonic development (White et al., 1994). To examine the requirement of this region in adult photoreceptor PCD, mitotic recombination was used to make homozygous Df(H99) mutant cells, hereafter called ΔH99 cells, in the retina. In our experimental design, these cells lacked the w+ gene and therefore could be identified by the absence of pigment granules. Light and electron microscopic analysis showed that most ΔH99 cells looked similar to wild-type cells at 4 d of age in norpA+ flies (Fig. 2A,C). A few ΔH99 cells showed a different morphology (arrow) but were easily discernable from degenerating norpA photoreceptors.
Figure 2.
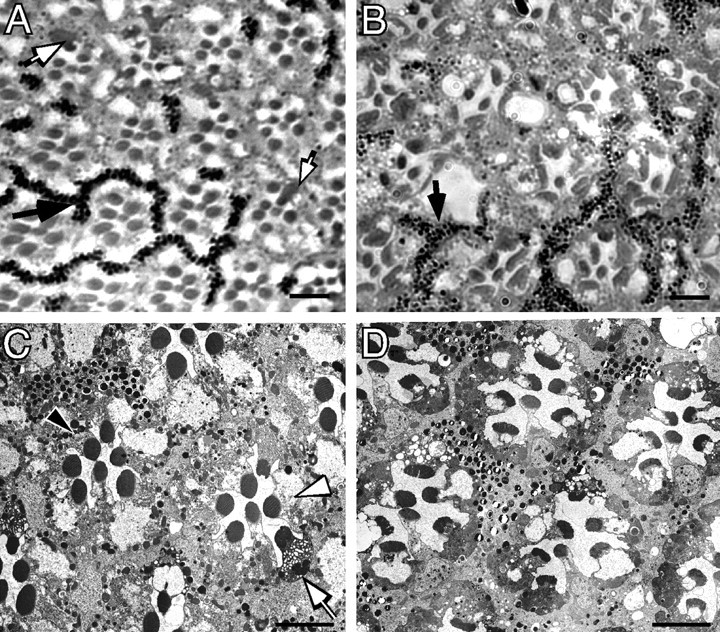
The effect of ΔH99 [homozygous Df(H99) deletion] on wild-type and norpA photoreceptor degeneration. Light (A, B) and electron micrographs (C, D) of 3-d-old ΔH99 mosaic norpA+ and norpA mosaic ommatidia. The areas of the eye containing photoreceptors and surrounding pigment cells with pigment granules contain ΔH99+ cells, the area of the eye lacking pigment granules contain ΔH99 photoreceptor cells. A, norpA+ mosaics at 3 d. Ommatidia appear normal in both wild-type (pigmented) and ΔH99 (nonpigmented) regions of the mosaic norpA+ genotype. One region of pigment granules is identified by a black arrow. Several ΔH99 photoreceptors show increased cytoplasmic staining; two of these are identified by white arrows. B, norpA mosaics at 3 d. In the norpA mutant, a region of the retina with both ΔH99+ and ΔH99 photoreceptors show that all cells are indistinguishable from each other and are undergoing degeneration. One region of pigment granules is identified by a black arrow. The dispersion of the pigment granules (compare with A) is a result of the degeneration process. C, norpA+ mosaics at 3 d. The ΔH99+ cells, containing pigment granules (1 cell is marked by a black arrowhead) appear wild type, and the nonpigmented ΔH99 cells (1 cell is marked with a white arrowhead) also are wild type. One ΔH99 photoreceptor with cytoplasmic vacuoles (white arrow) identifies one of the abnormally stained cells noted in A. D, norpA mosaics at 3 d. The field contains an approximately equal number of norpA; ΔH99+ and norpA; ΔH99 photoreceptor cells, as assessed by the distribution of pigmented-nonpigmented pigment cells. Individual photoreceptors cannot be unambiguously assigned as H99+ or ΔH99 because both types of cells are undergoing degeneration. Scale bars, ∼5 μm.
The viability of homozygous H99 photoreceptor cells allowed us to test for a role of genes within the H99 region in norpA degeneration. Figure 2, B and D, shows that ΔH99 cells (no pigment) undergo a similar time course of degeneration as the neighboring pigmented H99+ cells. We observed no difference in the timing and morphological correlates of norpA degeneration in H99+ and ΔH99 photoreceptors. Thus, unlike developmental PCD processes, the genes of the H99 region do not play a role in the cellular processes responsible for norpA degeneration.
Limited P35 suppression of norpA degeneration
We examined the ability of the baculoviral P35 protein to alter the degeneration process in norpA mutants by introducing GMRp35 into norpA flies (Fig. 3). To accentuate differences in rates of degeneration, flies were reared on a 12 hr light/12 hr dark cycle. As a result, the time course of degeneration in the control norpA flies is notably slower than in the experiment described in Figure 1. Figure 3A shows data collected from observation of the DPP structure in this experiment. We confirmed that GMRp35 was capable of suppressing photoreceptor degeneration triggered by hid expression under control of the ninaE rhodopsin promoter (Hsu et al., 2002). In contrast, the DPP was retained for only ∼1 d longer in norpA with P35 than seen in norpA flies lacking P35. These results show that P35, although active in suppressing Hid-induced degeneration, allows only limited protection from the loss of the DPP attributable to the norpA mutation.
Figure 3.
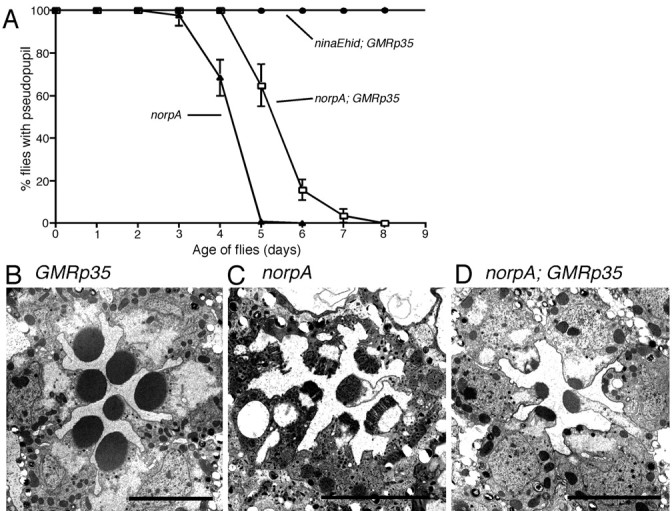
The influence of P35 on photoreceptor cell death in the norpA mutants. Flies were reared in a 12 hr light/dark cycle at 22°C. A, Rate of retinal degeneration assessed by DPP analysis of norpA and norpA; GMRp35. GMRp35 delayed the loss of the DPP in norpA by ∼1 d. The same GMRp35 transgene completely suppressed ninaEhid degeneration. B, Electron micrograph of 6-d-old GMRp35 retina without norpA from this experiment. Photoreceptors retain wild-type morphology. C, Electron micrograph of 6-d-old norpA retina without GMRp35 from this experiment. Photoreceptors show frayed rhabdomeres and are full of vesicles and electron-dense material. D, Electron micrograph of 6-d-old norpA; GMRp35 retina from this experiment. The photoreceptors show signs of retinal degeneration, notably the loss of rhabdomeric volume. There is less degeneration than in norpA lacking p35 (C), consistent with the DPP results. Scale bars, ∼5 μm.
Electron microscopy was used to examine the photoreceptor morphology of these flies at ages for which the DPP study suggested differences. Figure 3B-D shows electron micrographs from 6-d-old retina of GMRp35 alone (Fig. 3B), norpA alone (Fig. 3C), and norpA; GMRp35 (Fig. 3D). Expression of GMRp35 has no effect on retinal structure in norpA+ flies. norpA flies show an improved retinal structure attributable to GMRp35 expression (Fig. 3, compare C,D). These cells lack the frayed rhabdomere structure seen in the norpA flies, and the cytoplasm shows a near normal appearance, lacking electron-dense material and accumulation of vesicles, at these time points. On the other hand, the norpA; GMRp35 cells are clearly in the process of degeneration, notably showing significant loss of rhabdomeric material. Thus, the images are in agreement with DPP results showing that P35 expression has a modest effect on slowing, but not preventing, degeneration in the norpA mutant.
To confirm that P35 provided limited protection, we investigated the ability of P35 to suppress norpA under a variety of reduced light and temperature conditions expected to attenuate the rate of norpA degeneration. In all cases, we documented the small but reproducible capability of P35 to slow the rate of degeneration. Figure 4A shows the result of an experiment in which flies were raised at 18°C and under constant illumination of 2700 lux. In this experiment, P35 extended the DPP+ period in norpA by ∼5 d. Figure 4C-E shows the photoreceptor morphology of flies from this experiment. At the 13 d time point, the norpA photoreceptors are more degenerated than the norpA; P35 photoreceptors. However, by 20 d (Fig. 4E), the norpA; P35 flies show the same morphological changes, including cytoplasmic condensation and the engulfment of dying photoreceptor cells, seen in norpA flies at 13 d. We repeated this experiment using lower light conditions with similar results. These studies show that experimental conditions that better distinguish the protective effects of P35 are mild conditions that also dramatically slow the norpA degeneration rate. In no case, however, was P35 able to completely protect against norpA degeneration.
Figure 4.
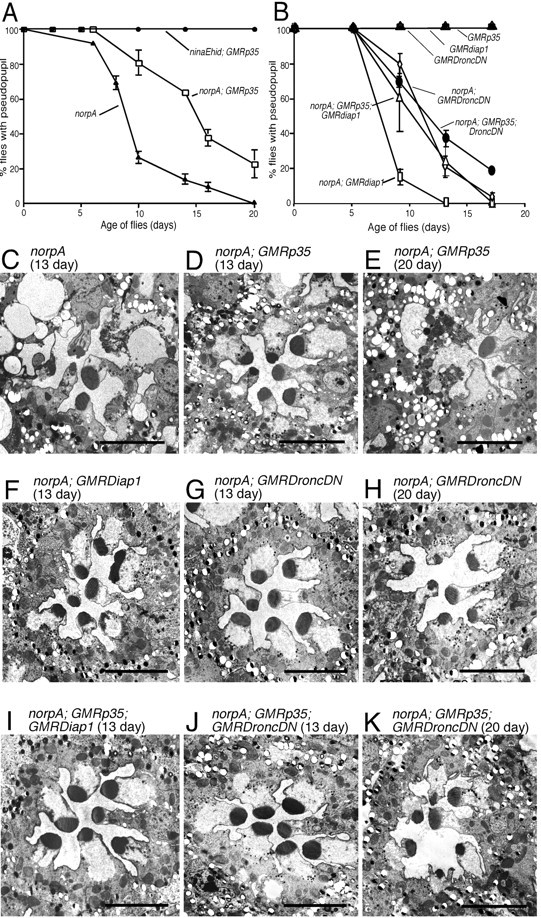
P35, Dronc, and Diap suppression of norpA in a low temperature environment. All flies were reared at 18°C under constant light of 2700 lux. A, Rate of retinal degeneration assessed by DPP analysis of norpA and norpA; GMRp35. GMRp35 delayed the loss of the DPP in norpA by ∼5 d. Under these conditions, and all others tested, the same GMRp35 transgene provided complete suppression of ninaEhid-triggered degeneration. B, Rate of norpA retinal degeneration assessed by DPP analysis in the presence of DroncDN, DroncDN, and P35, Diap1, and Diap1 and P35 cell death suppressors. Neither DroncDN nor Diap1 enhanced P35 suppression. C-E, Electron micrographs of photoreceptors from norpA and norpA; GMRp35 flies at 13 d (C, D) and norpA; GMRp35 flies at 20 d (E) from the same experiment shown in A. Consistent with the DPP analysis and previous experiments, GMRp35 provides some suppression at the earlier time point, with more extensive degeneration evident by 20 d. F-H, Electron micrographs of photoreceptors from norpA; GMRDroncDN and norpA; GMRdiap1 flies at 13 d (F, G) and norpA; GMRDroncDN flies at 20 d (H) from the same experiment shown in B. Consistent with the DPP analysis, initial signs of degeneration are seen at 13 d, with more extensive degeneration evident at 20 d (data not shown for GMRdiap1). I-K, Electron micrographs of norpA photoreceptors expressing P35 with either DroncDN or Diap1 at 13 d (I, J) and P35; DroncDN at 20 d. Consistent with the DPP analysis, no improvement in retinal structure is provided by simultaneous expression of two different caspase inhibitors. Scale bars, ∼5 μm.
Because of the limited protection of norpA by P35, two genes that have been implicated in P35-insensitive cell death pathways, diap1 and DroncDN (Meier et al., 2000; Igaki et al., 2002), were analyzed for effects on the rates of norpA degeneration. We first showed that, like P35, expression of diap1 or DroncDN does not cause retinal degeneration (Fig. 4B). GMR-driven expression of diap1 did not alter the norpA degeneration process. GMR-driven expression of DroncDN provided weak protection similar to that observed for P35. The pseudopupil analysis was confirmed by electron microscopic observations of these genotypes (Fig. 4F-H) in which 13-d-old photoreceptors show reduced rhabdomeres that further deteriorate by 20 d of age. Given the possibility that Dronc represents a P35-insensitive caspase (Meier et al., 2000), we also examined whether the combination of P35 and DroncDN, or alternatively P35 and Diap1, could suppress degeneration. These results, shown in Figure 4B, provided no evidence of synergistic suppression. The electron microscopic images of photoreceptors from these genotypes are consistent with this conclusion (Fig. 4I-K).
Limited P35 suppression of other retinal degeneration mutants
Other researchers have reported significant suppression of the rdgC and ninaE mutants by expression of P35 (Davidson and Steller, 1998; Alloway et al., 2000). The failure to document suppression of norpA by P35 caused us to reevaluate the effect of P35 on other retinal degeneration mutants. Figure 5A-C shows the effects of P35 expression on rdgB5 reared under constant light at 22°C. As with norpA, although we observed a subtle delay in the rate of degeneration, P35 was not able to prevent the degenerative process. Electron microscopic examination at an intermediate time point suggests that P35 may influence the rate of cellular condensation and other morphological attributes of the degeneration process (Fig. 5B,C). We performed similar experiments with ninaED1, a dominant rhodopsin mutant causing late onset of retinal degeneration (Kurada and O'Tousa, 1995). Again, P35 had a modest effect on the rate of degeneration but could not prevent degeneration (Fig. 5D-F). Similar results were also observed for the ninaERH27 and for rdgC1 (data not shown).
The GMR promoter is active in the adult eye and mutant photoreceptors
One explanation for the inability of GMRP35, GMRdiap1, and GMRDroncDN to suppress degeneration is that genes controlled by the GMR promoter are not expressed in adult photoreceptors or in norpA, rdgB, and ninaED1 mutant backgrounds. To study GMR promoter activity, the GFP content of wild-type and mutant norpA, rdgB, or ninaED1 flies expressing GFP under GMR control were analyzed by protein blot. Figure 6 shows that the GMR promoter is active in adult wild-type flies (lanes 1-3). In addition, before degeneration, mutant norpA, rdgB, and ninaED1 retinas have high GMR promoter activity (lanes 4-6). Therefore, lack of expression in adult and mutant photoreceptors is not the reason for ineffective suppression by P35, Diap1, and DroncDN.
Figure 6.

Western blot analysis of GFP driven by the GMR promoter in adult and mutant photoreceptors. Flies were raised in 12 hr light/dark cycle at 22°C. Flies carrying both GMR-Gal4 and UAS-GFP were crossed into norpA, rdgB, and ninaED1/+ mutant backgrounds. The protein equivalent of one fly head is loaded into each lane for GFP detection (top row); the same blot was also probed for actin (bottom row) to examine loading differences. The GFP protein is detected in wild-type 1-, 3-, and 5-d-old heads (lanes 1-3). Similar levels of GFP protein are detected in 1-d-old norpA (lane 4), rdgB (lane 5), and ninaED1 (lane 6) heads.
Discussion
norpA encodes the phospholipase C responsible for phototransduction and is representative of a large number of photoreceptor-specific genes active in phototransduction that, when mutated, trigger retinal degeneration (O'Tousa, 1997). Because of the lack of a receptor potential in norpA mutants, there is no rise in intracellular Ca2+ levels (Peretz et al., 1994). The failure to activate Ca2+-dependent enzymes required for rhodopsin recycling causes excessive endocytosis of rhodopsin (Orem and Dolph, 2002), which is thought to initiate the cell death process (Alloway et al., 2000; Kiselev et al., 2000). We show here, in confirmation of a previous report (Alloway et al., 2000), that norpA degeneration is accompanied by morphological changes consistent with PCD processes, such as loss of cell contact, cytoplasmic condensation, and engulfment of the dying cells (Arends and Wyllie, 1991). The neighboring retinal pigment cells perform the phagocytosis of the dying photoreceptor cells. The primary goal of this study was to determine the role of known PCD pathways in the retinal degeneration processes triggered by the norpA mutant. We present a series of results establishing that characterized developmental PCD components are not required for norpA retinal degeneration. Our results suggest that novel cell death processes, not those characterized from developmental PCD studies, play the major role in retinal degeneration syndromes.
We first showed that rpr, hid, and grim gene activity are not required for retinal degeneration triggered by norpA mutants. Our experimental design created a homozygous deficiency for the H99 region containing the rpr, hid, and grim genes in the norpA adult eye. This homozygous deficiency in the embryo eliminates nearly all developmental cell death (White et al., 1994). Thus, the underlying events responsible for norpA cell death cannot be identical to those responsible for developmental PCD. sickle, a fourth gene in the 75C region, maps just outside the H99 region (Christich et al., 2002; Srinivasula et al., 2002; Wing et al., 2002). In the adult Drosophila eye, sickle expression is not able to induce cell death on its own, but it can enhance the cell death triggered by an rpr or an rpr/grim chimera (Wing et al., 2002), and its action is antagonized by P35 and DIAP1. It is proposed to act by antagonizing the activity of IAPs, inhibitors of apoptosis proteins (Christich et al., 2002; Srinivasula et al., 2002). Because sickle is not removed by the H99 deletion, its role in the norpA cell death process was not addressed in the mosaic experiment. However, sickle is postulated to signal through IAPs to caspases. The P35 and DIAP1 studies described below suggest that this pathway does not play a major role in the norpA cell death process.
We then examined the ability of the anti-apoptotic baculoviral protein P35 to suppress norpA retinal degeneration. P35 is a broad-spectrum caspase inhibitor and hence blocks PCD subsequent to the action of Rpr, Hid, and Grim. P35 has a modest impact on the rate of degeneration in norpA. Electron microscopy showed that, at intermediate stages of degeneration, there are two notable effects of P35 on the norpA PCD process. First, although the rhabdomere volume still decreases dramatically in the presence of P35, most rhabdomeres do not show the severe unraveled structure seen in norpA in the absence of P35. Second, progression of the cell bodies to an electron-dense and particulate appearance takes longer in the presence of P35. The P35 cells become electron dense at later time points and are engulfed by the neighboring pigment cells. These results support the view that P35-sensitive caspase pathways are active in some processes of dying photoreceptors but are not pivotal in the cell death decision.
Our conclusion that the major cell death determinants in retinal degeneration mutants are not sensitive to P35 contrasts with two previous reports. Davidson and Steller (1998) reported that P35 is a strong suppressor of rdgC and ninaERH27 degeneration. In another report, Alloway et al. (2000) showed evidence of P35 suppression of the norpAEE5 mutant. The differences in our results cannot be explained by the use of different alleles or mutations, because we also observed no or limited rescue of rdgC1, ninaED1, ninaERH27, or rdgB5. In an effort to reconcile these results, we showed that reduced light and temperature conditions can accentuate morphological differences between the mutant with and without P35, giving the appearance of strong suppression at certain time points. However, we found no experimental situations for which P35 shows complete protection from degeneration.
In this and all previous reports examining the effect of P35 on retinal degeneration mutants, P35 was expressed under control of the GMR promoter. We showed that GMR does maintain high levels of gene expression throughout adult life in wild-type and mutant eyes through Western blot analysis using a GFP reporter construct. In addition, P35 expressed from the GMR and the major rhodopsin promoter, which is active throughout adult life, show similar suppression of Rpr- and Hid-triggered photoreceptor degeneration (Hsu et al., 2002). In these experiments, the rhodopsin promoter drove Rpr or Hid expression exclusively in photoreceptors after retinal cell type specification and continued to be expressed throughout the adult life.
We then sought evidence that P35-insensitive PCD processes are involved in norpA degeneration. The Dronc caspase is identified as a P35-insensitive caspase in one study (Meier et al., 2000) but as P35 sensitive from other results (Dorstyn et al., 1999; Colussi et al., 2000). Dronc is sensitive to the inhibitory effects of Diap1 (Meier et al., 2000). Diap1 is reported to inhibit a Dronc complex responsible for both caspase-dependent and caspase-independent cell death processes (Igaki et al., 2002). So, given the possibility that Dronc or Diap1 would affect P35-insensitive PCD processes, we examined their effect on norpA degeneration. We found that neither overexpression of a dominant-negative Dronc gene nor the diap1 gene in the adult eye prevented norpA degeneration. Furthermore, simultaneous expression of P35 and DroncDN, or P35 and Diap1, did not further suppress the rate of degeneration.
The failure to prevent norpA degeneration by known components of developmental PCD implies that other cellular events play the major role in the norpA degeneration process. It is not known whether these events constitute a separate cell death signaling pathway and whether similar events occur in other mutant-triggered retinal degenerations. Genetic approaches were successful in characterizing the components of developmental PCD, so we anticipate that identification and analysis of retinal degeneration suppressors will help answer these questions.
Footnotes
This work was supported by National Institutes of Health Grant EYO6808. We thank Drs. B. Hay and P. Kurada for supplying Drosophila stocks and Jonathan Piccini and Frank McDonald for technical assistance.
Correspondence should be addressed to Joseph E. O'Tousa, Department of Biological Sciences, University of Notre Dame, Notre Dame, IN 46556. E-mail: jotousa@nd.edu
C.-D. Hsu's present address: Department of Ophthalmology and Visual Science, Washington University School of Medicine, 660 South Euclid, Box 8096, St. Louis, MO 63110.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240500-08$15.00/0
C.-D.H and M.A.W. contributed equally to this work.
References
- Alloway PG, Howard L, Dolph PJ (2000) The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron 28: 129-138. [DOI] [PubMed] [Google Scholar]
- Arends MJ, Wyllie AH (1991) Apoptosis: mechanisms and roles in pathology. Int Rev Exp Pathol 32: 223-254. [DOI] [PubMed] [Google Scholar]
- Bloomquist BT, Shortridge RD, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak WL (1988) Isolation of a putative phospholipase C gene of Drosophila, norpA, and its role in phototransduction. Cell 54: 723-733. [DOI] [PubMed] [Google Scholar]
- Chang GQ, Hao Y, Wong F (1993) Apoptosis: final common pathway of photoreceptor death in rd, rds, and rhodopsin mutant mice. Neuron 11: 595-605. [DOI] [PubMed] [Google Scholar]
- Chen J, Flannery JG, LaVail MM, Steinberg RH, Xu J, Simon MI (1996a) bcl-2 overexpression reduces apoptotic photoreceptor cell death in three different retinal degenerations. Proc Natl Acad Sci USA 93: 7042-7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Nordstrom W, Gish B, Abrams JM (1996b) grim, a novel cell death gene in Drosophila. Genes Dev 10: 1773-1782. [DOI] [PubMed] [Google Scholar]
- Christich A, Kauppila S, Chen P, Sogame N, Ho SI, Abrams JM (2002) The damage-responsive Drosophila gene sickle encodes a novel IAP binding protein similar to but distinct from reaper, grim, and hid. Curr Biol 12: 137-140. [DOI] [PubMed] [Google Scholar]
- Clarke G, Heon E, McInnes RR (2000) Recent advances in the molecular basis of inherited photoreceptor degeneration. Clin Genet 57: 313-329. [DOI] [PubMed] [Google Scholar]
- Colussi PA, Quinn LM, Huang DC, Coombe M, Read SH, Richardson H, Kumar S (2000) Debcl, a proapoptotic Bcl-2 homologue, is a component of the Drosophila melanogaster cell death machinery. J Cell Biol 148: 703-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson FF, Steller H (1998) Blocking apoptosis prevents blindness in Drosophila retinal degeneration mutants. Nature 391: 587-591. [DOI] [PubMed] [Google Scholar]
- Dorstyn L, Colussi PA, Quinn LM, Richardson H, Kumar S (1999) DRONC, an ecdysone-inducible Drosophila caspase. Proc Natl Acad Sci USA 96: 4307-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MC, O'Neill EM, Rubin GM (1993) Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development 119: 855-865. [DOI] [PubMed] [Google Scholar]
- Franceschini N (1975) Sampling of the visual environment by the compound eye of the fly: fundamentals and applications. In: Photoreceptor optics (Snyder A, Menzel R, eds), pp 98-125. New York: Springer.
- Grether ME, Abrams JM, Agapite J, White K, Steller H (1995) The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev 9: 1694-1708. [DOI] [PubMed] [Google Scholar]
- Hao W, Wenzel A, Obin MS, Chen CK, Brill E, Krasnoperova NV, Eversole-Cire P, Kleyner Y, Taylor A, Simon MI, Grimm C, Reme CE, Lem J (2002) Evidence for two apoptotic pathways in light-induced retinal degeneration. Nat Genet 32: 254-260. [DOI] [PubMed] [Google Scholar]
- Hawkins CJ, Yoo SJ, Peterson EP, Wang SL, Vernooy SY, Hay BA (2000) The Drosophila caspase DRONC cleaves following glutamate or aspartate and is regulated by DIAP1, HID, and GRIM. J Biol Chem 275: 27084-27093. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM (1994) Expression of baculovirus P35 prevents cell death in Drosophila. Development 120: 2121-2129. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wassarman DA, Rubin GM (1995) Drosophila. homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83: 1253-1262. [DOI] [PubMed] [Google Scholar]
- Hsu CD, Adams SM, O'Tousa JE (2002) Rpr- and hid-driven cell death in Drosophila photoreceptors. Vision Res 42: 507-516. [DOI] [PubMed] [Google Scholar]
- Igaki T, Yamamoto-Goto Y, Tokushige N, Kanda H, Miura M (2002) Down-regulation of DIAP1 triggers a novel Drosophila cell death pathway mediated by Dark and DRONC. J Biol Chem 277: 23103-23106. [DOI] [PubMed] [Google Scholar]
- Joseph RM, Li T (1996) Overexpression of Bcl-2 or Bcl-XL transgenes and photoreceptor degeneration. Invest Ophthalmol Vis Sci 37: 2434-2446. [PubMed] [Google Scholar]
- Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R (2000) A molecular pathway for light-dependent photorecptor apoptosis in Drosophila. Neuron 28: 139-152. [DOI] [PubMed] [Google Scholar]
- Kurada P, O'Tousa JE (1995) Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron 14: 571-579. [DOI] [PubMed] [Google Scholar]
- Lolley RN, Rong H, Craft CM (1994) Linkage of photoreceptor degeneration by apoptosis with inherited defect in phototransduction. Invest Ophthalmol Vis Sci 35: 358-362. [PubMed] [Google Scholar]
- Meier P, Silke J, Leevers SJ, Evan GI (2000) The Drosophila caspase DRONC is regulated by DIAP1. EMBO J 19: 598-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsome TP, Asling B, Dickson BJ (2000) Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127: 851-860. [DOI] [PubMed] [Google Scholar]
- O'Tousa JE (1997) Normal physiology and retinal degeneration in the Drosophila visual system. In: Progress in retinal and eye research (Osborne NN, Chader GJ, eds), pp 691-703. New York: Pergamon.
- Orem NR, Dolph PJ (2002) Loss of the phospholipase C gene product induces massive endocytosis of rhodopsin and arrestin in Drosophila photoreceptors. Vision Res 42: 497-505. [DOI] [PubMed] [Google Scholar]
- Pearn MT, Randall LL, Shortridge RD, Burg MG, Pak WL (1996) Molecular, biochemical, and electrophysiological characterization of Drosophila norpA mutants. J Biol Chem 271: 4937-4945. [DOI] [PubMed] [Google Scholar]
- Peretz A, Suss-Toby E, Rom-Glas A, Arnon A, Payne R, Minke B (1994) The light response of Drosophila photoreceptors is accompanied by an increase in cellular calcium: effects of specific mutations. Neuron 12: 1257-1267. [DOI] [PubMed] [Google Scholar]
- Portera-Cailliau C, Sung CH, Nathans J, Adler R (1994) Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci USA 91: 974-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan R, Malicki DM, Zuker CS (1995) Signal transduction in Drosophila photoreceptors. Annu Rev Neurosci 18: 283-317. [DOI] [PubMed] [Google Scholar]
- Smith SB, Bora N, McCool D, Kutty G, Wong P, Kutty RK, Wiggert B (1995) Photoreceptor cells in the vitiligo mouse die by apoptosis. TRPM-2/clusterin expression is increased in the neural retina and in the retinal pigment epithelium. Invest Ophthalmol Vis Sci 36: 2193-2201. [PubMed] [Google Scholar]
- Song Z, Steller H (1999) Death by design: mechanism and control of apoptosis. Trends Cell Biol 9: M49-M52. [PubMed] [Google Scholar]
- Srinivasula SM, Datta P, Kobayashi M, Wu JW, Fujioka M, Hegde R, Zhang Z, Mukattash R, Fernandes-Alnemri T, Shi Y, Jaynes JB, Alnemri ES (2002) sickle, a novel Drosophila death gene in the reaper/hid/grim region, encodes an IAP-inhibitory protein. Curr Biol 12: 125-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark WS, Sapp R (1989) Retinal degeneration and photoreceptor maintenance in Drosophila: rdgB and its interaction with other mutants. Prog Clin Biol Res 314: 467-489. [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR, O'Tousa JE (1991) Isolation and characterization of the Drosophila retinal degeneration B (rdgB) gene. Genetics 127: 761-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn T, O'Tousa JE (1992) Nonsense suppression of the Drosophila rhodopsin gene. Genetics 130: 585-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H (1994) Genetic control of programmed cell death in Drosophila. Science 264: 677-683. [DOI] [PubMed] [Google Scholar]
- White K, Tahaoglu E, Steller H (1996) Cell killing by the Drosophila gene reaper. Science 271: 805-807. [DOI] [PubMed] [Google Scholar]
- Wing JP, Karres JS, Ogdahl JL, Zhou L, Schwartz LM, Nambu JR (2002) Drosophila sickle is a novel grim-reaper cell death activator. Curr Biol 12: 131-135. [DOI] [PubMed] [Google Scholar]
- Xu T, Rubin GM (1993) Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117: 1223-1237. [DOI] [PubMed] [Google Scholar]
- Zinkl GM, Maier L, Studer K, Sapp R, Chen DM, Stark WS (1990) Microphotometric, ultrastructural, and electrophysiological analyses of light-dependent processes on visual receptors in white-eyed wild-type and norpA (no receptor potential) mutant Drosophila. Vis Neurosci 5: 429-439. [DOI] [PubMed] [Google Scholar]