

Abstract
β-Amyloid (βA) peptide is strongly implicated in the neurodegeneration underlying Alzheimer's disease, but the mechanisms of neurotoxicity remain controversial. This study establishes a central role for oxidative stress by the activation of NADPH oxidase in astrocytes as the cause of βA-induced neuronal death. βA causes a loss of mitochondrial potential in astrocytes but not in neurons. The mitochondrial response consists of Ca2+-dependent transient depolarizations superimposed on a slow collapse of potential. The slow response is both prevented by antioxidants and, remarkably, reversed by provision of glutamate and other mitochondrial substrates to complexes I and II. These findings suggest that the depolarization reflects oxidative damage to metabolic pathways upstream of mitochondrial respiration. Inhibition of NADPH oxidase by diphenylene iodonium or 4-hydroxy-3-methoxy-acetophenone blocks βA-induced reactive oxygen species generation, prevents the mitochondrial depolarization, prevents βA-induced glutathione depletion in both neurons and astrocytes, and protects neurons from cell death, placing the astrocyte NADPH oxidase as a primary target of βA-induced neurodegeneration.
Keywords: Alzheimer, astrocyte, astroglia, calcium, mitochondria, NADPH, neuron
Introduction
The deposition of β-amyloid (βA) plays a central role in the pathogenesis of Alzheimer's disease (AD). Accumulation of βA in neuritic plaques is the defining feature for diagnosis of the disease, and the amyloid load correlates well with the degree of cognitive impairment (Naslund et al., 2000). The 1-40 and 1-42 amino acid forms of βA are neurotoxic, a property conserved in the shorter peptide form 25-35, whereas the reverse peptide, 35-25, is innocuous. We have found recently in neurons and astrocytes grown in coculture, in cultures of cortical astrocytes, or in organotypic hippocampal slices that βA causes a delayed rise in intracellular free calcium ([Ca2+]c) in astrocytes but not in neurons (Abramov et al., 2003). These data suggested that βA forms a pore in astrocyte (but not neuronal) membranes that permits Ca2+ influx from the extracellular space. The combined effect of high [Ca2+]c with oxidative stress may damage mitochondrial function and has been implicated as a pathophysiological mechanism in many systems (Duchen, 2000). Therefore, we have now investigated the connection between changes in [Ca2+]c and changes in mitochondrial function in βA neurotoxicity.
Mitochondrial degeneration is one of the earliest signs of Alzheimer pathology, appearing before neurofibrillary tangles are evident (Hirai et al., 2001). It is also clearly associated with the overexpression of the amyloid precursor protein (APP) (Askanas et al., 1996) or expression of the APP751 form in cultured cells (Grant et al., 1999). Recently, it has also been shown that APP may also be targeted to mitochondria, causing mitochondrial dysfunction (Anandatheerthavarada et al., 2003), in addition to established localization in the plasma membrane and endoplasmic reticulum. The mechanism of the mitochondrial involvement in βA-induced neurotoxicity, however, remains unclear. As well as being a target of oxidative damage, mitochondria may be a source of endogenous production of reactive oxygen species (ROS), and βA may increase mitochondrial ROS production (Sheehan et al., 1997), causing further impairment of mitochondrial function (Arias et al., 2002). Enzyme systems containing iron-sulfur centers, including several enzyme complexes of the respiratory chain, α-ketoglutarate dehydrogenase, and aconitase, are particularly vulnerable to damage by both βA and ROS (Blass and Gibson, 1991; Casley et al., 2002b; Longo et al., 2000). βA also has multiple direct effects on isolated mitochondria, causing alterations in enzyme activity, damage to the respiratory chain, and opening of the mitochondrial permeability transition pore (Canevari et al., 1999; Parks et al., 2001; Shevtzova et al., 2001; Kim et al., 2002; Moreira et al., 2002). The latter may trigger cell death, either promoting cytochrome c release and apoptosis or causing energetic failure and necrosis. Furthermore, βA-induced apoptosis requires an intact respiratory chain, because βA did not cause apoptosis in cells lacking mitochondrial DNA (Morais Cardoso et al., 2002). Even in intact cortical neurons, βA causes mitochondrial dysfunction, reducing ATP levels (Casley et al., 2002a), mostly through inhibition of complex I, causing both mitochondrial depolarization and a loss of mitochondrial mass (Casley et al., 2002b). Therefore, we have now investigated the nature and mechanism of βA-induced mitochondrial dysfunction.
Materials and Methods
Cell culture. Mixed cultures of hippocampal neurons and glial cells were prepared as described previously (Abramov et al., 2003) from Sprague Dawley rat pups 2-4 d postpartum [University College London (UCL) breeding colony]. Hippocampi were removed into ice-cold Gey's salt solution (Invitrogen, Paisley, UK) with 20 μg/ml gentamycin. The tissue was minced and trypsinized (0.1% for 15 min at 37°C), triturated, plated on poly-d-lysine-coated coverslips, and cultured in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and 2 mm l-glutamine. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, fed twice a week, and maintained for a minimum of 10 d before experimental use to ensure the expression of glutamate and other receptors. Neurons were easily distinguishable from glia: they appeared phase bright, had smooth rounded somata and distinct processes, and lay just above the focal plane of the glial layer. Cells were used at 10-20 d in vitro (DIV) unless stated otherwise.
Isolated cortical astrocytes were prepared as described previously (Abramov et al., 2003). Cerebra taken from adult Sprague Dawley rats (UCL breeding colony), were chopped and triturated until homogeneous, passed through a 297 μm mesh, and trypsinized (50,000 U/ml porcine pancreas; Sigma, Gillingham, UK) with 336 U/ml DNase 1 (bovine pancreas; Sigma), and 1.033 U/ml collagenase (Sigma, Gillingham, UK) at 37°C for 15 min. After addition of fetal bovine serum (10% of final volume) and filtering through 140 μm mesh, the tissue was centrifuged through 0.4 m sucrose (400 gm, 10 min), and the resulting pellet was transferred to Minimal Essential Medium supplemented with 5% fetal calf serum, 2 mm glutamine, and 1 mm maleate in tissue culture flasks precoated with 0.01% poly-d-lysine. The cells reached confluency at 12-14 DIV and were harvested and reseeded onto 24-mm-diameter glass coverslips (BDH, Poole, UK) precoated with 0.01% poly-d-lysine for fluorescence measurements and used during 2-4 d. Purity of cell type was confirmed by immunohistochemistry using antibodies directed to glial acidic fibrillary protein (GFAP). In excess of 99% of the cells examined stained positively for GFAP.
In all experiments we have used both hippocampal neurons and astrocytes in cocultures and preparations of purified astrocytes from hippocampus or cortex. The former was used routinely as a “physiological” model in which we could ensure the selective effects of agents on neurons or astrocytes. The astrocyte cultures were used because the predominant effects seen were in astrocytes. We need to be sure that these effects were not secondary to some action on neurons that was not being measured, and also we could exclude contributions to the response from other cell types, especially microglia, because we have ascertained that the microglial contamination of the astrocyte cultures is extremely small. In this paper, we have referred to data from the different preparations interchangeably because we have seen no difference in astrocytes responses from any of the cultures systems that we have used.
Peptides and treatments. βA 25-35, βA 1-42, and βA 35-25 (Bachem, St. Helens, UK) were dissolved at 1-5 mm in sterile ultrapure water (Milli-Q standard; Millipore, Watford, UK) and kept frozen until use. The peptides were added to cells during experimental recordings, except for the neurotoxicity measurements, in which they were added 24 hr before the assays of cell death (see below). βA 25-35 and 35-25 were used at concentrations of up to 50 μm to ensure that it was present in molar excess compared with inhibitors and so would exclude any direct interaction, and βA 1-42 was used at concentrations of 1-5 μm. The monoclonal antibody CD36 (10 μg/ml; clone FA6-152; Immunotech, Marseille, France) was incubated with cells 30 min before experiments at 37°C and was also added at the time of experiments. The 25-35 peptide is easy to use and relatively inexpensive, but the 1-42 peptide probably represents a more physiologically appropriate model, and so all experiments have been repeated with this peptide. All experiments were also repeated using the 35-25 reverse peptide as a control. We have never seen any significant response to this treatment.
Imaging [Ca2+]c and Δψm and ROS generation. Cells were loaded for 30 min at room temperature with 5 μm fura-2 AM (Molecular Probes, Eugene, OR) and 0.005% pluronic acid in a HEPES-buffered salt solution (HBSS) composed of (in mm): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.35 with NaOH. For simultaneous measurement of [Ca2+]i and Δψm, rhodamine (Rh) 123 (10 μm; Molecular Probes) was added into the cultures during the last 15 min of the fura-2 loading period. For measurements of rates of ROS generation, cells were incubated with 20 μm 2′,7′-dichlorodihydrofluorescein diacetate (Molecular Probes) in HBSS for 40 min at room temperature.
Fluorescence measurements were obtained on an epifluorescence inverted microscope equipped with a 20× fluorite objective. [Ca2+]i and Δψm were monitored in single cells using excitation light provided by a Xenon arc lamp with the beam passing sequentially through 10 nm bandpass filters centered at 340, 380, and 490 nm housed in a computer-controlled filter wheel (Cairn Research, Kent, UK). Emitted fluorescence light was reflected through a 515 nm long-pass filter to a frame transfer cooled CCD camera (Orca ER; Hamamatsu, Welwyn Garden City, UK). All imaging data were collected and analyzed using Kinetic Imaging software (Kinetic Imaging, Wirral, UK). The fluorescence data were acquired at intervals of 10-15 sec. The fura-2 data have not been calibrated in terms of [Ca2+]i because of the uncertainty arising from the use of different calibration techniques. Accumulation of Rh123 in polarized mitochondria quenches the fluorescent signal; in response, emission is dequenched. An increase in Rh123 signal therefore signals mitochondrial depolarization (Duchen and Biscoe, 1992). We have presented these data normalized between 0, representing the resting Rh123 fluorescence, and 100, representing the maximal increase in Rh123 fluorescence in response to complete mitochondrial depolarization by 1 μm carbonyl cyanide 4-(trifluoromethoxy)phenylhydazone (FCCP), which was done routinely at the end of every experiment (indicated by a label Rh123 fluorescence, scaled). In some experiments, cells showed a loss of Rh123 and so showed no, or only small, responses to FCCP. In such traces, this normalization was not possible, and so the data are shown normalized only to a baseline set at 100% (indicated as Rh123 fluorescence, percentage). All presented data were obtained from at least five coverslips and two to three different cell preparations.
In all experiments in the present paper, we have used Rh123 in “dequench: mode. We have also conducted equivalent experiments using the dye tetramethyl rhodamine methyl ester (TMRM) in “redistribution mode,” in which the dye was present continuously at 20 nm and allowed to equilibrate. Mitochondrial depolarization is then seen as the movement of dye from mitochondria into the cytosol. Responses to βA were seen showing a slow and progressive decline in mitochondrially localized dye on which were superimposed more dramatic and reversible decreases in mitochondrial-specific signal, but high-resolution imaging of mitochondria over this long period of time was confounded by movement of mitochondria and swelling of cells associated with the calcium transients. We therefore have limited the data presented to the Rh123 data set, although the TMRM experiments show qualitatively the same phenomenology.
Cells loaded with 2′,7′-dichlorodihydrofluorescein (DCF) were illuminated at 490 nm at low light levels to avoid auto-oxidation. Changes in the rate of rise of the signal were interpreted as changes in rates of ROS generation and could be fitted with a regression coefficient of >0.98 by a linear regression line (Origin; Microcal Software Inc., Northampton, MA).
Toxicity experiments. For toxicity assays cells were loaded simultaneously with 20 μm propidium iodide, which is excluded from viable cells but exhibits a red fluorescence after a loss of membrane integrity, and 4.5 μm Hoechst 33342 (Molecular Probes), which gives a blue staining to chromatin, to count the total number of cells. With use of phase-contrast optics, a bright-field image allowed identification of neurons, which look quite different from the flatter astrocytes and also lie in a different focal plane, above the astrocytic layer. A total number of 600-800 neurons or astrocytes were counted in 20-25 fields of each coverslip. Each experiment was repeated five or more times using separate cultures.
Statistical analysis. Statistical analysis and exponential curve fitting were performed using Origin 7 (Microcal Software Inc.) software. Results are expressed as means ± SEM (SEM).
Results
Application of the βA peptide fragment 25-35 (5-50 μm) or the full-length peptide 1-42 (0.5-5 μm) to hippocampal neurons and astrocytes in coculture caused changes in Δψm of astrocytes (n = 510 cells) after a delay of ∼3-10 min, whereas mitochondrial potential remained completely stable in adjacent neurons (n = 298 cells) (Fig. 1A). Neurons were positively identified by the application of 100 μm glutamate toward the end of the experiment, because glutamate routinely causes a collapse of mitochondrial potential in neurons at this stage in culture (Vergun et al., 1999), without altering mitochondrial potential in astrocytes. Remarkably, although glutamate caused the predicted collapse of potential in neurons, in astrocytes it reversed the mitochondrial depolarization caused by βA (Fig. 1A) (and see below), further accentuating the difference in the responses of these two cell types. Switching to a Ca2+-free saline at the end of the experiment revealed that the mitochondrial depolarization in the neurons was reversible at this stage (Khodorov et al., 1996; Vergun et al., 1999), despite the presence of βA, suggesting that under these conditions βA does not significantly enhance the neurotoxicity of glutamate.
Figure 1.
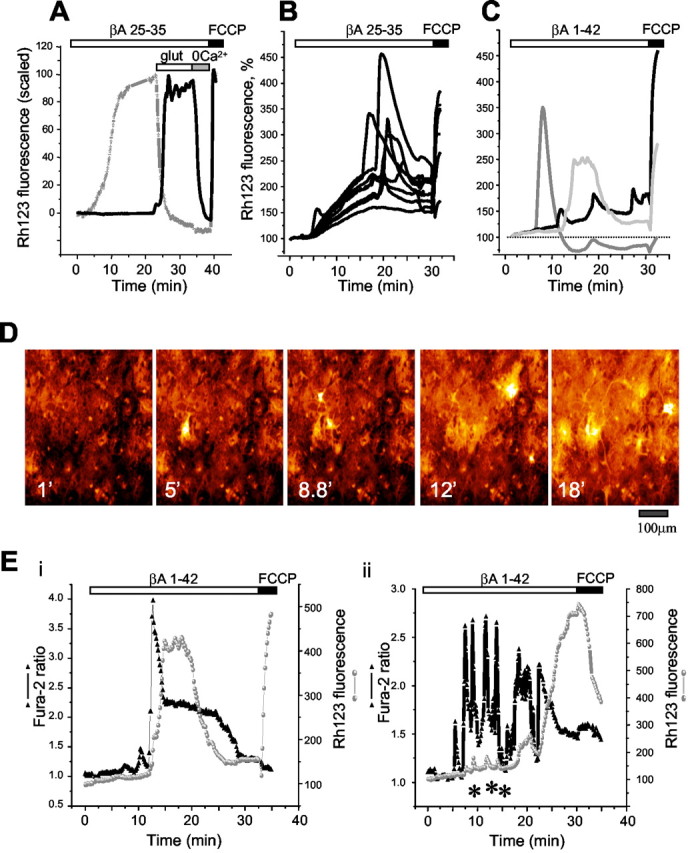
β-Amyloid causes depolarization of mitochondria in astrocytes but not in neurons. Changes in Δψm were measured using Rh123 in dequench mode; the loss of potential is seen as an increase in fluorescence. A, Changes in Δψm from a neuron (black line) and an astrocyte (gray symbols) in a mixed culture from rat hippocampus (15 DIV) after exposure to 50 μm βA 25-35. In the example shown, βA 25-35 caused a slow progressive collapse of Δψm in the astrocyte, whereas no change at all was seen in a nearby neuron. Application of 300 μm glutamate caused a rapid collapse of Δψm in the neuron but promoted recovery of Δψm in the astrocyte. On subsequent removal of external Ca2+, neuronal Δψm recovered, showing that mitochondrial injury was still reversible. In this and all subsequent records, the protonophore FCCP was added at the end of the experiment to determine the extent of the Rh123 signal in response to complete mitochondrial depolarization. The Rh123 fluorescence signal in these traces is normalized between 0, representing the resting Rh123 fluorescence, and 100, representing the maximal increase in Rh123 fluorescence in response to complete mitochondrial depolarization by 1 μm FCCP. In traces in which the Rh123 signal was lost in some cells during exposure to βA, this normalization was not possible, and so the data are shown normalized only to a baseline set at 100%. Thus, the trace shown in A is actually very similar to some of the traces in B, in which FCCP responses were maintained. In B is shown an example of an experiment using astrocytes in culture in response to the full peptide βA 1-42 (5 μm). The cells responded with a slow loss of mitochondrial potential on which were superimposed abrupt depolarizing transitions. Some of these were reversible, but the larger depolarizations were followed by loss of the dye, suggesting cell death. Some examples of these traces are extracted from another data set to illustrate these three types of response in C. The series of images shown in D illustrate examples from an extended time sequence showing the transient increases in signal and the gradual increase in basal signal in response to βA 25-35. The image after FCCP saturates the display at this range because it is much brighter than the rest of the sequence and so is not shown. The time of each extracted image is indicated in minutes. In Eii and Eii are shown records from two astrocytes co-loaded with fura-2 and Rh123 to measure [Ca2+]c (black triangles) and Δψm (gray lines and filled circles) simultaneously during exposure to 50 μm βA 25-35. As we have shown previously, βA caused fluctuations in [Ca2+]c in the astrocytes. Although the abrupt mitochondrial depolarizations were clearly associated with [Ca2+]c signals, they did not show a tight or fixed correlation in time or amplitude. Thus, large changes in Δψm followed large changes in [Ca2+]c, and some small transient depolarizations of Δψm could be seen associated with [Ca2+]c transients (asterisks in Eii).
Both the full-length peptide βA 1-42 and the peptide fragment 25-35 produced changes in Δψm in the vast majority (95.2 ± 1.3%) of cells in a culture of cortical astrocytes and in 89.6 ± 2.1% of astrocytes in coculture with neurons. The nontoxic reverse peptide βA 35-25 had no effect at all on Δψm either in hippocampal neurons (n = 123 cells) or in cortical or hippocampal astrocytes (n = 191 cells). The βA-induced changes in Δψm of astrocytes could be divided into three categories, some examples of which are also presented in Figure 1, B and C: (1) a slow and progressive mitochondrial depolarization; (2) large, transient, and reversible abrupt mitochondrial depolarizations, such as we have never witnessed before under any conditions, often superimposed on a slow progressive depolarization; and (3) abrupt mitochondrial depolarizations that failed to recover and were followed by the complete release of Rh123 from the mitochondria (and showed no further response to FCCP application), probably signifying cell death. The traces shown in Figure 1B show a range of examples of cell responses from one experiment, and in Figure 1C we have extracted three examples from another experiment to illustrate these three patterns of response. For the majority of traces that were used as illustrative material we have chosen traces that show both sustained and transient components, because these show how the components are modulated separately. It should be clear from the distribution histograms, however, that a substantial proportion of cells routinely show only graded sustained responses. Examples of the images from which these data were taken are shown in Figure 1D, in which sample images from a time series have been extracted. Note how the Rh123 signal becomes bright in individual cells only to recover later. These cells usually responded again to FCCP, which was applied routinely at the end of the experiment.
Role of [Ca2+]c in amyloid-induced changes in mitochondrial potential
We have shown previously that βA induces, after a delay, the appearance of oscillatory [Ca2+]c signals in astrocytes (Abramov et al., 2003) and not in neurons. Because changes in [Ca2+]c may initiate changes in mitochondrial membrane potential (for review, see Duchen, 1999), we conducted experiments in which Δψm and [Ca2+]c were measured simultaneously. These experiments revealed an association between the transient increases in [Ca2+]c and the transient mitochondrial depolarizations. Close scrutiny of traces from individual cells (Fig. 1Ei,Eii) shows that the transient mitochondrial responses are clearly routinely associated with [Ca2+]c transients, although not all [Ca2+]c transients cause mitochondrial depolarizations.
We have shown previously that βA-induced changes in [Ca2+]c are dependent on the presence of external calcium, reflecting βA-induced Ca2+ influx from the extracellular space (Abramov et al., 2003). In keeping with this observation, changes in [Ca2+]c were abolished on the removal of external Ca2+ (Fig. 2Ai,Bi). In the absence of external Ca2+, βA 25-35 or 1-42 still induced a slow mitochondrial depolarization in hippocampal and cortical astrocytes (n = 231) (Fig. 2Aii), but transient depolarizations were much reduced and irreversible abrupt depolarizations were abolished. This is further emphasized by the analysis of each component of the responses illustrated in Figure 2C. Washing the cells with a saline containing Ca2+ after a period in Ca2+-free conditions caused an abrupt [Ca2+]c increase in the astrocytes (Fig. 2Bi) and triggered a rapid loss of mitochondrial potential in many cells (Fig. 2Bii). Thus, the slow progressive mitochondrial depolarization is apparently relatively independent of [Ca2+]c, whereas the rapid transient changes in potential appear to be more closely associated with βA-induced changes in [Ca2+]c (Fig. 2C). The average change in Rh123 signal caused by 30 min of exposure to βA in the absence of Ca2+ was significantly smaller than seen in the standard Ca2+-containing saline (p < 0.05; from 64.1 ± 13.7% in standard buffer to 29.2 ± 6.3% in Ca2+-free) (Fig. 2D). These differences in signal were primarily attributable to the absence of large rapid transients or irreversible depolarizations in the Ca2+-free saline.
Figure 2.

Transient mitochondrial depolarizations are dependent on βA-induced Ca2+ influx. Simultaneous measurements of [Ca2+]c and Δψm were made from astrocytes in mixed hippocampal culture (14 DIV) co-loaded with fura-2 (Ai, Bi) and Rh123 (Aii, Bii). Traces are shown from two cells in each case. A, As shown previously (Abramov et al., 2003), the [Ca2+]c signals were abolished in the absence of external Ca2+ (a saline with no added Ca2+ and with the addition of 500 μm EGTA). The transient mitochondrial depolarizations were also suppressed, but a significant slow depolarization was still evident as shown by the analysis of different components of the response shown in C. Note that the appearance of irreversible abrupt depolarizations was abolished and that of transient reversible depolarizations much reduced, whereas the slow increase in signal was unaffected. It may appear to increase simply because cells are not undergoing abrupt loss of signal. B, When βA was added in the absence of external Ca2+, as in A, no change in [Ca2+]c and only a modest mitochondrial depolarization were seen. Washing with a Ca2+ containing saline then caused the abrupt appearance of [Ca2+]c fluctuations and the appearance of large mitochondrial depolarizations. The data shown in D summarize the mean changes in the normalized Rh123 signal measured at 30 min exposure to 50 μm βA 25-35 in the presence (Control) or absence of calcium and in the presence of 1 mm Zn2+ and 2 μm clioquinol, which inhibit completely the [Ca2+]c response to βA (Abramov et al., 2003). In all three cases, the mitochondrial depolarization was reduced significantly. *p < 0.01; **p < 0.001.
Zn2+ (1 mm) and the heavy metal chelator clioquinol (2 μm) both block the βA-induced [Ca2+]c fluctuation in astrocytes (Abramov et al., 2003). We therefore examined their actions on the mitochondrial response and found that both of the agents also completely blocked the effect of βA on mitochondria (Fig. 2D). The most probable explanation is that Zn2+ and clioquinol may prevent the aggregation of βA and its incorporation into membranes, thereby preventing its action either on calcium signaling or on mitochondrial membrane potential.
Mitochondrial substrates protect mitochondria from the effects of βA
As shown in Figure 1A, the slow progressive mitochondrial depolarization seen in astrocytes could surprisingly be reversed by the application of glutamate (from a mean change in Rh123 signal of 64.1 ± 13.7 to 4.9 ± 3.2%; n = 108 cells). This action was unaffected by (S)-(+)-α-methyl-4-carboxyphenylglycine (50 μm; n = 178) or (+)-5-methyl-10,11-dihydroxy-5H-dibenzo(a,d)cyclohepten-5,10-imine (15 μm; n = 97), inhibitors of the metabotropic and ionotropic glutamate receptors, respectively (data not shown). Glutamate was most effective at concentrations of 1-5 mm, whereas it had smaller effects at the lower (1-300 μm) concentrations expected to saturate receptors, suggesting that glutamate is acting as a metabolic substrate rather than as a neurotransmitter. We therefore explored the actions of substrates for mitochondrial complex I (1 mm glutamate; also pyruvate and malate, 5 mm each), complex II [10 mm methyl-succinate (me-succinate)], a membrane-permeant metabolizable form of succinate (Maechler and Wollheim, 1999), and complex IV [2,3,5,6-tetramethyl-p-phenylenediamine (TMPD) (200 μm) with 1 mm ascorbate]. These all increased the mitochondrial potential at rest (seen as a decrease, an increased quench, of the Rh123 signal) when added before the βA (Fig. 3A,B) (TMPD trace not shown). Glutamate, pyruvate/malate, and me-succinate all suppressed the slow mitochondrial depolarization in response toβA, as seen in the traces of Figure 3, A and B. Some images extracted from a sequential image series in the presence of me-succinate are in Figure 3D to show the remaining transient mitochondrial depolarizations. Note that cells that “light up” intermittently during the exposure to βA also become bright with FCCP, showing that recovery of signal must represent recovery of potential and reaccumulation of dye into the mitochondria. Additional mitochondrial substrate also dramatically reduced the percentage of cells that failed to recover from abrupt mitochondrial depolarization shown in an analysis of the components of the response for glutamate in Figure 3E. Glutamate (500 μm-1 mm,), pyruvate/malate (5 mm of each), and me-succinate (p < 0.01; n = 99) all significantly reduced the βA-induced mitochondrial depolarization measured as the mean increase of Rh123 signal after 30 min of exposure to βA from an increase of Rh123 signal by 64.1 ± 13.7 to 7.12 ± 2.34% for glutamate (n = 194 cells; p < 0.01), to 6.1 ± 2.8%, for pyruvate/malate (n = 78 cells; p < 0.01), and to 18.6 ± 4.17% for me-succinate, (n = 99; p < 0.01) (Fig. 3F) in cortical and hippocampal astrocytes. In contrast, and perhaps surprisingly, TMPD/ascorbate (n = 87) had no significant effect on the changes in Δψm signal induced by βA in either cortical or hippocampal astrocytes (Fig. 3F) (this will be discussed later). The combined effect of Aβ causing calcium influx associated with fast transient mitochondrial depolarizations, and a metabolic effect that causes a slow loss of potential, is emphasized by the experiment illustrated in Figure 3C, in which Aβ was applied in the presence of 1 mm glutamate and in the absence of external calcium. Under these conditions, the effect of Aβ on the mitochondria was abolished almost completely (Fig. 3C) (n = 79).
Figure 3.
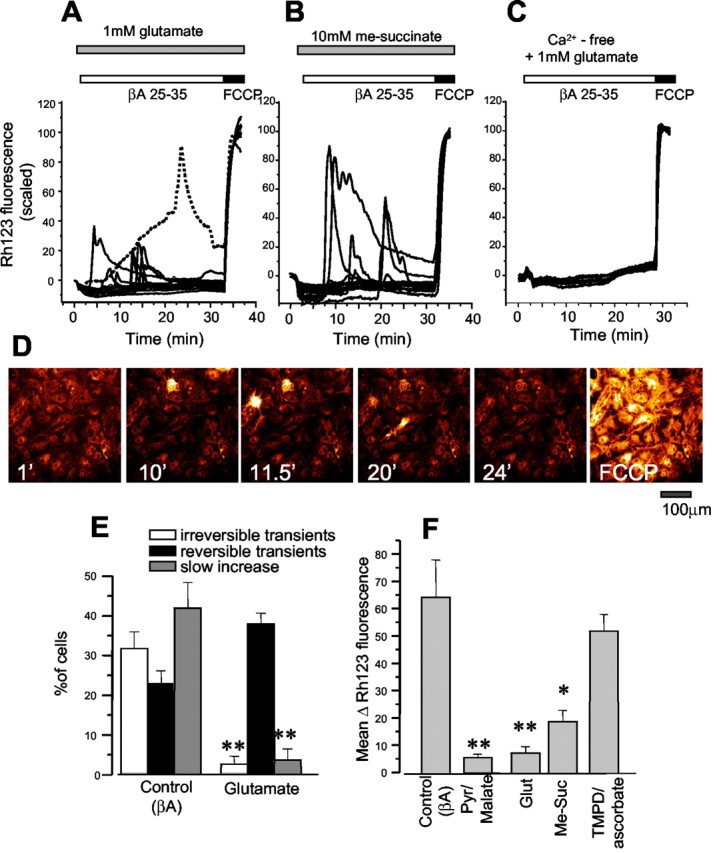
The slow mitochondrial depolarization induced by βA is reversed by mitochondrial substrates. Changes in Rh123 signal were measured in a culture of cortical astrocytes in response to βA 25-35 (50 μm) in the presence of 1 mm glutamate (A), 10 mm methyl-succinate (B), and 1 mm glutamate in a Ca2+-free saline with the addition of 500 μm EGTA (C). In the presence of either glutamate or me-succinate, fast reversible transient mitochondrial depolarizations were still seen, but the slow progressive mitochondrial depolarization was abolished almost completely; compare with the day-matched control trace shown in A as a dashed line. In the presence of glutamate and the absence of external Ca2+, the entire response was abolished. In D are shown a series of images extracted from a time sequence showing changes in Δψm in response to βA in the presence of methyl succinate. Note that the Rh123 signal in some cells becomes transiently very bright but then is restored and shows a further increase with FCCP in the final image. The final image with FCCP shows the first image acquired immediately after application of FCCP, because the signal continued to get even brighter with a saturation of the display at this range. The time of each image in the sequence is indicated (minutes). An analysis of the effect of glutamate on different components of the response is shown in E, and the measurements of the mean increase in Rh123 fluorescence at 30 min in response to βA in the presence of glutamate, methyl succinate, and TMPD/ascorbate are shown in F. *p < 0.01; **p < 0.001.
In all of these experiments, [Ca2+]c and mitochondrial potential were measured simultaneously. The [Ca2+]c signal was not altered significantly by the presence of glutamate or other mitochondrial substrates.
Role of ROS in the mitochondrial depolarization induced by βA
As discussed above, there is ample evidence to implicate ROS in the toxicity of Aβ. We wished to explore whether the change in Δψm was a response to or a cause of increased ROS generation. To investigate the role of ROS in the collapse of Δψm of cortical or hippocampal astrocytes produced by βA, we used the antioxidant compound TEMPO. TEMPO is a cell-permeable nitroxide that acts to catalyze O2- dismutation (Samuni et al., 1988), and it has been shown to remove oxidant species and protect cells from oxidative damage in some model systems (Grinberg and Samuni, 1994; Vergun et al., 2001). To avoid the toxic action of H2O2 generated by O2- dismutation, TEMPO (500 μm) was used in combination with catalase (250 U/ml), a scavenger of H2O2. The cells were incubated with TEMPO and catalase for 30 min and then exposed to βA 1-42 or 25-35 in the presence of these antioxidants. Although the catalase acts extracellularly, this combination has been shown to be effective in scavenging superoxide generated by xanthine/xanthine oxidase in neurons (Vergun et al., 2001). Figure 4A shows the changes in Δψm in response to βA 25-35 in the presence of TEMPO plus catalase. The major effect of the antioxidants was to suppress the slow mitochondrial depolarization (Fig. 4B), similar to the action of glutamate or methyl-succinate. The average change in Rh123 signal measured after 30 min of exposure to βA in astrocytes pretreated with TEMPO and catalase was significantly smaller (p < 0.01) than in control cells (from an increase by 64.1 ± 13.7% in controls to an increase of just 14.5 ± 5.2%; n = 156 cells) (Fig. 4C). We have shown previously that antioxidants do not alter the [Ca2+]c response of astrocytes to βA (Abramov et al., 2003), and so this effect seems specific to the metabolic response to Aβ.
Figure 4.

The mitochondrial response to βA is suppressed by antioxidants. A, Cortical astrocytes co-loaded with Rh123 and fura-2 were preincubated for 30 min with 500 μm TEMPO plus catalase (250 U/ml) before exposure to 50 μm βA 25-35. Only the Rh123 trace is shown; the fura-2 record was indistinguishable from controls. The antioxidants remained in the chamber during the exposure to βA. All aspects of the response were much reduced, as indicated in the analysis in B, and the mean mitochondrial depolarization measured after 30 min in the presence of βA was reduced significantly, as shown in C *p < 0.01; **p < 0.001.
Role of the mitochondrial permeability transition pore in the mitochondrial response to βA
The fast large transient mitochondrial depolarizations seen in cortical and hippocampal astrocytes during the βA exposure were remarkable and unprecedented. We wondered whether they might be induced by transient opening of the mitochondrial permeability transition pore (MPTP) or whether βA might be internalized and form a pore directly in the mitochondrial inner membrane, as it does in the plasma membrane (Lin et al., 2001; Abramov et al., 2003). The hippocampal astrocytes were incubated with cyclosporin A (CsA) (0.5 μm), the classic inhibitor of MPTP opening, for 30 min and then exposed to βA 25-35, still in the presence of the inhibitor. The data shown in Figure 5, A and D, show that CsA did not significantly alter the slow mitochondrial depolarization in βA-treated cells and only suppressed slightly the transient depolarizations, but it completely blocked the large abrupt and irreversible depolarizations (Fig. 5D). CsA has no significant effect on the calcium signal (data not shown), which was always measured simultaneously with the mitochondrial signals. The average amplitude of the mitochondrial depolarization decreased from 64.1 ± 13.7% in control to 38.43 ± 7.3% in CsA-treated astrocytes (n = 147) (Fig. 5E).
Figure 5.

Role of the mitochondrial permeability transition pore in the mitochondrial depolarization induced by βA. A, Hippocampal astrocytes in coculture were loaded with Rh123 and preincubated with 0.5 μm cyclosporin A before exposure to 50 μm βA 25-35. Large irreversible mitochondrial depolarizations were suppressed completely, and the incidence of smaller transient and reversible depolarizations was also reduced, whereas the slow mitochondrial depolarization was not affected, as indicated by the analysis of the components of the response shown in D. The traces in B show that combined treatment of cells with CsA and the antioxidants TEMPO/catalase almost completely suppressed any response to βA, as did a combination of TEMPO and catalase combined with the removal of extracellular Ca2+, as illustrated in C. E summarizes measurements of the normalized Rh123 signal at 30 min of exposure to βA with each of these manipulations. These data strongly suggest that the mitochondrial response to βA involves the synergistic action of Ca2+ and oxidative stress leading together to the activation of the MPTP. *p < 0.01; **p < 0.001.
The major mechanism that triggers MPTP opening is the synergistic action of raised intramitochondrial calcium with oxidative stress (for review, see Duchen, 2000; Crompton, 2000). When CsA was combined with the antioxidant combination of TEMPO with catalase (n = 107), the action of βA on Δψm was suppressed almost completely (Fig. 5B,E), again without altering the calcium response (data not shown). Similarly, removing both triggers to MPTP opening by using TEMPO plus catalase in the absence of extracellular Ca2+ (n = 67 cells) also completely blocked the βA-triggered changes in Δψm (Fig. 5C). Thus, the full effect of βA on mitochondrial potential appears to be produced by the combined action of ROS and Ca2+.
Sources of βA-induced ROS: a role for the NADPH oxidase
Many mechanisms have been proposed to account for increase in ROS formation induced by β-amyloid. We have recently found that the plasmalemmal NADPH oxidase, an enzyme expressed predominantly in neutrophils and macrophages (for review, see Droge, 2002), is expressed also in astrocytes (Shimohama et al., 2000; A. Abramov and J. Jacobson, unpublished observations). The NADPH oxidase inhibitor DPI (0.5 μm) completely prevented the βA-induced mitochondrial depolarization, although it had no apparent effect on the βA-induced changes in [Ca2+]c (Fig. 6). Because DPI is not fully selective for the NADPH oxidase but also inhibits mitochondrial NADH dehydrogenases and therefore may inhibit mitochondrial respiration, we examined the effect of this concentration of the drug on Δψm of untreated astrocytes and found no significant effect (n = 79 cells). Furthermore, because the mitochondrial ATPase could maintain the potential in the face of mitochondrial inhibition by DPI, we tested the effect of oligomycin (2.5 μg/ml) on the Rh123 signal in the presence of DPI, and this had no detectable effect (n = 43 cells). This demonstrates that DPI had no significant effect on the mitochondrial dehydrogenases at this concentration and appears to act only on the plasma membrane. Another specific inhibitor of NADPH oxidase, 4-hydroxy-3-methoxy-acetophenone (apocynin), used at 1 mm (Gao et al., 2002), which binds to a different site on the enzyme from DPI, also completely inhibited βA-induced mitochondrial depolarization (n = 63 cells), further confirming the involvement of NADPH oxidase in βA-induced mitochondrial depolarization.
Figure 6.
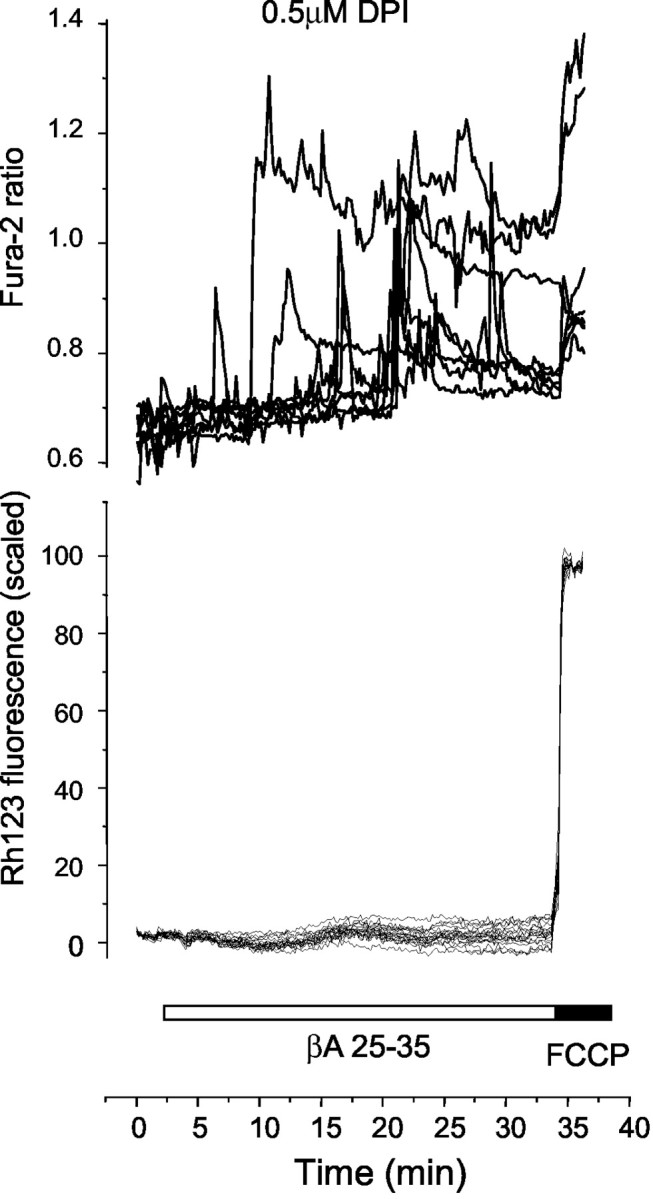
The mitochondrial response to βA but not the change in [Ca2+]c is mediated by the NADPH oxidase. Astrocytes were co-loaded with fura-2 and Rh123 for simultaneous measurement of [Ca2+]c and Δψm. Cells were preincubated with 0.5 μm DPI for 30 min. [Ca2+]c transients were seen as usual after application of 50 mm βA, whereas the mitochondrial response was suppressed completely.
Effect of βA on ROS generation
To explore further the increase in ROS induced by βA, we used DCF to examine directly the rate of ROS generation. DCF is oxidized to a fluorescent product by ROS, and so we measured the rate of increase of signal as a measure of the rate of ROS generation. βA 25-35 (50 μm) or βA 1-42 (2.5 μm) caused a rapid and substantial increase in the rate of rise of the DCF signal with a slope of 3.19 ± 0.18 arbitrary fluorescence units per minute, in both cortical and hippocampal astrocytes (n = 88 cells) (Fig. 7A,D). It is interesting that although there was a 5-10 min delay before the onset of either [Ca2+]c signals or mitochondrial depolarization, the DCF signal increased almost immediately after the application of βA. The rate of rise of the DCF signal induced by βA was significantly reduced when cells were preincubated in Ca2+-free saline (from 3.19 ± 0.18 arbitrary fluorescence units per minute to 1.18 ± 0.11; p < 0.001; n = 101 cells) (Fig. 7B). The βA-stimulated ROS production in astrocytes was inhibited almost completely by 0.5 μm DPI (from 3.19 ± 0.18 arbitrary fluorescence units per minute to 0.61 ± 0.14; n = 114 cells; p < 0.001) (Fig. 7C) or 1 mm apocynin (from 3.19 ± 0.18 arbitrary fluorescence units per minute to 0.97 ± 0.11;. n = 68), strongly suggesting that the response is mediated mainly by the activation of the NADPH oxidase.
Figure 7.

βA-increased ROS generation in astrocytes is dependent on Ca2+ and activation of the NADPH oxidase. Hippocampal cultures were loaded with DCF. A, Addition of 5 μm βA 1-42 caused a clear increase in the rate of appearance of the fluorescent product, suggesting increased ROS generation. The line shown was fitted by a linear regression with a correlation coefficient of 0.98. The rate of increase of ROS production was reduced significantly in the absence of external Ca2+ (B) and was blocked almost completely by 0.5 μm DPI (C). The data are summarized in D, which shows the mean rate of rise of DCF signal under these conditions and also after exposure to 1 mm apocynin, another inhibitor of the NADPH oxidase. *p < 0.01.
It has been reported that, in microglia, βA activates the NADPH oxidase through an interaction of fibrillar peptide with the scavenger receptor CD36 (Coraci et al., 2002). To test this, we preincubated cells with an antibody to CD36 (5 μg/ml FA6-152) for 30 min. We saw no significant difference in the rate of rise of the DCF signal in cortical astrocytes on three separate cultures (54 cells) or in hippocampal astrocytes (two cultures, 40 cells).
We have shown previously that βA causes a loss of glutathione (GSH) in astrocytes and also in neurons in coculture (Abramov et al., 2003), but we had not previously identified the source of βA-induced oxidative stress. We therefore asked whether the depletion of GSH in astrocytes and neurons was related to activation of the NADPH oxidase. Using monochlorobimane (MCB) (Keelan et al., 2001) to measure GSH levels in adjacent neurons and astrocytes in coculture, we found as before that βA caused GSH depletion both in astrocytes and in neurons in hippocampal cocultures. This response was abolished almost completely by treatment with DPI (n = 127 neurons and n = 198 astrocytes; p < 0.05 for both types of cells) (Fig. 8A,B), which had no significant effect on the resting GSH levels in the cells. Furthermore, GSH levels could be better maintained in the face of βA exposure by provision of the GSH precursor 1 mm γ-glutamyl cysteine (γ-GluCys), which significantly increased MCB fluorescence in βA-treated astrocytes from 497 ± 21 to 847 ± 63 (n = 295) and in neurons from 415 ± 23 to 556 ± 27 (n = 136; p < 0.05) (Fig. 8A,B).
Figure 8.

GSH depletion and cell death induced by βA are dependent on free radical generation by NADPH oxidase. βA 25-35 or 1-42 caused GSH depletion in neurons (A) and astrocytes (B) measured in hippocampal cocultures loaded with monochlorobimane. This was prevented almost completely by 0.5 μm DPI and by 1 mm apocynin and was reversed by provision of 1 mm γ-glutamyl-cysteine. βA 25-35 or 1-42 also increased cell death of neurons (C) to ∼40% and astrocytes (D) to ∼15% above background. In both cell types, cell death was reduced significantly by all manipulations that also suppressed the mitochondrial depolarization in astrocytes: in the absence of external Ca2+, with the addition of antioxidants, and in the presence of 0.5 μm DPI or 1 mm apocynin. The most effective protection was afforded by the combination of antioxidants and CsA. The addition of 1 mm glutamate suppressed βA-induced cell death in astrocytes and made no significant difference to cell death in the neurons. Both neurons and astrocytes were also substantially protected from death induced by βA by previous incubation with 1 mm γ-GluCys (see Results). *p < 0.01; **p < 0.001.
Effect of βA on cell viability and rescue by GSH precursors
We then examined the effect of incubation with βA on cell viability of neurons and astrocytes. Exposure of mixed hippocampal cultures to βA 25-35 for 24 hr caused neuronal cell death of 38.4 ± 9.3% above that seen in untreated cells (which was 9.8 ± 3.2%; n = 13 experiments) (Fig. 8C). βA caused significantly less cell death among the astrocytes, with an increase of 15.4 ± 4.2% above a background of 7.8 ± 2.9% (n = 13 experiments) (Fig. 8D). Interestingly, two compounds that are normally highly neurotoxic, βA 25-35 and glutamate (500 μm), did not have a synergistic effect on neuronal cell death. In fact, glutamate was significantly protective in astrocytes, reducing βA-induced cell death to 4.9 ± 2.2% above background (p < 0.05) (Fig. 8D). The antioxidant combination of TEMPO and catalase afforded significant protection from βA-induced cell death in both cell types [to 16.8 ± 4.6% for neurons (p < 0.05) and 4.6 ± 2.1% for astrocytes (p < 0.05) for both cell types; n = 5 experiments], but the most effective protection was given by TEMPO and catalase combined with CsA (to 4.7 ± 2.9% for neurons and 0.3 ± 0.3% for astrocytes; p < 0.001 for neurons and astrocytes; n = 5). DPI also dramatically protected both classes of cells (to 11.1 ± 4.3% for neurons and to 5.1 ± 1.1% for astrocytes; p < 0.05 for both type of the cells; n = 4) but was less effective than the antioxidant/CsA combination, suggesting that DPI, despite inhibiting βA-induced ROS generation very effectively, may have a marginal toxicity.
The importance of neuronal glutathione is underscored by some experiments in which GSH in the cultures was enriched with the provision of the GSH precursor 1 mm γ-GluCys (and see above). Cultures were preincubated with γ-GluCys for 6 hr and then exposed to βA still in the presence of the γ-GluCys for 24 hr. This treatment significantly reduced cell death in neurons (from 40.1 ± 8.5 to 20.4 ± 3.2%; p < 0.05; n = 4 experiments) and in astrocytes (from 15.4 ± 4.2 to 7.1 ± 1.3%; p < 0.05; n = 4 experiments) (Fig. 8C,D).
Discussion
We have found that βA treatment causes dramatic changes in mitochondrial potential in cultures of neurons and astrocytes from rat hippocampus. There are several remarkable features of these responses. First, the mitochondrial responses were restricted to the astrocytes, whereas no significant changes in mitochondrial function were seen in neurons over the time frame investigated. The mitochondrial response appears to be a complex response to several different processes taking place within the astrocytes that are triggered by Aβ and seems to reflect changes in several different but related aspects of mitochondrial function, with oxidative stress as a common underlying factor.
The slow mitochondrial depolarization, occurring over a time scale of minutes, is attributable to the effect of oxidative stress on enzymes that ensure mitochondrial substrate supply. The response could be reversed or blocked by a supply of substrates to complex I (glutamate) or complex II (me-succinate), by antioxidants, by DPI, and by apocynin. These observations show that the response is caused by an effect of oxidative stress as a consequence of the activation of NADPH oxidase. Enzymes of the TCA cycle, aconitase and α-ketoglutarate dehydrogenase, have been shown to be impaired both by βA and by oxidative stress in various model systems (Blass and Gibson, 1991; Chinopoulos et al., 1999; Casley et al., 2002b). The observation that the mitochondrial potential could be recovered almost completely by substrates of complex I argues that that this effect was not caused by impaired activity of the respiratory complexes and that it must lie upstream of mitochondrial metabolism. Both glucose uptake and glycolysis are impaired by exposure to βA (Mark et al., 1997; Pereira et al., 1999), providing the most plausible mechanism for these observations. TMPD/ascorbate should bypass other components of the respiratory chain but failed to reverse the effect. It has been shown previously that the Km of cytochrome oxidase for cytochrome c may be raised by βA (Casley et al., 2002b). Because the TMPD/ascorbate pair supplies electrons to cytochrome c, this may account for the failure of the TMPD/ascorbate combination to restore potential. It seems that βA-induced oxidative stress causes impairments of metabolism at multiple sites through intermediary metabolism, but the failure of the glycolytic supply of mitochondrial substrate appears to be the first to become functionally significant. We have also shown previously that βA causes impaired activity of several complexes of the mitochondrial respiratory chain (Canevari et al., 1999; Casley et al., 2002a). This in turn may act as an auto-amplifying mechanism, because impaired mitochondrial respiration may act as yet another source of intramitochondrial free radical generation.
The other dramatic feature of the mitochondrial response was the occurrence of profound abrupt and reversible mitochondrial depolarizations. These effects were remarkable and unprecedented. There seem to be two probable explanations for such abrupt and reversible changes in potential, both based on transient openings of a large conductance pore. One is that the βA is internalized and inserts into the mitochondrial membrane where it acts as a pore much as it does at the plasma membrane. This is difficult to test, but seems unlikely, because the protein would have to cross the outer mitochondrial membrane and insert itself into the inner membrane. The other, perhaps more probable explanation is that the combination of transient pulses of high [Ca2+]c with an underlying oxidative stress suffices to cause transient openings of the MPTP. CsA suppressed the large irreversible mitochondrial depolarizations almost completely, suggesting that these reflect MPTP opening and cell death. The effect of CsA on the smaller, reversible transient depolarizations was unimpressive, however, suggesting that these might still reflect another process. The observation that the transients were suppressed so completely by those manipulations that suppressed oxidative stress—DPI, apocynin, TEMPO/catalase—without altering the [Ca2+]c signal argues strongly that the events cannot be a “simple” consequence of Ca2+ influx and also argues strongly against the insertion of βA as a pore in the mitochondrial membrane. The events were also inhibited effectively in the absence of external calcium and appeared to be temporally related to [Ca2+]c transients, again suggesting a possible role for the MPTP, although the calcium dependence could also reflect the calcium dependence of the rate of ROS generation. These experiments show clearly that the transient depolarizations are strongly dependent both on calcium and on oxidative stress and that, although both are required, neither alone is sufficient to cause them.
Thus, the underlying key to the response seems to be an oxidative stress generated by βA, and the various effects on mitochondria are attributable to the consequences of oxidative stress on different aspects of mitochondrial function in the astrocytes. The key question then becomes what is the mechanism of oxidative stress, of the selectivity of the βA action for astrocytes, and finally of neuronal cell death. We have found recently that astrocytes selectively express the plasmalemmal NADPH oxidase (our unpublished observations). It is not clear what normally regulates the activity of this enzyme or what its role in the CNS might be. Nevertheless, the dramatic protection afforded by DPI, at concentrations that do not affect other potential targets (mostly mitochondrial dehydrogenases), and by apocynin, an agent that binds at a separate site on the target enzyme, strongly suggests that this enzyme represents a major source of radical species in response to βA and that the selective expression of the enzyme might account in some measure for the selective effects of βA on astrocyte, and not neuronal, mitochondria. The importance of the NADPH oxidase in βA toxicity has already been suggested but with reference to the expression of the enzyme in microglia (Bianca et al., 1999; Qin et al., 2002). In the present study, however, astrocytic ROS generation seems to play a critical role in βA neurotoxicity, suggesting that the astrocyte may have been overlooked as a source of ROS. The imaging of DCF appears to us to provide unequivocal evidence identifying the astrocytes as the source of ROS. Thus, we measure ROS generation from astrocytes in pure astrocyte cultures (>99% GFAP positive). In mixed cultures, an increase in DCF signal is seen over astrocytes and not over nearby neurons, showing that the response cannot arise as some diffusion (itself unlikely) of ROS from occasional contaminating microglia.
We do not yet understand how βA might activate the NADPH oxidase. The dependence of the rate of rise of ROS generation on calcium suggests that the βA-induced increase in [Ca2+]c might play a role in activating the enzyme. It also seemed possible that the βA might interact with the enzyme through the CD36 scavenger receptors as reported for microglia (Coraci et al., 2002). We did not see any effect of antibodies to CD36 on the βA-induced increase in astrocyte ROS generation in these experiments, again highlighting the importance of expression of the enzyme in astrocytes as well as in microglia. One isoform of NADPH oxidase, nox5, expressed in testis, spleen, and lymph nodes (Banfi et al., 2001), is activated by a rise in [Ca2+]c. Our data do not clearly resolve this issue. That the rate of ROS generation tends to increase almost immediately on βA application whereas the [Ca2+]c changes occur with a delay of 5-10 min strongly argues against a Ca2+ dependence of NADPH oxidase. However, the Ca2+ dependence of the βA-induced ROS generation and of GSH oxidation is consistent with Ca2+-dependent modulation of the NADPH oxidase. This issue will require additional work to clarify the specific relationship between changes in [Ca2+]c and ROS generation. These data nevertheless suggest that these two “arms” of the response to βA, the influx of Ca2+ and the activation of NADPH oxidase and generation of oxidative stress, and their impact on intracellular metabolic pathways and mitochondrial function are coupled.
Why do neurons die more than the astrocytes even though the apparent pathophysiology takes place in astrocytes? We have shown previously that βA reduces astrocyte glutathione concentrations. Astrocytes supply amino acid precursors for neuronal GSH synthesis, and neurons depleted of GSH die through an inability to withstand endogenous pro-oxidants. In turn, astrocytes can sustain themselves adequately with glycolytic metabolism, seem not to be significantly affected by loss of mitochondrial function, and are also far more resistant to oxidative stress than are the neurons. The importance of GSH as a protective mechanism is again emphasized by the protection afforded by the GSH precursor γ-glutamyl cysteine. It remains possible that neuronal death is further mediated by other mechanisms, perhaps through cytokines released from damaged astrocytes or perhaps through increased vulnerability of neurons to other stimuli. These data suggest, however, that a major part of βA neurotoxicity reflects the neuronal dependence on astrocyte support and results from a complex interplay between actions of the peptide on astrocytes, altered astrocyte [Ca2+]c signaling, free radical generation, and resultant oxidative stress. This in turn impairs the ability of astrocytes to maintain neuronal integrity and results eventually in neuronal attrition.
Footnotes
This work was supported by the Wellcome Trust, The Medical Research Council, and the Miriam Marks Fund. We especially thank Dr. Jake Jacobson for invaluable suggestions and discussion while the work was in progress, and we thank Professors John Clark, Stephen Bolsover, and Rosario Rizzuto for helpful discussion and comments on this manuscript.
Correspondence should be addressed to Michael R. Duchen, Department of Physiology, University College London, Gower Street, London WC1E 6BT, UK. E-mail: m.duchen@ucl.ac.uk.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240565-11$15.00/0
References
- Abramov AY, Canevari L, Duchen MR (2003) Changes in [Ca2+]c and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23: 5088-5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananda theertha varada HK, Biswas G, Robin MA, Avadhani NG (2003) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impair mitochondrial function in neuronal cells. J Cell Biol 161: 1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias C, Montiel T, Quiroz-Baez R, Massieu L (2002) beta-Amyloid neurotoxicity is exacerbated during glycolysis inhibition and mitochondrial impairment in the rat hippocampus in vivo and in isolated nerve terminals: implications for Alzheimer's disease. Exp Neurol 176: 163-174. [DOI] [PubMed] [Google Scholar]
- Askanas V, McFerrin J, Baque S, Alvarez RB, Sarkozi E, Engel WK (1996) Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc Natl Acad Sci USA 93: 1314-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH (2001) A Ca2+-activated NADPH oxidase in testis, spleen, and lymph nodes. J Biol Chem 276: 37594-37601. [DOI] [PubMed] [Google Scholar]
- Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F (1999) beta-Amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem 274: 15493-15499. [DOI] [PubMed] [Google Scholar]
- Blass JP, Gibson SA (1991) The role of oxidative abnormalities in the pathophysiology of Alzheimer's disease. Rev Neurol (Paris) 147: 513-525. [PubMed] [Google Scholar]
- Canevari L, Clark JB, Bates TE (1999) β-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett 457: 131-134. [DOI] [PubMed] [Google Scholar]
- Casley C, Land J, Sharpe M, Clark J, Duchen M, Canevari L (2002a) β-Amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol Dis 10: 258-267. [DOI] [PubMed] [Google Scholar]
- Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA (2002b) β-Amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80: 91-100. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Tretter L, Adam-Vizi V (1999) Depolarization of in situ mitochondria due to hydrogen peroxide-induced oxidative stress in nerve terminals: inhibition of alpha-ketoglutarate dehydrogenase. J Neurochem 73: 220-228. [DOI] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB (2002) CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol 160: 101-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M (2000). Mitochondrial intermembrane junctional complexes and their role in cell death. J Physiol (Lond) 529: 11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82: 47-95. [DOI] [PubMed] [Google Scholar]
- Duchen MR (1999) Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signaling and cell death. J Physiol (Lond) 516: 1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR (2000) Mitochondria and calcium: from cell signaling to cell death. J Physiol (Lond) 529: 57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ (1992) Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol (Lond) 450: 33-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao HM, Hong JS, Zhang W, Liu B (2002) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22: 782-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SM, Shankar SL, Chalmers-Redman RM, Tatton WG, Szyf M, Cuello AC (1999) Mitochondrial abnormalities in neuroectodermal cells stably expressing human amyloid precursor protein (hAPP751). NeuroReport 10: 41-46. [DOI] [PubMed] [Google Scholar]
- Grinberg LN, Samuni A (1994) Nitroxide stable radical prevents primaquine-induced lysis of red blood cell. BBA 1201: 284-288. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RM, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen PB, Perry G, Smith MA (2001) Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 21: 3017-3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelan J, Allen NJ, Antcliffe D, Pal S, Duchen MR (2001) Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res 66: 873-884. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N (1996) Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett 397: 230-234. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee JH, Lee JP, Kim EM, Chang KA, Park CH, Jeong CJ, Wittendorp MC, Seo JH, Choi SH, Suh YH (2002) Amyloid β peptide induces cytochrome c release from isolated mitochondria. NeuroReport 13: 1989-1993. [DOI] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R (2001) Amyloid β protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J 15: 2433-2444. [DOI] [PubMed] [Google Scholar]
- Longo VD, Viola KL, Klein WL, Finch CE (2000) Reversible inactivation of superoxide-sensitive aconitase in Abeta1-42-treated neuronal cell lines. J Neurochem 75: 1977-1985. [DOI] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB (1999) Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402: 685-689. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP (1997) Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 17: 1046-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais Cardoso S, Swerdlow RH, Oliveira CR (2002) Induction of cytochrome c-mediated apoptosis by amyloid beta 25-35 requires functional mitochondria. Brain Res 931: 117-125. [DOI] [PubMed] [Google Scholar]
- Moreira PI, Santos MS, Moreno A, Rego AC, Oliveira C (2002) Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J Neurosci Res 69: 257-267. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283: 1571-1577. [DOI] [PubMed] [Google Scholar]
- Parks JK, Smith TS, Trimmer PA, Bennett JP, Parker WD (2001) Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem 76: 1050-1056. [DOI] [PubMed] [Google Scholar]
- Pereira C, Santos MS, Oliveira C (1999) Involvement of oxidative stress on the impairment of energy metabolism induced by A beta peptides on PC12 cells: protection by antioxidants. Neurobiol Dis 6: 209-219. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong JS (2002) Microglia enhance beta-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J Neurochem 83: 973-983. [DOI] [PubMed] [Google Scholar]
- Samuni A, Krishna CM, Riesz P, Finkelstein E, Russo A (1988) A novel metal-free low molecular weight superoxide dismutase mimic. J Biol Chem 263: 17921-17924. [PubMed] [Google Scholar]
- Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB (1997) Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer's disease. J Neurosci 17: 4612-4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtzova EF, Kireeva EG, Bachurin SO (2001) Effect of beta-amyloid peptide fragment 25-35 on nonselective permeability of mitochondria. Bull Exp Biol Med 132: 1173-1176. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S (2000) Activation of NADPH oxidase in Alzheimer's disease brains. Biochem Biophys Res Commun 273: 5-9. [DOI] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR (1999) Glutamate-induced mitochondrial depolarization and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol (Lond) 519: 451-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR (2001) Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol (Lond) 531: 147-163. [DOI] [PMC free article] [PubMed] [Google Scholar]