

Abstract
Glutamate transporters are operative at an early developmental stage well before synapse formation, but their functional significance has not been determined. We now report that blockade of glutamate transporters in the immature neocortex generates recurrent NMDA receptor-mediated currents associated with synchronous oscillations of [Ca2+]i in the entire neuronal population. Intracerebroventricular injections of the blocker to pups generate seizures that are prevented by coinjections of NMDA receptor blockers. Therefore, the early expression of glutamate transporters plays a central role to prevent the activation by local glutamate concentrations of NMDA receptors and the generation of seizures that may alter the construction of cortical networks. A dysfunction of glutamate transporters may be a central event in early infancy epilepsy syndromes.
Keywords: cortex, epilepsy, network, synchrony, transport, N-methyl-d-aspartate
Introduction
In the adult brain, high-affinity Na+-dependent glutamate transporters control the concentration of glutamate in the synaptic cleft and terminate its actions (Danbolt, 2001). A tight control of the extracellular concentration of glutamate is of critical importance because an appropriate level of the transmitter is required for several physiological processes such as induction of synaptic plasticity (Katagiri et al., 2001), whereas its accumulation is potentially excitotoxic (Olney et al., 1969) and may generate epileptic seizures (Tanaka et al., 1997; Meldrum et al., 1999). In addition, by preventing diffusion of glutamate to distal sites, glutamate transporters enable a topic inflow of information and limit the activation of other excitatory synapses and a cooperative interaction between excitatory synapses (Diamond, 2001; Arnth-Jensen et al., 2002). Glutamate transporters also contribute to the synthesis of GABA via the supply of glutamate to GABAergic terminals (Sepkuty et al., 2002; Mathews and Diamond, 2003)
A very different situation prevails in the immature brain. Glutamate transporters are functional at an early stage when there are little or no synaptic connections (Juraska and Fifkova, 1979; Blue and Parnavelas, 1983a,b; Furuta et al., 1997; Owens and Kriegstein 2001; Demarque et al., 2002), suggesting that they may serve other roles than the modulation of synaptic transmission. In keeping with this, glutamate receptors, in particular NMDA receptors, are operative on migrating neurons that have no synaptic connections, and their activation or blockade modulate the speed of migration (Komuro and Rakic, 1993). To gain further insights on the function of glutamate transporters, we have now studied the effects of transporters blockade on the activity of immature neocortical neurons. We report that the application of the selective glutamate transporters inhibitor dl-threo-β-benzyloxyaspartate (dl-TBOA) generates in immature neocortical slices large NMDA receptor-mediated currents and [Ca2+]i oscillations in the entire neuronal population. A similar intracerebroventricular application to pups generates NMDA receptor-mediated seizures. Therefore, a disruption of glutamate transport may play a central role in the high incidence of seizures during childhood and infancy.
Materials and Methods
Wistar rats obtained from the day of birth [postnatal day (P) 0] to P5 were anesthetized with chloral hydrate, then decapitated, and brains were removed rapidly and placed in oxygenated ice-cooled artificial CSF (ACSF) with the following composition (in mm): 126 NaCl, 3.5 KCl, 2 CaCl2, 1.3 MgCl2, 25 NaHCO3 1.2 NaHPO4, and 10 glucose (95% O2 and 5% CO2, pH 7.4). Transverse neocortical slices (350–400 μm) were obtained with a vibratome (Micron HM 650V, Walldorf, Germany) and kept in oxygenated ACSF at room temperature at least 1 hr before use. Individual slices were then transferred to the recording chamber, where they were fully submerged and superfused with ACSF at 32–34°C at a rate of 2–3 ml/min.
Neurons were recorded under visual control with an axioscope (Zeiss, Oberkochen, Germany) or a DM LFS microscope (Leica, Nussloch, Germany) using the patch-clamp technique in the whole cell-configuration. Microelectrodes had a resistance of 5–10 MΩ and filled in the majority of the experiments with a solution containing (in mm): 120 KM2SO4, 20 KCl, 0.1 CaCl2, 1.1 EGTA, 10 HEPES, 4 Mg2+ATP, and 0.3 Na+GTP, pH 7.25; 270–280 MΩ. QX-314 was sometime added to the pipette solution to intracellularly block voltage-dependent Na+ channels. Some experiments have been performed with a CsCl-filled pipette solution (in mm): 120 CsCl, 10 CsGlu, 0.1 CaCl2, 1.1 EGTA, 10 HEPES, 4 Mg2+ATP, and 0.3 Na+GTP. Whole-cell measurements in the voltage-clamp or current-clamp mode were filtered at 3 kHz using an EPC-9 amplifier (Heka Elektronik, Lambrecht, Germany). All data were digitized (1–2 kHz) with a digidata (Axon Instruments, Foster City, CA) interface card to a personal computer and analyzed with a minianalysis program (Synaptosoft). To obtain a current–voltage curve (I/V), voltage ramps were applied for 2 sec from –70 to 20 mV. The I/V curves of slow oscillations (SOs) were constructed by subtracting the ramp response in control from that during the maximal current response. Extracellular recordings were performed with a glass pipette (<1 MΩ) filled with ACSF and placed in the same layer as the patch electrode, and the signal was recorded with a DAM80 amplifier (WPI).
Similar solutions and procedures were used for experiments performed on the entire neocortex, although the perfusion was increased to 8–10 ml/min (for more details, see Khalilov et al., 1997). Patch-clamp and extracellular recordings were performed with electrodes positioned at a depth of 150–200 μm from the surface.
In vivo experiments. Injection of TBOA was performed in P5 rats anesthetized with domitor (0.5 mg/kg; Orion Pharma, Espoo, Finland). The animal was then placed in a stereotaxic frame, and 0.5–1 μl of TBOA (20 ng/μl) was injected in the right lateral ventricle with a needle via a peristaltic pump (250 nl/min; WPI) at stereotaxic coordonates adapted from the atlas of Paxinos et al. (1991): anterior, –1; lateral, –1.8; H, –2.5. The animal was awakened with the antidote antisedan (1.25 mg/kg; Orion Pharma). The animal was immediately awakened after the injection of the antidote.
Ca2+ imaging. Cells were loaded with the Ca2+ indicator dye Fura-2 by bath application of Fura-2AM (Molecular Probes, Eugene, OR). For AM loading, slices were incubated in a small vial containing 2.5 ml of oxygenated ACSF with 25 μl of Fura2-AM (1 mm) solution (in 100% dimethylsulphoxyde; Molecular Probes) for 20–30 min. Slices were incubated in the dark, and incubation solution was maintained at 35–37°C. Fura-2-loaded slices were transferred to a recording chamber on the stage of a fluorescent upright microscope (Axioscope; Zeiss) equipped with a 380 nm excitation filter. Recordings of [Ca2+]i were imaged with 40× water immersion objectives at 32–34°C. Loaded slices were excited by an UV lamp connected to a personal computer at a rate of 0.5 Hz using Axobench software (Axon Instruments) at a wave-length of 380 nm. Fluorescent-emitted were collected through a reflecting pathway by an UV camera (Till Photonics). Images were analyzed on line with Axobench software. Pseudocolor images were coded so that blue indicated high level and red indicated low level of Ca2+. For each image, fluorescence intensity (F) was averaged over a delimited area of the soma. Changes in the fluorescence (ΔF) were measured as changes in baseline fluorescence and expressed as: %ΔF/F = [(Fpost – Frest)/] × 100.
Results
The experiments were performed on layer IV–V pyramidal cells in neocortical slices from P0–P5 rats. At that stage, synaptic connectivity is poorly developed (Juraska and Fifkova, 1979; Blue and Parnavelas, 1983a,b). Nevertheless, as described previously, spontaneous activity comprised spontaneous fast postsynaptic currents (PSCs) with a frequency of 0.95 ± 0.35 Hz (n = 50) that were sensitive to glutamatergic and GABAergic receptors antagonists (for review, see Owens and Kriegstein, 2001).
dl-TBOA (30 μm) was used to reduce the activity of the main glutamate transporters present in the neocortex of postnatal rat pups: the neuronal EAAC1 and glial GLT1 and glutamate-aspartate transporter (GLAST) transporters (Furuta et al., 1997, Ullensvang et al., 1997). TBOA has several advantages compared with other glutamate transporters inhibitors: (1) it is a nontransportable inhibitor and, thus, does not induce an artificial release of the transmitter through hetero-exchange; and (2) it does not act as a partial glutamate receptor agonist (Shimamoto et al., 1998; Jabaudon et al., 1999).
Blockade of glutamate transporters generates synchronized SOs
Bath application of TBOA (30 μm) generated within a few seconds (126 ± 23 sec) slow current oscillations (in the voltage-clamp mode) or depolarizations associated with action potentials (in the current-clamp mode) in 44 of 50 cells (Fig. 1A). SOs presented the following features: mean amplitude of 99 ± 16 pA (in the voltage-clamp mode at Vh =–65 mV); mean duration, 21 ± 2 sec (range, 5–70); mean oscillation frequency, 0.015 ± 0.003 Hz. They were associated with a large significant increase in the frequency of PSCs (14.3 ± 1.8 Hz; p < 0.001; ANOVA) (Fig. 1A,C). SOs washed out when TBOA application was stopped and reinstated with additional applications. When recorded with extracellular field electrodes, the frequency of events within SOs ranged between 7 and 30 Hz (mean, 15.8 ± 3 Hz; n = 20) (Fig. 1A), indicating that the blockade of glutamate transporters synchronized the activity of immature neurons in a theta–low gamma range. SOs were network driven as they were blocked by TTX (1 μm) (n = 11 of 11; data not shown) but not by QX-314 the intracellular Na+ channel blocker Lower concentrations of TBOA (<10 μm) did not generate SOs, whereas higher concentrations of TBOA (>100 μm) to fully block transporters (Shimamoto et al., 1998) generated one or two oscillations, followed by a long-lasting NMDA-mediated inward current (n = 11) (Fig. 1D,E). These effects were prevented by the addition of Cd2+ (100–200 μm), the voltage-dependent Ca2+ blocker (n = 7of7), suggesting that these actions are mediated by transmitter release and not by a direct activation of NMDA receptors. In keeping with this observation, applications of NMDA generates a current in the presence of Cd2+ (n = 4 of 4; data not shown). Therefore, TBOA generates network-driven oscillations in a dose-dependent manner.
Figure 1.

TBOA generates SOs mediated by NMDA receptors. A1, Whole-cell recording of a P3 pyramidal neuron from layer V of the somatosensory cortex combined with an extracellular recording of the same layer. Bath application of TBOA (30 μm) generates current oscillations (top trace) or voltage oscillations (middle trace) that are associated with extracellular activities (E.C.; bottom trace). SOs did not persist after the washout of TBOA and reappeared with additional application of the blocker. A2, Magnification of SOs indicated by letters in A1. B, Relationship of SOs to the voltage (I/V). Voltage ramps were performed from –70 to 20 mV (with a pipette filled with KM2SO4 plus QX-314 to block action potential in the recorded cell) before and at the peak of SOs (top trace). The graph represents the difference of the two I/V curves (indicated by numbers). C, Representative histogram in which the number of synaptic events was plotted versus time. d-APV (80 μm) blocks the SOs and the associated increase in the frequency of synaptic events. D, Long-lasting inward current induced by TBOA (100 μm). E, Pyramidal neuron recorded in the current-clamp mode (the pipette contained QX-314) from another experiment showing that the long-lasting depolarization generated by 100 μm TBOA is sensitive to APV.
To further emphasize that SOs are generated by a synchronous activation of cortical neurons, we then performed Ca2+ imaging to visualize the activity of individual neurons simultaneously. Slices were incubated with the Ca2+-sensitive dye Fura2-AM, and fluctuations of [Ca2+]i levels were monitored simultaneously with patch-clamp recordings of neurons. As shown in Fig. 2, application of TBOA induced a synchronous rise in [Ca2+]i in all imaged cells. These calcium events were synchronous with the SOs recorded in the voltage-clamp mode or current-clamp mode (n = 4 of 4 slices). Therefore, partial blockade of glutamate transporters periodically synchronizes cortical neuron activity.
Figure 2.
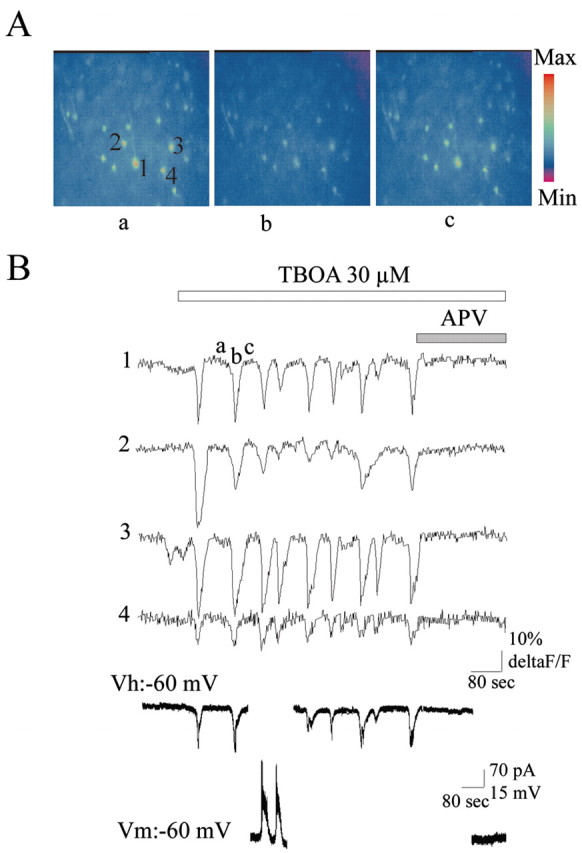
SOs are associated with an APV-sensitive synchronous rise of [Ca2+]i in pyramidal neurons. A, Fura-2AM Ca2+ imaging of layer V of a P5 somatosensory cortex. The three images (objective, 40×) correspond to the Fura-2 Ca2+ imaging before an oscillation (a), at the peak of the oscillation (b), and at the end of the oscillation (c). B, Traces of the time course of the fluorescence intensity of the four cells selected by numbers and concomitant with the whole-cell recording of a pyramidal neuron (in the same layer but out of focus) in the voltage clamp and current clamp. Ca2+ rise is shown as a decrease in the fluorescence.
SOs are mediated by NMDA receptors
The following observations indicate that SOs are mediated by NMDA receptors: (1) d-APV (80 μm), a specific NMDA receptor antagonist, blocked the oscillations observed in intracellular, as well as in extracellular, recordings or in Ca2+ imaging (Figs. 1C, 2B) (n = 5); and (2) the current-voltage relationship of SOs showed a region of negative slope at hyperpolarized membrane potentials and reversed polarity close to 0 mV (3 ± 2 mV; n = 5) (Fig. 1B). However, AMPA/kainate receptors were also involved in their generation because SOs were blocked by the AMPA/kainate receptor antagonist CNQX (20 μm) (n = 5; data not shown). In contrast, the GABAA receptor antagonist bicuculline (50 μm) blocked neither SOs nor the associated Ca2+ rise, suggesting that GABAA receptors do not contribute to their generation (n = 5; data not shown).
Bath application of NMDA generates oscillations
If SOs are triggered by the activation of glutamate receptors, then bath applications of their agonists would produce similar effects. Bath application of NMDA (10 μm) generated current or voltage oscillations, albeit with a higher frequency (0.43 ± 0.07 Hz; n = 15) and smaller duration and amplitude (2 ± 0.4 sec and 48.9 ± 18.3 pA; n = 15) than SOs (Fig. 3A,B). These oscillations were generated by the synchronous activation of neurons as demonstrated by the presence of concomitant extracellular field activity. These oscillations were mediated by glutamate receptors as they reversed polarity at 0 mV (Fig. 3C) and were blocked by APV or CNQX but not by bicuculline (Fig. 3D). Moreover, as SOs, these oscillations were driven by the network because they were blocked by TTX (Fig. 3E). NMDA failed to generate oscillations at lower concentrations (3 μm), whereas higher concentrations of NMDA (30 μm) generated a large inward current without intracellular and extracellular field oscillations (Fig. 3A). This was reminiscent of the results obtained with TBOA and suggested that the synchronization of cortical network activity required appropriated levels of NMDA receptor activation. Applications of AMPA (1–3 μm), kainate (100–300 nm), and (±)-1-Aminocyclopentane-trans-1,3-dicarboxylic acid (100–200 μm), which activate AMPA, kainate, and metabotropic glutamate receptors, respectively, did not generate any oscillation even when applied together (n = 15; data not shown). Altogether, these data reinforce the predominant role played by NMDA receptors in TBOA-induced SOs.
Figure 3.

NMDA generates synchronous network-driven oscillations. A1, Bath application of NMDA (10 μm) generated current oscillations (top traces) associated with extracellular activities (bottom traces). At 30 μm, NMDA generated a large inward current but no oscillations. A2, Magnification of the three intracellular and extracellular recordings indicated by letter sinA1. B, Oscillations recorded in the current-clamp mode. C, Oscillations recorded at a different membrane potential with a pipette filled with KM2SO4. D, E, Oscillations were blocked by CNQX and TTX but not by bicuculline.
TBOA generates SOs in the intact neocortex in vitro and seizures in pups
To determine whether SOs were region specific, we used the intact neocortex preparation developed by Khalilov et al. (1997) (Fig. 4A). Bath applications of TBOA (30 μm) generated SOs in the frontal, parietal, and occipital cortices with similar features as those recorded in slices [inter-SO frequency, 0.011 ± 0.001 Hz; duration, 18 ± 3 sec (range, 7–54); frequency of events within the SOs, 15 ± 2Hz and full blockade by APV; n = 8) (Fig. 4B1). There is no preferential generating site because SOs could be initiated in any cortical region (Fig. 4B2). Therefore, the networks present in any region of the neocortex are capable of generating SOs that may propagate to other regions.
Figure 4.

SOs are generated in the entire intact neocortex. A, Photograph of the experimental device. The entire cortex of one hemisphere from a P5 rat is placed in the recording chamber with the three recording electrodes. The whole-cell pipette (W.C.) is in the occipital cortex, the extracellular electrode E.C1 is in the frontal cortex, and E.C2 is in the parietal cortex. B1, Bath application of TBOA-induced SOs at the three recordings sites. These oscillations were blocked by the addition of d-APV. B2, Magnification of two sets of oscillations. Note that SOs could be initiated in any cortical region and occurred in the three regions within the seconds range.
Finally, we tested the effects of intracerebroventricular injection of TBOA to pups to determine the consequences of reducing transporters activity in freely moving animals. TBOA (20 ng in 1 μl of ACSF) was injected under deep anesthesia into the right lateral ventricle of P5 rats (see Materials and Methods). The animals were then monitored by videotape. This treatment consistently generated in 12 of 13 pups recurrent stereotyped seizures during a period of 2 hr after the injection (see supplemental video at http://www.inmed.univ-mrs.fr/resources/Demarque2004.avi). Immediately after arousal, the animals displayed myoclonus of anterior or posterior limbs, followed by a tonic extension of the four limbs frequently associated with head bobbing. This type of seizure occurred every 3–7 min and lasted for 30–90 sec. Animals injected with the vehicle alone (n = 4) did not display seizures. Parallel injections of APV (15 ng in 1 μl) fully prevented the effects of TBOA, because the pups either showed no seizures (n = 6 of 8 pups) or displayed a single episode during the 2 hr after the injection (n = 2 of 8 pups). Therefore, in vivo, reduction of glutamate transporter activity generates seizures through the activation of NMDA receptors. These seizures are reminiscent of those reported by Kabova et al. (1999) with direct intraperitoneal injection of NMDA.
Discussion
Our results suggest that high-affinity glutamate transporters play an important role in the regulation of intercellular communication at an early developmental stage when neurons have few functional synapses. In vitro, blockade of the transporters persistently altered the ongoing neuronal activity and replaced it with SOs. SOs are network-driven events and are associated with Ca2+ oscillations in the entire neuronal population. They are triggered by glutamate and by activation of NMDA receptors. In vivo, blockade of glutamate transporters generates seizures also triggered by NMDA receptor activation. Thus, the early function of glutamate transporters is an important developmental feature that allows the expression of physiological activities while preventing abnormal synchronization of neuronal activities and seizures.
Mechanism of TBOA-generated SOs
The activation of NMDA receptors is a central feature in the synchronization of the maturing network, because SOs are fully blocked by NMDA receptor antagonists and bath application of NMDA produces oscillations. Furthermore, SOs and NMDA-induced oscillations share similar pharmacological properties, including their sensitivity to TTX but not to intracellular blockade of Na+ channels. This indicates that oscillations are driven by the network and not by intrinsic oscillatory mechanisms. This contrasts with the effect of the sustained NMDA receptor activation reported in other studies (Hu and Bourque, 1992; Tell and Jean, 1993; Hochmann et al., 1994). Thus, the mechanism leading to the synchronization of the network we describe would involve an elevation of extracellular glutamate concentration and the activation of synaptic (and possibly extrasynaptic) NMDA receptors leading to the generation of bursts of action potentials. These bursts may in turn lead to the release of glutamate and the activation of glutamate receptors of target neurons. Mechanisms implicated in the generation of the oscillatory pattern remain to be clarified. A periodic desensitization of NMDA receptors is unlikely because applications of higher concentrations of TBOA or NMDA generated a long-lasting inward current. We propose that SOs are triggered by a cyclic modulation of glutamate levels in the extracellular space by mechanisms that could include: (1) a presynaptic inhibition of the release of glutamate (MacDermott et al., 1999); (2) a transient release of glutamate by the reversal of transporters unaffected by TBOA (30 μm). This reversion may occur as a consequence of a sustained uptake and the dissipation of electrochemical gradients (Danbolt, 2001); or (3) a cyclic pattern of glutamate release. Glial cells may contribute to this pattern because they display spontaneous Ca2+ oscillations with frequency and duration in the range of SOs and they release glutamate (for review, see Auld and Robitaille, 2003).
TBOA induces epilepsy
There is abundant, but quite contradictory, literature concerning the possible role of transporters in the epilepsies. Thus, in surgical resections from human epileptic structures as well as in animal models, an increase or a decrease of transporter expression has been reported (Meldrum et al., 1999; Crino et al., 2002; Proper et al., 2002). Experiments using knock-out mice for transporters or chronic treatment with antisense have also led to conflicting observations because in some studies, but not others, seizures were generated. Thus, GLT1 knock-out mice display spontaneous recurrent seizures or are more susceptible to pentamethylenetetrazole, which has been explained by an elevation of extracellular glutamate concentration (Tanaka et al., 1997), whereas chronic administration of antisense nucleotide of GLT1 or GLAST in adult rats produced elevated extracellular levels of glutamate associated with excitotoxicity but not seizures (Rothstein et al., 1996). These contradictory results also applied to the neuronal EAAC1 transporter [i.e., whereas nucleotide antisense treatment resulted on epileptic seizures as a possible consequence of a decrease in GABA synthesis (Sepkuty et al., 2002), the EAAC1 knock-out mice have a reduced spontaneous locomotor activity but do not develop seizures (Peghini et al., 1997)]. Also, cortical or hippocampal slices obtained from these mice did not show synchronous network activity that is the hallmark of epilepsy. In the present study, we used an acute treatment that reduces the three main transporters. Our data suggest that seizures observed in vivo are related to the blockade of transporters and to SOs that may represent ictal-like activity. Thus, (1) the clinical manifestation is immediate, like the generation of SOs observed in slices or in the entire neocortex, (2) the range of SO duration matches the seizure manifestation observed in vivo, (3) SOs are observed in all cortical areas and are generalized like seizures, and (4) SOs and seizures are blocked or prevented by APV. Our data also indicate that the synchronization of network activity and seizures may not be related to a decrease of GABA synthesis or GABA efficiency (Sepkuty et al., 2002) because an increase of spontaneous PSCs was observed during SOs that were also resistant to bicuculline. A fundamental property of the developing network is its capacity to oscillate (Ben-Ari, 2001), and the accumulation of glutamate that results from blockade of transporters leads to the activation of NMDA receptors and the synchronization of networks. These combined factors underlie the low threshold of seizure generation in the developing cortex. Therefore, dysfunction of glutamate transporters and the resulting increase of glutamate may play an important role in infantile epilepsies possibly by the activation of NMDA receptors. Additional studies are required to evaluate the clinical relevance of these observations in view of the excitotoxicity produced by the activation of NMDA receptors and the apoptotic damage produced by NMDA receptor antagonists (Ikonomidou et al., 1999; Olney, 2002).
Footnotes
This work was supported by Fondation Cino and Simone Del Duca. M.D. is a fellow of the Ministère de la Recherche and of the Fondation de la Recherche Médicale. We thank Drs. Valérie Crépel and Rosa Cossart for helpful discussion.
Correspondence should be addressed to Dr. Laurent Aniksztejn, Institut de Neurobiologie de la Méditerranée/Institut National de la Santé et de la Recherche Médicale U29, Parc Scientifique de Luminy Boilé Postale 13, 13273 Marseille cedex 09, France. E-mail: anik@inmed.univ-mrs.fr.
Dr. N. Villeneuve's present address: Hôpital de la Timone, Service de Neurophysiology Clinique, 264 rue Saint-Pierre, 13005 Marseille, France.
DOI:10.1523/JNEUROSCI.5338-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243289-06$15.00/0
References
- Arnth-Jensen N, Jabaudon D, Scianziani M (2002) Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci 5: 325–331. [DOI] [PubMed] [Google Scholar]
- Auld DS, Robitaille R (2003) Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron 40: 389–400. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y (2001) Developing networks play a similar melody. Trends Neurosci 24: 353–360. [DOI] [PubMed] [Google Scholar]
- Blue ME, Parnavelas JG (1983a) The formation and maturation of synapses in the visual cortex of the rat. II Qualitative analysis. J Neurocytol 12: 599–616. [DOI] [PubMed] [Google Scholar]
- Blue ME, Parnavelas JG (1983b) The formation and maturation of synapses in the visual cortex of the rat. II Quantitive analysis. J Neurocytol 12: 697–712. [DOI] [PubMed] [Google Scholar]
- Crino PB, Jin H, Shumate MD, Robinson MB, Coulter DA, Brooks-Kayal AR (2002) Increased expression of the neuronal glutamate transporter (EAAT3/EAAC1) in hippocampal and neocortical epilepsy. Epilepsia 43: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1–105. [DOI] [PubMed] [Google Scholar]
- Demarque M, Represa A, Becq H, Khalilov I, Ben-Ari Y, Aniksztejn L (2002) Paracrine intercellular communication by a Ca2+and SNARE independent release of GABA and glutamate prior to synapse formation. Neuron 36: 1051–1061. [DOI] [PubMed] [Google Scholar]
- Diamond JS (2001) Neuronal glutamate transporter limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci 21: 8328–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17: 8363–8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmann S, Jordan LM, Schmidt BJ (1994) TTX resistant NMDA receptor mediated voltage oscillations in mammalian lumbar motoneurons. J Neurophysiol 72: 2559–2562. [DOI] [PubMed] [Google Scholar]
- Hu B, Bourque CW (1992) NMDA receptor-mediated rythmic bursting activity in rat supraoptic nucleus neurones in vitro. J Physiol (Lond) 458: 667–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283: 70–74. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler B, Gerber U (1999) Inhibition of uptake unmasks rapid extracellular turnover of glutamate non vesicular origin. Proc Natl Acad Sci USA 96: 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juraska JM, Fifkova E (1979) An electron microscope study of the early postnatal development of the visual cortex of the hooded rat. J Comp Neurol 183: 257–267. [DOI] [PubMed] [Google Scholar]
- Kabova R, Liptakova S, Slamberova R, Pometlova M, Velisek L (1999) Age-specific N-methyl-d-aspartate-induced seizures: perspectives for the West syndrome model. Epilepsia 40: 1357–1369. [DOI] [PubMed] [Google Scholar]
- Katagiri H, Tanaka K, Manabe T (2001) Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci 14: 547–553. [DOI] [PubMed] [Google Scholar]
- Khalilov I, Esclapez M, Medina I, Aggoun D, Lamsa K, Leinekugel X, Khazipov R, Ben-Ari Y (1997) A novel in vitro preparation: the intact hippocampal formation. Neuron 19: 743–749. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P (1993) Modulation of neuronal migration by NMDA receptors. Science 260: 95–97. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA (1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu Rev Neurosci 22: 443–485. [DOI] [PubMed] [Google Scholar]
- Mathews GS, Diamond JS (2003) Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci 23: 2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS, Akbar MT, Chapman AG (1999) Glutamate receptors and transporters in genetic and acquired models of epilepsy. Epilepsy Res 36: 189–204. [DOI] [PubMed] [Google Scholar]
- Olney JW (2002) New insights and new issues in developmental neurotoxicology. Neurotoxicology 23: 659–668. [DOI] [PubMed] [Google Scholar]
- Olney JW, Sharpe LG (1969) Brain lesions in an infant rhesus monkey treated with monosodium glutamate. Science 166: 386–388. [DOI] [PubMed] [Google Scholar]
- Owens DF, Kriegstein AR (2001) Maturation of channels and receptors: consequences for excitability. Int Rev Neurobiol 45: 43–87. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Törk I, Tecott LH, Valentino KL (1991) Atlas of the developing rat brain. San Diego: Academic.
- Peghini P, Janzen, Joffel W (1997) Glutamate transporter EAAC-1 deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J 16: 3822–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN, et al. (2002) Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 125: 32–43. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxycity and clearance of glutamate. Neuron 16: 675–686. [DOI] [PubMed] [Google Scholar]
- Septuky JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, Coulter DA, Rothstein JD (2002) A Neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci 22: 6372–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998) dl-threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195–201. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 267: 1699–1702. [DOI] [PubMed] [Google Scholar]
- Tell F, Jean A (1993) Ionic basis for endogenous rythmic pattern induced by activation of NMDA receptors in neurons of the rat nucleus tractus solitarii. J Neurophysiol 70: 2379–2390. [DOI] [PubMed] [Google Scholar]
- Ullensvang K, Lehre KP, Storm-Mathisen J, Danbolt NC (1997) Differential developmental expression of the two rat brain glutamate transporter proteins GLAST and GLT. Eur J Neurosci 9: 1646–1655. [DOI] [PubMed] [Google Scholar]