

Abstract
The non-inactivating potassium M-current exerts a strong influence on neuronal excitability. The channels responsible for this current are made up of KCNQ subunits, and mutations in most of these produce human pathologies. Notably, in terms of excitation, mutations in either KCNQ2 or KCNQ3 lead to benign neonatal familial convulsions. Although a mere reduction of 25% in KCNQ2/3 function can increase excitability to epileptogenic levels, the potentiation of these subunits has anti-epileptogenic effects. After KCNQ2/3 heteromerization, current levels can augment as much as 10-fold, and we have discovered that there are three processes underlying this potentiation. First, there is an increase in the number of channels inserted in the membrane after heteromerization of the C-terminal region. Second, the N-terminal domain from KCNQ2 exerts a negative influence on the current level. Finally, Ala 315 of KCNQ3, a residue located in the inner vestibule after the selectivity filter, plays a critical role in preventing current flow in KCNQ3 homomeric channels, whereas it is permissive in heteromers in combination with Thr at the equivalent 276 position of KCNQ2.
Keywords: Kv7, KCNQ, M-current, epilepsy, pore, surface expression
Introduction
Although there is a remarkable redundancy on the function “K+ permeation,” with >75 genes encoding potassium channels in the human genome (Gutman et al., 2003), mutations on some K+ channels provoke diseases (potassium channelopathies), indicating that their precise function cannot be completely replaced by other channels (Hubner and Jentsch, 2002). Approximately 10 potassium channel genes are known to be mutated in human disease, and, interestingly, four of these genes encode potassium channels of the Kv7 family, also known as KCNQ (Jentsch, 2000). Mutations in KCNQ1 (Kv7.1) lead to cardiac arrhythmia (Wang et al., 1996) and congenital deafness (Neyroud et al., 1997). Mutations in KCNQ4 (Kv7.4) produce a dominant form of progressive hearing loss (Kubisch et al., 1999). Finally, mutations in either KCNQ2 (Kv7.2) or KCNQ3 (Kv7.3) cause the same phenotype, benign familial neonatal convulsions (BFNC), a dominantly inherited epilepsy of the newborn (Biervert et al., 1998). Furthermore, some mutations in KCNQ2 produce a syndrome in which BFNC is followed later in life by myokimia (Dedek et al., 2001).
KCNQ2 and KCNQ3 can form heterotetramers that are expressed mainly in the nervous system (Biervert et al., 1998). They give rise to the “M-current” (Wang et al., 1998), which is active near the threshold of action potential firing and is inhibited by stimulating muscarinic acetylcholine receptors and other receptors that activate phospholipase C (Brown and Yu, 2000). Voltage dependency, slow kinetics, and non-inactivation are key properties that allow the M-type current to control neuronal excitability (Marrion, 1997). Furthermore, much attention has been paid to M-channel openers as possible targets for the treatment of neuronal disorders such as epilepsies, acute pain, neuropathic pain, migraine pain, and some neurodegenerative and psychiatric disorders (Gribkoff, 2003).
The importance of this current in neuronal excitability is illustrated by the fact that a functional impairment of <25% of KCNQ2/KCNQ3 heteromers appears to be sufficient to drive neurons to epileptogenic levels of excitability and causes BFNC in affected families (Schroeder et al., 1998). This observation also highlights the physiological importance of maintaining the activity of M-channels above a certain functional threshold. However, to date, limited information is available regarding the mechanisms that regulate the levels of M-channel activity. It is known that the number of channels at the membrane increases when KCNQ2 and KCNQ3 are coexpressed together in heterologous systems. This increase in the number of channels is paralleled by a concomitant increase in the current levels, leading to the proposal that it is at the basis of current potentiation (Schwake et al., 2000). We have investigated this issue in detail and show here that there are at least three mechanisms at work. Surprisingly, the increase in the number of channels plays only a secondary role.
Materials and Methods
Chimeric KCNQ2/KCNQ3 channels. Human KCNQ2 (Y15065) and KCNQ3 (NM004519) cDNAs were kindly provided by Thomas Jentsch (Zentrum für Molekulare Neuropathobiologie, Hamburg, Germany). Silent restriction sites were introduced, and chimeric KCNQ2/KCNQ3 subunits were constructed by exchanging regions in KCNQ3 for the equivalent ones in KCNQ2 by site-directed cloning or by overlapping PCR. A SacII restriction site was introduced within the codons for amino acids P89/R90 of KCNQ2 and P118/R119 of KCNQ3 and a BamH I site within the codons for G310-L312 of KCNQ2 and G349-L351 of KCNQ3, at the beginning and at the end of the transmembrane region, respectively. When no restriction sites were available, the chimeras were constructed using a multistep PCR protocol using both subunits as templates. Point mutants were generated by PCR-based mutagenesis changing KCNQ3 residues for the equivalent ones in KCNQ2 and inversely. The amplified chimeric cassettes were confirmed by sequencing and replaced in a KCNQ3 backbone. Silent sites were introduced to facilitate the screening of positive clones.
Channel expression and electrophysiology. Channel subunits were flanked by the non-coding 5′ region of the globin gene of Xenopus and the 3′ polyA of SV40. After linearization of the cDNAs with ApaL I, the cRNAs were synthesized in vitro with the T7 transcription kit (Ambion, Austin, TX). Oocyte preparation and electrophysiological recordings were performed as described previously (Villarroel and Schwarz, 1996). Stage V or VI oocytes were defolliculated enzymatically with collagenase (C9891; Sigma, Madrid, Spain), 1 mg/ml in Ca2+-free OR2, made with (in mm) 82.5 NaCl, 2.5 KCl, 1 MgCl2, and 5 HEPES, pH 7.5, and then were transferred to a Ca2+-containing solution, ND96, consisting of (in mm) 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.5. The oocytes were injected with 50 nl of KCNQ2 or KCNQ3 cRNA solution, containing ∼10 ng of KCNQ2 or KCNQ3 cRNA or 10 ng of a 1:1 mixture in coexpression experiments.
Three days after injection, whole-cell currents were recorded in oocytes at room temperature (∼22°C) with a two-electrode voltage clamp using a virtual-ground Geneclamp 500B amplifier (Axon Instruments, Foster City, CA). Borosilicate electrodes were filled with 3 m KCl and had resistances of ∼1 MΩ. The oocytes were perfused continuously in Xenopus saline made with (in mm) 100 NaCl, 2.5 KCl, 1 MgCl2, 2 MnCl2, and 5 HEPES, pH 7.5. All chemicals, except stated otherwise, were from Sigma. Data were acquired at a sampling rate of 1 KHz and filtered at 100 Hz; voltage-step protocols and current analysis were performed with pCLAMP 8.1 software (Axon Instruments).
Current values represent the average (±SE) of the maximal conductance obtained from the fit of the g-V relationship to a Boltzmann equation:
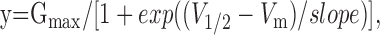 |
where Gmax = Imax/(Vstep - Vrev), Imax is the maximal current, Vrev is the reversal potential (-87.3 ± 0.34 mV; n = 387; in our recording conditions), y is the tail current amplitude in an 800 msec voltage step to -20 mV (Vstep) elicited after 800 msec depolarizing voltage pulses from -120 to +50 mV in 10 mV increments (Vm) evoked every 10 sec, and V1/2 is the membrane potential at which the current is half Imax. To get the absolute amplitude, the tail current values were subtracted with the amplitude of the tail current preceded by a voltage step to -120 mV. The Gmax value was normalized to the averaged Gmax obtained the same day using five or more oocytes expressing KCNQ2/KCNQ3 heteromers.
To monitor the assembly of different subunits, the sensitivity to the blockage by tetraethylammonium (TEA) was measured (Hadley et al., 2003). Currents were elicited by an 800 msec depolarizing step from -60 to -20 mV every 20 sec. The percentage of inhibition was represented against the log [TEA], and curves were fitted with the Hill equation:
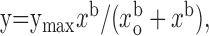 |
where y is the percentage of inhibition, ymax is full inhibition, x is the concentration of the blocker, xo is the IC50 value (the concentration at which inhibition reaches 50%), and b is the power equivalent to the Hill slope. Data were normalized in Excel and plotted in SigmaPlot. For statistical evaluation, unpaired Student's t test was applied.
Surface expression. We followed the method described by Schwake et al. (2000), using the KCNQ3 subunit tagged with a hemagglutinin (HA) epitope in the extracellular loop that connects transmembrane domains S1 and S2 (kindly provided by T. Jentsch). Oocytes were injected with 20 ng of a 1:1 cRNA mixture that always included 10 ng of KCNQ3-HA. After 3 d at 19°C, the oocytes were placed in ND96 with 1% BSA (ND96/BSA) at 4°C for 30 min to block unspecific binding. Subsequently, the oocytes were incubated in ND96/BSA for 60 min at 4°C with 1 μg/ml rat monoclonal anti-HA antibody (3F10; Roche Diagnostics, Barcelona, Spain), washed with ND96/BSA, and incubated in ND96/BSA with HRP-coupled secondary antibody for 60 min (goat anti-rat antigen binding fragments; Jackson ImmunoResearch, West Grove, PA). Finally, the oocytes were washed thoroughly with ND96/BSA and then with ND96 alone to eliminate the background signal that BSA produces. Individual oocytes were placed in 50 μl Power signal ELISA solution (Pierce, Rockford, IL), and chemiluminescence was quantified in a Sirius luminometer (Berthold, Pforzheim, Germany).
Nomenclature. In the figures representing the chimeric channels, black is used for regions originating from KCNQ2 and white for those from KCNQ3 (see Fig. 1 B). The transmembrane region is depicted as a box between two lines, which represent the N and C termini. These regions are referred to as T, N, or C, followed by the number of the original subunit, 2 or 3. Bold characters are used for segments originated from KCNQ2, whereas characters are underlined to indicate regions from KCNQ3. For example, KCNQ2 will be represented as N2T2C2 or often as Q2 to abbreviate. Similarly, KCNQ3 will be referred to as N3T3C3 or Q3. In some chimeras, the pore region was exchanged (aa 253-291 and 282-330 in Q2 and Q3, respectively), and it is referred to as P. For instance, when the pore region of KCNQ3 is replaced, the resulting construct is denominated N3T3P2C3.
Figure 1.

The transmembrane region is critical for Q2/Q3 potentiation. A, Current records evoked by 800 msec voltage steps to +50 mV from a holding potential of -50 mV of Q2, Q3, and Q2/Q3 channels. B, Schematic representation of the subunits. The figure indicates the amino acid boundaries for the three portions: N terminus (N), C terminus (C), and transmembrane region (T). The transmembrane region is depicted as a box; segments from Q2 are shaded in black, and those from Q3 are shaded in white. C, Left, Schematic representation of chimeric Q2/Q3 constructs. The averaged normalized maximal conductance obtained after fitting a Boltzmann distribution to g-V relationships (E) from tail currents measured at -20 mV (G) of the constructs indicated were coexpressed with Q3 in a 1:1 ratio (n ≥ 10 from 2 or more batches of oocytes). D, Normalized surface expression of HA-tagged Q3 subunits after coexpression with the constructs indicated on the left (n ≥ 15). Heteromerization of the C terminus increased the number of Q3 subunits at the membrane. The differences in the mean surface expression of the first four groups were not statistically significant. E, Representative conductance-voltage relationship of tail currents measured in 800 msec current traces at -20 mV for Q2/Q3 heteromers, Q3 homomers, and selected chimeric constructs coexpressed with Q3. Boltzmann distributions were fitted to the data (continuous lines). The averaged Boltzmann parameters were: Q2+Q3 (gray circles): V1/2 = -28.68 ± 0.53 mV, slope = 10.4 ± 0.15 mV (n = 118); N3T2C2+Q3 (open triangles): V1/2 =-32.79 ± 2.26 mV, slope = 10.3 ± 0.63 mV (n = 11); N2T2C3+Q3 (open squares): V1/2 = -22.02 ± 1.07 mV, slope = 11.5 ± 0.69 mV (n = 11); N3T2C3+Q3 (filled triangles): V1/2 =-19.80 ± 1.50 mV, slope = 12.6 ± 0.72 mV (n = 9). F, Dose-response curves for TEA blockage of Q2 (filled circles; n = 6), Q3 (empty circles; n = 4), Q2/Q3 (gray circles; n = 15). The solid lines are the result of fitting the Hill equation to the data with an IC50 value of ∼0.16 mm for Q2, ∼4.1 mm for Q2+Q3, and >200 mm for Q3. The data for Q3 was obtained with the A315T mutant to clearly evoke measurable currents (see Fig. 2C). G, Representative collection of 800 msec tail currents recorded at -20 mV evoked after 800 msec voltage steps from -120 to +50 mV evoked in oocytes held at -50 mV. The vertical bar represents 2.0 μA, except for N3T2C2+Q3 (open triangle), where it represents 6.0 μA.
Results
The transmembrane domain is critical for potentiation
When KCNQ2 and KCNQ3 form heteromers, an increase in current has been detected in many expression systems (Wang et al., 1998; Selyanko et al., 2001). This potentiation is particularly dramatic in Xenopus oocytes, leading us to examine this phenomenon in this expression system in which only modest currents are obtained when KCNQ3 or KCNQ2 subunits are expressed alone, and there is a remarkable 10-fold increase when both subunits are expressed together (Fig. 1A) (Wang et al., 1998). To investigate the mechanisms underlying this potentiation, we used chimeric KCNQ2/3 channels to identify the molecular determinants involved.
The topology of KCNQ (Kv7) channels is similar to that of other Kv channels. It consists of intracellular N- and C-terminal regions, six transmembrane segments, and a loop between S5 and S6 that forms the pore (P). We conceptually divided the channel into three portions: N terminus, transmembrane, and C terminus (Fig. 1B). We will refer to these regions as N, T, or C, followed by the number of the original subunit, 2 or 3 (see Materials and Methods).
Chimeric subunits of the six possible combinations of these three segments were constructed and coexpressed with KCNQ3 (Fig. 1C). Because KCNQ3 alone does not generate currents above background levels, any current recorded will flow through heteromeric channels or homomeric chimeric channels. Both alternatives can be distinguished based on the differential sensitivity to the potassium channel blocker TEA (see below and Fig. 1F). The maximal conductance (Gmax) was estimated by fitting a Boltzmann distribution to the g-V relationships, which was averaged and normalized to Q2+Q3 control values obtained from the amplitudes of the tail currents measured at -20 mV (Fig. 1E,G). In this way, the analysis became insensitive to shifts in the voltage dependency caused by the mutations.
Only three chimeras yielded currents that were clearly above background levels, and all contained the Q2 transmembrane segment (N3T2C2, N2T2C3, N3T2C3) (Fig. 1C,G). The sensitivity of these three chimeras to TEA, which depends on a key pore residue located within the transmembrane segment, was intermediate to that of homomeric Q2 or homomeric Q3 channels and was indistinguishable to that of Q2/Q3 heteromers (Fig. 1F). This indicates that these three chimeras efficiently assembled with Q3, forming heteromeric channels (Hadley et al., 2003). The failure to obtain significant current with the remaining three chimeras (N2T3C3, N3T3C2, N2T3C2) was not attributable to a gross defect in their biogenesis because currents were obtained when they were coexpressed with Q2 (see Fig. 3B) with intermediate TEA sensitivity (data not shown). Thus, a heteromeric transmembrane configuration is required for potentiation.
Figure 3.

The N-terminal region regulates the degree of potentiation. A, Normalized maximal conductance of the chimeras indicated on the left coexpressed with Q3. The data from Figure 1C is replotted here to highlight the effect of the N terminus. B, Normalized maximal conductance of the chimeras indicated on the left coexpressed with Q2 (n > 8). Statistically significant differences are indicated by asterisks: *p < 0.05; **p < 0.01; ***p < 0.001.
This result was unexpected because previous studies have suggested that an increase in surface expression mediated by the C terminus is the most likely basis for current potentiation (Schwake et al., 2000). We therefore examined how the different segments affect the number of KCNQ3 subunits at the membrane and found that the three chimeras containing the C terminus from Q2 stimulated Q3 surface expression, although to a lesser extent than Q2 itself (Fig. 1D). This small difference suggests that other regions play a minor role in the export of the channels to the membrane. Remarkably, the results show that the number of channels at the membrane does not correlate with the current flow and that the transmembrane segment is the most decisive region for current potentiation.
A residue in the inner pore vestibule is critical for the potentiation
We generated additional chimeras to further analyze the transmembrane domain. The chimera N3T3P2C3 (Q3/Q2253-291), in which the pore region (P) was exchanged, potentiated the current even more than Q2, whereas N2T2P3C2 (Q2/Q3282-349) and other chimeras with a Q3 pore produced little or no current. Hence, the smallest region of KCNQ2 needed for potentiation was that around the pore region (Fig. 2A). The functional channels again showed intermediate sensitivity to TEA, indicating the formation of heteromeric channels (data not shown).
Figure 2.

A residue located in the inner vestibule after the selectivity filter is critical for potentiation. A, Left, Schematic representation of the chimeras. The boxes indicate transmembrane segments, and the loop represents the pore between S5 and S6. Right, Normalized maximum conductances after coexpression with Q3 (n > 10 from 2 different batches). Chimeras containing the pore region from Q2 were able to increase the current, and the heteromeric nature of the channels was verified with TEA (see Fig.1 F). Incontrast, those containing the pore from Q3 produced little or no current. B, Top, Alignment of the pore region of Q2 and Q3. The residues that differ between both subunits are indicated in bold. The gray box encompasses an extracellular loop, and the open box encompasses the pore. Bottom, Different residues were mutated in a Q3/Q2253-291 background to the equivalent ones in Q3. The normalized maximal conductance after coexpression with Q3 is plotted (n > 4). When the residue equivalent to threonine 276 of Q2 was changed to alanine, present at the equivalent position 315 in Q3, the current was reduced to Q3 homomeric levels. C, Normalized maximum conductance levels of different pore point mutants, expressed in pairs as indicated on the left (n ≥ 8). D, Backbone of a homology model of the pore region of Q2/Q3 heteromers drawn to scale based on the structure of KcsA (Doyle et al., 1998), indicating the position of T276 and A315. A Q3 subunit located in the front view has been removed for clarity. K+ ions are represented as gray circles.
In this region, a nonconserved domain forms an extracellular loop (Fig. 2B, gray box), which is followed by a highly conserved segment forming the selectivity filter (white box). When mutations were introduced into the conserved region on a N3T3P2C3 background, a residue critical for potentiation was identified in the inner pore, corresponding to T276 in Q2 or A315 in Q3 (Fig. 2B). In addition, potentiation was abolished when the equivalent residue in Q2, threonine 276, was mutated to alanine (underlined). The mutant Q2 T276A produced a channel yielding small currents reminiscent of homomeric Q3 channels, even when coexpressed with KCNQ3 (Fig. 2C). Furthermore, the Q3A315T mutant yielded currents that were even larger than the reference Q2/Q3 currents when coexpressed with Q3A315T (i.e., homomers), or with Q3, but comparable with the reference when coexpressed with Q2 (Fig. 2C). This difference can be at least partially accounted for by the negative influence of the N2 segment (see below).
KCNQ3 homomeric channels show a delayed decline in the currents activated at voltages greater than +20 mV. This may be attributable to an inactivation process as observed for KCNQ1, but not in KCNQ2. Interestingly, a critical residue for KCNQ1 inactivation is located one position before the equivalent of the T/A position (Seebohm et al., 2001). Similarly, the KCNQ3 A315T mutant channel did not inactivate, but inactivation appeared in the homomeric KCNQ2 T276A channels. These data imply that this residue plays an important role in the gating process associated with channel inactivation (data not shown).
The N-terminal region controls current amplitude
The data in Figure 1C from functional channels differing only in the N-terminal region are re-plotted in Figure 3A showing that those subunits with the N terminus from Q2 gave rise to significantly smaller conductances than those with the Q3 N terminus (Fig. 3A). Hence, we further explored the role of the N terminus by coexpressing these chimeras with Q2 (Fig. 3B). Again, when the N terminus from Q3 was replaced with the N terminus of Q2, the conductance clearly diminished, regardless of the transmembrane or the C-terminal configuration. The homomeric Q3 N-terminal configuration produced the greatest conductance levels and the homomeric Q2 configuration the least.
The three regions control current levels through quasi-independent mechanisms
By coexpressing pairs of the different chimeras, we could analyze heteromeric (h) or homomeric (Q2 or Q3) configurations of each region (N, T, or C) to determine whether the effect of the three regions on the current level was independent or not. Every combination within a group gave rise to comparable maximum conductance levels, as expected, if the control exerted by each of the three regions was independent of the others (Fig. 4). However, the data from set four indicate that the three mechanisms are not completely independent.
Figure 4.

The N-terminal, transmembrane, and C-terminal segments control maximal current levels through quasi-independent mechanisms. Pairs of chimeras represented below each bar were coexpressed, creating seven configurations that were either homomeric or heteromeric at the N-terminal, transmembrane, or C-terminal segments. The resulting configuration is color coded in the schematic representation of the channel above the bars, using gray for the heteromeric configuration, black for the Q2 homomeric configuration, and white for the Q3 homomeric configuration of each segment. The configurations were grouped into four sets: (1) NhetThetChet; (2) NhetThomoChet; (3) NhomoThetChet; (4) NhetThetChomo (het, heteromeric; homo, homomeric). In sets two to four, there were two groups differing in the homomeric configuration (Q2 or Q3) of one segment (N, T, or C). Each combination within a group gave rise to comparable maximum conductance levels (n ≥ 6), revealing that each region (N, T, and C) controls the maximum conductance level through a mechanism that is independent of the other segments. The difference of the means of the pair NhThC3 in set 4 was not statistically significant. The mean conductance for all constructs and the reference mean conductance were statistically different (p < 0.01), except for the chimeras in set 1 and the pairs that from the configuration NhThC3 in set 4.
Set four shows the effect of having a homomeric C-terminal configuration, a region that controls the number of channels at the surface (Fig. 1D). Because the surface expression of the homomeric Q2 or Q3 channels is inefficient (Schwake et al., 2000), it was expected that the NhThC2 or NhThC3 channels should have given rise to lower current levels than Q2/Q3 (NhThCh), and this was the case for two of the combinations (N3T2C2+ N2T3C2 and N3T3C2+N2C2T2; p < 0.01). Although the mean conductance value for the pair N2T3C3+N3T2C3 was smaller than the Q2/Q3 reference value, the difference did not reach statistical significance. However, we found that coexpression of N2T2C3 with Q3 (NhThC3) consistently gave conductance levels indistinguishable from those obtained after coexpression of Q2 with Q3. Hence, apart from controlling the number of available channels, the C terminus may also be affecting the current levels by another mechanism (Maljevic et al., 2003; Li et al., 2004). In line with this idea, as has been also noted by others (Maljevic et al., 2003), we observed that there was a systematic shift in the V1/2 of the g-V relationship depending on the C-terminal configuration (data not shown). Because the configuration of the C terminus changed from Q2, to heteromeric, to Q3, the V1/2 shifted to a more depolarized potential (data not shown), suggesting that the C terminus of Q2 might stabilize the open configuration or make the closed conformation more unstable.
Discussion
We show here that three mechanisms underlie the potentiation after heteromerization of KCNQ2/KCNQ3 channels and provide additional evidence for the modular design of K+ channels, with different regions serving separate functions in an almost independent manner. The current flow depends on the product of three parameters: N (number of channels); Po (probability of the channel being open); and γ (single-channel conductance). In KCNQ channels, N clearly depends on the intracellular C terminus (Schwake et al., 2000). However, we surprisingly found that in Xenopus oocytes there is no correlation between the number of channels at the membrane and the current, and both the N-terminal and the transmembrane regions play prominent roles in the potentiation process.
The C-terminal domain controls the number of channels at the membrane
Although the number of channels at the membrane increases after heteromerization of the C-terminal region, Schwake et al. (2000) have shown that the total protein levels stay relatively constant, ruling out stabilization of the channel to explain the increase in surface expression. Thus, retention/retrieval or export signals may be masked or unmasked after heteromerization. In the C terminus, there are four regions with high probability of forming an α helix (helices A-D) (Yus-Najera et al., 2002). Clusters of positively charged residues reminiscent of retention/retrieval signals (Teasdale and Jackson, 1996; Ma and Jan, 2002) are present at the end (Lerche et al., 1999) and close to helix C. This is intriguing because helix C makes up the assembly domain (Maljevic et al., 2003; Schwake et al., 2003), and it could easily be envisaged that putative retention signals close to this helix could be concealed after channel assembly.
Surface expression appears to be also regulated by other regions, because all chimeras were less efficient than Q2 at promoting Q3 traffic to the membrane (Fig. 1D). In fact, residues located in the transmembrane region of Kv1.1 channels (Manganas et al., 2001; Zhu et al., 2001) and the distantly related GluR2 receptor (Greger et al., 2003) are involved in the folding and therefore in the trafficking of the channel. Nevertheless, although it appears that the overall structure of the pore loop of GluR2 is critical for tetramerization and exit from the endoplasmic reticulum (Greger et al., 2003), it remains to be determined whether the T/A site in the pore loop of KCNQ2/3 plays a similar role.
Other parameters that influence current levels also appear to be affected by the C terminus (Maljevic et al., 2003; Li et al., 2004), because when KCNQ3 was coexpressed with the N2T2C3 chimera, the number of channels at the surface was very small (Fig. 1D), yet the maximal current was similar to that obtained after coexpression with N2T2C2 (Figs. 1C, 4).
The N-terminal domain may affect the maximal open probability
In the presence of a heteromeric pore arrangement, the N terminus had a greater influence on the maximal conductance than the C terminus. The largest conductance levels were obtained with a homomeric Q3 N-terminal configuration, and the homomeric Q2 N-terminal configuration produced the lowest current.
The small impact of the N terminus in the number of channels at the membrane contrasts with its effect on the maximal conductance. For instance, we found that by switching from a heteromeric to homomeric Q3 N-terminal configuration, the 10-40% reduction in the number of channels at the membrane was accompanied by a sixfold increase in maximal conductance. Thus, the N terminus must mainly affect Po*γ.
We favor the idea that the N terminus affects Po, rather than γ, because the differences of single-channel conductance measured for homomeric KCNQ3 and heteromeric channels cannot account for the increase in maximal current (Schwake et al., 2000; Selyanko et al., 2001). This idea is in agreement with the reduction in Po observed after heteromerization in Chinese hamster ovary cells (Selyanko et al., 2001) but clashes with the increase estimated by nonstationary noise analysis in oocytes (Schwake et al., 2000). The different estimates for maximal Po of homomeric Q3 channels ranges from 0.4 to 0.9 (Schwake et al., 2000; Selyanko et al., 2001; Li et al., 2004). Changes in maximal Po could, at best, lead to a 2.5-fold increase in conductance. In contrast, the estimates for maximal Po for homomeric Q2 ranges from 0.15 to 0.61. The lower estimate of Po is more in line with our results and permits a 6.7-fold increase in conductance. Thus, the Po dynamic range is larger for Q2 channels than for Q3 subunits. It has been proposed that the C terminus is responsible for this differential property (Li et al., 2004). We propose that the N terminus is also contributing significantly to the maximal Po attainable. Given the weak conservation in the N terminus across the Kv7 family and its important effect on KCNQ channel function, this region may be a good target for developing more specific therapeutic drugs for the treatment of diverse neurological conditions (Gribkoff, 2003).
Role of the pore residue in potentiation
We have identified A315 as a residue in the inner pore vestibule of KCNQ3 subunits that critically controls channel activity, and that exerts a strong negative effect on current flow. When the four subunits of the tetrameric channel have an alanine at this position, either in a KCNQ3 or a KCNQ2 backbone, the current levels are close to the background, even when a considerable number of channels are in the membrane. In heteromers, this negative effect is neutralized by the threonine at the equivalent 276 position in Q2. Interestingly, the other functional partners of KCNQ3 (Q4 and Q5) also have a threonine at the equivalent position.
A comparison of the 75 known human potassium channels (Gutman et al., 2003) reveals a high degree of conservation at this position, it being occupied by threonine in 66 subunits. The only other subunit that has an alanine is Kir3.1 (A142), which belongs to the G-protein-gated inwardly rectifying channels (also named GIRK). Interestingly, this subunit does not produce functional homomeric channels, and it associates with other family members to enhance their activities (Chan et al., 1996). Nevertheless, the role of alanine 142 in Kir3.1 remains unclear. In Shaker channels, the mutation of the equivalent threonine 441 to alanine does not impair channel function, and this position is not sensitive to gating in double-cycle mutation analysis (Yifrach and MacKinnon, 2002). Mutations at this position in KCNQ2 (T276S) and other potassium channels alter ionic selectivity, increasing the conductance of some ions (Prole and Marrion, 2004). However, various single-channel analyses indicate that there is no change or only a mild increase in conductance after heteromerization (Schwake et al., 2000; Selyanko et al., 2001), which is insufficient to explain the potentiation of the current.
One intriguing possibility is that this residue shapes a binding site for an endogenous blocking molecule. Mutational analysis in KCNQ2, Kir2.1 and Shaker is consistent with the involvement of T276/A315 in an intracellular ionic block (Yellen et al., 1991; Zhou et al., 1996; Alagem et al., 2001; Slesinger, 2001; Prole and Marrion, 2004). Indeed, the corresponding T623 is critical for anti-arrhythmic drug blockage of human ether-à-go-go related gene channels (Mitcheson et al., 2000), and in KCNQ1 there are important determinants for benzodiazepine channel block in the neighborhood of this residue (Seebohm et al., 2001). In addition, the Q/R site that participates in the blockage of AMPA receptors by the naturally occurring polyamines is located in the vicinity of the residue equivalent to T276/A315 of KCNQs (Bowie and Mayer, 1995). In summary, there is ample evidence to suggest that this residue might contribute to a binding pocket. Hence, we would expect that homomeric KCNQ3 channels could be suppressed to varying degrees depending on the concentration of the putative blocker. Should a natural ligand exist for this site, Xenopus oocytes may provide an abundant source for its identification, because relative KCNQ3 homomeric current levels are much lower than in other expression systems.
In summary, compared with homomeric KCNQ3 channels, after heteromerization there is an increase in the number of KCNQ3 channels at the membrane and a reduction in Po because of the presence of the Q2 N-terminal segment. In addition to a possible effect on single-channel conductance, two possible roles should be considered for the site in the inner vestibule of the pore. It may affect assembly and membrane expression and/or the formation of a binding pocket for a hypothetical endogenous blocker.
Footnotes
This work was supported by grants from the European Union (LSFM-CT-2004-50303), Fondo de Investigaciones Sanitarias (FIS01/1136), Comunidad de Madrid (08.5/0011/2001.1), and Ministerio de Educación y Ciencia (BFI2003-00693). A.E. was supported by a fellowship from the Basque Government. We thank Dr. Mark Sefton for critical comments and editorial assistance.
Correspondence should be addressed to Dr. Alvaro Villarroel, Unidad de Biofísica, Consejo Superior de Investigaciones Científicas-Universidad del País Vasco (UPV)/Euskal Herriko Unibersitatea, UPV, Barrio Sarriena s/n, 48940 Leioa, Spain. E-mail: gbxvimua@lg.ehu.es.
M. Paz Regalado's present address: Department of Psychiatry and Behavioral Sciences, Stanford University School of Medicine, 1201 Welch Road, Room P154, Palo Alto, CA 94304-5485.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249146-07$15.00/0
References
- Alagem N, Dvir M, Reuveny E (2001) Mechanism of Ba2+ block of a mouse inwardly rectifying K+ channel: differential contribution by two discrete residues. J Physiol (Lond) 534: 381-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK (1998) A potassium channel mutation in neonatal human epilepsy. Science 279: 403-406. [DOI] [PubMed] [Google Scholar]
- Bowie D, Mayer ML (1995) Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron 15: 453-462. [DOI] [PubMed] [Google Scholar]
- Brown BS, Yu SP (2000) Modulation and genetic identification of the M channel. Prog Biophys Mol Biol 73: 135-166. [DOI] [PubMed] [Google Scholar]
- Chan KW, Sui JL, Vivaudou M, Logothetis DE (1996) Control of channel activity through a unique amino acid residue of a G protein-gated inwardly rectifying K+ channel subunit. Proc Natl Acad Sci USA 93: 14193-14198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK (2001) Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci USA 98: 12272-12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais CJ, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280: 69-77. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB (2003) AMPA receptor tetramerization is mediated by Q/R editing. Neuron 40: 763-774. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK (2003) The therapeutic potential of neuronal KCNQ channel modulators. Expert Opin Ther Targets 7: 737-748. [DOI] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE, Covarriubias M, Desir GV, Furuichi K, Ganetzky B, Garcia ML, Grissmer S, Jan LY, Karschin A, Kim D, Kuperschmidt S, Kurachi Y, Lazdunski M, Lesage F, Lester HA, et al. (2003) International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev 55: 583-586. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Passmore GG, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA (2003) Stoichiometry of expressed KCNQ2/KCNQ3 channels and subunit composition of native ganglionic M-currents deduced from block by tetraethylammonium (TEA). J Neurosci 23: 5012-5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, Jentsch TJ (2002) Ion channel diseases. Hum Mol Genet 11: 2435-2445. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21-30. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96: 437-446. [DOI] [PubMed] [Google Scholar]
- Lerche H, Biervert C, Alekov AK, Schleithoff L, Lindner M, Klinger W, Bretschneider F, Mitrovic N, Jurkat-Rott K, Bode H, Lehmann-Horn F, Steinlein OK (1999) A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol 46: 305-312. [DOI] [PubMed] [Google Scholar]
- Li Y, Gamper N, Shapiro MS (2004) Single-channel analysis of KCNQ K+ channels reveals the mechanism of augmentation by a cysteine-modifying reagent. J Neurosci 24: 5079-5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Jan LY (2002) ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol 12: 287-292. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Lerche C, Seebohm G, Alekov AK, Busch AE, Lerche H (2003) C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J Physiol (Lond) 548: 353-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganas LN, Wang Q, Scannevin RH, Antonucci DE, Rhodes KJ, Trimmer JS (2001) Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc Natl Acad Sci USA 98: 14055-14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV (1997) Control of M-current. Annu Rev Physiol 59: 483-504. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC (2000) A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 97: 12329-12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P (1997) A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 15: 186-189. [DOI] [PubMed] [Google Scholar]
- Prole DL, Marrion NV (2004) Ionic permeation and conduction properties of neuronal KCNQ2/KCNQ3 potassium channels. Biophys J 86: 1454-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ (1998) Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396: 687-690. [DOI] [PubMed] [Google Scholar]
- Schwake M, Pusch M, Kharkovets T, Jentsch TJ (2000) Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem 275: 13343-13348. [DOI] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T (2003) A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep 4: 76-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebohm G, Scherer CR, Busch AE, Lerche C (2001) Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J Biol Chem 276: 13600-13605. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Brown DA (2001) Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol (Lond) 534: 15-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger PA (2001) Ion selectivity filter regulates local anesthetic inhibition of G-protein-gated inwardly rectifying K+ channels. Biophys J 80: 707-718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teasdale RD, Jackson MR (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the golgi apparatus. Annu Rev Cell Dev Biol 12: 27-54. [DOI] [PubMed] [Google Scholar]
- Villarroel A, Schwarz TL (1996) Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci 16: 2522-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT (1996) Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12: 17-23. [DOI] [PubMed] [Google Scholar]
- Yellen G, Jurman ME, Abramson T, MacKinnon R (1991) Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science 251: 939-942. [DOI] [PubMed] [Google Scholar]
- Yifrach O, MacKinnon R (2002) Energetics of pore opening in a voltage-gated K+ channel. Cell 111: 231-239. [DOI] [PubMed] [Google Scholar]
- Yus-Najera E, Santana-Castro I, Villarroel A (2002) The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem 277: 28545-28553. [DOI] [PubMed] [Google Scholar]
- Zhou H, Chepilko S, Schutt W, Choe H, Palmer LG, Sackin H (1996) Mutations in the pore region of ROMK enhance Ba2+ block. Am J Physiol 271: C1949-C1956. [DOI] [PubMed] [Google Scholar]
- Zhu J, Watanabe I, Gomez B, Thornhill WB (2001) Determinants involved in Kv1 potassium channel folding in the endoplasmic reticulum, glycosylation in the Golgi, and cell surface expression. J Biol Chem 276: 39419-39427. [DOI] [PubMed] [Google Scholar]