

Abstract
Mutations in the CLN2 gene, which encodes a lysosomal serine protease, tripeptidyl-peptidase I (TPP I), result in an autosomal recessive neurodegenerative disease of children, classical late-infantile neuronal ceroid lipofuscinosis (cLINCL). cLINCL is inevitably fatal, and there currently exists no cure or effective treatment. In this report, we provide the characterization of the first CLN2-targeted mouse model for cLINCL. CLN2-targeted mice were fertile and apparently healthy at birth despite an absence of detectable TPP I activity. At ∼7 weeks of age, neurological deficiencies became evident with the onset of a tremor that became progressively more severe and was eventually accompanied by ataxia. Lifespan of the affected mice was greatly reduced (median survival, 138 d), and extensive neuronal pathology was observed including a prominent accumulation of cytoplasmic storage material within the lysosomal-endosomal compartment, a loss of cerebellar Purkinje cells, and widespread axonal degeneration. The CLN2-targeted mouse therefore recapitulates much of the pathology and clinical features of cLINCL and represents an animal model that should provide clues to the normal cellular function of TPP I and the pathogenic processes that underlie neuronal death in its absence. In addition, the CLN2-targeted mouse also represents a valuable model for the evaluation of different therapeutic strategies.
Keywords: cerebellum, degeneration, enzyme, epilepsy, lysosome, neuropathology, proteolysis
Introduction
The neuronal ceroid lipofucinoses (NCLs) are a group of inherited progressive neurodegenerative diseases characterized by an accumulation of autofluorescent material in lysosomes within the brain and other tissues of affected individuals (Goebel et al., 1999). Mutations in at least seven different genes result in NCL (Williams et al., 1999; Hofmann and Peltonen, 2001).
Classical late-infantile NCL (cLINCL) is typically diagnosed between 2 and 4 years of age with the onset of seizures. Disease progression is relentless with clinical symptoms that include seizures, blindness, ataxia, mental retardation, and dementia before affected individuals become unresponsive and eventually die between 7 and 15 years of age (Lyon et al., 1996; Goebel et al., 1999). The defective gene in cLINCL, CLN2, was mapped to 11p15 (Sharp et al., 1997) and subsequently identified using a proteomic approach that compared the complement of lysosomal proteins within the brain of cLINCL patients with that of normal controls (Sleat et al., 1997). CLN2 encodes a lysosomal serine protease (Lin et al., 2001), tripeptidyl-peptidase I (TPP I) (Rawlings and Barrett, 1999; Vines and Warburton, 1999), and to date, 52 CLN2 mutations have been associated with disease (Mole, 2002). In some rare cases, compound heterozygosity for a null allele and an Arg447His missense mutation caused a late-onset (∼8 years) protracted form of disease with some patients surviving into the fourth decade of life (Sleat et al., 1999).
To date, no animal model has been described for cLINCL, although there are models for other forms of human NCLs. For infantile NCL and juvenile NCL, mouse models have been generated by targeting genes CLN1 (Gupta et al., 2001) and CLN3 (Greene et al., 1999; Katz et al., 1999), respectively. Spontaneous mouse mutants nclf and mnd have defects in CLN6 (Gao et al., 2002; Wheeler et al., 2002) and CLN8 (Ranta et al., 1999), respectively, providing models for a variant late-infantile neuronal ceroid lipofuscinosis and epilepsy, progressive, with mental retardation. Lipofuscinoses have also been identified in a number of other species (for review, see Hofmann and Peltonen, 2001) including sheep, cattle, and dogs (English setter, border collie, miniature schnauzer, and Tibetan terrier). Although the actual gene defects remain to be identified in nonmurine lipofuscinoses, the possibility that CLN2 defects underlie disease has been eliminated in most by measurement of the enzymatic activity of the CLN2 gene product (Sohar et al., 1999).
NCL animal models, murine and nonmurine alike, have not only provided detailed insights into the neurobiology of their respective diseases but also represent invaluable model systems for testing and evaluating potential therapies. In this report, we describe the first animal model for cLINCL, a mouse in which CLN2 is disrupted by gene targeting. CLN2 targeting results in a loss of detectable TPP I activity, a progressive neurologic phenotype, and a greatly shortened lifespan. Similarities with the human disease are also observed at the cellular level, in which progressive accumulation of fluorescent material and neuronal death are both evident.
Materials and Methods
Targeted disruption of CLN2 in mice. The complete sequence of the murine CLN2 gene and proximal regions was determined using a combination of standard molecular methods (plasmid subcloning of λ genomic clones and exon-exon PCR using a gene structure inferred from the human CLN2 gene) and database searching. Mouse chromosome 7 clone RP23-466I14 (accession number AC121823) contains this sequence. Long and short arms were amplified from 129SvEv genomic DNA. The 1.9 kb short arm, corresponding to nucleotides 119277-121171 of mouse chromosome 7 clone AC121823, was cloned as a BglII fragment into the BamHI site of pTK-LNL (obtained from Richard Mortensen, University of Michigan, Ann Arbor, MI), a targeting vector containing thymidine kinase for negative selection and a loxP-flanked neo cassette for positive selection. The 4.8 kb long arm, corresponding to nucleotides 114520-119276 of AC121823, was engineered to contain a point mutation (G119096A of AC121823) that was predicted to change amino acid 446 of the primary murine CLN2 gene product from arginine to histidine, corresponding to the late-onset human allele Arg447His (Sleat et al., 1999). The long arm was cloned into the SalI site of pTK-LNL, and the targeting plasmid was linearized with NotI, eliminating the bacterial plasmid-derived DNA (see Fig. 1 A). W9.5 embryonic stem (ES) cells (derived from 129SvEv mice) were electroporated with linear DNA. ES cells growing in the presence of neomycin and gancyclovir were isolated on day 10 postelectroporation and subcultured in gelatinized 96-well plates. DNA was purified from duplicate plates grown in the absence of feeders, and screening was achieved by Southern blotting using a 32P-labeled probe generated using random hexamers and PCR-amplified genomic DNA (primer sequences, TCCTGGTCTATATAAAGAGTTCCA and TTGGGCTGGGACCTAGGAATACAAAG) that detected 5.0 and 9.0 kb EcoRI fragments from targeted and wild-type alleles, respectively (see Fig. 1 A). Targeted ES cell clones were identified, expanded, and confirmed by Southern blotting using a series of restriction enzymes and probes upstream and downstream of the targeted region. Targeted ES cells were injected into C57BL/6 blastocytes that were implanted into pseudopregnant Swiss-Webster females using standard techniques. Chimeric males were bred with C57BL/6 females generating a number of offspring that were confirmed to be heterozygotes by Southern blotting. Subsequent genotyping was achieved using a set of three primers (ATCTGATGGCTACTGGGTGG, CCCGGTAGAATTCCGATCAT, and CCCCCAAACACTGGAGTAGA) that generated 402 nt and 328 nt products from the wild-type and targeted alleles, respectively.
Figure 1.

Targeted disruption of the mouse CLN2 gene. A, Structure of CLN2 and proximal genes, the targeting construct, and the targeted CLN2 locus (boxes: black shading, exons; gray shading, CLN2 3′-untranslated sequence; unshaded, thymidine kinase and neo selection markers). Murine CLN2 transcripts terminate at two different polyadenylylation sites (indicated by arrows above CLN2 gene), and in some cases, intron 3 is not spliced from the mature mRNA (the structure of the corresponding transcript is indicated by an extended exon box above the CLN2 gene). The targeted CLN2 gene is disrupted by the insertion of neo into intron 11 and by the incorporation of an Arg446His missense mutation into exon 11 immediately upstream of the neo insertion (indicated by *). The integrin linked kinase gene is set lower than TAF10 because they are overlapping. B, TPP I enzyme assays in wild-type, heterozygote, and homozygous CLN2-targeted mice; each point represents an individual mouse. C, Detection of CLN2 and TAF10 mRNAs in duplicate mutant and control mice by Northern blotting. Note the CLN2 transcripts of ∼500 nt greater length than the two wild-type mRNAs that are weakly detectable in the -/- mouse. Full-length TAF10 mRNA is ∼0.8 kb, and this size is consistent with the major transcript detected (arrow). D, Schematic of the aberrant splicing event that introduces 498 additional nucleotides derived from the neo selection marker to the CLN2 mRNA.
Mice were in a mixed C57BL/6:129Sv genetic background unless stated otherwise. All experiments and procedures involving live animals were conducted in compliance with approved Institutional Animal Care and Use Committee protocols. Unless stated otherwise, mice were routinely killed for study using 98 mg/ml sodium pentobarbital containing 12.5 mg/ml phenytoin (a 1:4 dilution of Euthasol; Delmarva Laboratories, Midlothian, VA).
Preparation and analysis of RNA. Total RNA was purified from mouse tissues that were snap frozen and stored in liquid nitrogen using an RNeasy kit (Qiagen, Valencia, CA), and poly(A) mRNA was purified using the Oligotex Direct mRNA kit (Qiagen). RNA [15 μg total or 5 μg poly(A)] was fractionated by denaturing agarose gel electrophoresis and transferred to nylon membrane. CLN2 mRNA was detected using a 32P-labeled probe generated using random hexamers from a 1.8 kb EcoRI-PstI fragment from mouse TPP I expressed sequence tag (EST); AA087886 and TATA-binding protein-associated factor (TAF)10 mRNA was detected using a probe synthesized from an EcoRI-NotI fragment of TAF10 EST BQ748216. Quantitation of CLN2 and TAF10 transcription by duplicate independent real-time PCR experiments was achieved with the Brilliant SYBR Green QRT-PCR kit (Stratagene, La Jolla, CA) and an MX4000 Multiplex Quantitative PCR System (Stratagene). Primers were designed to generate a 293 nt CLN2 reverse transcription (RT)-PCR product (CACCATCCAGTTACTTCAATGC and CTGACCCTCCACTTCTTCATTC) and a 282 nt TAF10 RT-PCR product (TGATGCAGTTGGAGGATTACAC and GCTTCTTCACATTGATGCCATA), both of which were confirmed by sequencing.
TPP I assay. TPP I was measured using a modification (Sohar et al., 2000) of a previously described method (Vines and Warburton, 1998).
Behavioral assessment. CLN2 knock-out and wild-type littermate mice were evaluated using a standardized functional observation battery of tests once a week from 8 to 19 weeks of age. Animals were videotaped over a 2 min period, and behavior was scored across a number of parameters including general appearance, gait, posture, and overt seizure activity (convulsions). Open-field motor activity was measured in a Plexiglas cage surrounded by two arrays of photobeams to capture movement and rearing activity (Opto-Micro Animal Activity System; Columbus Instruments, Columbus, OH). Total beam breaks were recorded over a 60 min period captured in 5 min intervals. Motor coordination and balance were measured using a rotating rod apparatus (SmartRod; Accuscan Instruments, Columbus, OH). Animals were tested in two ways: first, the rod accelerated to a final speed of 30 rpm over the course of 60 sec; and second, the rod was programmed to rock backwards and forwards for up to 2.5 sec duration with the overall acceleration in either direction increasing to 25 rpm. Cutoff times were 60 sec for the accelerating test and 54 sec for the rocking test. Animals were tested three times with each version of the test with a rest period of at least 5 min between measurements. Average fall latency from the rod (or cutoff time) was recorded for each animal and used for statistical analysis.
Analyses of locomotor function were performed using Student's t test (Prism; GraphPad, San Diego, CA). Data are mean ± SEM. p < 0.05 was considered as a statistically significant difference.
Perfusion and fixation of mouse tissues. Homozygous mutants (-/-), heterozygotes (+/-), and wild-type (+/+) littermate mice ranging in age from 35 to 150 d were available for study. Mice were anesthetized and perfused transcardially at room temperature with 0.9% NaCl, followed by PBS, pH 7.4-7.6, containing aldehyde fixatives (2% paraformaldehyde-0.03% glutaraldehyde or 10% formalin). The brain was removed and postfixed overnight at 4°C, then prepared for histological evaluation as described below.
Histology. Six micrometer paraffin sections were stained with hematoxylin and eosin (H&E), Luxol fast blue-cresyl violet (LFB-CV), and periodic acid-Schiff (PAS)-hematoxylin. Unstained sections were examined for autofluorescence. Selected paraffin sections were also stained immunohistochemically with antibodies to lysosome-associated membrane protein-1 (LAMP-1) (BD Biosciences, San Jose, CA) (Chen et al., 1985). Antibody binding was visualized with an avidin-biotin peroxidase detection kit (Vector Laboratories, Burlingame, CA) and stained with diaminobenzidine.
As a marker for cerebellar Purkinje cells, 50 μm Vibratome sections were stained with an antibody against calbindin (Sigma, St. Louis, MO) using fluorescent secondary antibody detection.
For silver neurodegeneration staining, brains were embedded together in a gelatin-based matrix and sectioned serially in the coronal plane on a freezing microtome at 35 μm thickness (Neuroscience Associates, Knoxville, TN). Every 12th section was stained by the amino cupric silver method of DeOlmos for degenerating neurons with a neutral red counterstain (DeOlmos and Ingram, 1971; Switzer, 2000). Alternate sets of matrix sections were stained with H&E.
PAS staining was also conducted on plastic-embedded samples. Tissue slices were postfixed with 1% osmium tetroxide for 4 hr at 4°C, dehydrated through a graded series of ethanol washes (70-100%), then infiltrated with catalyzed JB-4 Plus Solution A (EMS, Fort Washington, PA) for 24 hr and placed into embedding molds containing infiltration solution mixed with hardener. After polymerization, sections of 1 μm were cut with a microtome and processed for PAS staining. Briefly, sections were oxidized in 0.4% periodic acid for 30 min at 57°C and then incubated in Schiff's Reagent for 15 min after thorough washing. Sections were then counterstained with Gill's hematoxylin for 10 min and coverslipped.
Electron microscopy. Dehydrated samples of 1 mm3 were infiltrated in 100% Spurr's resin for 24 hr, then placed in Beem capsules and baked at 60°C for 16 hr. Ultra-thin 50 nm sections were cut with a diamond knife and placed on grids, stained with uranyl acetate and lead citrate, and examined with a Hitachi (Tokyo, Japan) H7000 transmission electron microscope.
Results
Targeted disruption of CLN2
The murine CLN2 gene was targeted as shown in Figure 1A, generating a mutant mouse with undetectable levels of TPP I activity (Fig. 1B). Designed to eventually be a hypomorph after Cre (cAMP response element)-mediated excision of neo, the mutant characterized in this report retains the intronic neo insertion in addition to the Arg446His mutation, and is therefore designated neoinsArg446His.
In designing our targeting strategy, examination of the chromosomal region proximal to the murine CLN2 gene revealed a gene-rich region of the genome (Liu et al., 1998) (Fig. 1A); thus, our approach was designed to minimize potential positional effects on other genes. This was achieved in several ways. First, we used a loxP-flanked neo selection marker to allow removal of this cassette if necessary. Second, we decided to disrupt CLN2 with a point mutation rather than a major deletion or replacement. Third, we chose an intronic region of low homology between human and mouse to insert neo. The rationale for the latter is that there may exist a transcript of unknown function that overlaps CLN2 and that is transcribed in the opposite direction (Liu et al., 1998); intronic sequences of low conservation between humans and mice are less likely to represent exonic sequences for this transcript than those of higher homology. We made three targeting constructs: a null, a hypomorph, and a control, which were designed to introduce point mutations into exon 11 to generate Arg446Stop, Arg446His, or Arg446Arg446, respectively, into the targeted allele, in addition to insertion of the neo selection marker into intron 11. More than 3600 G418-resistant clones arising from ES cells electroporated with the three constructs were screened by Southern blotting, and a single clone incorporating the Arg446His missense mutation was identified, from which the CLN2-targeted mouse containing a neo insertion and Arg446His mutation (neoinsArg446His) was subsequently generated.
The reason underlying the low frequency for homologous recombination at the CLN2 locus is not apparent. However, given that the Arg446His mutation, in conjunction with the severe splicing defect caused by the neo insertion, appeared to result in a null phenotype, and that fact that there were no positional effects on the closely neighboring TAF10 gene, we chose to characterize the neoinsArg446His mutant further. The results presented here indicate that it provides an appropriate model for cLINCL.
The combined effect of the neo insertion within intron 11 and the Arg446His mutation on CLN2 expression was investigated at both RNA and protein levels. Northern blotting of total RNA from brain and epididymis of wild-type mice revealed two transcripts of ∼3 and 4 kb (Fig. 1C), representing transcription termination at two different polyadenylylation sites within the 3′-untranslated region of exon 13. In the CLN2-targeted mice, neither of these transcripts was detectable, but low levels of two transcripts of ∼3.5 and 4.5 kb were present, suggesting that the presence of the neo insertion within intron 11 was causing missplicing of the CLN2 mRNA. We investigated this possibility by sequencing RT-PCR products generated from the CLN2-targeted mice using forward and reverse primers within exons 11 and 12, respectively. We found that the CLN2 mRNA transcribed from the targeted allele contains Arg446His and is correctly spliced at the exon 11-intron 11 and intron 11-exon 12 boundaries, but that cryptic splice donor and acceptor sites within the antisense neo transcript are also used, resulting in a 498 nt insertion within the mRNA (Fig. 1D). This antisense neo insertion is in frame with the CLN2 coding region and contains no stop codons and is therefore predicted to add an additional 166 amino acids to the final protein product. Given the sensitivity of this protein to even single amino acid changes (Sleat et al., 1999), we predicted that such an insertion would completely disrupt the structure of the final protein and thus result in a nonfunctional and probably unstable product (Fig. 1B).
Although correctly spliced CLN2 mRNA lacking the neo insertion could not be detected by Northern blotting, we did observe a minor PCR product that corresponded to correctly spliced mRNA by RT-PCR. Using real-time PCR, we determined that the amount of correctly spliced CLN2 mRNA corresponds to ∼4% of the normal levels in brain and epididymis (data not shown). Taken together, these results indicate that the insertion of neo into intron 11 disrupts CLN2 gene transcription and/or mRNA stability such that only 4% of normally spliced mRNA levels is present. This residual CLN2 mRNA contains the Arg446His mutation and encodes no detectable TPP I by Western blotting and immunohistochemistry (data not shown) and by enzyme assay (Fig. 1B). The neoinsArg446His CLN2-targeted mouse therefore represents either an extreme hypomorph or a null mutant and for brevity is therefore referred to as -/-. The transcription start site of CLN2-proximal gene, transcription factor TAF10, is ∼480 nts downstream of the polyadenylylation site of the longest CLN2 transcript. Using real-time PCR, we determined levels of TAF10 mRNA in the CLN2 mutant to be 85 ± 10% of control in brain. Northern blot analysis revealed that the TAF10 mRNA appeared identical in both mutant and control mice (Fig. 1C). Taken together, these data indicate that positional effects of the CLN2 disruption on TAF10 expression do not contribute to the observed phenotype.
Phenotypic manifestations of CLN2 disruption
Homozygous CLN2-targeted mice were born at the expected frequency (24.4% homozygous targeted from heterozygote matings) and appeared healthy at birth. Growth was normal, both male and female mice were fertile, and mice presented no outward signs of disease until ∼7 weeks of age when a slight, constant tremor became detectable. Mice continued to grow normally and the tremor became progressively more severe. As disease approached end stage, tremor was accompanied by locomotor difficulties including abnormal hunched gait with feet pointed outward (duck walk) and splayed hind limbs, side-to-side shaking while walking, and ataxia.
Preliminary studies of general activity and locomotor performance were conducted on CLN2 mutant mice between 9 and 18 weeks of age (Fig. 2). In measurements taken during a 1 hr period in a novel (clean) Plexiglas cage, the CLN2 mutants were found to be as or more active than the wild-type controls. In contrast, the CLN2 mutants reared less, and thus the ratio of rearing to total activity was significantly decreased. Coordination, measured in terms of performance on the accelerating version of the rotating rod test, was normal until 14 weeks, then dropped precipitously thereafter. For the rocking rotating rod, performance of the CLN2 mutant was impaired from 10 weeks onwards. In conclusion, the CLN2 mutant mice remain quite ambulatory late into their disease but show significant impairment on the rotating rod that is consistent with underlying neuropathology and not simply the inability to perform the task because of gross motor impairment.
Figure 2.

Survival and motor function phenotype in the CLN2 mutant. A, Open-field motor activity and rearing measurements on wild-type (filled symbols; n = 8-11) and CLN2(-/-) (open symbols; n = 8-11) animals. B, Motor coordination and balance measurements on wild-type (n = 10) and CLN2(-/-) (n = 9) animals using accelerating and rocking versions of the rotating rod test. C, Survival analysis of CLN2 mutants and controls in a mixed 129Sv/C57BL6 background and in an isogenic 129Sv background. For the mixed 129Sv/C57BL6 background (-/-) (n = 286), control mice were heterozygous (n = 226) and CLN2 (+/+) (n = 124) mice in both mixed-strain and isogenic backgrounds. In the mixed and isogenic backgrounds, 286 and 37 -/- mice were analyzed, respectively. For A and B, data are mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Epileptic seizures are a characteristic of NCLs; thus, we have examined the CLN2 mutant carefully for signs of such a phenotype. In several late-stage mice, sudden brief seizures or violent myoclonic bursts that propelled the mice vertically were observed, but seizures do not appear to be a generalized feature of the phenotype. However, additional observation and EEG studies of both early and end-stage CLN2 mutants are needed to comprehensively define the epileptic status of this model.
Lifespan of the CLN2-deficient mouse was considerably shortened, and this is consistent with most other mouse models of human lysosomal storage diseases. Survival of the CLN2 mutant was 138 d, with 90% of the animals being found dead between days 108 and 169 (Fig. 2A). Survival of the CLN2 mutants in an isogenic 129Sv background was longer (median, 164 d; 90% death between 136 and 185 d), possibly indicating the presence of a strain-specific modifier(s) of the CLN2 phenotype.
Cytoplasmic storage in CLN2-targeted mice
The histological hallmark of cLINCL is the accumulation of autofluorescent lipopigments (ceroid lipofuscin) within the cells of affected individuals. By 154 d, some autofluorescent material was observed in heterozygote (Fig. 3A) and wild-type (data not shown) neocortex samples. Autofluorescent material accumulated in a punctate cytoplasmic distribution suggestive of lysosomal localization, which is consistent with normal lipofuscin accumulation. In the CLN2-targeted mice of the same age, autofluorescent material increased in amount to such an extent that it essentially filled the cytoplasm of the neuronal cell body (Fig. 3B). Autofluorescent storage was observed as early as 48 d and progressively accumulated as the mice aged. Fluorescent storage material was present in most brain regions including the neocortex, hippocampus, thalamus, and extending down through the hindbrain and into the spinal cord with significant involvement of motor neurons. Sensory neurons within dorsal root ganglion also had a prominent accumulation of lysosomes visible under epifluorescence.
Figure 3.

Unstained paraffin-processed sections of neocortex photographed under epifluorescence from +/- control (A) and -/- (B) at 154 d survival and demonstrating a significant increase in autofluorescent lysosomes within the mutant brain.
In CLN2-targeted mice, brainstem storage was evident from PAS stains of plastic-embedded brains at as early as 35 d of age (Fig. 4A). PAS-positive material was localized to the cytoplasm and increased in intensity and amount as the mice aged. Sections from other regions of the brain, including the cortex, hippocampus, striatum, and cerebellum showed similar PAS-positive inclusions, again with age-dependent increase in number and size (data not shown). Storage material was also visible in H&E-stained paraffin sections through the brain as a distinct accumulation of eosinophilic inclusions within the neuronal cytoplasm of -/- animals (Fig. 4B). At the light microscopic level, storage pathology was readily visible within mid- to large-sized neurons, particularly in magnocellular regions of the hindbrain. Inclusions were visible in some neurons by 48 d of age (the earliest time point studied by H&E), although cellular changes were generally quite subtle at this time point with the cytoplasm taking on a faint pink hue without overt storage pathology. By 90-100 d of age, distinct clusters of red inclusions-lysosomes were present within neurons. In older animals (>110 d), the amount of storage material continued to increase, often distorting the cell margin. In some cases, the storage material appeared to change from a tight cluster of individual inclusions-lysosomes to a unitary mass or concretion within a neuron.
Figure 4.
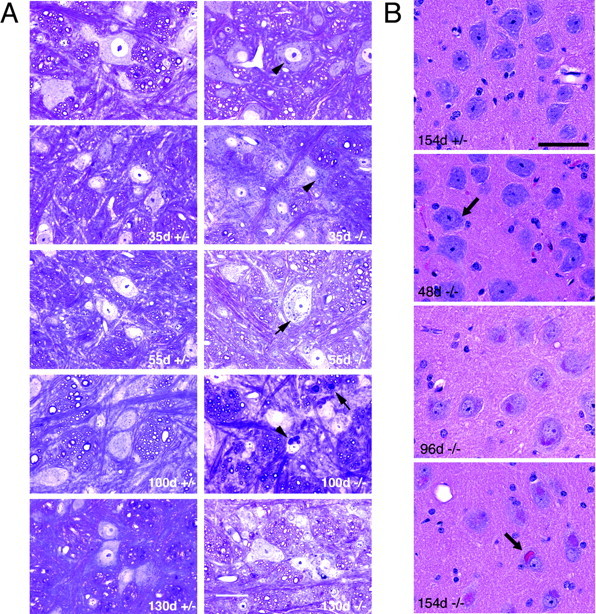
Progressive accumulation of cytoplasmic storage material in CLN2-targeted mouse brain. Ages of CLN2 mutants and age-matched littermate heterozygote controls are indicated on each panel. A, PAS-stained sections from the brainstem at the level of the hypoglossal nucleus. Brains were processed through JB-4 plastic embedding media. Positive inclusions in the cell cytoplasm are evident (punctate granules; arrowheads). Arrow in 100 d -/- panel indicates the presence of inclusions in neural fibers. Scale bar, 20 μm. B, H&E-stained sections of large neurons in the reticular nucleus of the medulla. Neuronal eosinophilic inclusions are barely visible at 48 d (arrow) but increase significantly in number and size with survival time. By 154 d survival, the inclusions appear to have aggregated into a single mass in some neurons (arrow). Neuronal eosinophilic inclusions were not present in tissue from littermate controls (+/+ or +/-) (non-neuronal staining derives from residual erythrocytes not removed during perfusion). Scale bar, 50 μm.
Storage within the brain of the CLN2-targeted mice was also clearly resolved with LFB-CV staining as bluish-green deposits within the cytoplasm along with a loss of stainable Nissl substance (Fig. 5). The lysosomal membrane protein LAMP-1 was variably increased within the cytoplasm of neurons and white matter possibly as a secondary response to lysosomal storage as found in other lysosomal storage diseases (Meikle et al., 1997).
Figure 5.
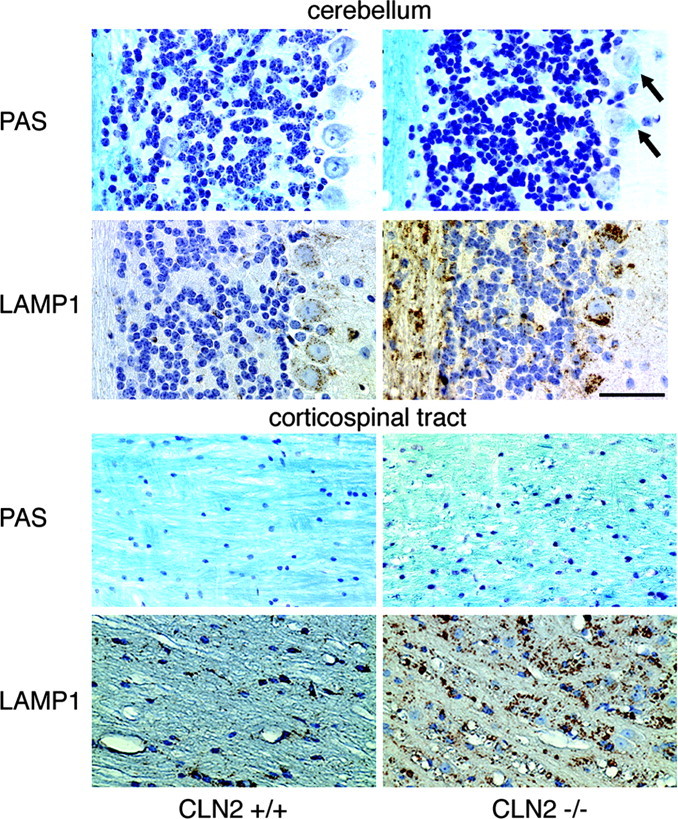
Matched sections from the cerebellum and corticospinal tract stained for LFB-CV and immunohistochemically for LAMP-1. In the cerebellum, there is a loss of Nissl substance from -/- cells, and storage pathology is visible as bright blue deposits within the cytoplasm of Purkinje cells (arrows). In both the corticospinal tract and cerebellum, lysosomal staining for LAMP-1 is markedly increased. In the latter, staining in increased in both Purkinje cells of -/- animals and in the underlying cerebellar white matter. Control mice (+/+) were 104 or 154 d old, and mutants (-/-) were 115 or 154 d old. Scale bar, 50 μm.
Pathological changes in the white matter were also observed in CLN2-targeted mice. In the oldest animals examined (∼150 d), there was a clear disruption of myelin in the white matter, notably the corticospinal tracts, as visualized by LFB-CV staining (Fig. 5). In normal brain, axons were closely packed and individually enveloped in LFB-stained myelin sheaths. In CLN2-targeted mice, the myelin sheaths were disorganized with prominent dilatations throughout the tract, indicative of degeneration. As noted previously, LAMP-1 staining of the corticospinal tract was markedly increased in the CLN2-targeted animals (Fig. 5) compared with the control animals. This may reflect infiltration of microglia that are intensely positive for LAMP-1 immunoreactivity and/or increased neuronal LAMP-1 levels as a secondary response to storage (see above).
Curvilinear storage bodies are an ultra-structural hallmark of cLINCL (Goebel et al., 1999), and similar inclusions were observed in the CLN2-deficient mice at as early as 35 d of age, the earliest time point evaluated. Figure 6 shows representative storage material in cortex, but storage density and size were similar in other brain regions examined (data not shown). Curvilinear storage bodies were large and not always surrounded by a discernable membrane, an observation that has also been made with cLINCL patients (Williams et al., 1999).
Figure 6.

Electron photomicrographs of cells in sections of cortex from CLN2-targeted mice at different ages and a 100 d +/- control littermate. Lysosomal curvilinear bodies are indicated by asterisks. Tissues were processed in Spurr's media, and 0.05 μm sections analyzed by transmission electron microscopy. Scale bar, 500 nm.
The presence of storage material outside of the brain was also examined in the CLN2-targeted mice (data not shown). Increased levels of autofluorescent material were present in a variety of organs from mutants, including the liver, kidney, spleen, and heart, although no gross cytopathology associated with storage was observed. The structure of the retina, including photoreceptors, was fairly well preserved in the CLN2 mutants, although there was evidence for vacuolization of the retinal ganglion cell layer along with accumulation of autofluorescent material in some cells
Neuropathology in CLN2-targeted mice
In addition to storage pathology, brains from CLN2-targeted mice showed mild to moderate atrophy in forebrain structures, such as the hippocampus and overlying neocortex, consistent with loss of neurons. At the electron microscopic level, there was a disorganization of the neuropil and shrunken, misshapen neuronal cell bodies containing pyknotic or karyolytic nuclei (data not shown).
Within the cerebellum, degenerative changes in Purkinje cells were evident in PAS-stained material (data not shown) and by immunohistochemical staining for calbindin (Fig. 7). Globally, Purkinje cell loss was patchy and tended to be variable in extent with respect to the age of the mice, reflecting the variability in progression of disease and lifespan. However, neuronal loss was generally more prevalent in the anterior lobules of the cerebellum, particularly in lobules III and VI (Fig. 7).
Figure 7.
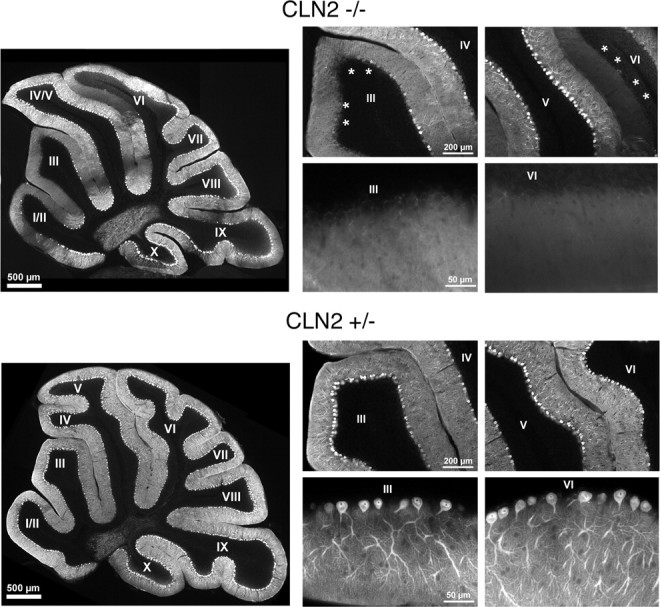
Degenerative changes affecting cerebellar Purkinje cells. Cerebellar sections from a 77-d-old CLN2-targeted mouse (top set) and heterozygote controls (bottom set) were stained for calbindin and photographed at low magnification (large left panel of each set) showing a patchy loss of Purkinje cells and associated dendrites within the molecular layer. Lobes III and VI, which showed significant loss of Purkinje cells in the mutant (asterisks), were photographed at medium magnification (top right panels of each set), and the boundaries between the molecular and granular layers were photographed at high magnification (bottom right panels of each set).
Sections from -/- brains prepared with the DeOlmos silver method for the selective identification of damaged or degenerating neurons (DeOlmos and Ingram, 1971) were intensely stained, demonstrating more widespread damage and neurodegeneration throughout the brain than was first appreciated on the basis of the staining results described previously. At all levels of the brain, there was dense impregnation of axons-fiber tracts, terminal projections, and occasional neuronal cells bodies (Fig. 8). Neocortical regions were noticeably thinner in the CLN2-targeted mice compared with controls (Fig. 9A,B) with prominent fiber staining, but the limbic cortex was relatively spared, with a sharp line of demarcation at the rhinal fissure (Fig. 8A). Motor neurons in the hindbrain and cerebellar Purkinje cells, along with their degenerating dendritic trees, were heavily stained (Figs. 8D, 9C). Although neuronal degeneration was widespread, it was not random, an observation that has also been in a number of pathogenic states (Sarna and Hawkes, 2003). In many areas, the DeOlmos silver stain indicated that the -/- brain was affected with remarkable anatomic precision. For example, auditory pathways were heavily involved at all levels, but the optic tract and ascending visual projections were not (Figs. 8B,9D). Storage pathology did not typically overlap or appear to react with the silver stain, although the cell bodies of motor neurons in the hindbrain were intensely stained. The overall pattern of silver staining was similar in all four mutant mice examined. Parallel studies of other mouse models of lysosomal storage diseases (Pompe and Krabbe's diseases, infantile NCL, and Niemann Pick type A) suggest that the pattern of neurodegeneration in the CLN2 mutant is specific to this model.
Figure 8.
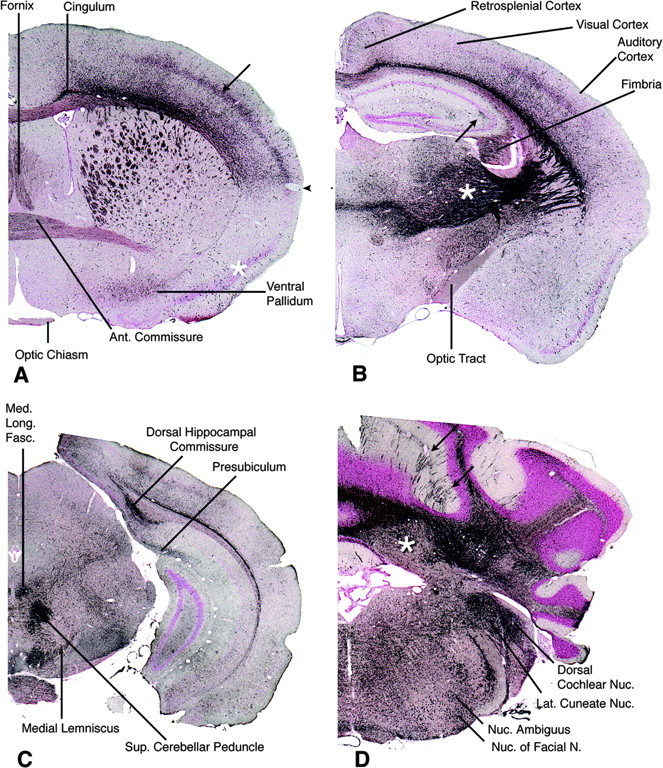
Rostrocaudal series of sections through the brain of a male -/- animal at 132 d survival stained by the DeOlmos silver degeneration method. A, Rostrally, at the level of the decussation of the anterior commissure, impregnation of structures is anatomically discrete. The anterior limb of the anterior commissure is moderately stained. The cingulum and fiber bundles crossing the caudate-putamen are strongly stained, as is the fornix. The ventral pallidum, but not the adjacent pyriform cortex (asterisk), is moderately positive. In the cerebral neocortex, the somatosensory cortex is distinctly positive (arrow), but ventral allocortex is relatively negative with a fairly sharp boundary between them (arrowhead). The optic chiasm is unstained. B, At the level of the diencephalon, the hypothalamus is unstained, but the lateral thalamus is strongly positive (asterisk), as are fiber pathways connecting thalamus with cerebral cortex. The optic tract, in contrast, is virtually unstained. A discrete area of mossy fiber degeneration is present in the hippocampus (arrow). Within the cerebral cortex, the auditory cortex is positive, particularly in layer 4 and deeper layer 6, whereas the adjacent visual cortex is almost negative. Staining of the retrosplenial cortex is slight. C, At the midbrain level, several tracts are intensely stained including the dorsal hippocampal commissure, medial longitudinal fasciculus, medial lemniscus, and superior cerebellar peduncle. D, Section through cerebellum and medulla. Groups of stained Purkinje cells in parasagittal bands are visible in some cerebellar cortical lobules such as the simplex (arrows; see also Fig. 9C). Heavily stained Purkinje axons fill the cerebellar white matter and terminate on mostly unstained neurons in the cerebellar nuclei (asterisk). Additional strong staining, mainly of axon terminals, is seen in the lateral cuneate and dorsal cochlear nuclei. Cell bodies, but not their axons, are strongly stained in the motor nucleus of the facial nerve and nucleus ambiguus. Representative results are shown from silver staining of four CLN2 mutants and two age-matched controls.
Figure 9.
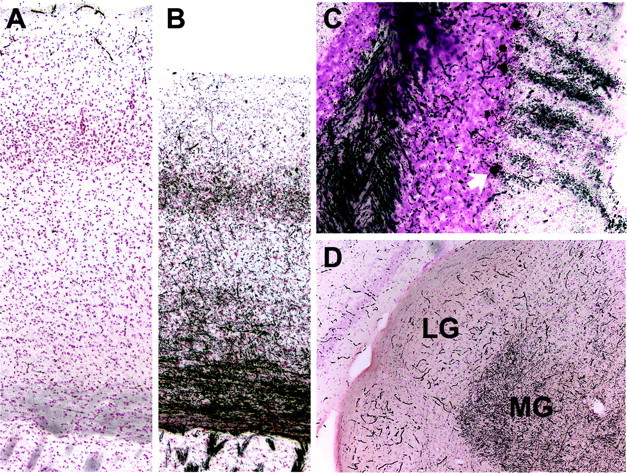
Higher-magnification illustrations from mouse brain sections stained by the DeOlmos silver degeneration method. A, Somatosensory cortex in a +/+ control. Staining is limited to a light, nonspecific “dusting” overlying cell nuclei and intense reaction within poorly perfused blood vessels. B, The corresponding somatosensory regions of a CLN2 mutant. Fibers throughout the striatum, callosum, and neocortex are intensely impregnated. The neocortex is also noticeably thinner at this level compared with control. C, In the cerebellar cortex of a CLN2 mutant, a cluster of Purkinje cells is densely stained (cell bodies along with their dendritic trees extending through the molecular layer), as well as fibers in the underlying white matter. The cell body of the lowermost Purkinje cell is arrowed. D, As an example of anatomical selectivity in silver staining, fibers coursing into the medial geniculate (MG) are strongly positive in a CLN2 mutant, as are most other components in the auditory circuitry, whereas the adjacent lateral geniculate (LG) that receives visual projections is unstained (except for nonperfused blood vessels). Mice were male and 128-132 d of age.
Discussion
Neurodegeneration in the CLN2-targeted mouse
Like cLINCL patients, the CLN2-targeted mouse is apparently healthy at birth but develops signs of progressive neurological deficits as it ages, developing tremors and becoming ataxic. Survival of the CLN2 mutant is greatly reduced (median survival, 138 d). This represents a significantly shorter lifespan than seen in other mouse NCL models, which was unexpected given that survival of cLINCL patients is similar to or longer than that of infantile neuronal ceroid lipofuscinosis patients (Table 1). Although it is conceivable that positional effects on neighboring genes may be conferred by the insertion of the neo cassette, we have excluded positional effects on proximal transcription factor TAF10 that might contribute to the relatively rapid demise of the CLN2 mutant, and so consider it likely that relative difference in longevity between the human and mouse NCL diseases can be attributed to species-specific metabolic differences. Such apparent discrepancies are not uncommon in mouse models of lysosomal storage diseases in general.
Table 1.
Life expectancy for classical forms of NCLs and their corresponding mouse null mutants
|
Gene |
Type |
Lifespan of patients (in years) |
Lifespan of mouse model (in months) |
|---|---|---|---|
| CLN1 | Infantile | 8-13 (EEG flat by ∼3 years) (Santavuori et al., 1999b) | 5 to >9 (Gupta et al., 2001) |
| CLN2 | Late infantile | 6-13 (Sleat et al., 1999) | 3-6 |
| CLN3 | Juvenile | ∼14-26 (Wisniewski et al., 1988) | 18-20 (Mitchison et al., 2004) |
| CLN4 | Adult | >15-50 (Martin et al., 1999) | n/a |
| CLN5 | Variant late infantile | 14-32 (Santavuori et al., 1999a) | n/a |
| CLN6 | Variant late infantile | 5-12 (Elleder et al., 1997) | ∼9 (Bronson et al., 1998) |
| CLN8 |
Epilepsy with mental retardation |
Normal (Haltia et al., 1999) |
6.5-12 (Messer et al., 1999) |
The severe neurological impairment and shortened lifespan of the CLN2-targeted mouse was matched by an underlying progressive, global neuropathology that involved the brain, spinal cord, and also peripheral sensory neurons. At the light microscopic level, the primary cellular feature was an accumulation of small, rounded cytoplasmic inclusions, identified as distended lysosomes, that were clearly resolved by staining with PAS, H&E, and LFB-CV, were brightly autofluorescent, and colocalized with immunohistochemical staining for the lysosomal marker LAMP-1. Distended lysosomal pathology is a hallmark of many metabolic storage diseases that affect the brain. Although the identity of the stored material in the mouse brain remains to be determined, eosinophilic staining of the lysosomal compartment is consistent with a proteinaceous nature. Subunit c of mitochondrial ATP synthase is a major component of the storage material in NCLs (Palmer et al., 1989a,b), and future studies will investigate storage of this protein and other material in the CLN2-targeted mouse. The presence of curvilinear deposits at the electron microscopic level is of particular interest because these represent a pathognomonic feature of cLINCL relative to other forms of Batten disease that bear a different signature of cytoplasmic inclusion (Goebel et al., 1999).
The extensive, severe loss of neurons in the cerebral cortex of human cLINCL patients is not seen to the same degree in the CLN2-targeted mouse, although the neocortex was noticeably thinner and there was widespread impregnation of axons and axonal endings demonstrated with the DeOlmos silver degeneration stain (see below). These findings suggest that neuronal degenerative disease has begun in the mouse cerebral neocortex but is much milder than in human cLINCL cases, perhaps because the mouse lifespan is so much shorter. Similarly, human cases also show loss of granule cell neurons in the cerebellar cortex that may be secondary to the Purkinje cell loss (Goebel et al., 1999), but in the mouse, death of the animal may be occurring too soon after Purkinje cell loss for this secondary abnormality to have time to evolve.
Although retinal degeneration is a prominent feature in cLINCL (Goebel et al., 1999), photoreceptors and other cell layers in the mutant retina appeared primarily intact and there was only light silver staining of ascending visual pathways in the brain. In contrast, there was extensive degeneration at all central levels in the mouse auditory pathway, including the dorsal cochlear nucleus, trapezoid nucleus and tract, superior olivary nucleus, lateral lemniscus, central nucleus of the inferior colliculus, medial geniculate nucleus, and primary auditory neocortex. Hearing defects have not been described in human patients. Similarly, the -/- mouse brains show silver-stained degeneration in the somatosensory pathway (gracile and cuneate nuclei, medial lemniscus, nucleus ventralis posterolateralis and posteromedialis in the thalamus), in the taste pathway (rostral part of the solitary nucleus), and in the olfactory pathway (lateral olfactory tract), whereas abnormalities in these sensory modalities are not significant features in human cases. The functional significance of these specific and localized patterns of neurodegeneration in the CLN2 mutant mouse will require a detailed characterization of the model in terms of sensory acuity.
The CLN2-targeted mouse as a therapeutic model
There currently exists a pressing need for an effective therapy for cLINCL. Given the lack of other animal models, we anticipate that the CLN2-targeted mouse will prove to be an invaluable and immediate resource for the development and evaluation of approaches such as enzyme replacement therapy, gene therapy (Haskell et al., 2003), and stem cell therapy. In this report, we have provided a detailed description of neurodegeneration in the CLN2 mutant that provides a firm basis for future studies aimed toward establishing a detailed time course for pathology. Such studies, together with behavioral tests to measure the progression of locomotor and sensory dysfunction, will provide essential quantitative parameters for evaluating potential therapies. In addition, additional study of the CLN2-targeted mouse and characterization of the storage material should shed light on the pathogenic processes that underlie neuronal death and may provide new avenues for therapeutic investigation.
Note added in proof. We observed several instances of apparent fatal startle seizures in the CLN2 mutant mouse that are induced by strong environmental stimuli such as loud noise and that strain differences in survival may reflect differences in sensitivity to such stimuli. Although the induction of fatal seizures in the CLN2 mutant requires additional investigation, environmental factors clearly need to be carefully considered in future studies of this model.
Footnotes
This work was supported by National Institutes of Health Grants NS37918 (P.L.) and HD42837 (M.M.S.). We thank Rosalie Gin, Vivienne Halili, Holly Collins, Jie Bu, and Tatyana Taksir for technical assistance and Istvan Sohar, Li Lin, Thomas Ludwig, John Pintar, Alwin Schuller, and Jixiang Ding for helpful discussions, reagents, and other assistance.
Correspondence should be addressed to Peter Lobel or David Sleat, Center for Advanced Biotechnology and Medicine, 679 Hoes Lane, Piscataway, NJ 08854. E-mail: lobel@cabm.rutgers.edu or sleat@cabm.rutgers.edu.
G. R. Stewart's present address: Medtronic Neurological, Columbia Heights, MN 55421.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249117-10$15.00/0
References
- Bronson RT, Donahue LR, Johnson KR, Tanner A, Lane PW, Faust JR (1998) Neuronal ceroid lipofuscinosis (nclf), a new disorder of the mouse linked to chromosome 9. Am J Med Genet 77: 289-297. [DOI] [PubMed] [Google Scholar]
- Chen JW, Pan W, D'Souza MP, August JT (1985) Lysosome-associated membrane proteins: characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch Biochem Biophys 239: 574-586. [DOI] [PubMed] [Google Scholar]
- DeOlmos JS, Ingram WR (1971) An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res 33: 523-529. [DOI] [PubMed] [Google Scholar]
- Elleder M, Franc J, Kraus J, Nevsimalova S, Sixtova K, Zeman J (1997) Neuronal ceroid lipofuscinosis in the Czech Republic: analysis of 57 cases. Report of the `Prague NCL group'. Eur J Paediatr Neurol 1: 109-114. [DOI] [PubMed] [Google Scholar]
- Gao H, Boustany RM, Espinola JA, Cotman SL, Srinidhi L, Antonellis KA, Gillis T, Qin X, Liu S, Donahue LR, Bronson RT, Faust JR, Stout D, Haines JL, Lerner TJ, MacDonald ME (2002) Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet 70: 324-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel HH, Mole SE, Lake BD (1999) The neuronal ceroid lipofuscinoses (Batten disease). Washington, DC: IOS.
- Greene ND, Bernard DL, Taschner PE, Lake BD, de Vos N, Breuning MH, Gardiner RM, Mole SE, Nussbaum RL, Mitchison HM (1999) A murine model for juvenile NCL: gene targeting of mouse Cln3. Mol Genet Metab 66: 309-313. [DOI] [PubMed] [Google Scholar]
- Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL (2001) Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc Natl Acad Sci USA 98: 13566-13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haltia M, Tyynela J, Hirvasniemi A, Herva R, Ranta US, Lehesjoki AE (1999) CLN8 northern epilepsy. In: The neuronal ceroid lipofuscinoses (Batten disease). In: Biomedical and health research (Goebel HH, Mole S, Lake BD, eds), pp 117-124. Amsterdam: IOS.
- Haskell RE, Hughes SM, Chiorini JA, Alisky JM, Davidson BL (2003) Viral-mediated delivery of the late-infantile neuronal ceroid lipofuscinosis gene, TPP-I to the mouse central nervous system. Gene Ther 10: 34-42. [DOI] [PubMed] [Google Scholar]
- Hofmann SL, Peltonen L (2001) The neuronal ceroid lipofuscinoses. In: The metabolic and molecular bases of inherited disease (Scriver CR, Beaudet AL, Sly WS, Childs B, Vogelstein B, eds), pp 3877-3894. New York: McGraw-Hill.
- Katz ML, Shibuya H, Liu PC, Kaur S, Gao CL, Johnson GS (1999) A mouse gene knockout model for juvenile ceroid-lipofuscinosis (Batten disease). J Neurosci Res 57: 551-556. [PubMed] [Google Scholar]
- Lin L, Sohar I, Lackland H, Lobel P (2001) The human CLN2 protein/tripeptidyl-peptidase I is a serine protease that autoactivates at acidic pH. J Biol Chem 276: 2249-2255. [DOI] [PubMed] [Google Scholar]
- Liu CG, Sleat DE, Donnelly RJ, Lobel P (1998) Structural organization and sequence of CLN2, the defective gene in classical late infantile neuronal ceroid lipofuscinosis. Genomics 50: 206-212. [DOI] [PubMed] [Google Scholar]
- Lyon G, Raymond AD, Kolodny EH (1996) Neurology of hereditary metabolic diseases of children, Ed 2. New York: McGraw-Hill.
- Martin J-J, Gottlob I, Goebel HH, Mole S (1999) CLN4 adult NCL. In: The neuronal ceroid lipofuscinoses (Batten disease). In: Biomedical and health research (Goebel HH, Mole S, Lake BD, eds), pp 77-90. Amsterdam: IOS.
- Meikle PJ, Brooks DA, Ravenscroft EM, Yan M, Williams RE, Jaunzems AE, Chataway TK, Karageorgos LE, Davey RC, Boulter CD, Carlsson SR, Hopwood JJ (1997) Diagnosis of lysosomal storage disorders: evaluation of lysosome-associated membrane protein LAMP-1 as a diagnostic marker. Clin Chem 43: 1325-1335. [PubMed] [Google Scholar]
- Messer A, Manley K, Plummer JA (1999) An early-onset congenic strain of the motor neuron degeneration (mnd) mouse. Mol Genet Metab 66: 393-397. [DOI] [PubMed] [Google Scholar]
- Mitchison HM, Lim MJ, Cooper JD (2004) Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol 14: 86-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole S (2002) Neuronal ceroid lipofuscinoses (NCL) mutations. http://www.ucl.ac.uk/ncl/.
- Palmer DN, Martinus RD, Cooper SM, Midwinter GG, Reid JC, Jolly RD (1989a) Ovine ceroid lipofuscinosis. The major lipopigment protein and the lipid-binding subunit of mitochondrial ATP synthase have the same NH2-terminal sequence. J Biol Chem 264: 5736-5740. [PubMed] [Google Scholar]
- Palmer DN, Fearnley IM, Medd SM, Walker JE, Martinus RD, Bayliss SL, Hall NA, Lake BD, Wolfe LS, Jolly RD (1989b) Lysosomal storage of the DCCD reactive proteolipid subunit of mitochondrial ATP synthase in human and ovine ceroid lipofuscinoses. Adv Exp Med Biol 266: 211-222. [DOI] [PubMed] [Google Scholar]
- Ranta S, Zhang Y, Ross B, Lonka L, Takkunen E, Messer A, Sharp J, Wheeler R, Kusumi K, Mole S, Liu W, Soares MB, Bonaldo MF, Hirvasniemi A, de la Chapelle A, Gilliam TC, Lehesjoki AE (1999) The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat Genet 23: 233-236. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ (1999) Tripeptidyl-peptidase I is apparently the CLN2 protein absent in classical late-infantile neuronal ceroid lipofuscinosis. Biochim Biophys Acta 1429: 496-500. [DOI] [PubMed] [Google Scholar]
- Santavuori P, Rapola J, Haltia M, Tyynela J, Peltonen L, Mole S (1999a) CLN5 Finnish variant late infantile NCL. In: The neuronal ceroid lipofuscinoses (Batten disease) (Goebel HH, Mole S, Lake BD, eds), pp 16-36. Amsterdam: IOS.
- Santavuori P, Gottlob I, Haltia M, Rapola J, Lake BD, Tyynela J, Peltonen L (1999b) CLN1 infantile and other types of NCL with GROD. In: The neuronal ceroid lipofuscinoses (Batten disease). Amsterdam: IOS.
- Sarna JR, Hawkes R (2003) Patterned Purkinje cell death in the cerebellum. Prog Neurobiol 70: 473-507. [DOI] [PubMed] [Google Scholar]
- Sharp JD, Wheeler RB, Lake BD, Savukoski M, Jarvela IE, Peltonen L, Gardiner RM, Williams RE (1997) Loci for classical and a variant late infantile neuronal ceroid lipofuscinosis map to chromosomes 11p15 and 15q21-23. Hum Mol Genet 6: 591-595. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P (1997) Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science 277: 1802-1805. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S, Pullarkat RK, Palmer DN, Lerner TJ, Boustany RM, Uldall P, Siakotos AN, Donnelly RJ, Lobel P (1999) Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am J Hum Genet 64: 1511-1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohar I, Sleat DE, Jadot M, Lobel P (1999) Biochemical characterization of a lysosomal protease deficient in classical late infantile neuronal ceroid lipofuscinosis (LINCL) and development of an enzyme-based assay for diagnosis and exclusion of LINCL in human specimens and animal models. J Neurochem 73: 700-711. [DOI] [PubMed] [Google Scholar]
- Sohar I, Lin L, Lobel P (2000) Enzyme-based diagnosis of classical late infantile neuronal ceroid lipofuscinosis: comparison of tripeptidyl peptidase I and pepstatin-insensitive protease assays. Clin Chem 46: 1005-1008. [PubMed] [Google Scholar]
- Switzer III RC (2000) Application of silver degeneration stains for neurotoxicity testing. Toxicol Pathol 28: 70-83. [DOI] [PubMed] [Google Scholar]
- Vines D, Warburton MJ (1998) Purification and characterisation of a tripeptidyl aminopeptidase I from rat spleen. Biochim Biophys Acta 1384: 233-242. [DOI] [PubMed] [Google Scholar]
- Vines DJ, Warburton MJ (1999) Classical late infantile neuronal ceroid lipofuscinosis fibroblasts are deficient in lysosomal tripeptidyl peptidase I. FEBS Lett 443: 131-135. [DOI] [PubMed] [Google Scholar]
- Wheeler RB, Sharp JD, Schultz RA, Joslin JM, Williams RE, Mole SE (2002) The gene mutated in variant late-infantile neuronal ceroid lipofuscinosis (CLN6) and in nclf mutant mice encodes a novel predicted transmembrane protein. Am J Hum Genet 70: 537-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RE, Gottlob I, Lake BD, Goebel HH, Winchester BG, Wheeler R (1999) CLN2 classic late infantile NCL. In: The neuronal ceroid lipofuscinoses (Batten disease) (Goebel HH, Mole S, Lake BD, eds), pp 37-54. Amsterdam: IOS.
- Wisniewski KE, Rapin I, Heaney-Kieras J (1988) Clinico-pathological variability in the childhood neuronal ceroid-lipofuscinoses and new observations on glycoprotein abnormalities. Am J Med Genet [Suppl] 5: 27-46. [DOI] [PubMed] [Google Scholar]