

Abstract
Neurite outgrowth is influenced by positive and negative signals that include the semaphorins, an important family of axonal outgrowth inhibitors. Here we report that the Rac GTPase activating protein (GAP)α2-chimaerin is involved in Semaphorin 3A (Sema 3A) signaling. In dorsal root ganglion neurons, Sema 3A-induced growth cone collapse was inhibited by α2-chimaerin mutated to eliminate GAP activity or interaction with phosphotyrosine. Activation of α2-chimaerin by phorbol ester caused growth cone collapse. Active α2-chimaerin interacts with collapsin response mediator protein-2 (CRMP-2) and cyclin-dependent kinase (Cdk) 5/p35 kinase through its SH2 and GAP domains, respectively. Cdk5 phosphorylates CRMP-2 at serine 522, possibly facilitating phosphorylation of serine 518 and threonine 514 by glycogen synthase kinase 3β (GSK3β), a kinase previously implicated in Sema 3A signaling. Phosphorylation of CRMP-2 serine 522 was essential for Sema 3A-induced growth cone collapse, which is dependent on Cdk5 but not Rho kinase activity. α2-chimaerin, like CRMP-2, can associate with the Sema 3A receptor. These results indicate that active α2-chimaerin Rac GAP, Cdk5/p35, and its substrate CRMP-2, are implicated in the dynamics of growth cone guidance initiated through Sema 3A signaling.
Keywords: Sema 3A, RacGTPase, CRMP-2, α2-chimaerin, Cdk5/p35, growth cone collapse
Introduction
In neuronal development, axonal outgrowth is guided by attractive and repulsive signals that stimulate Rho GTPase pathways regulating actin dynamics in the growth cone. RhoA and Rho kinase (ROK) are activated in response to lysophosphatidic acid (LPA), ephrin A5, and Nogo, causing increased contractility and neurite retraction (Tigyi et al., 1996; Wahl et al., 2000; Niederost et al., 2002). NGF, netrins, and other attractive guidance signals activate Rac and Cdc42, promoting neurite outgrowth through effector proteins including p21-activated kinase (PAK) and IRSp53 (insulin receptor tyrosine kinase substrate) (Kozma et al., 1997; Daniels et al., 1998; Govind et al., 2001; Li et al., 2002). Paradoxically, Rac is also required for growth cone collapse and retraction induced by Semaphorin 3A (Sema 3A) (Jin and Strittmatter, 1997; Vastrik et al., 1999). The Sema 3A receptor is a neuropilin-1/plexin-A complex (Takahashi et al., 1999) that may signal through an associated tyrosine kinase, Fyn (Sasaki et al., 2002), Fps/fes (Mitsui et al., 2002), or possibly off-track (Winberg et al., 2001). Molecule interacting with CasL oxidoreductases also associate with plexin (Terman et al., 2002), and redox signaling can play a role downstream of Sema 3A neurite outgrowth in PC12 cells (Schwamborn et al., 2004). In the Sema 3A pathway, actin polymerization is influenced by phosphorylation of cofilin by LIM kinase (Aizawa et al., 2001) that can act downstream of either PAK or ROK (Edwards et al., 1999; Maekawa et al., 1999). Another pathway component is collapsin response mediator protein-2 (CRMP-2) (Goshima et al., 1995), a neuronal phosphoprotein involved in axonal outgrowth (Minturn et al., 1995; Inagaki et al., 2001) and microtubule dynamics (Gu and Ihara, 2000; Fukata et al., 2002). CRMP-2 can reversibly switch RhoA and Rac morphological phenotypes, regulated by ROK (Hall et al., 2001a), which phosphorylates CRMP-2 at threonine 555 (Arimura et al., 2000). CRMP-2 associates with ROK, potentially attenuating its activity (Leung et al., 2002). Rho GTPases are downregulated by GTPase activating proteins (GAPs), including phospholipid-activated chimaerins, which act on Rac (Diekmann et al., 1991; Manser et al., 1992; Ahmed et al., 1993; Caloca et al., 1997). α2-Chimaerin, containing an Src homology 2 domain (SH2) domain in addition to C1 and GAP domains, is highly expressed in the developing nervous system (Hall et al., 1993, 2001b). Cyclin-dependent kinase (Cdk) 5 and its neuronal regulator p35 play an important role in neuronal migration, neurite outgrowth, and guidance (Nikolic et al., 1996; Chae et al., 1997; Sasaki et al., 2002). In the Sema 3A pathway, Fyn phosphorylates and activates Cdk5 (Sasaki et al., 2002). Here we show that α2-chimaerin Rac GAP activity is necessary for Sema 3A-induced growth cone collapse in dorsal root ganglion (DRG) neurons. Active α2-chimaerin associates with CRMP-2 and Cdk5/p35 via different domains. Cdk5 phosphorylates CRMP-2 at serine 522; this phosphorylation is required for Sema 3A-induced growth cone collapse and may facilitate subsequent phosphorylation of CRMP-2 at serine 518 and threonine 514 by the important kinase glycogen synthase kinase 3β (GSK3β). Both CRMP-2 and α2-chimaerin can associate with the Sema 3A receptor. These findings indicate that α2-chimaerin and its partners, CRMP-2 and Cdk5/p35, participate in Sema 3A signaling in which activation of α2-chimaerin is required.
Materials and Methods
Antibodies used were as follows: rabbit polyclonal α2-chimaerin and CRMP-2 (Hall et al., 2001a,b); agarose-coupled mouse anti-Flag (M2-FLAG-AC), rabbit polyclonal FLAG, mouse β-tubulin (Sigma-Aldrich, Poole, UK); rabbit polyclonal Cdk5 (C-8), agarose-coupled Cdk5-AC, mouse monoclonal p35 (C-19), anti-myc and glutathione S-transferase (GST) (Z-5) antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), mouse monoclonal anti-haemagglutinin (HA) tag (Babco, Richmond, CA), mouse monoclonal green fluorescent protein (GFP) (Clontech, Oxford, UK), agarose-coupled anti-myc antibody (Abcam, Cambridge, UK); and mouse monoclonal anti-Rac and anti-phosphotyrosine RC20 (Transduction Labs, Oxford, UK).
Protein purification and overlay assay. Rat brain homogenate (20%) in 25 mm MES, pH 6.5, 0.5 mm MgCl2, 0.05% Triton X-100, 50 μm ZnCl2, 0.1 mm EGTA, 5 mm DTT, 0.5 mm PMSF, 1 μg/ml pepstatin, 1 μg/ml aprotinin was centrifuged at 100,000 × g for 1 hr at 4°C, and p64 (CRMP-2) was purified by fractionation of the soluble extract by chromatography on S-Sepharose, Q-Sepharose, Zinc chelating Sepharose, and phenyl-Sepharose columns, sequentially, as described for purification of α2-chimaerin (Hall et al., 1993). Fractions were eluted from S-Sepharose in a salt gradient, and 0.2 m NaCl fraction (see Fig. 1 A, lane 10) was diluted and fractionated on Q-Sepharose. P64 eluted with 0. 2 m NaCl (see Fig. 1 A, lane 13) was adjusted to pH 7.5 with 1 m Tris-HCl, pH 7.5, and applied to a Zinc chelating column precharged with ZnCl2 (Hall et al., 1993). P64 was eluted in two separate fractions, at pH 6.0 and with 50 mm imidazole. The latter fraction (which also contained α2-chimaerin) was adjusted to 0.4 m NH4SO4, applied to a phenyl-Sepharose column, and p64 was eluted in 0.2 m NH4SO4 [α2-chimaerin is eluted by 0.2 m deoxycholate, pH 8.5 (Hall et al., 1993)]. Proteins in this fraction were separated by two-dimensional (2-D) gel electrophoresis. Column fractions were analyzed by overlay assay (Manser et al., 1992) using recombinant α2-chimaerin or α2-chimaerin SH2 domain γ 32P-labeled by phosphorylation with PKC (0.05 μg) in [γ 32P]ATP (Amersham Biosciences, Little Chalfont, UK), 50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 0.25 mm CaCl2, 2.5 μg of phosphatidyl serine, 0.005 μg of PDBu (Sigma-Aldrich), and 0.012 mm ATP.
Figure 1.

Purification of CRMP-2 from rat brain extracts. A, Rat brain extracts (see Materials and Methods) fractionated on S-Sepharose were analyzed by Western blot and overlay asay with 32P-labeled α2-chimaerin SH2 (lanes 1-3) or GAP domain (lanes 4-5) (left). A 64 kDa protein was purified by sequential chromatography of brain soluble fraction (lane 7) on S-Sepharose (lanes 8-11), Q-Sepharose (lanes 12-15), Zinc chelating Sepharose (lanes 16-19), and phenyl Sepharose (lanes 21-23) (Materials and Materials) (Hall et al., 1993). Fractions were eluted from S-Sepharose in a salt gradient, and 0.2 m NaCl fraction (lane 10) was diluted and fractionated on Q-Sepharose. P64 eluted with 0.2 m NaCl (lane 13) and applied to a Zinc chelating column, eluted at pH 6.0 (lane 17) or with 50 mm imidazole (lane 19). The latter, adjusted to 0.4 m, was bound to phenyl-Sepharose and eluted with 0.2 m NH4S04. Eluted fractions were analyzed by overlay assay for α2-chimaerin interaction. Fractions shown are as follows: 0.1 m NaCl, lanes 2, 5, 9, 12; 0.2 m NaCl, lanes 3, 6, 10, 13; 0.3 m NaCl, lanes 11 and 14; 0.4 m NaCl, lane 15; Zn chelate unbound, lane 16; pH 6.0 elution, lane 17; 20 mm imidazole, lane 18; 50 mm imidazole, lane 19; phenyl-Sepharose unbound, lane 20; 0.2 m NH4SO4, lane 21. The final fraction was analyzed by 2-D gel electrophoresis, stained with Coomassie blue, and two p64 spots identified by overlay were cut out for peptide sequence analysis. The peptide sequences that were obtained are underlined in B. B, CRMP-2 sequence. Peptides isolated and sequenced are underlined. C, Western analysis of CRMP-2 and α2-chimaerin in embryonic and neonatal rat brain.
Site-directed mutagenesis. pXJ40 CRMP-2 and pXJ40 α2-chimaerin mammalian cDNA constructs were as described previously (Hall et al., 2001a,b). Cdk5 and p35 cDNA constructs were as described (Qi et al., 1998) or cloned in pXJ40. Myc-tagged Plexin-A construct was generously provided by Stephen Strittmatter (Yale University, New Haven, CT), and myc-tagged neuropilin-1 cDNA was provided by Rabinder Prinjha (GlaxoSmithKline, Harlow, UK).
Putative phosphorylation sites in CRMP-2 at serine 522, 518, threonine 509, 514 were individually mutated in pXJ40 Flag CRMP-2 to alanine residues using the QuikChange XL Site-Directed Mutagenesis Kit (Stratagene Europe, Amsterdam, The Netherlands) and sequenced (Cytomix, Cambridge, UK). The following mutagenic primers were used: S522A: 5′-CAGCTAAGACAGGCCCTGCCAAGCAG-5′; T509A: 5′-GAG GTG TCT GTG CGG CCC AAG ACG GTC-3′; T514A: 5′-CCC AAG ACG GTC GCT CCG GCC TCA TC-3′; S518A: 5′-C ACT CCG GCC TCA GCA GCT AAG ACA TCC-3′.
Cell treatment. N1E-115 cell lines cultured in DMEM, 10% fetal calf serum, and plated on laminin as described previously (Hall et al., 2001b) were transfected using Lipofectamine 2000 (Invitrogen, Paisley, UK). COS-7 cells were transfected with Lipofectamine (Invitrogen). For inhibition of tyrosine phosphatases, pervanadate (100 μm) was freshly prepared by treating 20 mm Na3VO4 in PBS with 0.05% H2O2 for 5 min at room temperature and excess H2O2 was removed by catalase (200 μg/ml), and cells in DMEM were treated for 20 min at 37°C.
Preparation of rat dorsal root ganglia neurons. Dorsal root ganglia were removed from the spinal cord of newborn Wistar rats (day 0-1) and treated with 0.125% collagenase (Worthington Biochemical Corporation, Freehold, NJ) in F12 Ham's media (Sigma-Aldrich), 5% FCS/F12 for 1-2 hr, washed in Hank's buffered saline (Sigma-Aldrich) at 37°C, and treated with 1% trypsin for 10 min at 37°C. After washing in 5% FCS/F12, DRG neurons were triturated in F12 media and centrifuged through a 15% BSA/F12 gradient at 800 rpm for 10 min. Cells were either electroporated (Amaxa Biosystems, Koln, Germany) or plated directly in F12 media with N2 supplement, 3% BSA, 10 nm cytosine arabinoside (Sigma-Aldrich), antibiotic/antimycotic (Invitrogen) plus NGF 100 ng/ml (Calbiochem, La Jolla, CA; Merck Biosciences, Nottingham, UK). DRG neurons, plated on acid-washed coverslips precoated with 10 μg/ml mouse laminin (Invitrogen), were maintained at 37°C in 5% CO2.
Collapse assays. DRG neurons transferred to 20 ng/ml NGF for 4 hr before collapse assays in the presence or absence of the Cdk5 inhibitor roscovitine 10 μm (Sigma-Aldrich) or Rho kinase inhibitor Y-27632 (10 μm) (Calbiochem), or N1E-115 cells permanently expressing α2-chimaerin (Hall et al., 2001b), were treated with either purified Sema 3A-Fc protein (time-lapse analysis) or Sema 3A-Fc enriched supernatant at 1:50 dilution (Eickholt et al., 2002) for 30 min before the cells were fixed for quantification. Phorbol ester, phorbol 12,13-myristate acetate (PMA; 100 nm) (LC Labs, Woburn, MA), was added to DRG neurons in the presence or absence of the PKC inhibitor bisindolylmaleimide (10 μm) (LC Labs). Immunostaining of DRG neurons was as described previously (Hall et al., 2001b). For time-lapse microscopy, an N1E-115 cell line permanently overexpressing α2-chimaerin was used (Hall et al., 2001b). Cells were plated on laminin-coated coverslip-bottom dishes (World Precision Instruments, Hertfordshire, UK) and transfected (16 hr) or treated with inhibitors before analysis 10 min before and 30 min after addition of Sema 3A through a micropipette placed in the vicinity of the growth cone, using a CCD camera and MetaMorph software to take images every 30 sec.
Immunoprecipitation. For immunoprecipitations, cleared cell lysates in 1% Triton X-100, 25 mm HEPES, pH 7.5, 0.3 m NaCl, 1 mm MgCl2, 1 mm EGTA, 20 mm β glycerophosphate, 5% glycerol were incubated with agarose-conjugated antibody (FLAG-M2 or Cdk5-AC) at 4°C for 2-3 hr and washed in lysis buffer, and proteins eluted in Laemmli SDS sample buffer were analyzed by Western blotting. Protein A agarose (Sigma-Aldrich) was used to isolate α2-chimaerin immunoprecipitates.
In vitro kinase assays. Assays were performed on recombinant protein substrates or proteins immunoprecipitated from COS-7 cells. Substrate protein (2-5 μg) in 120 mm MOPS, 5 mm MgCl2, recombinant Cdk5/p25 kinase, ATP (100 μm), 5 μCi of [32P]γATP was incubated at 30°C for 30 min. GSK3β (Upstate, Milton Keynes, UK) kinase assays were according to manufacturer's protocol.
Rac-GTP assays were performed using GST-PAK Cdc42/Rac binding region as described (Sander et al., 1999).
Results
CRMP-2 interacts with α2-chimaerin SH2 domain
The selective expression of α2-chimaerin in the embryonic nervous system and its effects on cell morphology indicated that its isoform-specific SH2 domain might interact with proteins involved in neurite outgrowth (Hall et al., 2001b). To identify such proteins, rat brain extracts were probed with 32P-labeled α2-chimaerin in an overlay assay (Fig. 1A). A 64 kDa protein in S-Sepharose-fractionated brain extracts was detected by either full-length α2-chimaerin or its SH2 domain (Fig. 1A), but not its GAP domain. The protein was purified by further chromatography and two-dimensional gel electrophoresis (Fig. 1A). Peptide sequence analysis revealed its identity with CRMP-2 (Minturn et al., 1995) (Fig. 1B). CRMP-2 and α2-chimaerin have similar expression profiles in embryonic and neonatal rat brain (Fig. 1C).
Recombinant CRMP-2 bound to the SH2 domain of α2-chimaerin in vitro in Western and dot blots (Fig. 2A). Binding did not occur on substitution of the important asparagine 94 by histidine (Fig. 2A). Binding still occurred with α2-chimaerin R73L and R56L. These mutations of the invariant or highly conserved arginines R73 and R56 (in the SH2 domain βB5 and αA2 positions) abolish interaction with phosphotyrosine (Waksman et al., 1993; Hall et al., 2001b). Substitution of the atypical glutamic acid by the almost invariant βA1 tryptophan also did not prevent the interaction of α2-chimaerin with CRMP-2 (Fig 2A).
Figure 2.

CRMP-2 interacts with α2-chimaerin SH2 domain. A, In vitro association of recombinant CRMP-2 with α2-chimaerin. Top, Western blot of recombinant CRMP-2 probed with 32P-labeled SH2 domain, SH2 domain mutants E49W, R56L, R73L, N94H, and full-length α2-chimaerin (FL). Bottom, Dot blot of recombinant CRMP-2 and p67phox control protein, probed with 32P-labeled α2-chimaerin SH2 domain or GST control. B, In vivo interaction of CRMP-2 with α2-chimaerin. Full-length Flag-α2-chimaerin (WT) and N-terminal truncations Flag-α2-chimaerin(196 - 459) and Flag-α2-chimaerin (268 - 459) were coexpressed in COS-7 cells with HA-CRMP-2 in the presence and absence of PMA (10 μm). Western blots of immunoprecipitated (IP) Flag-chimaerin were probed with anti-HA antibody to detect associated CRMP-2. C, Western blots of HA-α2-chimaerin associated with immunoprecipitated Flag-CRMP-2 after coexpression in COS-7 cells treated with PMA 0-10 μm as specified. D, Western blots of α2-chimaerin, α2-chimaerin E49W, α2-chimaerin R56L, and α2-chimaerin R73L immunoprecipitated with Flag-CRMP-2 after coexpression in COS-7 cells treated with PMA (10 μm). E, Western blots of α2-chimaerin (top), phosphotyrosine-containing proteins (detected with RC20 antibody; middle) immunoprecipitated with Flag-CRMP-2 (bottom) from cells of a permanent α2-chimaerin N1E-115 cell line transiently transfected with Flag-CRMP-2.
Activation of α2-chimaerin by phorbol ester promotes its interaction with CRMP-2 in vivo
CRMP-2 was strongly associated with α2-chimaerin when coexpressed in COS-7 cells only after cell treatment with phorbol ester (Fig. 2B,C), with the association showing a phorbol ester concentration dependence (Fig. 2C). This association required the SH2 domain of α2-chimaerin (Fig. 2B) but was independent of phosphotyrosine interactions, because R73L or R56L mutants still associated (Fig. 2D). In neuroblastoma cells but not in COS-7 cells (data not shown), however, treatment with pervanadate to inhibit tyrosine phosphatases promoted a slight association between CRMP-2 and α2-chimaerin (Fig. 2E), suggesting that neuronal cell-specific tyrosine phosphorylation may be capable of promoting α2-chimaerin association with CRMP-2 and/or its activation.
p35 and Cdk5 interact with α2-chimaerin GAP domain, potentiated by phorbol ester
Chimaerin was recently reported to interact with p35, the neuronal regulator of Cdk5, in a yeast two-hybrid screen (Qi et al., 2004). We confirmed this interaction in COS-7 cells, in which p35 could be coimmmunoprecipitated with Flag/α2-chimaerin for which the GAP domain was sufficient (Fig. 3A). Cdk5 also interacted with α2-chimaerin, and as with p35, the GAP domain was sufficient (Fig. 3B). When coexpressed with both Cdk5 and p35, which form a kinase-active complex (Fig. 3C), α2-chimaerin did not coimmunoprecipitate with Flag/p35. When coexpressed with p35 and kinase-inactive Cdk5N144, α2-chimaerin coimmunoprecipitated with Flag/p35 only in small and variable amounts (Fig. 3C). In all cases Cdk5 coprecipitated with Flag/p35, suggesting that the interaction of p35 and Cdk5 is of higher affinity than of either for α2-chimaerin; however, treatment with phorbol ester to activate α2-chimaerin resulted in increased association with either p35 or Cdk5, both in a concentration-dependent manner (Fig. 3D,E). This treatment also resulted in coimmunoprecipitation of all three proteins, using Flag/p35 (Fig. 3F). No phosphorylation of α2-chimaerin by Cdk5 was detected.
Figure 3.

Cdk5 and its neuronal regulator p35 interact with α2-chimaerin GAP domain. A, In vivo interaction of p35 with full-length Flag-α2-chimaerin (WT) and N-terminal truncations Flag-α2-chimaerin (196 - 459) and Flag-α2-chimaerin (268 - 459) immunoprecipitated after their coexpression in COS-7 cells with p35. B, In vivo interaction of Cdk5 with full-length Flag-α2-chimaerin (WT) and N-terminal truncations Flag-α2-chimaerin (39 - 459), Flag-α2-chimaerin (196 - 459), and Flag-α2-chimaerin (268 - 459) immunoprecipitated after coexpression in COS-7 cells with Cdk5. C, Immunoprecipitation of Flag-p35 from COS-7 cells after coexpression with the indicated combinations of Cdk5, kinase inactive Cdk5 N144, and/or GFP-α2-chimaerin. Western blots of α2-chimaerin, Cdk5, and Flag p35 in immunoprecipitates are shown. D, Coimmunoprecipitation of α2-chimaerin with Flag-p35 after coexpression in COS-7 cells treated with increasing amounts of PMA (0-10 μm) as indicated. E, Coimmunoprecipitation of Cdk5 with Flag-α2-chimaerin after coexpression in COS-7 cells treated with PMA (0-10 μm) as indicated. F, Coimmunoprecipitation with anti-Flag-p35 of Cdk5 andα2-chimaerin from COS-7 cells expressing all three proteins and treated with PMA (0-10 μm) as indicated. G, Immunoprecipitation with anti-Flag-p35 of GFP-α2-chimaerin (WT), α2-chimaerin N94H, α2-chimaerin R304G, and α2-chimaerin Δ303-305 after coexpression in COS-7 cells.
α2-Chimaerin may be in an autoinhibited conformation in cytosol
Mutation of either the SH2 or GAP domain can promote membrane translocation of α2-chimaerin. The SH2 domain mutant α2-chimaerin N94H is membrane associated and constitutively active (Hall et al., 2001b). The α2-chimaerin Δ303-305 (YRV) mutant deleted in the GAP domain was also translocated to the membrane (Fig. 4A). These two α2-chimaerin mutants, N94H and Δ303-305, also showed an increased association with p35 (Fig. 3G) or other targets (data not shown) compared with wild type, whereas the point-mutant α2-chimaerin R304G did not translocate to the membrane and showed no increased binding of p35 (Figs. 3G, 4A). Because these mutations in either the N or C terminal can both lead to membrane association and increased target interaction, this implies that these disparate activities of α2-chimaerin are unmasked through its conformational alteration. Unless activated, it appears that α2-chimaerin exists in an autoinhibited state in the cytosol.
Figure 4.

Phorbol ester activation and membrane translocation of α2-chimaerin, autoinhibited in cytosol. A, PMA activation of GFP-α2-chimaerin expressed in COS-7 cells (A, B) and of α2-chimaerin in N1E-115 cell lines (C-E). A, Cell morphology and distribution of α2-chimaerin, α2-chimaerin R304G, and α2-chimaerin Δ303-305 (2 GAP in active mutants) (top) in the presence and absence of PMA treatment. Phalloidin-stained actin is shown for the PMA-treated cells (bottom). B, Quantitation of COS-7 cells showing ruffling (lamellipodia; top) and collapse (bottom) in response to increasing concentrations of PMA after transfection with α2-chimaerin (filled columns) or GAP inactive α2-chimaerin Δ303-305 (open columns). C, Western analysis of α2-chimaerin from permanent α2-chimaerin N1E-115 cell line treated with increasing concentrations of PMA and fractionated into soluble, membrane, and 1% Triton-insoluble fraction (Kozma et al., 1996). D, Rac activity, determined by Rac-GTP pull-down with GST PAK GTPase binding region, in α2-chimaerin permanent cells treated with PMA at the indicated concentrations; Western blot with anti-Rac antibody shows total Rac and Rac-GTP bound to a GST-PAK CRIB (Cdc42/Rac interactive binding) affinity matrix. E, Effect of PMA treatment on neurites of N1E-115 cells, differentiated by serum starvation and transiently transfected with GFP-α2-chimaerin or GFP-α2-chimaerin GAP inactive mutant. Results show the average of three experiments (SEM). F, DRG neurons treated with PMA 100 nm (filled columns) for 30 min, in the presence or absence PKC inhibitor bisindolylmaleimide (10 μm) after inhibitor pretreatment of 20 min or 4 hr, or after transfection with GAP mutant GFP-α2-chimaerin R304G (see Materials and Methods). Cells were fixed and stained with phalloidin tetramethylrhodamine isothiocyanate, and GFP-transfected cells with collapsed growth cones were quantitated. PMA-induced collapse of α2-chimaerin R304G-transfected cells was significantly less than control cells plus or minus PKC inhibition; p > 0.008 and p > 0.003, respectively.
α2-Chimaerin is activated by PMA
Chimaerins are diacylglycerol/phorbol ester receptors (Ahmed et al., 1993), and β2-chimaerin is membrane translocated by phorbol esters (Caloca et al., 2001). In COS-7 cells and fibroblasts, PMA treatment promotes membrane ruffling, possibly through a Ras-Rac activation pathway (Fig. 4) (Kozma et al., 1996). In COS-7 cells, PMA treatment-induced ruffling was abolished on expression of exogenous α2-chimaerin, and instead cells showed a PMA-dependent collapse. GAP-inactive mutant α2-chimaerin had no effect on PMA-induced ruffling, and cells did not collapse, showing that the effects of α2-chimaerin in PMA-treated cells are attributable to its GAP activity (Fig. 4A,B). In N1E-115 cell lines permanently transfected with α2-chimaerin, PMA treatment promoted translocation of α2-chimaerin to cell membrane and cytoskeletal fractions (Fig. 4C) and also reduced endogenous Rac-GTP levels (Fig. 4D).
α2-Chimaerin activation induces neurite retraction
PMA ester treatment caused retraction of neurites in NIE-115 neuroblastoma cells differentiated in low serum (Fig. 4E). This PMA-induced retraction was inhibited by expression of GAP-inactive α2-chimaerin R304G (Fig. 4E), indicating that activation of endogenous α2-chimaerin GAP is sufficient to promote neurite retraction. α2-chimaerin is highly expressed in embryonic and adult dorsal root ganglia neurons (see Fig. 8A) (Hall et al., 2001b). Phorbol ester is known to promote growth cone collapse in these primary neurons (Fournier et al., 2000), an effect attributed to PKC activation. In DRG neurons, PMA-induced collapse of neuronal growth cones was unaffected by the specific PKC inhibitor bisindolylmaleimide, but partially inhibited by expression of GAP-inactive α2-chimaerin R304G (Fig. 4F). This suggests that PMA-induced activation of α2-chimaerin rather than of PKC is responsible for this collapse response. Together, these results indicate that PMA induces membrane translocation and activation of α2-chimaerin GAP, enhancing association with target proteins, and that active α2-chimaerin can promote neurite retraction.
Figure 8.

α2-chimaerin is involved in growth cone collapse in DRG neurons. A, DRG neurons in culture stained with α2-chimaerin antibody and with Cy5-conjugated anti-rabbit IgG secondary antibody (red) and phalloidin-FITC (green). B, DRG neurons transfected with GFP, GFP α2-chimaerin, GFP-α2-chimaerin R304G, and α2-chimaerin R73L were treated plus or minus Sema 3A as described in Materials and Methods, and collapsed growth cones were quantified. **p < 0.01; ***p < 0.001; growth cone collapse significantly different in presence of either PMA (B) or Sema 3A (C) than in its absence. C, DRG neurons were transfected with GFP, GFP plus ArhGAP15, or GFP plus ArhGAP15 GAP inactive. Neurons were fixed and stained for actin, and collapsed growth cones were quantified. All three demonstrated an equally significant increase in Sema 3A-induced growth cone collapse.
Cdk5 phosphorylates CRMP-2 at S522
CRMP-2 is tyrosine and serine phosphorylated in different collapse pathways (Arimura et al., 2000; Mitsui et al., 2002). In DRG growth cone collapse, CRMP-2 T555 is transiently phosphorylated by ROK in response to LPA but not Sema 3A (Arimura et al., 2000). In the Sema 3A pathway, because CRMP-2 may act downstream of Cdk5/p35, we tested whether CRMP-2 was a Cdk5 target. CRMP-2 synthesized in COS-7 cells could indeed be phosphorylated in vitro by recombinant Cdk5/p25 (Fig. 5A). Selected mutations were made in CRMP-2 at Cdk5 kinase consensus sequences, including T509 and S522. These two sites are among those hyperphosphorylated in Alzheimer's disease, where CRMP-2 is associated with neurofibrillary tangles (Yoshida et al., 1998; Gu et al., 2000) and Cdk5/p35 activity is disregulated (Patrick et al., 1999). CRMP-2 T509A but not CRMP-2 S522A was phosphorylated by Cdk5 (Fig. 5A), identifying S522 in the optimal consensus sequence S/TPXK/R as the single Cdk5 phosphorylation site in CRMP-2.
Figure 5.

CRMP-2 serine 522 is a novel substrate of Cdk5 and required for phosphorylation by GSK3 β. A,B, Flag-CRMP-2 (WT) and mutants of the sites indicated were synthesized in COS-7 cells, purified, and subjected to in vitro kinase assay with [32P]ATP and recombinant Cdk5/p25 (A) or GSK3β (B). C, Western blot analysis of CRMP-2 or CRMP-2 mutants as indicated (top) coexpressed in COS-7 cells with GSK3β (bottom); roscovitine treatment 10 μm (+Rosc). D, Western blot analysis showing effect of roscovitine treatment (1, 5, 20 μm) on GST-CRMP-2 phosphorylated in COS-7 cells in the presence or absence of GSK3β. Phosphorylated GST-CRMP-2, purified using glutathione agarose (middle; anti-GST) from cell lysates (bottom; anti-GST), was detected by anti-phosphothreonine antibody (top), which recognized the lower mobility band. E, Alignment of CRMP-2 C-terminal sequence with CRMP family members, indicating identified phosphorylation sites.
GSK3β phosphorylates CRMP-2 at S518 and T514 primed by phosphorylation at S522
GSK3β is activated in Sema 3A-induced growth cone collapse (Eickholt et al., 2002), and potential GSK3β phosphorylation sites are present in CRMP-2. We observed that coexpression of CRMP-2 with GSK3β in COS cells induced a mobility shift in CRMP-2 in SDS gels (Fig. 5C). Interestingly, this was abolished by mutation of the Cdk5 site, CRMP-2 S522A, and inhibited by treatment with roscovitine, which inhibits cyclin-dependent kinases (Fig. 5C,D). GSK3β phosphorylation of certain substrates is primed by previous phosphorylation by other kinases (Frame and Cohen, 2001). Potential GSK3β sites in CRMP-2 at S518 and T514 are spaced at four amino acid intervals upstream of S522 (Fig. 5E). To determine whether GSK3β phosphorylated CRMP-2 at any of these sites, appropriate mutant proteins were synthesized and subjected to in vitro kinase assays (Fig. 5B). CRMP-2 was indeed phosphorylated by GSK3β, with S518 being one of the phosphorylation sites (Fig. 5B). Mutant CRMP-2 S522A was not phosphorylated by GSK3β, suggesting that previous phosphorylation of S522 may be necessary for phosphorylation of S518 by GSK3β. Although the possibility that S522 is another GSK3β site cannot be excluded, this is unlikely because CRMP-2 S518A is not phosphorylated at all. CRMP-2 T514A showed reduced GSK3β phosphorylation (Fig. 5B) and no increased mobility (Fig. 5C), suggesting that T514 is also a GSK3β site. Mutation of any of these three sites, S518, T514, and S522, abolished the GSK3β-induced mobility shift of CRMP-2. Treatment of COS cells with the specific Cdk5 inhibitor roscovitine at an appropriate range of concentrations (Meijer et al., 1997; Bain et al., 2003) inhibited GSK3β phosphorylation in the lower mobility species, detected using an anti-phosphothreonine antibody (Fig. 5D). CRMP-1 and CRMP-4 also contain a potential Cdk5 site corresponding to CRMP-2 S522, but only CRMP-2 and CRMP-4 contain all three potential sites (Fig. 5E). Together, these results are consistent with initial phosphorylation of CRMP-2 S522 (possibly by Cdk5 in vivo) and subsequently of S518 and T514 by GSK3β.
Sema 3A-induced growth cone collapse requires phosphorylation of CRMP-2 S522
N1E-115 cells are responsive to Sema 3A (van Horck et al., 2002), and we initially used α2-chimaerin permanent transfectants of N1E-115 cells to investigate the in vivo effects of CRMP-2 and Cdk5 on the Sema 3A response. The time-lapse analysis is shown in Figure 6A. In response to Sema 3A, neurite retraction occurred after of 10-15 min and was complete within 30 min. Ruffles retracted with a redistribution of actin as shown in GFP actin-transfected cells. In the presence of ROK inhibitor Y-27632, Sema 3A still induced growth cone collapse and neurite retraction, indicating that ROK was not involved; however, Sema 3A-induced retraction was inhibited by the Cdk5 inhibitor roscovitine. This retraction was also inhibited by transfection of CRMP-2 S522A (nonphosphorylatable by Cdk5) but not CRMP-2 (Fig. 6A,B).
Figure 6.
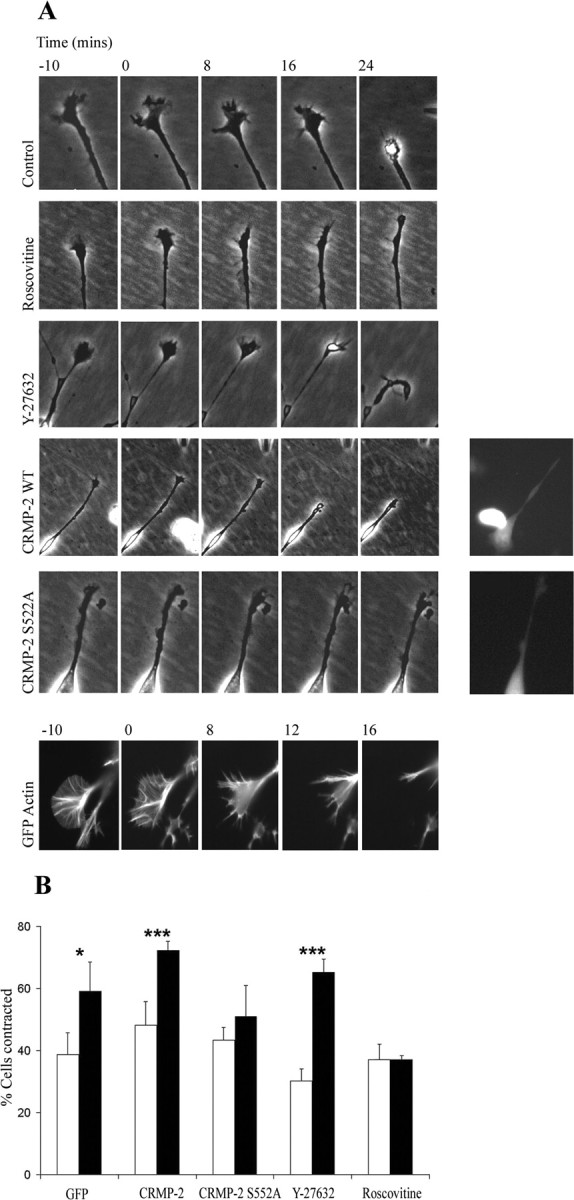
Sema 3A-induced growth cone collapse requires Cdk5, but not Rho kinase, and is blocked by CRMP-2S522A. A, Time-lapse phase images of α2-chimaerin N1E-115 cell growth cone response to Sema 3A, 10 min before and for 24 min after addition of purified Sema 3A; Sema 3A alone (6 of 10 collapsed); with roscovitine (2 of 10); with Y-27632 Rho kinase inhibitor (9 of 10); cells transfected with Flag-CRMP-2 plus GFP (8 of 10), or cells transfected with Flag-CRMP-2 S522A plus GFP (4 of 10). Insets show fluorescent images of transfected cells. Fluorescent image of GFP-actin in Sema 3A-induced collapse is also shown. B, Graph shows quantitation of α2-chimaerin permanent N1E-115 cells contracted in the presence (filled columns) or absence (empty columns) of Sema 3A when transfected with GFP, GFP plus CRMP-2, or GFP plus CRMP-2 S522A or treated with roscovitine (10 μm) or Y-27632 (10 μm). Cells were treated with Sema 3A for 30 min, fixed, and stained with phalloidin before counting (∼450 cells). *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t test); Sema 3A-treated cells significantly different from absence of Sema 3A.
The Sema 3A response is well characterized in DRG neurons. Rat DRG neurons express CRMP-2 and α2-chimaerin (Figs. 7A, 8A), with both occurring throughout the cell, neurites, and in growth cones. In DRG neurons in culture, growth cone collapse induced by Sema 3A was unaffected by the ROK inhibitor Y27632 but inhibited by the Cdk5 inhibitor roscovitine (Fig. 7B). Growth cone collapse induced by LPA treatment, which involves ROK phosphorylation of CRMP-2 (Arimura et al., 2000), was unaffected by roscovitine. LPA treatment resulted in similar values for collapse (55%), in the presence or absence of roscovitine, in duplicate experiments.
Figure 7.

In DRG neurons, growth cone collapse induced by Sema 3A requires Cdk5 and is blocked by CRMP-2S522A. A, DRG neurons in culture stained withαCRMP-2 antibody (red; left) and with Cy5-conjugated anti-rabbit IgG secondary antibody, and phalloidin-FITC (green) and analyzed with Zeiss LSM 410 confocal microscope. B, C, Cultures of rat DRG neurons either transfected with CRMP-2 (or CRMP-2 mutants) or treated with inhibitors as in Figure 6 (see Materials and Methods) and after incubation in the presence (filled columns) or absence (empty columns) of Sema 3A for 30 min were fixed and actin stained with phalloidin. Collapsed growth cones in GFP-transfected cells (or in the presence of inhibitors) were counted; 100-200 growth cones were scored in three experiments. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t test); Sema 3A-treated cells significantly different from absence of Sema 3A. In the presence of Sema 3A, collapse was significantly inhibited by roscovitine (p < 0.0001; p < 0.0003) and by CRMP-2S522A (p < 0.005 vs GFP; p < 0.0008 vs CRMP-2). Inset, Phalloidin-stained growth cone plus or minus Sema 3A treatment. D, Rat DRG cultures expressing GFP plus CRMP-2, GFP plus CRMP-2 T514A, or GFP plus CRMP-2 S518A were treated with Sema 3A for 30 min, before being fixed and stained, and collapsed growth cones were quantified. Sema 3A significantly increased the number of collapsed growth cones (*p < 0.05; **p < 0.01; ***p < 0.001), although expression of CRMP-2 T514A or CRMP-2 S518A significantly decreased the proportion of collapsed growth cones after Sema 3A treatment, compared with CRMP-2 (p < 0.0003 and p > 0.005, respectively).
Sema 3A-induced collapse was inhibited, however, by expression of CRMP-2 S522A but not of wild-type CRMP-2 in DRG neurons (Fig. 7C).
Because GSK3β plays a role in Sema 3A-induced collapse in DRG neurons, we then investigated whether inactivation of GSK3β phosphorylation sites in CRMP-2 also affected this collapse response. In contrast to CRMP-2 S522A, expression of either CRMP-2 T514A or S518A mutants did not completely abolish Sema 3A-induced collapse (Fig. 7D). Nevertheless, the Sema 3A-induced response was attenuated, consistent with an involvement of GSK3β after the pivotal phosphorylation at S522 by Cdk5.
α2-chimaerin is required for Sema 3A-induced growth cone collapse in DRG neurons
Because α2-chimaerin can induce neurite retraction and interacts with components of Sema 3A signaling, we examined its participation in Sema 3A-induced growth cone collapse, which is known to be Rac dependent. In rat DRG neurons, transfection with α2-chimaerin appeared to result in a very slight increase, but transfection with GAP-inactive mutant α2-chimaerin R304G abolished Sema 3A-induced growth cone collapse (Fig. 8B). A single mutation of the SH2 domain, R73L, was also sufficient to inhibit Sema 3A-induced collapse (Fig. 8B). This mutation corresponds to the essential arginine βB5 required for phosphotyrosine interaction (Waksman et al., 1993; Hall et al. 2001b), suggesting that α2-chimaerin may participate in the Sema 3A pathway through a phosphotyrosine-dependent association. Transfection with another specific Rac-GAP, ArhGAP15, or its GAP-inactive mutant ArhGAP15 Δ315-318 (Seoh et al., 2003), had no effect on Sema 3A-induced growth cone collapse (Fig. 8C). These results indicate that α2-chimaerin GAP activity is involved in the Sema 3A pathway.
α2-Chimaerin interacts with the Sema 3A receptor
The Sema 3A receptor is a complex of neuropilin-1 and plexin-A. CRMP-2 has recently been shown to interact with plexin-A1 in COS cells (Deo et al., 2004); the interaction is enhanced by expression of neuropilin only on Sema 3A treatment. Thus far, we have been unable to detect in COS cells a Sema 3A-induced association of α2-chimaerin with its binding partners Cdk5/p35 and CRMP-2, which are all necessary components of the Sema 3A pathway in DRG neurons. Nevertheless, we have been able to detect in COS cells CRMP-2 interaction with plexin-A and with neuropilin-1 (Fig. 9), as well as α2-chimaerin interaction with plexin-A and variably with neuropilin-1 (Fig. 9) (and data not shown). No interaction of α2-chimaerin with either receptor component occurred in the absence of PMA, indicating that activation and/or membrane translocation of α2-chimaerin is required.
Figure 9.

CRMP-2 andα2-chimaerin interact with plexin-A and neuropilin-1. A, COS-7 cells were transfected with Myc-tagged neuropilin-1 or plexin-A with either Flag-CRMP-2 or Flag-α2-chimaerin, in the presence or absence of PMA (5 μm). Myc-plexin-A or myc-neuropilin-1 was immunoprecipitated, and the presence of CRMP-2 or α2-chimaerin was analyzed by Western blotting.
Discussion
These results show that Sema 3A-induced growth cone collapse in DRG neurons requires both active α2-chimaerin and CRMP-2 phosphorylated at serine 522, a Cdk5 site. Cdk5/p35 kinase and its novel substrate CRMP-2 can associate with separate domains of active α2-chimaerin, which suggests that these Rac-interactive proteins may be part of a functional complex. Rac is a required component of the Sema 3A pathway, but its role has remained elusive, especially because it also participates in axonal outgrowth. The key to these dual and conflicting activities may lie in Rac being able to interact with a different set of effector proteins, including kinases. Different collapse pathways themselves involve separate kinases. ROK is necessary for ephrin A5- and Nogo-induced neurite retraction (Wahl et al., 2000; Niederost et al., 2002) but not Sema 3A-induced retraction (Fig. 7) (Arimura et al., 2000), whereas activity of Cdk5/p35 is required for Sema 3A-induced growth cone collapse (Fig. 7) (Sasaki et al., 2002).
Cdk5 phosphorylation of CRMP-2 in collapse
Here we identify a single Cdk5 phosphorylation site in CRMP-2, serine 522, which when mutated is sufficient to block Sema 3A-induced collapse. CRMP-2, an early identified component of the Collapsin 1/Sema 3A response (Goshima et al., 1995), is also phosphorylated in LPA-induced growth cone collapse in DRG neurons (Arimura et al., 2000) at a nearby site, T555. Phosphorylation of the C-terminal region appears to be linked with the role of CRMP-2 in collapse rather than outgrowth. CRMP-2 S522 phosphorylation may prime for subsequent phosphorylation at serine 518 and at threonine 514 by GSK3β, a kinase also activated in response to Sema 3A (Eickholt et al., 2002). In vivo phosphorylation of these sites by GSK3β was inhibited by mutation of serine 522, further indicating that the Cdk5 site at S522 is a pivotal phosphorylation site in CRMP-2. Mutation at either of these GSK3β sites attenuated but did not abolish collapse, consistent with a role of GSK3β downstream of Cdk5 in the Sema 3A pathway. Cdk5 and GSK3β activities are also coordinated in netrin signaling where MAP1B is a substrate (Del Rio et al. 2004). CRMP-2 promotes axon formation (Inagaki et al., 2001) binds tubulin heterodimers, and is involved in microtubule polymerization (Fukata et al., 2002). CRMP-2 may function in a heteromeric structure with CRMP-1 (Deo et al., 2004) and affect ROK (Leung et al., 2002). CRMP-2 C terminal plays an important regulatory role in axon outgrowth (Inagaki et al., 2001); however, this region of the protein does not directly interact with the identified CRMP-2 partners phospholipase D (Lee et al., 2002), Numb (Nishimura et al., 2003), or tubulin (Fukata et al., 2002). These C-terminal sites in CRMP-2 are hyperphosphorylated in Alzheimer's disease, in which CRMP-2 becomes associated with neurofibrillary tangles (Yoshida et al., 1998; Gu et al., 2000), suggesting that as with Tau, CRMP-2 function may be compromised in neurodegeneration.
Both Cdk5 and p35 kinase complex will interact with α2-chimaerin when it is activated by phorbol esters/diacylglycerol (DAG). Our results and those of Qi et al. (2004) indicate that GAP and kinase activities are present in the chimaerin/Cdk5 complexes. It thus appears that the active form of α2-chimaerin can target the p35/Cdk5 kinase complex. Because Cdk5/p35 is activated by Rac-GTP (Nikolic et al., 1998), Cdk5 activity may be subject to modulation by α2-chimaerin acting as a Rac GAP.
α2-chimaerin participation in collapse
Activation of α2-chimaerin by phorbol ester, an analog of diacylglycerol, itself can promote collapse in DRG neurons. This phorbol ester-induced growth cone collapse in neurons could be inhibited by GAP-inactive α2-chimaerin but did not appear to involve PKC. Phorbol ester-induced growth cone collapse and endocytosis in neurons was previously attributed entirely to its activation of PKC, which has a related C1 domain and an involvement in G-protein signaling (Fournier et al., 2000; Xiang et al., 2002).
The Sema 3A-induced collapse also appeared to require α2-chimaerin with a GAP-inactive mutant blocking this collapse. Mutation of the essential SH2 domain βB5 arginine R73 also blocked the collapse, suggesting that α2-chimaerin may participate in the Sema 3A collapse pathway through an SH2-phosphotyrosine interaction (Waksman et al., 1993; Hall et al., 2001b). Whether phosphotyrosine-SH2 interaction activates α2-chimaerin or DAG interaction is first required to release an autoinhibited conformation and cause its membrane translocation remains to be determined. The phosphotyrosine target may well be CRMP-2, because its interaction with α2-chimaerin was induced by tyrosine phosphorylation in pervanadate-treated N1E-115 cells. Although α2-chimaerin can interact with CRMP-2 independently of phosphotyrosine in COS-7 cells when treated with phorbol ester, specific tyrosine phosphorylation of CRMP-2 in response to Sema 3A may enable a high-affinity SH2 domain interaction in DRG neurons. CRMP-2 is known to be tyrosine phosphorylated by Fes/Fps in response to receptor signaling (Mitsui et al., 2002). Alternative candidates for phosphotyrosine interaction are components of the receptor complex. Active α2-chimaerin can also associate with plexin-A and neuropilin-1 (Fig. 9), as does CRMP-2 (Fig. 9) (Deo et al., 2004), where the interaction is sensitive to Sema 3A (Deo et al., 2004). Sasaki et al. (2002) have proposed that neuropilin-1/plexin-A2 that binds Fyn is linked to Cdk5 through its active state. α2-Chimaerin could be an intermediary in linking the receptor complex to Cdk5. We have been unable thus far to show Sema 3A dependence on α2-chimaerin associations, and this may reflect the absence in COS cells of a signaling component that translocates and activates α2-chimaerin. Sema 3A-induced growth cone collapse involves Rac-mediated endocytosis (Fournier et al., 2000; Jurney et al., 2002). The chimaerin partners CRMP-2 and Cdk5 have also been associated with vesicle function (Nishimura et al., 2003; Tan et al., 2003). There is a transient decrease in Rac activity preceding collapse (Jurney et al., 2002), which may well reflect α2-chimaerin Rac GAP activity at this early stage.
The requirement for Rac both in neurite outgrowth and in Sema 3A-induced collapse (Jin and Strittmatter, 1997; Kozma et al., 1997) suggests that Rac may be a common component of the different protein assemblies that effect these dynamic changes at the growth cone. It was reported that active Rac 1 interacts with plexin-B but not with plexin-A, the Sema 3A coreceptor (Rohm et al., 2000; Vikis et al., 2000; Driessens et al., 2001; Zanata et al., 2002). A more recent report, however, indicates that Rac1 can interact with the cytoplasmic domain of plexin-A and regulate receptor activity (Turner et al., 2004). The interaction of Sema 3A with neuropilin-1/plexin-A1 receptor, through an associated tyrosine kinase such as Fyn (Sasaki et al., 2002), may recruit and/or activate Rac GAP α2-chimaerin (possibly through local generation of diacylglycerol), enabling its association with binding partners. In conclusion, our results implicate Cdk5/p35 kinase, substrate CRMP-2, and active Rac GAP, whether they function together or separately in this pathway, in the dynamics of microtubule and actin reorganization and endocytosis in Sema 3A signaling.
Footnotes
This research was supported in part by the GSK/Singapore Research Fund. We thank Dr. Greg J. Michael, Neuroscience Section, Division of Biomedical Sciences, Queen Mary London University for helpful practical advice.
Correspondence should be addressed to Christine Hall, Department of Molecular Neuroscience, Institute of Neurology, University College London, 1 Wakefield Street, London WC1N 1PJ, UK. E-mail: c.hall@ion.ucl.ac.uk.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248994-11$15.00/0
M.B. and T.J. contributed equally to this work.
References
- Ahmed S, Lee J, Kozma R, Best A, Monfries C, Lim L (1993) A novel functional target for tumor-promoting phorbol esters and lysophosphatidic acid. The p21rac-GTPase activating protein n-chimaerin. J Biol Chem 268: 10709-10712. [PubMed] [Google Scholar]
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I (2001) Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci 4: 367-373. [DOI] [PubMed] [Google Scholar]
- Arimura N, Inagaki N, Chihara K, Menager C, Nakamura N, Amano M, Iwamatsu A, Goshima Y, Kaibuchi K (2000) Phosphorylation of collapsin response mediator protein-2 by Rho-kinase. Evidence for two separate signaling pathways for growth cone collapse. J Biol Chem 275: 23973-23980. [DOI] [PubMed] [Google Scholar]
- Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. Biochem J 371: 199-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caloca MJ, Fernandez N, Lewin NE, Ching D, Modali R, Blumberg PM, Kazanietz MG (1997) Beta2-chimaerin is a high affinity receptor for the phorbol ester tumor promoters. J Biol Chem 272: 26488-26496. [DOI] [PubMed] [Google Scholar]
- Caloca MJ, Wang H, Delemos A, Wang S, Kazanietz MG (2001) Phorbol esters and related analogues regulate the subcellular localization of β2-chimaerin, a non-protein kinase C phorbol ester receptor. J Biol Chem 276: 18303-18312. [DOI] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18: 29-42. [DOI] [PubMed] [Google Scholar]
- Daniels RH, Hall PS, Bokoch GM (1998) Membrane targeting of p21 activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO J 17: 754-764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio JA, Gonzalez-Billault C, Urena JM, Jimenez EM, Barallobre MJ, Pascual M, Pujadas L, Simo S, La Torre A, Wandosell F, Avila J, Soriano E (2004) MAP1B is required for Netrin 1 signaling in neuronal migration and axonal guidance. Curr Biol 14: 840-850. [DOI] [PubMed] [Google Scholar]
- Deo RC, Schmidt EF, Elhabazi A, Togashi H, Burley SK, Strittmatter SM (2004) Structural bases for CRMP function in plexin-dependent semaphorin 3A signaling. EMBO J 23: 9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diekmann D, Brill S, Garrett MD, Totty N, Hsuan J, Monfries C, Lim L, Hall C, Hall A (1991) bcr encodes a GTPase activating protein for p21Rac. Nature 351: 400-402. [DOI] [PubMed] [Google Scholar]
- Driessens MH, Hu H, Nobes CD, Self A, Jordens I, Goodman CS, Hall A (2001) Plexin-B semaphorin receptors interact directly with active Rac and regulate the actin cytoskeleton by activating Rho. Curr Biol 11: 339-344. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM, Gill GN (1999) Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signaling to actin cytoskeletal dynamics. Nat Cell Biol 1: 253-259. [DOI] [PubMed] [Google Scholar]
- Eickholt BJ, Walsh FS, Doherty P (2002) An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J Cell Biol 157: 211-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, Nakamura F, Kawamoto S, Goshima Y, Kalb RG, Strittmatter SM (2000) Semaphorin 3A enhances endocytosis at sites of receptor-F-actin colocalization during growth cone collapse. J Cell Biol 149: 411-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001) GSK3 takes center stage more than 20 years after its discovery. Biochem J 359: 1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4: 583-591. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM (1995) Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature 376: 509-514. [DOI] [PubMed] [Google Scholar]
- Govind S, Kozma R, Monfries C, Lim L, Ahmed S (2001) Cdc42Hs facilitates cytoskeletal reorganization and neurite outgrowth by localizing the 58-kD insulin receptor substrate to filamentous actin. J Cell Biol 152: 579-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Ihara Y (2000) Evidence that collapsin response mediator protein-2 is involved in the dynamics of microtubules. J Biol Chem 275: 17917-17920. [DOI] [PubMed] [Google Scholar]
- Gu Y, Hamajima N, Ihara Y (2000) Neurofibrillary tangle-associated collapsin response mediator protein-2 (CRMP-2) is highly phosphorylated on Thr-509, Ser-518, and Ser-522. Biochemistry 39: 4267-4275. [DOI] [PubMed] [Google Scholar]
- Hall C, Sin WC, Teo M, Michael GJ, Smith P, Dong JM, Lim HH, Manser E, Spurr NK, Jones TA, Lim L (1993) Alpha 2-chimerin, an SH2-containing GTPase-activating protein for the ras-related protein p21rac derived by alternate splicing of the human n-chimerin gene, is selectively expressed in brain regions and testes. Mol Cell Biol 13: 4986-4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Brown M, Jacobs T, Ferrari G, Cann N, Teo M, Monfries C, Lim L (2001a) Collapsin response mediator protein switches RhoA and Rac1 morphology in N1E-115 neuroblastoma cells and is regulated by Rho kinase. J Biol Chem 276: 43482-43486. [DOI] [PubMed] [Google Scholar]
- Hall C, Michael GJ, Cann N, Ferrari G, Teo M, Jacobs T, Monfries C, Lim L (2001b) alpha2-chimaerin, a Cdc42/Rac1 regulator, is selectively expressed in the rat embryonic nervous system and is involved in neuritogenesis in N1E-115 neuroblastoma cells. J Neurosci 21: 5191-5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K (2001) CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci 4: 781-782. [DOI] [PubMed] [Google Scholar]
- Jin Z, Strittmatter SM (1997) Rac1 mediates collapsin-1-induced growth cone collapse. J Neurosci 17: 6256-6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurney WM, Gallo G, Letourneau PC, McLoon SC (2002) Rac1-mediated endocytosis during ephrin A2 and semaphorin 3A-induced growth cone collapse. J Neurosci 22: 6019-6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Ahmed S, Best A, Lim L (1996) The GTPase-activating protein n-chimaerin cooperates with Rac1 and Cdc42Hs to induce the formation of lamellipodia and filopodia. Mol Cell Biol 16: 5069-5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R, Sarner S, Ahmed S, Lim L (1997) Rho family GTPases and neuronal growth cone remodeling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol 17: 1201-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kim JH, Lee CS, Kim JH, Kim Y, Heo K, Ihara Y, Goshima Y, Suh P-G, Ryu SH (2002) Collapsin response mediator protein-2 inhibits neuronal phospholipase D2 activity by direct interaction. J Biol Chem 277: 6542-6549. [DOI] [PubMed] [Google Scholar]
- Leung T, Ng Y, Cheong A, Ng CH, Tan I, Hall C, Lim L (2002) p80 ROKalpha binding protein is a novel splice variant of CRMP-1 which associates with CRMP-2 and modulates RhoA-induced neuronal morphology. FEBS Lett 532: 445-449. [DOI] [PubMed] [Google Scholar]
- Li X, Saint-Cyr-Proulx E, Aktories K, Lamarche-Vane N (2002) Rac1 and Cdc42 but not RhoA or Rho kinase activities are required for neurite outgrowth induced by the Netrin-1 receptor DCC (deleted in colorectal cancer) in N1E-115 neuroblastoma cells. J Biol Chem 277: 15207-15214. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuna K, Narumiya S (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and Lim-kinase. Science 285: 895-898. [DOI] [PubMed] [Google Scholar]
- Manser E, Leung T, Monfries C, Teo M, Hall C, Lim L (1992) Diversity and versatility of GTPase activating proteins for the p21rho subfamily of ras G proteins detected by a novel overlay assay. J Biol Chem 267: 16025-16028. [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP (1997) Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem 243: 527-536. [DOI] [PubMed] [Google Scholar]
- Minturn JE, Fryer HJL, Geschwind DH, Hockfield S (1995) TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J Neurosci 15: 6757-6766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui N, Inatome R, Takahashi S, Goshima Y, Yamamura H, Yanagi S (2002) Involvement of Fes/Fps tyrosine kinase in semaphorin 3A signaling. EMBO J 21: 3274-3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE (2002) Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. J Neurosci 22: 10368-10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH (1996) The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev 10: 816-825. [DOI] [PubMed] [Google Scholar]
- Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH (1998) The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature 395: 194-198. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Fukata Y, Kato K, Yamaguchi T, Matsuura Y, Kamiguchi H, Kaibuchi K (2003) CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat Cell Biol 5: 819-826. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Mont S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. [DOI] [PubMed] [Google Scholar]
- Qi RZ, Ching YP, Kung HF, Wang JH (2004) Alpha-chimaerin exists in a functional complex with the Cdk5 kinase in brain. FEBS Lett 12: 177-180. [DOI] [PubMed] [Google Scholar]
- Qi Z, Tang D, Zhu X, Fujita DJ, Wang JH (1998) Association of neurofilament proteins with neuronal Cdk5 activator. J Biol Chem 273: 2329-2335. [DOI] [PubMed] [Google Scholar]
- Rohm B, Rahim B, Kleiber B, Hovatta I, Puschel AW (2000) The semaphorin 3A receptor may directly regulate the activity of small GTPases. FEBS Lett 486: 68-72. [DOI] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG (1999) Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behaviour. J Cell Biol 147: 1009-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Cheng C, Uchida Y, Nakajima O, Ohshima T, Yagi T, Taniguchi M, Nakayama T, Kishida R, Kudo Y, Ohno S, Nakamura F, Goshima Y (2002) Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 35: 907-920. [DOI] [PubMed] [Google Scholar]
- Schwamborn JC, Fiore R, Bagnard D, Kappler J, Kaltschmidt C, Puschel AW (2004) Semaphorin 3A stimulates neurite extension and regulates gene expression in PC12 cells. J Biol Chem 279: 30923-30926. [DOI] [PubMed] [Google Scholar]
- Seoh ML, Ng CH, Yong J, Lim L, Leung T (2003) ArhGAP15, a novel human RacGAP protein with GTPase binding property. FEBS Lett 539: 131-137. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM (1999) Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell 99: 59-69. [DOI] [PubMed] [Google Scholar]
- Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RA, Larsen MR, Cousin MA, Robinson PJ (2003) Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol 5: 701-710. [DOI] [PubMed] [Google Scholar]
- Terman JR, Mao T, Pasterkamp RJ, Yu H-H, Kolodkin AL (2002) MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109: 887-900. [DOI] [PubMed] [Google Scholar]
- Tigyi G, Fischer DJ, Sebok A, Yang C, Dyer DL, Miledi R (1996) Lysophosphatidic acid induced neurite retraction in PC12 cells: control by phosphoinositide-Ca2+ signaling and Rho. J Neurochem 66: 537-547. [DOI] [PubMed] [Google Scholar]
- Turner LJ, Nicholls S, Hall A (2004) The activity of the plexin-A1 receptor is regulated by Rac. J Biol Chem, 279: 33199-33205. [DOI] [PubMed] [Google Scholar]
- van Horck FP, Lavazais E, Eickholt BJ, Moolenaar WH, Divecha N (2002) Essential role of type I(alpha) phosphatidylinositol 4-phosphate 5-kinase in neurite remodeling. Curr Biol 12: 241-245. [DOI] [PubMed] [Google Scholar]
- Vastrik I, Eickholt BJ, Walsh FS, Ridley A, Doherty P (1999) Sema 3A-induced growth-cone collapse is mediated by Rac1 amino acids 17-32. Curr Biol 9: 991-998. [DOI] [PubMed] [Google Scholar]
- Vikis HG, Li W, He Z, Guan KL (2000) The semaphorin receptor plexin-B1 specifically interacts with active Rac in a ligand-dependent manner. Proc Natl Acad Sci USA 97: 12457-12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl S, Barth H, Ciossek T, Aktories K, Mueller BK (2000) Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J Cell Biol 149: 263-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J (1993) Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72: 779-790. [DOI] [PubMed] [Google Scholar]
- Winberg ML, Tamagnone L, Bai J, Comoglio PM, Montell D, Goodman CS (2001) The transmembrane protein Off-track associates with plexins and functions downstream of semaphorin signaling during axon guidance. Neuron 32: 53-62. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Li Y, Zhang Z, Cui K, Wang S, Yuan X, Wu C, Poo M, Duan S (2002) Nerve growth cone guidance mediated by G protein-coupled receptors. Nat Neurosci 5: 843-848. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Watanabe A, Ihara Y (1998) Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer's disease. J Biol Chem 273: 9761-9768. [DOI] [PubMed] [Google Scholar]
- Zanata SM, Hovatta I, Rohm B, Puschel AW (2002) Antagonistic effects of Rnd1 and RhoD GTPases regulate receptor activity in semaphorin 3A-induced cytoskeletal collapse. J Neurosci 22: 471-477. [DOI] [PMC free article] [PubMed] [Google Scholar]