

Abstract
There is a high correlation between pediatric epilepsies and neuronal migration disorders. What remains unclear is whether there are intrinsic features of the individual dysplastic cells that give rise to heightened seizure susceptibility, or whether these dysplastic cells contribute to seizure activity by establishing abnormal circuits that alter the balance of inhibition and excitation. Mice lacking a functional p35 gene provide an ideal model in which to address these questions, because these knock-out animals not only exhibit aberrant neuronal migration but also demonstrate spontaneous seizures.
Extracellular field recordings from hippocampal slices, characterizing the input-output relationship in the dentate, revealed little difference between wild-type and knock-out mice under both normal and elevated extracellular potassium conditions. However, in the presence of the GABAA antagonist bicuculline, p35 knock-out slices, but not wild-type slices, exhibited prolonged depolarizations in response to stimulation of the perforant path. There were no significant differences in the intrinsic properties of dentate granule cells (i.e., input resistance, time constant, action potential generation) from wild-type versus knock-out mice. However, antidromic activation (mossy fiber stimulation) evoked an excitatory synaptic response in over 65% of granule cells from p35 knock-out slices that was never observed in wild-type slices. Ultrastructural analyses identified morphological substrates for this aberrant excitation: recurrent axon collaterals, abnormal basal dendrites, and mossy fiber terminals forming synapses onto the spines of neighboring granule cells. These studies suggest that granule cells in p35 knock-out mice contribute to seizure activity by forming an abnormal excitatory feedback circuit.
Keywords: epilepsy, dentate granule cell, p35, recurrent excitation, intracellular labeling, neuronal migration disorder
Introduction
Disorders of brain development, involving abnormal migration of immature neurons along glial fibers, are a frequent cause of cortical malformations found in patients with epilepsy (Meencke and Veith, 1992, 1999). It remains unclear, however, whether the epileptogenicity of these malformations reflects abnormalities intrinsic to the aberrant cells (e.g., altered potassium conductances) (Beck et al., 1997; Castro et al., 2001) or arise from the disrupted organization of neuronal circuitry (Sutula, 1998). To address these questions, investigators have turned to animal models, including mouse mutants such as Lis-1, Cdk 5, doublecortin, and reeler (for review, see Allen and Walsh, 1999; Chevassus-au-Louis et al., 1999; Walsh, 1999; Ross, 2002). Unfortunately, although these animal models often have structural malformations characteristic of human migration disorders, they rarely present with spontaneous, recurrent seizures. An exception is the p35 knock-out mouse, which exhibits both structural malformations and spontaneous seizures (Chae et al., 1997; Wenzel et al., 2001).
p35, a neuron-specific activator of cyclin-dependent kinase 5 (Cdk5), has been studied as a key element in cortical development, where it plays a central role in neuronal migration and the formation of neuronal processes (Paglini and Caceres, 2001). Activation of Cdk5 requires direct binding with p35, which promotes its kinase activity (Tsai et al., 1994). Cdk5 knock-out mice display severe cortical lamination defects but die in the early perinatal period (Ohshima et al., 1996; Gilmore et al., 1998; Tanaka et al., 2001). In contrast, whereas the p35 knock-out mouse also demonstrates cortical lamination defects, it suffers from only sporadic adult lethality and exhibits spontaneous limbic-like seizures (Tsai et al., 1994; Ohshima et al., 1996; Gilmore et al., 1998; Tanaka et al., 2001; Wenzel et al., 2001; Gupta et al., 2003).
Many brain regions of p35-/- animals exhibit structural abnormalities (Chae et al., 1997; Kwon and Tsai, 1998; Kwon et al., 1999). Our studies have focused on specific disorganizational patterns observed in the hippocampal dentate gyrus (Wenzel et al., 2001). Similar abnormalities have been observed in other animal models of chronic seizure activity (for review, see Ribak and Dashtipour, 2002) and resemble pathologies observed in human patients with mesial temporal lobe epilepsy (MTLE) (Houser et al., 1990; Houser, 1999). It has been hypothesized that dentate reorganization, recurrent axonal projection back into the granule cell (GC) and molecular layers, affects the overall balance between excitation and inhibition in the dentate gyrus (Tauck and Nadler, 1985; Nadler, 2003), a region often viewed as the “gateway” for information entering the hippocampus (Lothman et al., 1992; Gloveli et al., 1998).
The goal of the current study was to evaluate the functional implications of dentate gyrus disorganization in the p35-/- mouse. Extracellular electrophysiological analyses of the p35-/- dentate GC region revealed a greater susceptibility to bicuculline-induced excitability than seen in wild-type slices. Intracellular recordings in p35-/- GCs showed no significant differences (compared with wild-type cells) in passive properties, intrinsic firing properties, or response to a general (high potassium) excitability challenge. However, many GCs from p35-/- animals demonstrated an aberrant excitatory synaptic response to antidromic stimulation of the mossy fibers (MFs), a functional correlate consistent with the excitatory feedback circuit (involving recurrent MF axon collaterals) characteristic of the p35-/- dentate.
Materials and Methods
Animals
A complete description of the gene deletion protocol and initial characterization of p35 knock-out mice has been published (Chae et al., 1997). Morphological analyses of hippocampal neurons, and observations of seizure behavior using EEG monitoring, were described by Wenzel et al. (2001). The p35-/- colony was maintained via brother-sister matings, and wild-type controls (+/+) were derived from the same stock as originally used to generate -/- breeding pairs (129 Sv × C57BL/6 background). All animal care and use conformed to the NIH Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of California, Davis.
Tissue preparation for light microscopy/histochemistry
General processing. Mice used for light-microscopic and histochemical studies were anesthetized with Nembutal (100 mg/kg, i.p.), then perfused with isotonic saline with heparin (500 U/ml), followed by a solution of 4% paraformaldehyde in 0.1 m sodium phosphate buffer (PB), pH 7.4. The brains were immediately removed from the skull cavity and placed in the same fixative for 4 hr at 4°C, cryoprotected with 10 and 30% sucrose, and then frozen on dry ice. Thirty-micrometer transverse serial sections were cut on a sliding microtome equipped with a freezing stage, then selected for additional processing. Sections were mounted on slides and stained with cresyl violet.
Timm histochemistry. The Timm method for staining heavy metals was used for the detection of synaptic vesicular zinc [particularly enriched in MF boutons (MFBs)]. After initial fixation with 4% paraformaldehyde as described above, the brains were transferred to a solution containing 3-4% glutaraldehyde, 0.1% Na2S, and 0.136 mm CaCl2 in 0.12 m Millonig's PB, pH 7.3, for 48 hr at 4°C, followed by cryoprotection in 30% sucrose. Frozen sections were cut at 30 μm and then mounted on slides, air dried, and transferred to a fresh developer solution containing 30 ml of gum Arabic (50%), 5 ml of 2 m citrate buffer, 15 ml of hydroquinone (5.76%), and 250 μl of silver nitrate (0.73%) for 1 hr in the dark. Sections were dehydrated, cleared in toluene, and coverslipped.
Electrophysiology
General tissue preparation. For in vitro slice electrophysiology, hippocampal slices were prepared as described previously (Wenzel et al., 2001). Mice were anesthetized with halothane, decapitated, and the brains were rapidly transferred to cold, oxygenated artificial CSF (ACSF) containing (in mm) 124 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 26 NaCO3, 2 CaCl2, and 10 dextrose. Brains were then blocked to contain the hippocampus; a Vibratome (Vibratome Company, St. Louis, MO) was used to cut 400-μm-thick slices (transverse to the longitudinal axis of the hippocampus) in a bath of oxygenated ACSF at 4°C. Individual slices, from the middle third of the hippocampus, were placed in a holding chamber at room temperature and allowed to equilibrate for at least 1 hr before transfer to a slice interface recording chamber (Fine Science Tools, Foster City, CA) for electrophysiological experiments. Slices rested on a nylon mesh over a well perfused with warmed (32-35°C), oxygenated ACSF; the slice surface was exposed to a warmed, humidified 95% O2/5% CO2 environment.
Extracellular field electrophysiology. Field EPSPs (fEPSPs) were recorded in the dentate gyrus of both p35-/- and wild-type mice with extracellular electrodes made from borosilicate glass pulled on a horizontal puller (Sutter Instruments, Novato, CA) and filled with ACSF (5-10 MΩ resistance). Evoked potentials in response to perforant path stimulation were recorded. ACSF with 6 mm K+ or 20 μm bicuculline methiodide (BMI; Sigma, St. Louis, MO) was exchanged with normal ACSF in the recording chamber to obtain recordings in the presence of elevated potassium or a GABAA antagonist, respectively.
Intracellular electrophysiology. Intracellular electrophysiological recordings were obtained from individual GCs in the superior blade of the dentate gyrus in both p35-/- and wild-type mice, using sharp microelectrodes filled with 4 m potassium acetate (80-150 MΩ). A bipolar stimulating electrode was used to stimulate either the perforant path fibers in the dentate molecular layer (“orthodromic stimulation”) or the MF axons in the CA3 stratum lucidum (“antidromic stimulation”), and evoked potentials were recorded and stored. Data analysis was performed only on cells that met the following criteria for cell “health”: stable resting membrane potential, more negative than -60 mV, for a minimum of 10 min before experimental manipulations; a smooth charging curve (in response to DC step current injection) that reflected a non-leaky recording configuration; and action potentials of a minimum 60 mV amplitude.
Data acquisition and analysis. All electrophysiological data were recorded using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA) with a bridge circuit (bridge balanced before cell impalement), digitized using a Digidata 1200 (Axon Instruments), and stored on a personal computer-based computer system. Data were analyzed with Axoscope software (Axon Instruments); graphical and statistical assistance was provided by SigmaPlot (Systat, Point Richmond, CA). All statistical values are reported as a mean ± SEM, unless noted otherwise.
Intracellular labeling with biocytin and reconstruction
Biocytin labeling. Intracellular electrodes were filled with 2% biocytin (Molecular Probes, Eugene, OR) dissolved in 1 m potassium acetate (80-230 MΩ). Neurons were impaled, and biocytin was injected iontophoretically with 700 msec duration, 0.5-1.0 nA hyperpolarizing current pulses delivered every second for 10-30 min. After biocytin injection, the electrode was withdrawn, and slices were left in the chamber for an additional 30 min before immersion fixation in 4% paraformaldehyde and 0.1-1% glutaraldehyde in 0.1 m sodium PB, pH 7.4, for 2-4 hr at 4°C.
Tissue processing and morphological analysis (light and electron microscopy). Tissue slices containing biocytin-labeled cells were then rinsed in 0.1 m PB and infiltrated for cryoprotection with 10% sucrose in 0.1 m PB for 1 hr, followed by 30% sucrose for 8-12 hr. Frozen sections were cut (60 μm) and further processed with a histochemical procedure for light microscopy and electron microscopy (EM), as outlined by Wenzel et al. (2001). Biocytin-filled GCs (including their dendrites and axonal arborizations) were visualized at the light-microscopic level and photographed before remounting and additional sectioning for EM. Camera lucida drawings were made of each biocytin-filled GC, and its dendrites and axonal arborizations were reconstructed by superimposing all sections of the hippocampal slice. Serial thin sections from various slices, containing different portions of the biocytin-filled GCs, were stained with uranyl acetate and Reynold's lead citrate and examined on a Philips CM 120 electron microscope. Electron micrographs of segments of aberrant MF collaterals (located within the GC and the molecular layers) and of basal dendrites of biocytin-labeled GCs (located within the hilus) were used to generate reconstructions. Three-dimensional reconstructions of axon segments containing varicosities and synaptic contacts were generated through serial alignment, tracing, and rendering of electron micrographs using the following software packages, respectively: SEM Align, IGL Trace (courtesy of Dr. J. C. Fiala, Boston University, Boston, MA; http://www.synapses.bu.edu/), and 3D Studio Max (Discreet, Montreal, Quebec, Canada). All chemicals were purchased from Fisher Scientific (Hampton, NH) unless noted otherwise.
Results
Disorganization in the dentate gyrus of p35-/- mice
Histological examination of brains from p35-/- mice revealed striking differences from brains of wild-type mice, including loss of neocortical lamination and dispersion of the principal cell layers in the hippocampus (with frequent interruptions in cell layers and displaced cell somata) (Wenzel et al., 2001). Nissl staining also demonstrated significant dispersion of dentate GCs, particularly in the superior blade, with GC somata displaced into the molecular layer and into the hilus (Fig. 1, compare A, B). Timm staining, to label zinc in synaptic vesicles of MFBs, demonstrated MF sprouting in the p35-/- dentate. In wild-type mice, Timm staining showed a characteristic pattern of MFs in the hilus (Fig. 1C), with a compact projection into the CA3 stratum lucidum. In p35-/- animals, however, numerous Timm-stained MF axons infiltrated the GC layer (GCL) and the inner molecular layer (IML), obscuring what is normally a distinct hilar-GCL border (Fig. 1D, arrows). As reported previously (Wenzel et al., 2001), the degree of GC dispersion and the amount of Timm staining in the GCL and IML was variable across p35-/- animals; these features were not observed in wild-type mice.
Figure 1.

Nissl-stained sections of the dentate gyrus in wild-type (A) and p35-/- (knock-out) (B) mice show dispersion of GC somata into the molecular layer and into the hilus (black arrows). Timm-stained sections of the dentate gyrus from wild-type (C) and knock-out (D) mice illustrate the characteristic MF terminal invasion into the granular layer and IML (D; black arrows). Scale bars, 50 μm.
Relative excitability of the p35 knock-out animal
In light of the epileptogenicity of p35-/- mice and the related structural abnormalities of the dentate GC region, we obtained a measure of the general excitability of the dentate gyrus subregion in p35-/- hippocampal slices. Extracellular field recordings were used to construct input-output (I/O) curves, under normal bathing medium conditions and when the tissue was challenged by the application of either an elevated concentration of extracellular potassium (Rutecki et al., 1985; Hardison et al., 2000) or introduction of BMI, a GABAA antagonist (Wuarin and Dudek, 1996; Patrylo and Dudek, 1998; Patrylo et al., 1999). A bipolar stimulating electrode was placed on the surface of the slice over the outer molecular layer of the superior blade of the dentate near the apex. Field potentials were recorded from the GCL in response to stimuli (50 μsec duration) of increasing amplitude (0.2-1.0 mA) at a frequency of 0.1 Hz. Slices were perfused initially in normal (control) ACSF and allowed to equilibrate for 15 min before determining an I/O curve. Slices were then perfused with ACSF containing either elevated potassium or BMI; 15-20 min was allowed to ensure exchange of perfusate before a new I/O curve was constructed. The rise time of the fEPSP (normalized to percentage of maximum) was plotted versus the stimulus intensity, for wild-type and knock-out animals, in control (3 mm K+) and elevated K+ (6 mm) conditions (Fig. 2). Evoked extracellular field responses to moderate amplitude stimulation (0.4 mA) of the perforant path in 3 mm K+ showed little difference between the wild-type and p35-/- groups (Fig. 2A, top; representative traces), and I/O relationships were virtually superimposable. Elevation of extracellular potassium concentration to 6 mm (Fig. 2B) revealed little difference in the I/O relationship between wild-type and knock-out animals, although a slight rightward shift in the curve (not statistically significant) was observed for the knock-out group (Fig. 2B, bottom). Perfusion of elevated potassium quickly led to the appearance of population spikes in the wild-type dentate but, surprisingly, not in the tissue from p35-/- mice (Fig. 2B, top); p35-/- fEPSP amplitude increased in 6 mm K+, but population spikes were not elicited at moderate stimulus intensities.
Figure 2.
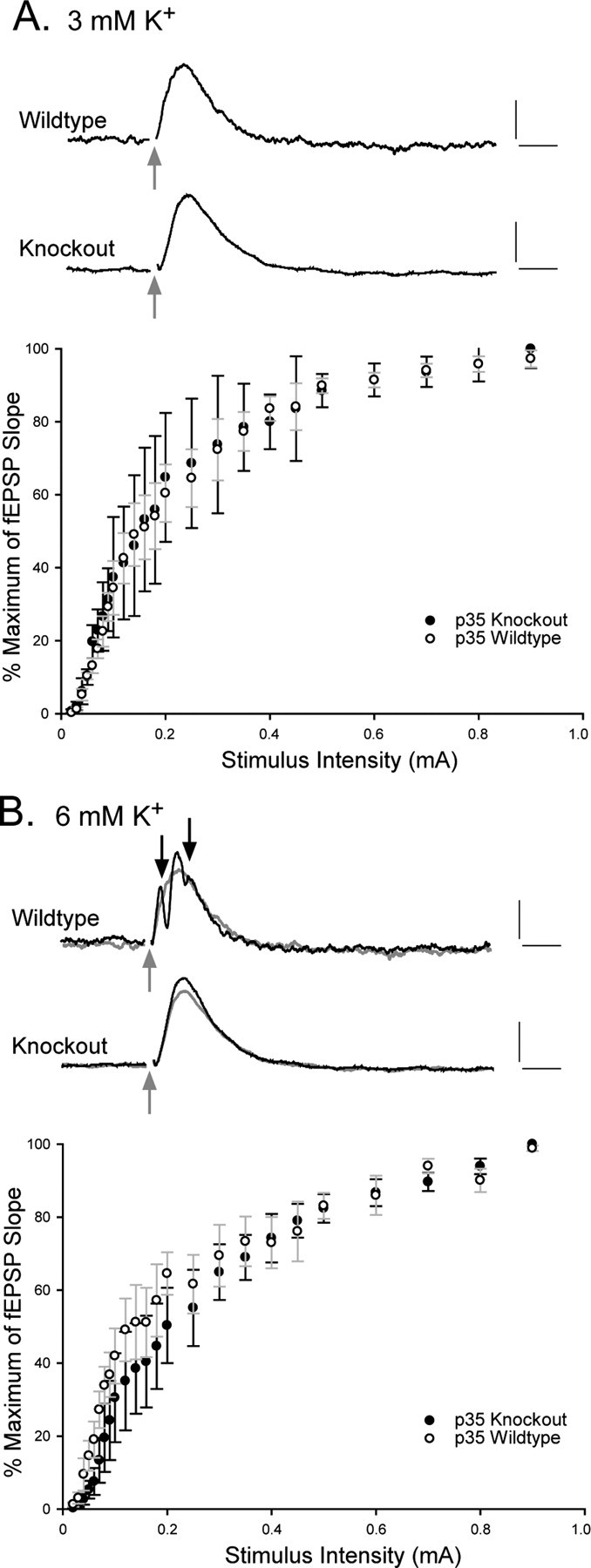
Field recordings in slices from p35-/- mice show little increased dentate excitability when extracellular potassium concentration is increased from 3 to 6 mm. A, Top, Representative field responses to perforant path stimulation (gray arrows) (moderate stimulation intensity, 0.4 mA) in wild-type and knock-out slices in 3 mm extracellular potassium. A, Bottom, Graph plots the percentage of the maximum of the slope of the fEPSP versus the stimulus intensity (milliamperes), under control (3 mm) conditions. B, Top, Representative evoked field responses (stimulation intensity of 0.4 mA) in the presence of 6 mm extracellular potassium (overlaid on gray traces from A). B, Bottom, Graph plots the rise of the fEPSP slope as a percentage of the maximum response versus the stimulus intensity. Calibration: 0.5 mV (vertical), 5 msec (horizontal). The error bars in bottom graphs represent the SEM (n = 5 in each group).
Slices from wild-type and p35-/- mice were also compared under conditions of disinhibition, through the introduction of the GABAA antagonist bicuculline. Stimulus-evoked field potentials were first obtained in normal ACSF (Fig. 3A), and then 20 μm BMI was introduced to the bath for a subsequent set of recordings; washout with normal bathing medium was used to demonstrate recovery of normal responsiveness. Representative traces illustrate the dramatic difference between wild-type and p35-/- in sensitivity to BMI (Fig. 3B). BMI-induced hyperexcitability in p35-/- slices was reflected, particularly, in the appearance of a long-duration positive component in the evoked response after the stimulus. No such event was seen in wild-type slices (n = 8), although BMI did enhance population spike occurrence in response to stimulation (Fig. 3B, wild type). In p35-/- slices, the initial short-latency response increased in amplitude, but population spike activation was rarely seen (as was the case for the high K+ challenge). The long-latency, prolonged discharge was observed in response to moderate stimulus intensity in all p35-/- slices (n = 8) in the presence of BMI, although the duration and the latency was variable. To quantify this response, we measured the shift from baseline at a long poststimulus interval (150 msec) (Fig. 3C). This response pattern was normalized with subsequent perfusion of normal ACSF (data not shown).
Figure 3.

The GABAA antagonist BMI unmasks hyperexcitability in p35-/- slices. A, Under controlconditions, stimulation of the perforant path (0.4 mA) elicits similar evoked responses in wild-type and knock-out animals. B, The addition of BMI increases the amplitude of the fEPSP and induces population spiking (black arrow) in the wild-type slices. An expansion of the early part of the response is shown beneath the slow time sweep; the bicuculline condition is superimposed over the control (gray) trace. In knock-out slices, the initial evoked response is similar to that seen under control conditions (only a small increase in the fEPSP amplitude); however, there is also a long-latency, long-duration, high-amplitude event (gray shading). Calibration: 0.5 mV (vertical), 25 msec (horizontal). C, Field potential amplitude under bicuculline conditions, measured at 150 msec after the stimulus, shows the consistent occurrence of the late discharge in p35-/- slices but not in wild-type slices.
Intrinsic excitability of individual dentate GCs
One hypothesis that links structural abnormalities in the dentate to the functional consequence of seizure activity focuses on the properties of dentate GCs: that there are intrinsic abnormalities in the aberrant GCs that give rise to hyperexcitability in the dentate circuit. We used intracellular electrophysiology, therefore, to characterize and compare the intrinsic properties of GCs from wild-type and knock-out mice. Step DC current pulses were injected intracellularly to determine the input resistance, time constant, input conductance, and spike frequency features of these GCs. Action potential properties, including spike amplitude, width, and thresholds were also quantified and compared. Table 1 provides a summary of these properties. The resting membrane potential, input resistance, time constant, and action potential properties showed no statistically significant differences between cells from wild-type versus p35-/- animals. Figure 4A shows representative sets of responses to a series of depolarizing (0.1-0.5 nA) and hyperpolarizing (-0.1 to -0.5 nA) current step injections (inset). The current-voltage relationship was plotted, the slope of which is a measure of the input conductance (Fig. 4B); these plots showed little difference between the experimental groups. The average number of action potentials generated as a function of depolarizing current magnitude was also plotted (Fig. 4C)(n = 20); although no significant difference between p35-/- and wild-type cells was observed, the graph does suggest a less linear relationship in the firing frequency of GCs from knock-out slices, i.e., a slightly higher threshold for eliciting the first action potential but a greater number of action potentials at high-amplitude depolarizing steps (consistent with our data from the high-potassium experiments).
Table 1.
Passive properties of p35 wild-type and knock-out GCs
|
|
p35 wild type |
p35 knock-out |
|---|---|---|
| Number | 33 | 38 |
| Resting membrane potential (mV) | −75.5 ± 1.27 | −77.2 ± 1.04 |
| Input resistance (MΩ) | 81.9 ± 5.77 | 82.9 ± 4.39 |
| Time constant (msec) | 7.48 ± 0.49 | 8.38 ± 0.67 |
| Action potential Amplitude (mV) | 77.5 ± 1.49 | 76.0 ± 1.14 |
| Threshold (mV) | −46.7 ± 1.8 | −44.3 ± 1.86 |
| Duration (msec) |
2.11 ± 0.06 |
2.37 ± 0.05 |
Figure 4.

Intrinsic GC properties are similar in wild-type and knock-out dentate. A, A representative family of voltage responses to step current injections (from -0.5 to +0.5 nA; steps shown in inset) from a wild-type and a knock-out GC. Calibration: 10 mV (vertical), 25 msec (horizontal); inset, 0.2 nA (vertical), 25 msec (horizontal). B, Plots of the current-voltage relationship for the cells presented in A. C, Plots of the number of action potentials elicited at given current step amplitudes. Each point shows mean ± SEM for 20 cells in the wild-type and knock-out groups.
Recurrent excitation of p35-/- GCs in response to antidromic stimulation
To determine the functional consequence of the MF sprouting (observed with Timm staining) (Fig. 1D), intracellular recordings were obtained from GCs, and responses were evaluated to stimulation of the MF axons in the CA3 stratum lucidum (Fig. 5A). This stimulation should directly activate the axons of dentate GCs, generate antidromic action potential invasion of GCs, and activate any GC feedback circuits. In normal dentate (Fig. 5B), this stimulus led to an antidromic spike in GCs (identified by its short latency to the stimulus), followed by an inhibitory synaptic response mediated by MF synapses onto local interneurons (Fricke and Prince, 1984; Freund and Buzsaki, 1996). This postsynaptic potential is characterized as an IPSP by its responsiveness to hyperpolarizing current. In contrast, 68% of GCs from knock-out animals showed evidence of an orthodromic (i.e., synaptically mediated) excitatory response to antidromic stimulation; these responses did not invert when the membrane potential of the cell was depolarized above the reversal potential of GABA-mediated IPSPs (Fig. 5B, black arrow).
Figure 5.

A, Schematic shows the stimulation (in the stratum lucidum) and recording (in GC layer) configuration used in these experiments. B, Families of evoked responses, from a wild-type GC and a knock-out GC, illustrate commonly seen response patterns for these groups (19 wild-type GCs; 25 p35-/- GCs). DC current injection was used to vary the membrane potential of the GC during its response to MF stimulation in the CA3 stratum lucidum. Inversion of a short-latency component of the response, after the initial antidromic spike, was taken to reflect a GABAA-mediated IPSP. This IPSP was absent (black arrow) in a significant proportion of p35-/- cells. Dashed lines represent the resting membrane potential of the cells; gray arrows identify the time of stimulation. Calibration: 5 mV (vertical), 25 msec (horizontal). C, Representative responses from dentate granule cells from knock-out slices, illustrating the variable patterns of evoked responses. D, Hypothetical schematic suggesting potential circuitry configurations that may explain the variability illustrated in C. 1, A cell with both an antidromic and an orthodromic action potential (6 of 25 cells); 2, a cell with no antidromic response but an orthodromically driven action potential (11 of 25 cells); 3, a cell with an antidromic response but no subsequent orthodromic response component (3 of 25 cells); 4, a cell with an antidromic action potential, followed by a relatively normal IPSP (5 of 25 cells). Calibration: 5 mV (vertical), 25 msec (horizontal).
GCs from p35-/- animals showed a variety of responses to MF stimulation, often involving both an antidromic spike and an orthodromic/synaptic potential (Fig. 5C). Aberrant organization of the dentate circuitry that may account for the variety of responses is illustrated in Figure 5D. In this schematic, the number on the GC body corresponds to the pattern of evoked response illustrated in Figure 5C. For example, in cell 1, stimulation in the p35-/- stratum lucidum (identified by the gray stimulation zone in Fig. 5D) evokes both an antidromic response (the “normal” propagation of the action potential down the MF toward its parent cell body) (Fig. 5C, asterisks) and, because cell 1 receives synaptic input from excitatory axon collaterals (from other GCs) that project back into the GCL and molecular layer, an orthodromic excitatory response. Cell 2 generates only an orthodromic response (EPSP giving rise to an action potential) without an antidromic action potential because its axon “missed” the stimulation site; it does, however, receive recurrent excitation from neighboring GCs (e.g., from MF axons making contacts in the GCL or onto an abnormal basal dendrite projecting into the hilus; see below). Cell 3 generates an antidromic action potential response without an inhibitory response, seen in several GCs in p35-/- slices. Finally, cell 4 demonstrates a normal GC response to MF stimulation, suggesting that some proportion of GCs in p35-/- dentate are embedded in relatively normal circuitry.
Ultrastructural analysis of p35-/- biocytin-labeled MF collaterals
We used intracellular biocytin labeling and ultrastructural analyses to determine whether recurrent MF collaterals in the GCL and IML do indeed make excitatory synaptic contacts on neighboring GCs, as suggested by our electrophysiological data. Individual GCs were injected with 2% biocytin; the tissue was fixed, processed for biocytin labeling, and further processed with EM techniques as described in Materials and Methods. Segments of biocytin-labeled axons, which appeared at the light-microscopic level to be recurrent MF collaterals projecting back into the GC and molecular layers of the dentate (Fig. 6A), were serially sectioned, stained, and examined on an electron microscope. Two-dimensional reconstructions of serial electron micrographs were performed on axonal segments within the GC and molecular layers (a total axon length of 99.4 μm). These reconstructions (Fig. 6B) revealed multiple sites of synaptic contacts between MFBs and dendritic spines (90% of total synapses) and between MFBs and dendritic shafts (the remaining 10% of synapses). Individual electron micrographs identified synaptic contacts between biocytin-filled profiles with characteristic MF features (large varicosities with a high density of clear round synaptic vesicles and occasional dense-core vesicles). These MFBs establish asymmetric contacts with dendritic spines of presumed GCs (Fig. 6C-F). These reconstructions provided detailed descriptive information about the location of the postsynaptic densities, as well as quantitative information about the number of axonal varicosities (0.22/μm), intervaricosity distances (average of 3.8 μm), and the number, location, and types of synaptic contacts. A quantitative summary of these data (from two segments of axonal collaterals) is presented in Table 2. It is important to note that only 65% of these MF varicosities were associated with synaptic specializations.
Figure 6.

Light- and electron-microscopic analyses of an aberrant axon collateral, from a biocytin-labeled dentate GC in a p35 knock-out slice. A, Camera lucida drawing of the GC, showing a MF collateral (black arrows) projecting back into the GCL and molecular layer. Scale bar, 100 μm. B, Two-dimensional reconstruction (axon collateral identified in A, from serial section electron micrographs. Scale bar, 3 μm. The boxed regions of the reconstructed axon segment are magnified (C-F) and shown with representative electron micrographs (c-f) that illustrate the ultrastructural features of synaptic contacts (arrowheads) between the mossy fiber boutons (MFBs) and the postsynaptic spines (S) of presumed neighboring GCs (arrow identifies an axonal extension without a synapse). Three-dimensional reconstructions of two MF contacts (c2, d2) were implemented to demonstrate the tertiary structure of the axon and MFBs in relation to their postsynaptic contacts. Asterisks identify C-shaped synapses, and arrowhead in c2 points to a dendritic shaft synapse.
Table 2.
Quantitative summary of aberrant axon collaterals
|
|
|
|---|---|
| Two-dimensional length of axon (μm) | 99.4 |
| Number of axon varicosities | 26 |
| With synapses | 17 (65%) |
| With multiple synapses | 4 (15.4%) |
| Without synapses | 9 (35%) |
| Average intervaricosity distance (μm) | 3.8 |
| Number of synapses | 22 |
| Synapses per micrometer | 0.22 |
| Synapses per varicosity | 0.65 |
| Frequency of synaptic targets | |
| Dendritic spine | 19 (86.4%) |
| Dendritic shaft | 3 (13.6%) |
| Soma |
0 |
To obtain more detailed information about the precise location, shape, and size of synaptic active zones on presynaptic boutons, and to determine whether MFBs on aberrant axon collaterals at different locations (e.g., molecular layer and GCL vs hilus) were morphologically different, we examined two short axonal segments (each containing two varicosities) in three-dimensional reconstructions. The varicosities of the molecular layer axonal segment (Fig. 6C) formed asymmetric synapses (multiple and single contacts) with dendritic spines of presumed GC dendrites, and with a dendritic shaft (Fig. 6c2, arrowhead). The three-dimensional reconstruction of presynaptic boutons revealed three oval/round synapses and one C-shaped synapse (Fig. 6c2, asterisk) (see also supplemental material, available at www.jneurosci.org). The synaptic active zones were slightly invaginated into the bouton surface. In addition, one of the varicosities was associated with an axonal extension (without a synapse) (Fig. 6C, arrow) characteristic of large MFBs within the hilus. The three-dimensional reconstructed varicosities located within the GCL (Fig. 6d2) (supplemental material, available at www.jneurosci.org) exhibited a similar morphology, characterized by contacts with multiple spines and both round/oval and C-shaped active zones. These features are consistent with our two-dimensional reconstruction data on MFBs located within the hilus of normal animals (data not shown). However, none of the varicosities associated with aberrant axon collaterals were similar to the giant, complex MFBs that have been described in the stratum lucidum and hilus (Claiborne et al., 1986).
Ultrastructural analysis of p35-/- GC basal dendrites
Another abnormal morphological feature observed in p35-/- dentate was the presence of GC basal dendrites that project into the hilus, providing a potential postsynaptic target for MF collaterals of neighboring GCs. To characterize the features of this abnormal dendritic structure, and specifically to determine whether these basal dendrites receive excitatory synaptic input, we used intracellular biocytin labeling and ultrastructural reconstructions (as described above for axon collaterals). Reconstruction of a representative basal dendrite projecting into the hilus (Fig. 7A) revealed a structure that is typical of GC dendrites within the molecular layer. These basal dendrites gave rise to a variety of spine morphologies (e.g., both complex and simple spines) (Fig. 7B). The majority of spines formed asymmetric synapses with axonal terminals of presumed excitatory (based on synaptic morphology) neurons, and particularly with the terminals of GC axons (MFBs) (Fig. 7b1, shaded regions). The basal dendrites also formed symmetric synapses with unidentified interneurons (Fig. 7b2, black synapses in the schematic). Selected segments from the reconstruction in Figure 7B, with representative electron micrographs, illustrate these complex and perforated spine synapses, as well as dendritic shaft synapses, onto basal dendrites (Fig. 7C-F,c-f2). A quantitative summary of the features of five electron microscopically reconstructed basal dendrite segments is reported in Table 3.
Figure 7.
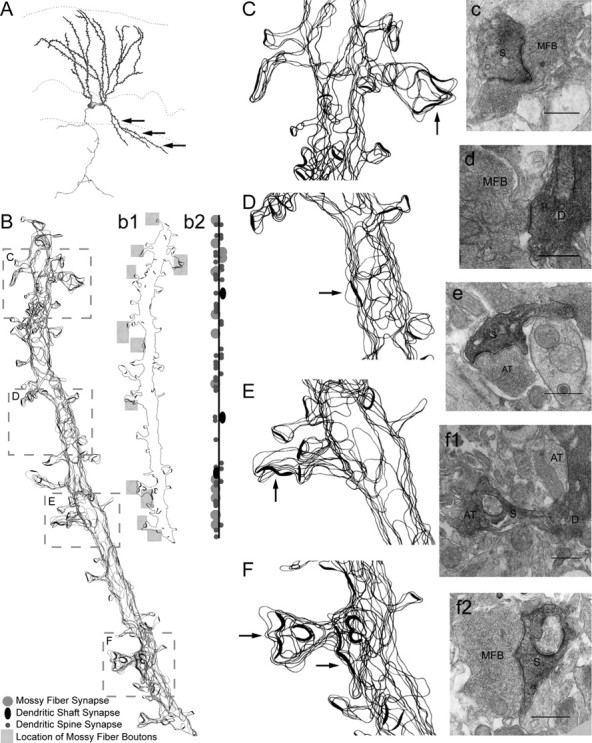
Light- and electron-microscopic analyses of a GC basal dendrite projecting into the hilus. A, Camera lucida drawing of a p35-/- GC, intracellularly labeled with biocytin, and observed at the light-microscopic level. Black arrows identify an abnormal basal dendrite. B, Reconstructed basal dendrite segment, as identified in A. The schematic (b1) identifies those areas of the basal dendrite that contact MFBs (gray shaded boxes); the adjacent schematic (b2) shows the location of not only MFB contacts but also other dendritic spine and shaft synpases. Representative regions of the basal dendrite (B; boxed areas) are magnified to show two-dimensional reconstructions (C-F) and associated representative electron micrographs (c-f), including mossy fiber boutons (MFBs) synapsing onto the spine (S) or dendrite (D) of the biocytin-labeled GC. Black arrows in C-F identify synapses in adjacent electron micrographs (c-f). AT, Axon terminals. Scale bars, 500 nm.
Table 3.
Quantitative summary of basal dendrite features
|
|
|
|---|---|
| Two-dimensional length of basal dendrite (μm) | 99.25 |
| Total number of synapses onto basal dendrites | 178 |
| Number of dendritic spines with synapses | 168 (90.4% of total) |
| Dendritic spines with MFBs | 23 |
| Complex spines (with perforated synapses) | 21 |
| Number of dendritic spines per micrometer | 1.62 |
| Number of dendritic shaft synapses | 17 (9.6%) |
| Number of dendritic shaft synapses with MFBs | 2 |
| Number of dendritic shaft synapses per micrometer |
0.17 |
Discussion
Clinical studies have long identified a strong correlation between abnormal cortical structure and epilepsy (Meencke and Veith, 1992; Schwartzkroin and Walsh, 2000). This correlation has become even more obvious with the introduction of state-of-theart brain imaging techniques that are more sensitive to these structural abnormalities (Barkovich et al., 1992). Among the key questions arising from this structure-function relationship is whether there are intrinsic features of individual cells that give rise to the seizure phenomenon or whether the epileptogenicity associated with structural abnormalities arises from abnormal circuits (note that these are obviously not mutually exclusive possibilities). Because the p35-/- mouse shows morphological features similar to those seen in many cases of MTLE, analysis of the structure-function relationships in this model may help us to address these important issues. Our principal findings suggest that the structurally aberrant GCs in the dentate of p35-/- mice show little evidence of intrinsic hyperexcitability but do contribute to an abnormal recurrent excitatory circuit that may provide the basis for seizure activity observed in these animals.
Increased excitability in the p35 knock-out animal
We used two well established experimental manipulations for demonstrating and/or unmasking hyperexcitability in a suspect circuit. In theory, increasing extracellular potassium depolarizes the resting membrane potential of a cell and decreases the potassium gradient associated with hyperpolarizing currents, thereby increasing the probability of neuronal discharge. This procedure does not discriminate between excitatory and inhibitory cells and may therefore result in a net effect that is relatively neutral. In our experiments, dentate wild-type tissue generated multiple population spikes when exposed to a modest extracellular potassium concentration increase. Surprisingly, this effect was not observed in the p35-/- slices (although synaptic drive did lead to an increased fEPSP amplitude), indicating some still-to-be-elucidated difference between p35-/- and wild-type dentate in their I/O relationship. The dispersion of the dentate GCL in p35-/- hippocampus may contribute to this difference because current summation and ephaptic interactions are certainly disrupted (Taylor and Dudek, 1984a,b). Interestingly, the higher population spike threshold of p35-/- dentate in high potassium is consistent with the higher spiking threshold of p35-/- GCs, as seen in our assessment of the current-discharge relationship.
The other approach to uncovering potential excitability, one used in several studies on other MTLE-like tissues (Patrylo and Dudek, 1998), was to partially block GABAergic inhibition with bicuculline. Under this disinhibition condition, perforant path stimulation had a differential effect on the short-latency response that was similar to the effect seen with high potassium (i.e., enhanced population spiking in wild-type dentate) but only a mild fEPSP increase in the p35-/- tissue. However, this bicuculline treatment also gave rise to a long-latency, long-duration discharge (depolarization) in p35-/- dentate that was never seen in wild-type slices. It is interesting to note that this long-duration discharge was somewhat different from the briefer burst responses reported by other laboratories that uncovered recurrent excitation by blocking GABAA-mediated inhibition. It is unclear whether the difference is a result of specific experimental procedure (e.g., bicuculline concentration), a function of species and species-specific dentate organization (mouse vs rat or human), and/or because of the specific nature of the abnormality found in the p35 knock-out animal (e.g., a genetically determined deficit that involves developmental reorganization). In light of the data from our experiments identifying a functional and structural feedback circuit, it is likely that the long-latency depolarizing potential observed in p35 knock-out dentate, in the absence of normal inhibition, is generated by aberrant excitatory feedback onto GCs that is usually suppressed by inhibitory mechanisms. It is likely from this result that inhibition is compromised in the p35-/- mouse. Certainly, the excitation-inhibition balance is significantly disrupted.
Structural and functional evidence of recurrent excitation
One of the remarkable features of the p35-/- animal is the striking similarity in structural abnormalities observed in the dentate gyrus when compared with many current animal models of mesial temporal lobe epilepsy (Sutula, 1990; Ribak and Dashtipour, 2002) as well as to pathologies observed in human tissue from patients with MTLE (Houser, 1990; Lurton et al., 1998; El Bahh et al., 1999; Thom et al., 2002). These studies have implicated GC dispersion, GC basal dendrites, and MF sprouting as structural features highly correlated to the epileptic phenomenology.
MF sprouting is perhaps the most studied of these phenomena; it involves a dramatic form of structural change and is often observed after various experimental manipulations resulting in robust and severe seizures (Scharfman, 2002; Nadler, 2003). Although MF sprouting is often thought to contribute to hippocampal excitability (Sutula et al., 1992; Wuarin and Dudek, 1996; Molnar and Nadler, 1999; Patrylo et al., 1999; Okazaki and Nadler, 2001; Nadler, 2003), there is also evidence to suggest that MF sprouting suppresses excitability by increasing excitation of inhibitory interneurons in the hippocampus (Sloviter, 1992; Buckmaster and Dudek, 1997), or that MF synapses back onto GCs are only mildly excitatory (Scharfman et al., 2003). To examine the functional relevance of MF sprouting to epileptogenicity, we used experimental manipulations that have been used to uncover hyperexcitable circuits, e.g., in the pilocarpine animal model (Cronin et al., 1992) as well as in human epileptic tissue (Franck et al., 1995). As was the case in these previous studies, the p35-/- dentate demonstrated little evidence of hyperexcitability under normal conditions but displayed epileptiform activity with partial (BMI-mediated) disinhibition; BMI-induced activity was not seen in similarly treated control tissue.
We also used in our p35-/- experiments techniques used by others to demonstrate recurrent excitation (e.g., in the kainic acid, kindling, and pilocarpine models of MTLE) (Tauck and Nadler, 1985; Cronin et al., 1992; Lynch and Sutula, 2000). Similar to the findings from those studies, our data support the hypothesis that recurrent MF collaterals provide abnormal feedback excitation onto dentate GCs and thereby promote excitability. Furthermore, the recurrent excitatory feedback circuit revealed in the p35-/- dentate is consistent with our ultrastructural data demonstrating the presence of aberrant MF collaterals and abnormal GC basal dendrites that form synaptic contacts with neighboring GCs. What remains unclear is why, at least in this in vitro environment, the consequences of this recurrent excitatory circuit are not seen until inhibitory controls are compromised. As shown by Miles and Wong (1987) in their work on CA3 of guinea pigs, extensive divergence of the inhibitory circuitry and powerful conductance changes that arise from activation of GABAA receptors may suppress excitability associated with recurrent excitatory circuitry, so that some compromise of this inhibitory effect is a prerequisite for the emergence of hyperexcitability. Our findings are consistent with this explanation and also suggest that the recurrent excitation in p35-/- dentate is superimposed on an abnormal inhibitory system.
Recurrent excitation in hippocampal dentate gyrus as the basis for epileptogenicity
Although our studies of the p35-/- brain have focused on the dentate gyrus, abnormalities have also been observed in other regions of the hippocampus (i.e., CA3 and CA1 subregions) as well as in the cortex. Our exclusive concentration on dentate was motivated by initial observations of dentate pathologies similar to those seen in the hippocampus of other models of MTLE (as described above). However, the widespread nature of the structural disorganization makes it difficult to identify the hippocampal dentate region as the source or generator of epileptiform activity. On what bases might one argue that expression of the recurrent excitatory circuit in the dentate gyrus is the key (causal?) factor in seizure initiation in the p35-/- mouse? The “limbic phenotype” of p35-/- seizures (behavioral stages including immobility, sniffing, and chewing, clonus, and finally generalized tonic/clonic convulsions) serves as an intriguing starting point (Wenzel et al., 2001). Furthermore, whereas cortical disorganization might certainly be involved in epileptogenicity in p35-/- mice, other animal models with comparable neocortical disorganization (e.g., the reeler mouse) do not characteristically exhibit spontaneous seizure activity (Ross, 2002). The CA3 region of the hippocampus also exhibits cell dispersion, but we have not yet explored the functional consequences of that structural abnormality with respect to, for example, enhanced recurrent excitation (certainly a characteristic feature of CA3 circuitry) (Swann et al., 1990, 1992). Although several different laboratories have demonstrated the development of abnormal synaptic reorganization, including recurrent excitation, in other areas of the hippocampus in human MTLE patients (Lehmann et al., 2000) and animal models of epilepsy (Perez et al., 1996; Lehmann et al., 2000, 2001; Shao and Dudek, 2004), similar widespread abnormalities (e.g., in the CA1 subfield) have not yet been investigated in the p35-/- mouse.
The p35-/- model provides a perhaps unique opportunity to test the hypothesis that formation of recurrent excitatory circuits (in the dentate) is primarily responsible for epileptogenesis. At least in theory, analysis of these circuits can be performed developmentally, to examine structural and electrophysiological features before observation of clinical seizures. Such an approach would bypass a complicating feature of most other animal models of MTLE (and certainly of human MTLE), a previous history of seizures/status epilepticus and related cell loss/injury, particularly in the hilus (Sundstrom et al., 1993; Dudek et al., 1994). The possibility that loss of p35 function could result, directly, in aberrant synaptic contacts is attractive, given that p35 is thought to play a critical role in axon formation and guidance, and that the p35 mRNA signal is present in axonal bundles and is associated with growth cone actin cytoskeleton (Paglini and Caceres, 2001). Furthermore, although in most brain regions p35 is expressed at high levels only during early stages of development (associated with migration of neurons to their final destination and their differentiation), it remains at high levels of expression in the pyriform cortex and in the pyramidal layers of the hippocampus, where its presence in axonal bundles may influence neuronal plasticity in the adult (Kwon et al., 1999; Paglini and Caceres, 2001).
Perhaps most appealing in the p35-/- dentate system, the related pathological structure suggests that MF “sprouting” may not be necessary for the formation of recurrent excitatory circuitry. GCs are dispersed into aberrant locations and form basal dendrites, presenting postsynaptic targets in the region in which normal MFs make synaptic contacts, the hilus (Austin and Buckmaster, 2004). Thus, the argument that MF sprouting (and/or cell loss in the hilus) is not necessary for epileptogenesis (Zhang et al., 2002; Raol et al., 2003) may not speak to the critical epileptogenic role played by recurrent excitatory circuitry in the dentate gyrus.
Conclusion
We found functional and structural evidence of a recurrent excitatory circuit that may contribute to the increased excitability in the dentate gyrus of the p35-/- mouse hippocampus. Our findings support the hypothesis that structural abnormalities arising from neuronal migration disorders contribute to hyperexcitability primarily through the creation of an abnormal neuronal circuitry, even when intrinsic electrophysiological properties of individual cells remain relatively normal. Additional studies are needed to relate these structural and functional changes directly to the occurrence of spontaneous seizure activity.
Footnotes
This work was supported by National Institutes of Health Grant NS18895. We thank Mareike Wenzel and Norma Anderson for excellent technical assistance with electron microscopy and Carol Robbins for assistance with initial electrophysiology studies.
Correspondence should be addressed to Dr. Philip A. Schwartzkroin, Department of Neurological Surgery, University of California, Davis, One Shields Avenue, Davis, CA 95616. E-mail: paschwartzkroin@ucdavis.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249005-10$15.00/0
References
- Allen KM, Walsh CA (1999) Genes that regulate neuronal migration in the cerebral cortex. Epilepsy Res 36: 143-154. [DOI] [PubMed] [Google Scholar]
- Austin JE, Buckmaster PS (2004) Recurrent excitation of granule cells with basal dendrites and low interneuron density and inhibitory postsynaptic current frequency in the dentate gyrus of macaque monkeys. J Comp Neurol 476: 205-218. [DOI] [PubMed] [Google Scholar]
- Barkovich AJ, Gressens P, Evrard P (1992) Formation, maturation, and disorders of brain neocortex. Am J Neuroradiol 13: 423-446. [PMC free article] [PubMed] [Google Scholar]
- Beck H, Clusmann H, Kral T, Schramm J, Heinemann U, Elger CE (1997) Potassium currents in acutely isolated human hippocampal dentate granule cells. J Physiol (Lond) 498: 73-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE (1997) Network properties of the dentate gyrus in epileptic rats with hilar neuron loss and granule cell axon reorganization. J Neurophysiol 77: 2685-2696. [DOI] [PubMed] [Google Scholar]
- Castro PA, Cooper EC, Lowenstein DH, Baraban SC (2001) Hippocampal heterotopia lack functional Kv4.2 potassium channels in the methylazoxymethanol model of cortical malformations and epilepsy. J Neurosci 21: 6626-6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai LH (1997) Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18: 29-42. [DOI] [PubMed] [Google Scholar]
- Chevassus-au-Louis N, Baraban SC, Gaiarsa JL, Ben-Ari Y (1999) Cortical malformations and epilepsy: new insights from animal models. Epilepsia 40: 811-821. [DOI] [PubMed] [Google Scholar]
- Claiborne BJ, Amaral DG, Cowan WM (1986) A light and electron microscopic analysis of the mossy fibers of the rat dentate gyrus. J Comp Neurol 246: 435-458. [DOI] [PubMed] [Google Scholar]
- Cronin J, Obenaus A, Houser CR, Dudek FE (1992) Electrophysiology of dentate granule cells after kainate-induced synaptic reorganization of the mossy fibers. Brain Res 573: 305-310. [DOI] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Schweitzer JS, Wuarin JP (1994) Functional significance of hippocampal plasticity in epileptic brain: electrophysiological changes of the dentate granule cells associated with mossy fiber sprouting. Hippocampus 4: 259-265. [DOI] [PubMed] [Google Scholar]
- El Bahh B, Lespinet V, Lurton D, Coussemacq M, Le Gal La Salle G, Rougier A (1999) Correlations between granule cell dispersion, mossy fiber sprouting, and hippocampal cell loss in temporal lobe epilepsy. Epilepsia 40: 1393-1401. [DOI] [PubMed] [Google Scholar]
- Franck JE, Pokorny J, Kunkel DD, Schwartzkroin PA (1995) Physiologic and morphologic characteristics of granule cell circuitry in human epileptic hippocampus. Epilepsia 36: 543-558. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G (1996) Interneurons of the hippocampus. Hippocampus 6: 347-470. [DOI] [PubMed] [Google Scholar]
- Fricke RA, Prince DA (1984) Electrophysiology of dentate gyrus granule cells. J Neurophysiol 51: 195-209. [DOI] [PubMed] [Google Scholar]
- Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K (1998) Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci 18: 6370-6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloveli T, Schmitz D, Heinemann U (1998) Interaction between superficial layers of the entorhinal cortex and the hippocampus in normal and epileptic temporal lobe. Epilepsy Res 32: 183-193. [DOI] [PubMed] [Google Scholar]
- Gupta A, Sanada K, Miyamoto DT, Rovelstad S, Nadarajah B, Pearlman AL, Brunstrom J, Tsai LH (2003) Layering defect in p35 deficiency is linked to improper neuronal-glial interaction in radial migration. Nat Neurosci 6: 1284-1291. [DOI] [PubMed] [Google Scholar]
- Hardison JL, Okazaki MM, Nadler JV (2000) Modest increase in extracellular potassium unmasks effect of recurrent mossy fiber growth. J Neurophysiol 84: 2380-2389. [DOI] [PubMed] [Google Scholar]
- Houser CR (1990) Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res 535: 195-204. [DOI] [PubMed] [Google Scholar]
- Houser CR (1999) Neuronal loss and synaptic reorganization in temporal lobe epilepsy. Adv Neurol 79: 743-761. [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, Delgado-Escueta AV (1990) Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J Neurosci 10: 267-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Tsai LH (1998) A novel disruption of cortical development in p35 (-/-) mice distinct from reeler. J Comp Neurol 395: 510-522. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Tsai LH, Crandall JE (1999) Callosal axon guidance defects in p35 (-/-) mice. J Comp Neurol 415: 218-229. [DOI] [PubMed] [Google Scholar]
- Lehmann TN, Gabriel S, Kovacs R, Eilers A, Kivi A, Schulze K, Lanksch WR, Meencke HJ, Heinemann U (2000) Alterations of neuronal connectivity in area CA1 of hippocampal slices from temporal lobe epilepsy patients and from pilocarpine-treated epileptic rats. Epilepsia 41 [Suppl 6]: S190-S194. [DOI] [PubMed] [Google Scholar]
- Lehmann TN, Gabriel S, Eilers A, Njunting M, Kovacs R, Schulze K, Lanksch WR, Heinemann U (2001) Fluorescent tracer in pilocarpine-treated rats shows widespread aberrant hippocampal neuronal connectivity. Eur J Neurosci 14: 83-95. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Stringer JL, Bertram EH (1992) The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res Suppl 7: 301-313. [PubMed] [Google Scholar]
- Lurton D, El Bahh B, Sundstrom L, Rougier A (1998) Granule cell dispersion is correlated with early epileptic events in human temporal lobe epilepsy. J Neurol Sci 154: 133-136. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sutula T (2000) Recurrent excitatory connectivity in the dentate gyrus of kindled and kainic acid-treated rats. J Neurophysiol 83: 693-704. [DOI] [PubMed] [Google Scholar]
- Meencke HJ, Veith G (1992) Migration disturbances in epilepsy. Epilepsy Res Suppl 9: 31-40. [PubMed] [Google Scholar]
- Meencke HJ, Veith G (1999) The relevance of slight migrational disturbances (microdysgenesis) to the etiology of the epilepsies. Adv Neurol 79: 123-131. [PubMed] [Google Scholar]
- Miles R, Wong RK (1987) Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol (Lond) 388: 611-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar P, Nadler JV (1999) Mossy fiber-granule cell synapses in the normal and epileptic rat dentate gyrus studied with minimal laser photostimulation. J Neurophysiol 82: 1883-1894. [DOI] [PubMed] [Google Scholar]
- Nadler JV (2003) The recurrent mossy fiber pathway of the epileptic brain. Neurochem Res 28: 1649-1658. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA 93: 11173-11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki MM, Nadler JV (2001) Glutamate receptor involvement in dentate granule cell epileptiform activity evoked by mossy fiber stimulation. Brain Res 915: 58-69. [DOI] [PubMed] [Google Scholar]
- Paglini G, Caceres A (2001) The role of the Cdk5-p35 kinase in neuronal development. Eur J Biochem 268: 1528-1533. [PubMed] [Google Scholar]
- Patrylo PR, Dudek FE (1998) Physiological unmasking of new glutamatergic pathways in the dentate gyrus of hippocampal slices from kainate-induced epileptic rats. J Neurophysiol 79: 418-429. [DOI] [PubMed] [Google Scholar]
- Patrylo PR, Schweitzer JS, Dudek FE (1999) Abnormal responses to perforant path stimulation in the dentate gyrus of slices from rats with kainate-induced epilepsy and mossy fiber reorganization. Epilepsy Res 36: 31-42. [DOI] [PubMed] [Google Scholar]
- Perez Y, Morin F, Beaulieu C, Lacaille JC (1996) Axonal sprouting of CA1 pyramidal cells in hyperexcitable hippocampal slices of kainate-treated rats. Eur J Neurosci 8: 736-748. [DOI] [PubMed] [Google Scholar]
- Raol YS, Budreck EC, Brooks-Kayal AR (2003) Epilepsy after early-life seizures can be independent of hippocampal injury. Ann Neurol 53: 503-511. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Dashtipour K (2002) Neuroplasticity in the damaged dentate gyrus of the epileptic brain. Prog Brain Res 136: 319-328. [DOI] [PubMed] [Google Scholar]
- Ross ME (2002) Brain malformations, epilepsy, and infantile spasms. Int Rev Neurobiol 49: 333-352. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D (1985) Epileptiform activity induced by changes in extracellular potassium in hippocampus. J Neurophysiol 54: 1363-1374. [DOI] [PubMed] [Google Scholar]
- Scharfman HE (2002) Epilepsy as an example of neural plasticity. Neuroscientist 8: 154-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Sollas AL, Berger RE, Goodman JH (2003) Electrophysiological evidence of monosynaptic excitatory transmission between granule cells after seizure-induced mossy fiber sprouting. J Neurophysiol 90: 2536-2547. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Walsh CA (2000) Cortical malformations and epilepsy. Ment Retard Dev Disabil Res Rev 6: 268-280. [DOI] [PubMed] [Google Scholar]
- Shao LR, Dudek FE (2004) Increased excitatory synaptic activity and local connectivity of hippocampal CA1 pyramidal cells in rats with kainate-induced epilepsy. J Neurophysiol 92: 1366-1373. [DOI] [PubMed] [Google Scholar]
- Sloviter RS (1992) Possible functional consequences of synaptic reorganization in the dentate gyrus of kainate-treated rats. Neurosci Lett 137: 91-96. [DOI] [PubMed] [Google Scholar]
- Sundstrom LE, Mitchell J, Wheal HV (1993) Bilateral reorganisation of mossy fibres in the rat hippocampus after a unilateral intracerebroventricular kainic acid injection. Brain Res 609: 321-326. [DOI] [PubMed] [Google Scholar]
- Sutula TP (1990) Experimental models of temporal lobe epilepsy: new insights from the study of kindling and synaptic reorganization. Epilepsia 31 [Suppl 3]: S45-S54. [DOI] [PubMed] [Google Scholar]
- Sutula TP (1998) A glimpse into abnormal cortical development and epileptogenesis at epilepsy surgery. Neurology 50: 8-10. [DOI] [PubMed] [Google Scholar]
- Sutula TP, Golarai G, Cavazos J (1992) Assessing the functional significance of mossy fiber sprouting. Epilepsy Res Suppl 7: 251-259. [PubMed] [Google Scholar]
- Swann JW, Smith KL, Brady RJ (1990) Neural networks and synaptic transmission in immature hippocampus. Adv Exp Med Biol 268: 161-171. [DOI] [PubMed] [Google Scholar]
- Swann JW, Smith KL, Gomez CM, Brady RJ (1992) The ontogeny of hippocampal local circuits and focal epileptogenesis. Epilepsy Res Suppl 9: 115-126. [PubMed] [Google Scholar]
- Tanaka T, Veeranna, Ohshima T, Rajan P, Amin ND, Cho A, Sreenath T, Pant HC, Brady RO, Kulkarni AB (2001) Neuronal cyclin-dependent kinase 5 activity is critical for survival. J Neurosci 21: 550-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV (1985) Evidence of functional mossy fiber sprouting in hippocampal formation of kainic acid-treated rats. J Neurosci 5: 1016-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CP, Dudek FE (1984a) Excitation of hippocampal pyramidal cells by an electrical field effect. J Neurophysiol 52: 126-142. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Dudek FE (1984b) Synchronization without active chemical synapses during hippocampal afterdischarges. J Neurophysiol 52: 143-155. [DOI] [PubMed] [Google Scholar]
- Thom M, Sisodiya SM, Beckett A, Martinian L, Lin WR, Harkness W, Mitchell TN, Craig J, Duncan J, Scaravilli F (2002) Cytoarchitectural abnormalities in hippocampal sclerosis. J Neuropathol Exp Neurol 61: 510-519. [DOI] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness Jr VS, Chae T, Harlow E (1994) p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371: 419-423. [DOI] [PubMed] [Google Scholar]
- Walsh CA (1999) Genetic malformations of the human cerebral cortex. Neuron 23: 19-29. [DOI] [PubMed] [Google Scholar]
- Wenzel HJ, Robbins CA, Tsai LH, Schwartzkroin PA (2001) Abnormal morphological and functional organization of the hippocampus in a p35 mutant model of cortical dysplasia associated with spontaneous seizures. J Neurosci 21: 983-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE (1996) Electrographic seizures and new recurrent excitatory circuits in the dentate gyrus of hippocampal slices from kainate-treated epileptic rats. J Neurosci 16: 4438-4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cui SS, Wallace AE, Hannesson DK, Schmued LC, Saucier DM, Honer WG, Corcoran ME (2002) Relations between brain pathology and temporal lobe epilepsy. J Neurosci 22: 6052-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]