

Abstract
The p53 family member, p73, is essential for the survival of sympathetic neurons during the developmental period of naturally occurring neuronal death. Here, we have asked whether ΔNp73, which is the only p73 isoform expressed in sympathetic neurons, mediates this survival by p53-dependent and/or p53-independent mechanisms. Initially, we used a genetic approach and crossed p53+/- and p73+/- mice. Quantitation of neurons in the sympathetic superior cervical ganglion during the period of naturally occurring cell death revealed that the loss of p53 partially rescued the death of neurons seen in p73-/- animals. Moreover, exogenous expression of ΔNp73 in cultured p53-/- sympathetic neurons rescued these neurons from apoptosis after NGF withdrawal. Biochemical studies asking how ΔNp73 inhibited NGF withdrawal-induced apoptosis in wild-type neurons demonstrated that it prevented the upregulation of the direct p53 targets p21 and Apaf-1 as well as cleavage of caspase-3. It also inhibited events at the mitochondrial apoptotic checkpoint, suppressing the induction of BimEL and the release of mitochondrial cytochrome c. Interestingly, ΔNp73 expression also inhibited one very early event in the apoptotic cascade, the activation of c-Jun N-terminal protein kinase (JNK), likely by binding directly to JNK. Finally, we show that neuronal cell size is decreased in p73-/- mice, and that this decrease is not rescued by the lack of p53, suggesting a role for p73 in regulating cell size that does not involve interactions with p53. Thus, ΔNp73 promotes neuronal survival via p53-dependent and -independent mechanisms, and it does so at multiple points, including some of the most proximal events that occur after NGF withdrawal.
Keywords: nerve growth factor, sympathetic neurons, Apaf-1, cytochrome c, JNK, Bim, cell size
Introduction
During development, the nervous system is confronted with the problem of establishing appropriate neuronal connectivity, a problem that is partially solved by overproducing neurons and then eliminating those cells that do not successfully compete for target territory (Oppenheim, 1991). This process of naturally occurring cell death is perhaps best understood in sympathetic neurons of the peripheral nervous system, where the ultimate survival of any given neuron is determined by its ability to sequester sufficient amounts of target-derived NGF. NGF then mediates a retrograde survival signal through interactions with the tyrosine receptor kinase A (TrkA)/NGF receptor on the terminal arbor (Miller and Kaplan, 2001). Interestingly, recent studies indicate that a second neurotrophin receptor (NTR), the p75 neurotrophin receptor (p75NTR), plays a functionally antagonistic role in this system and actually promotes the rapid elimination of those sympathetic neurons that do not compete successfully for target-derived NGF (Bamji et al., 1998; Majdan et al., 2001). Thus, the ultimate survival of any given neuron during this period is determined by the interaction between TrkA and p75NTR, a functional interaction that is best exemplified by studies in animals mutant in one or both of these genes (Smeyne et al., 1994; Bamji et al., 1998; Majdan et al., 2001).
The signaling pathways that underlie these interactions have been examined by focusing on cultured neonatal sympathetic neurons, which are dependent on NGF for their survival in culture as they are in vivo. The TrkA receptor mediates its prosurvival effects in these neurons by activating the phosphatidylinositol 3 (PI3)-kinase-Akt pathway, whereas p75NTR mediates its proapoptotic effects via a c-Jun N-terminal protein kinase (JNK)p53-Bax pathway (Kaplan and Miller, 2000). As would be predicted, this p75NTR-activated apoptotic pathway is similar to that induced when NGF is withdrawn from sympathetic neurons, although it is clear that there is a p75NTR-independent component to NGF withdrawal (Bamji et al., 1998; Majdan et al., 2001; Palmada et al., 2002), which likely involves cell cycle dysregulation (Freeman et al., 1994; Park et al., 1996, 1997).
The apoptotic pathway that is activated after NGF withdrawal in sympathetic neurons has been well characterized. Some of the earliest events involve activation of the JNKs and a robust increase in levels of and phosphorylation of the JNK target c-jun (Estus et al., 1994; Ham et al., 1995; Eilers et al., 1998). This early phase is followed by increases in the levels of the proapoptotic Bcl-2 family member BimEL (Putcha et al., 2001; Whitfield et al., 2001), translocation of Bax to the mitochondrion, and subsequent release of cytochrome c (Putcha et al., 1999). Cytochrome c, along with Apaf-1 and the initiator caspase-9, form the apoptosome that leads to activation of the downstream caspase-3 and ultimately neuronal apoptosis (Li et al., 1997). This whole process is relatively prolonged, taking ∼48 hr, and can be reversed by the readdition of NGF for time points up to 12 hr after initial NGF withdrawal (Deckwerth and Johnson, 1993).
We demonstrated previously a key role for the p53 family in these pathways. With regard to p53 itself, animals lacking even a single allele of p53 display a deficit in developmental sympathetic neuron death in vivo and the adenoviral protein early region 18 55K, which binds to and inhibits p53 (Kao et al., 1990; Yew and Berk, 1992), inhibits NGF withdrawal-induced apoptosis in culture (Aloyz et al., 1998). Surprisingly, a second member of the p53 family, p73 (Jost et al., 1997; Kaghad et al., 1997), plays a prosurvival role; p73-/- sympathetic neurons demonstrated enhanced apoptosis in vivo, and an N-terminal truncated isoform of p73, ΔNp73, completely inhibited NGF withdrawal-induced apoptosis (Pozniak et al., 2000). One of the ways that ΔNp73 may mediate this prosurvival effect is by acting as a naturally occurring dominant-inhibitory antagonist of p53, because we have shown previously that ΔNp73 binds to p53 and inhibits p53-mediated sympathetic neuron apoptosis (Pozniak et al., 2000). This prosurvival role for ΔNp73 may well generalize to other populations of neurons, because overexpression of ΔNp73 can rescue cortical neurons from apoptotic insults, and there is ongoing apoptotic death in the cortex of postnatal p73-/- animals (Pozniak et al., 2002).
In this study, we have asked, first, whether p73 mediates its prosurvival effects on sympathetic neurons by antagonizing the proapoptotic effects of p53 and, second, how ΔNp73 inhibits the proapoptotic cascade that is initiated after NGF withdrawal. Our data indicate that ΔNp73 mediates survival by p53-dependent and -independent mechanisms, and that it does so at multiple points in the mitochondrial apoptotic cascade, including the inhibition of JNK activation, one of the most proximal apoptotic events that occur after NGF withdrawal.
Materials and Methods
Animal analysis. Maintenance and genotyping of C57/Bl6 p53-/- and C129 p73-/- mice were performed as described previously (Aloyz et al., 1998; Pozniak et al., 2000). Initially, p53+/- and p73+/- mice were crossed to generate mice heterozygous for both genes. These mixed background double heterozygotes were then bred to produce progeny for analysis. Histological analysis was performed essentially as described previously (Aloyz et al., 1998; Majdan et al., 2001). Briefly, ganglia were drop-fixed in fresh 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, cryoprotected in 30% sucrose, and then cryosectioned at 7 μm on a Leica (Nussloch, Germany) cryostat. Sections were stained with cresyl violet and analyzed with a Zeiss (Thornwood, NY) Axioplan microscope using a 20× objective and the Northern Eclipse computer-based image analysis software (Empix, Ontario, Canada). Neuronal numbers were determined by counting all neuronal profiles with nucleoli on every third section and multiplying the obtained number by three, as per Coggeshall et al. (1984). This method does not correct for split nucleoli. Neuronal soma size was determined by measuring the mean cross-sectional area of 50-100 neurons with nucleoli per animal. Superior cervical ganglia (SCG) volume was determined by summing the SCG cross-sectional areas of every third section, multiplying by the section thickness (7 μm) to obtain the volume for one-third of the SCG, and then multiplying by three to obtain the whole SCG volume. Statistical results were expressed as the mean ± SEM and were tested for significance by Student's t test.
Immunohistochemistry was performed on alternate sections to those used for neuronal counts. Sections were washed with PBS, blocked and permeabilized for 1 hr at room temperature with 3% normal goat serum (Jackson ImmunoResearch, West Grove, PA), and 0.25% Triton X-100 in PBS. Sections were immunostained overnight at room temperature with rabbit anti-tyrosine hydroxylase (TH) (1:1000; Chemicon, Temecula, CA), washed three times in PBS, and then incubated for 1 hr at room temperature with goat anti-rabbit antibody conjugated with Alexa 488 (1:1000; Molecular Probes, Eugene, OR) before coverslipping.
Primary neuronal cultures. Mass cultures of neonatal rat sympathetic neurons were cultured as described by Ma et al. (1992). Briefly, postnatal day 1 (P1) SCGs were dissected and treated with 0.1% trypsin for 30 min at 37°C, followed by treatment with DNase (0.01 mg/ml) for 1 min. Ganglia were dissociated into single cells in Ultraculture media and then plated onto rat tail collagen-coated four-well chamber slides or six-well biochemistry dishes in Ultraculture media (Cambrex, East Rutherford, NJ) containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 3% rat serum (Wisent, Saint Jean-Baptiste de Rouville, Quebec, Canada), 0.7% cytosine arabinoside, and 50 ng/ml mouse 2.5S NGF prepared from mouse salivary glands (Cedarlane, Hornby, Ontario, Canada).
Mass cultures of mouse sympathetic neurons were cultured by a modification of the above method, as described previously (Majdan et al., 2001). Specifically, after dissociation of ganglia, neurons were plated in Ultraculture medium containing 2 mm glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 3% FBS, and 50 ng/ml mouse salivary gland 2.5S NGF. The next day, neurons were fed with the same media plus 0.5% cytosine arabinoside.
Primary cortical neuron cultures were obtained from embryonic day 16 (E16) to E17 mouse embryos, dissociated enzymatically with 10 U/ml papain and 0.1% DNase for 30 min at 37°C, and then dissociated mechanically with a glass Pasteur pipette. Cells were plated in Neurobasal medium containing penicillin/streptomycin (1%), l-Gln (0.25%), and B-27 supplement (2%) on poly-d-lysine-coated biochemistry dishes. On day 3 in vitro (DIV3), one-quarter of the media was replaced with fresh media containing cytosine arabinoside (final concentration, 3 μm) to eliminate non-neuronal cells. On DIV7, cells were harvested for Western blot analysis.
Adenoviral gene transfer. Established cultures of neonatal sympathetic neurons were infected overnight with 50 or 100 multiplicity of infection of replication-deficient recombinant adenovirus in the presence of Ultraculture media, 3% rat serum, 50 mm KCl, and 50 ng/ml NGF. Twenty-four hours later, media was replaced with fresh Ultraculture containing 10 ng/ml NGF but no KCl. Established cortical neurons were infected in a similar manner to sympathetic neurons, except the media used was Neurobasal medium containing penicillin/streptomycin (1%), l-Gln (0.25%), and B-27 supplement (2%), and no KCl. Adenoviruses used in these experiments have been described previously and included Ad5p53 (Slack et al., 1996), which expresses human p53, Ad5LacZ (Toma et al., 2000), and Ad5ΔNp73α or Ad5ΔNp73β (Pozniak et al., 2000), the latter two of which coexpress green fluorescent protein (GFP).
p53-/- sympathetic neuron cultures. SCGs from P1 progeny of C57/Bl6 p53+/- matings were individually dissected, dissociated, and maintained as described previously for mass mouse sympathetic neuron cultures. Each SCG was plated into a separate well of a 96-well collagen-coated culture dish. After performing PCR on tail DNA to confirm p53 genotype (Aloyz et al., 1998), p53+/+ or p53-/- neurons were infected on DIV4 overnight with Ad5GFP or Ad5ΔNp73β·GFP. On DIV6, neurons were withdrawn from NGF for 48 hr. Control neurons were maintained in NGF for the entirety of the experiment. Cells were fixed with 4% paraformaldehyde for 15 min, washed three times with PBS, stained with Hoechst dye for 10 min (1:3000), and washed three additional times with PBS. Nuclear morphology was then analyzed with a Zeiss Axiovert inverted microscope (20× objective) and the Northern Eclipse image analysis software (Empix).
Biochemical analysis. For Western blot analysis of cultured neurons, neurons were lysed and analyzed as described previously (Aloyz et al., 1998; Vaillant et al., 1999). The following primary antibodies were used for these analyses: rabbit polyclonal anti-phospho-JNK (1:1000; Bio-source, Camarillo, CA), rabbit polyclonal anti-JNK1 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-Blm (Bcl-2 interacting mediator of cell death) (1:500; StressGen, Victoria, British Columbia, Canada), rabbit anti-cleaved caspase-3 (1:1000; Cell Signaling Technology, Beverly, MA), rabbit anti-ERK-1 (extracellular signal-regulated kinase) (1:5000; Santa Cruz Biotechnology), rabbit anti-β-galactosidase (1:1000; ICN Biochemicals, Costa Mesa, CA), rat monoclonal anti-Apaf-1 (1: 1000; Chemicon, Boronia, Australia), and mouse anti-pan-p73 (clone ER-15, 1:200; NeoMarkers, Fremont, CA). As secondary antibodies, goat anti-mouse HRP (Bio-Rad, Hercules, CA), goat anti-rabbit HRP (Chemicon), or goat anti-rat HRP (Jackson ImmunoResearch) were used at a dilution of 1:5000. The blots were visualized using the ECL system (Amersham Biosciences, Arlington Heights, IL) and XAR X-OMAT AR film (Kodak, Rochester, NY). The films were digitized, and densitometry analysis was performed with NIH ImageJ 1.31v software. Specifically, the background was subtracted, and the density of each band was measured. The values of each band were normalized to loading control before calculating fold induction (i.e., the ratio of band densities between two conditions).
For JNK activity assays, cells were lysed in 1% NP-40 lysis buffer with protease inhibitor mixture (Roche, Hertforshire, UK), supplemented with 10 mm NaF, 1 mm PMSF, and 1 mm sodium orthovanadate, rocked for 10 min at 4°C, and then spun at 10,000 × g for 10 min to eliminate insoluble debris. Protein levels were standardized by BCA assay (Pierce, Rockford, IL). Ten percent of the lysate was kept for loading control, and 200 μg of total protein was incubated with rabbit anti-JNK1 C-17 antibody (1:100; Santa Cruz Biotechnology) for 1 hr at 4°C, followed by incubation with protein A Sepharose beads (Amersham Biosciences) for 1 hr. Immune complexes bound to beads were spun down and washed three times with NP-40 lysis buffer and two times with kinase buffer (25 mm HEPES, 25 mm MgCl2, 1 mm DTT). Beads were resuspended with 30 μl of kinase buffer containing 10 μCi32P-ATP, 50 μm ATP, and 1.5 μg of glutathione S-transferase (GST) c-Jun (1-169; Upstate Biotechnology, Lake Placid, NY) as a JNK substrate. Reactions were performed at 30°C for 30 min and then stopped with 4× Laemmli sample buffer. Reactions were boiled for 5 min and then loaded onto 7.5% SDS-PAGE. The gel was transferred to a nitrocellulose membrane, and the membrane was exposed to XAR x-ray film. The membrane was then probed with anti-JNK-1 antibody to confirm JNK immunoprecipitation. The original lysate was analyzed by Western blot analysis with rabbit anti-ERK-1 (1:3000; Santa Cruz Biotechnology) and anti-JNK-1 antibodies to demonstrate equal loading.
Cortical neuron lysates enriched for nuclear proteins were prepared by collecting cells in AT buffer (60 mm KCl, 14 mm β-mercaptoethanol, 2 mm EDTA, 15 mm HEPES, pH 7.9, and 0.3 m sucrose) containing 15 mm NaCl and protease inhibitors. Cells were sonicated and spun down, and the pellet was washed with AT buffer with 15 mm NaCl. The pellet was resuspended in AT buffer containing 150 mm NaCl, 0.1% NP-40, and protease inhibitors and then sonicated again. The samples were spun down, and the supernatants (enriched for nuclear proteins) were used for GST pull-down assays and immunoprecipitations.
GST pull-downs and immunoprecipitations. For GST pull-downs, cortical neuron lysates (0.5-1.0 mg) were mixed with 25 μg of recombinant GST-ΔNp73β bound to glutathione beads (Pozniak et al., 2000) and rocked for 1 hr at 4°C. As a control for specificity, lysates were also incubated with GST alone. After extensively washing the beads with lysis buffer, the beads were boiled for 5 min in 1× Laemmli sample buffer. The supernatant was then loaded onto 10% SDS-PAGE, and Western blot analysis was performed as above. To test for the direct interaction between GST-ΔNp73β and JNK, GST pull-downs were performed using 0.1-0.5 μg of purified recombinant active JNKα1 (14-327; Upstate Biotechnology) or unactive JNKα1 (14-328; Upstate Biotechnology) as substrates. For immunoprecipitations, samples were processed in a similar manner to the pull-downs, except Ad5ΔNp73α or Ad5ΔNp73β infected cortical neuron lysates were incubated with anti-p73 ER-15 mouse immunoclonal antibody bound to protein A-Sepharose beads.
Immunocytochemistry and confocal microscopy. For immunocytochemical analysis of cytochrome c, neurons were fixed in 4% paraformaldehyde for 15 min, washed with PBS three times, permeabilized with 0.5% Triton X-100 in PBS, and then blocked at room temperature for 1 hr with 0.5% BSA and 6% normal goat serum in PBS. Antibodies were diluted in 0.25% BSA and 3% NGS in PBS. The following primary antibodies were used in this study: rabbit polyclonal anti-β-galactosidase (1:1000; ICN Biochemicals), rabbit polyclonal anti-p53 FL-393, which is specific for the adenovirally driven human p53 (1:1000; Santa Cruz Biotechnology), chicken polyclonal anti-GFP, mouse monoclonal anti-cytochrome c (clone 6H2.B4; 1:500; PharMingen, San Diego, CA), and mouse monoclonal anti-cytochrome oxidase subunit IV (1:1000; Molecular Probes). After three washes with PBS, cells were incubated for 1 hr at room temperature in goat anti-rabbit antibody conjugated to Alexa 488, goat anti-chicken antibody conjugated to Alexa 488, or goat anti-mouse antibody conjugated to Alexa 555 (all 1:1000; Molecular Probes). Hoechst dye (1:3000) was added for 1 min to visualize nuclei. Cells were then washed three times with PBS before coverslipping. Cells were analyzed and quantitated with a Zeiss Axioplan microscope (40× water immersion objective) and the Northern Eclipse image analysis software.
For confocal microscopy, cells were treated as above and then analyzed with the Zeiss LSM5 PASCAL system and a 63× oil immersion lens. To capture red fluorescence, an excitation wavelength of 543 nm and a 560-615 nm bandpass filter were used. The images were then processed with the software provided by Zeiss.
Results
p53 and p73 interact genetically to partially determine sympathetic neuron survival in vivo
Our previous studies on p53-/- and p73-/- mice demonstrated that the two proteins had opposite roles in the nervous system. Mice lacking one or both p53 alleles had increased sympathetic neuronal number after naturally occurring cell death, consistent with the loss of a proapoptotic protein (Aloyz et al., 1998). In contrast, p73, found in the nervous system only as an anti-apoptotic isoform (ΔNp73), has a prosurvival role; mice lacking both alleles of p73 had decreased sympathetic neuron numbers (Pozniak et al., 2000). Because these two proteins can interact biochemically in some situations (Pozniak et al., 2000), these results suggested that their functionally antagonistic interactions might regulate naturally occurring sympathetic neuron death. We addressed this idea using the genetic approach of crossing p53+/- and p73+/- animals. In these experiments, if p73 supported neuronal survival only by inhibiting the p53 death pathway, then loss of p53 should completely rescue the enhanced death of p73-/- neurons in vivo. Conversely, if p73 supports neuronal survival via a p53-independent mechanism, then the genetic loss of p53 should have no effect on p73-/- neuronal death. A third alternative is that p73 inhibits apoptosis by both p53-dependent and -independent mechanisms, in which case loss of p53 would only partially rescue the p73-/- phenotype.
To perform these experiments, we crossed p53+/- mice in a C57/Bl6 background to p73+/- mice in a C129 background to generate p53+/-, p73+/- progeny in a mixed genetic background. We then crossed these double heterozygotes and analyzed their progeny by examining sympathetic neuron number in the SCG during the developmental death period. Genotyping of >100 animals resulting from these crosses revealed that the expected Mendelian ratio of progeny was obtained for males only; p53-/- females were rarely obtained regardless of p73 status, as reported previously (Armstrong et al., 1995; Sah et al., 1995). In addition, as reported previously (Pozniak et al., 2000; Yang et al., 2000), the majority of the p73-/- animals died within the first 2 weeks after birth, and this shortened lifespan was not rescued by loss of either one or both p53 alleles. We therefore analyzed the SCG of these mice at P10. At this time point, developmental neuron death is ongoing.
We initially asked whether the perturbations in neuronal number we had previously reported for p53-/- and p73-/- SCGs were maintained in this mixed genetic background. To perform this analysis, ganglia were dissected from mice at P10, cryostat sectioned, cresyl violet stained, and the number of neurons per section counted at 21 μm intervals throughout the entirety of the ganglion. As reported previously, p53+/+, p73-/- ganglia were smaller in size than ganglia from their wild-type littermates (Fig. 1A) with a mean ganglion volume of 0.057 ± 0.003 mm3 relative to a mean volume of 0.124 ± 0.005 mm3 for wild-type ganglia. Quantitation of sympathetic neurons also confirmed that numbers were decreased in p53+/+, p73-/- ganglia in this mixed genetic background (Fig. 2A), as they were in the pure C129 background (Pozniak et al., 2000); wild-type neuron numbers were 22,304 ± 2147 (n = 6), whereas p53+/+, p73-/- neuron numbers were 15,304 ± 1419 (n = 3) (p < 0.05). Interestingly, a careful morphological analysis revealed that the size of the p53+/+, p73-/- neurons was also decreased by ∼30% (Fig. 2B) (126.0 ± 13.5 vs 188.4 ± 12.4 μm2 for p53+/+, p73-/- vs p53+/+, p73+/+ neurons; p < 0.05), a decrease that, along with the lower neuron number, would explain the large decrease in ganglion size. Confirmation that these smaller cells were in fact sympathetic neurons was obtained by immunostaining ganglia for tyrosine hydroxylase, a marker for the noradrenergic neuron phenotype (Fig. 1C). Thus, the lack of p73 not only caused enhanced neuronal apoptosis but also caused decreased neuron size.
Figure 1.

The coincident absence of p53 partially rescues the phenotype of the p73-/- sympathetic superior cervical ganglion. A, Photomicrographs of cryosections of SCGs isolated from P10 progeny of different genotypes deriving from crosses of p53+/-, p73+/- animals. Representative cross-sections from the center of the SCGs were stained with cresyl violet and photographed at the same magnification. Scale bar, 100 μm. B, Photomicrographs of neurons within cryosections similar to those shown in A. The arrowheads indicate single neurons. Scale bar, 25 μm. C, Immunocytochemical analysis for TH in sections of the SCG from a p53+/-, p73-/- animal to demonstrate that sympathetic neurons have an appropriate noradrenergic phenotype in the absence of p73. The arrowheads indicate TH-positive neurons.
Figure 2.

Loss of p53 partially rescues the loss of sympathetic neurons in p73-/- ganglia. A, Quantitation of the mean sympathetic neuron number in P10 SCGs of different genotypes deriving from crosses of p53+/-, p73+/- animals. With the exception of p53-/-, p73-/- genotype, the bars represent the mean neuron numbers ± SEM (n > 3). For the p53-/-, p73-/- neurons, the bar indicates the average neuron number of two SCGs ± SEM (*p < 0.05 relative to p53+/+, p73+/+; **p < 0.05 relative to p53+/+, p73-/-). B, Quantitation of sympathetic neuron size in P10 SCGs of the progeny of p53+/-, p73+/- crosses. Loss of p53 does not rescue the reduced neuron size seen in p73-/- mice (*p < 0.05 relative to p53+/+, p73+/+)
We performed a similar analysis for p53+/-, p73+/+ and p53-/-, p73+/+ ganglia. We reported previously that sympathetic neuron number is increased to the same level in animals lacking either one or two p53 alleles (Aloyz et al., 1998). In the mixed genetic background used here, SCG size was approximately similar for p53+/+, p53+/-, and p53-/- animals (Fig. 1A). Moreover, neuron morphology (Fig. 1B) and size (Fig. 2B) were also similar. However, quantitation of neuron number confirmed that the lack of one or both alleles of p53 led to a similar increase in neuron number relative to p53+/+ littermates (Fig. 2B). More specifically, whereas wild-type littermates had 22,304 ± 2147 (n = 6) neurons in the P10 SCG, neuron numbers were significantly increased to 30,757 ± 553 (n = 4) in p53+/-, p73+/+ ganglia (p < 0.05). Similarly, p53-/-, p73+/+ neuron numbers were increased to 29,111 ± 2650 (n = 4), a number that was not significantly different from that obtained with p53+/-, p73+/+ (p > 0.05) ganglia.
Having established the baseline for our crosses, we then asked whether p53 and p73 interacted genetically during developmental sympathetic neuron death by quantifying neuron number in p73-/- animals lacking one or both p53 alleles. Analysis of two p53-/-, p73-/- animals that were obtained revealed that p53-/-, p73-/- ganglia were smaller than wild-type ganglia (Fig. 1A), and quantitation revealed that p53-/-, p73-/- neurons were also smaller than wild-type neurons but were similar in size to p53+/+, p73-/- neurons (Figs. 1B, 2B). However, whereas the size of neurons was similar, p53-/-, p73-/- ganglia contained more neurons than did the p53+/+, p73-/- ganglia (Fig. 2A). Specifically, p53-/-, p73-/- SCGs contained on average 17,894 ± 1130 neurons (n = 2), whereas p53+/+, p73-/- ganglia contained 15,304 ± 1419 (n = 3) neurons. Thus, the coincident absence of p53 had no effect on the smaller neuron size but partially rescued the loss of neurons seen in p73-/- animals.
Because sympathetic neuron number is similar in animals lacking one or both alleles of p53 (Fig. 2A), we examined p53+/-, p73-/- ganglia to confirm the numbers obtained in two p53-/-, p73-/- animals. This analysis revealed that, as seen with p53-/-, p73-/- ganglia, neurons in p53+/-, p73-/- ganglia were smaller than wild-type neurons but similar in size to the p53+/+, p73-/- neurons (Figs. 1B, 2B) (125.9 ± 11.4 vs 126.0 ± 13.5 μm2 for p53+/-, p73-/- vs p53+/+, p73-/- neurons). Moreover, p53+/-, p73-/- ganglia contained more neurons than did the p53+/+, p73-/- ganglia (Fig. 2A) (20,141 ± 999, n = 5, vs 15,304 ± 1419, n = 3, for p53+/-, p73-/- vs p53+/+, p73-/- neurons; p < 0.05). However, this number was significantly lower than that of p53+/-, p73+/+ animals (30,757 ± 553; n = 4; p < 0.05). Thus, the absence of one or both p53 alleles partially rescues the decreased neuron number seen in p73-/- ganglia, indicating that p73 must mediate neuronal survival by both p53-dependent and p53-independent mechanisms. In addition, the finding that neuronal cell size is decreased in p73-/- neurons and that this decrease is not rescued by the lack of p53 suggests a role for p73 in regulating cell size that does not involve interactions with p53.
ΔNp73 rescues p53-/- sympathetic neurons from NGF withdrawal-induced apoptosis
If, as indicated by the genetic data, p73 inhibits neuronal apoptosis by both p53-dependent and -independent mechanisms, ΔNp73 should be able to rescue p53-/- sympathetic neurons from apoptosis. To test this prediction, we cultured neonatal p53-/- neurons, infected them with recombinant adenoviruses that express either GFP alone (Ad5GFP) or ΔNp73β plus GFP (Ad5ΔNp73β), and asked whether they survived after NGF withdrawal. Initially, we characterized the time course of apoptosis of p53-/- neurons by culturing sympathetic neurons from p53+/+ and p53-/- littermates, establishing them in NGF for 5 d, and withdrawing them from NGF. After NGF withdrawal, we counted defined fields of phase-bright neurons daily for 4 d. This analysis revealed that, as reported previously (Vogel and Parada, 1998; Besirli et al., 2003), p53-/- neonatal sympathetic neurons were only modestly resistant to apoptosis after NGF withdrawal (data not shown). We then infected similar cultures with Ad5GFP or Ad5ΔNp73β, withdrew NGF, and quantified the number of surviving infected cells by examining nuclear morphology of the infected neurons 48 hr after NGF withdrawal (Fig. 3A). As seen in uninfected neurons, p53-/- sympathetic neurons that were transduced with GFP underwent apoptosis after NGF withdrawal. However, these neurons showed modest and somewhat variable enhanced survival relative to p53+/+ neurons that were transduced with GFP and withdrawn from NGF (Fig. 3B). In contrast, p53-/- sympathetic neurons that were transduced with ΔNp73β plus GFP were completely rescued from apoptosis induced by NGF withdrawal (Fig. 3B), supporting the conclusion that p73 can rescue neurons from apoptosis by a p53-independent mechanism(s).
Figure 3.

ΔNp73 rescues p53-/- neurons from NGF withdrawal-induced apoptosis. A, Fluorescence micrographs of p53-/- sympathetic neurons infected with a recombinant adenovirus expressing both ΔNp73β and GFP (Ad5ΔNp73β·GFP) withdrawn from NGF for 48 hr and then counterstained with Hoechst to show the nuclear morphology. The arrowhead indicates an uninfected neuron with a pyknotic, apoptotic nucleus, whereas the arrow denotes one of several infected GFP-positive neurons in the field that show round, healthy nuclei. B, Bar graphs showing quantitation of two representative experiments (of 3 in total) similar to that shown in A. p53+/+ and p53-/- neurons cultured from neonatal littermates were infected either with an adenovirus expressing only GFP (Ad5GFP) or with one expressing both ΔNp73β and GFP (Ad5ΔNp73β) withdrawn from NGF for 48 hr and analyzed as in A. Scale bar, 50 μm. Data were normalized so that the survival of GFP-infected p53+/+ neurons in NGF was 100%, and GFP-infected p53+/+ neurons withdrawn from NGF for 48 hr was 0%. Note that ΔNp73β completely rescued p53-/- neurons from apoptosis, and that survival of p53-/- neurons was higher than that of their wild-type counterparts, although the magnitude of this increase was variable.
p53 induces only some aspects of the NGF withdrawal-induced apoptotic cascade
To characterize the underlying p53-dependent and p53-independent mechanisms by which ΔNp73 mediates sympathetic neuronal survival, we initially characterized the apoptotic changes in sympathetic neurons induced by p53 itself. We demonstrated previously that p53 induction is sufficient to cause sympathetic neuron apoptosis in the presence of NGF (Slack et al., 1996). Moreover, we have shown that although p53 overexpression does not cause JNK activation, an early apoptotic signaling event after NGF withdrawal of sympathetic neurons, it does induce one of its direct gene targets, the cyclin-dependent kinase p21WAF1 (Aloyz et al., 1998). To extend this analysis, we asked whether increased p53 was sufficient to cause alterations in sympathetic neuron gene expression that were associated with neuronal apoptosis, focusing on Apaf-1, an apoptosome component that is a known downstream target of p53 in DNA damage-induced neuronal apoptosis (Fortin et al., 2001), and on BimEL, a proapoptotic BclII family member that is induced after NGF withdrawal (Putcha et al., 2001; Whitfield et al., 2001). Sympathetic neurons were infected with adenoviruses expressing either p53 (Ad5p53) or β-galactosidase (Ad5LacZ), maintained in NGF, and then analyzed at time points ranging from 27 to 56 hr after infection. Western blot analysis revealed that p53 caused a small but reproducible increase in Apaf-1 levels (Fig. 4A). In contrast, BimEL levels were unchanged by p53 overexpression, in contrast to the upregulation observed after NGF withdrawal (Fig. 4B). Thus, in sympathetic neurons, p53 induces expression of two of its known direct targets, Apaf-1 and p21WAF1 (Aloyz et al., 1998) but not BimEL.
Figure 4.

p53 induces increased levels of Apaf-1 as well as cytochrome c release from mitochondria but has no effect on expression of BimEL. A, B, Western blot analysis of cultured neonatal mouse (A) or rat (B) sympathetic neurons that were established in the presence of NGF, transduced with the recombinant adenoviruses Ad5p53 or Ad5LacZ as indicated, and maintained in NGF for various time points after infection. Controls were withdrawn from NGF for 24 hr. Western blots were then probed with antibodies specific for Apaf-1 (A) or BimEL (B). Blots were then reprobed for the ERKs to demonstrate equal loading (p42/p44 ERK). C, Confocal micrographs of sympathetic neurons that were either maintained in NGF (+NGF; left) or withdrawn from NGF for 24 hr (-NGF; right) and then fixed and immunostained for cytochrome c, demonstrating the patterns of staining that were defined as punctate (localized to mitochondria; left) or diffuse (released from mitochondria; right) for quantitation. Scale bar, 10 μm. D, Confocal micrographs of sympathetic neurons that were either maintained in NGF (+NGF; left) or withdrawn from NGF (-NGF; right) on DIV 5 and then immunostained 24 hr later for COX IV, a mitochondrial marker. Note that COX IV staining remains bright and punctate after NGF withdrawal. E, Quantitation of diffuse cytochrome c distribution, indicative of cytochrome c that has been released from mitochondria in sympathetic neurons 48 hr after infection with recombinant adenoviruses expressing either GFP or p53. Infected neurons were fixed and immunostained for p53 and cytochrome c, nuclei were visualized by Hoechstdye, and the percentage of infected neurons showing diffuse cytochrome c immunostaining was determined. Data represent the percentage of infected cells with diffuse cytochrome c staining in two independent experiments.
We next examined the mitochondrial apoptotic transition based on a number of recent reports indicating that p53 mediates apoptosis directly at the mitochondrial level (Schuler et al., 2000; Mihara et al., 2003; Chipuk et al., 2004). More specifically, we examined cytochrome c release, a critical cell death checkpoint in the process of NGF withdrawal-induced apoptosis. To assay for cytochrome c release, we used an immunocytochemical assay previously used extensively for sympathetic neurons (Neame et al., 1998; Deshmukh et al., 2000; Whitfield et al., 2001). By fluorescence microscopy, a bright, punctuate, cytochrome c staining pattern in these neurons corresponds to intact mitochondrial cytochrome c, whereas a faint, diffuse, staining pattern corresponds to cytochrome c that has been released from mitochondria (Fig. 4C). As a control for the specificity of this assay, we assessed mitochondrial structural integrity by immunostaining cells for the mitochondrial protein cytochrome oxidase subunit IV (COX IV) (Fig. 4D), which is not released during apoptosis. COX IV staining was bright and punctate in sympathetic neurons regardless of whether they were maintained in or withdrawn from NGF (Fig. 4D). Having established the assay, we then infected sympathetic neurons with recombinant adenoviruses expressing either p53 or GFP. Forty-eight hours postinfection, we performed double immunostaining for cytochrome c and for either p53 or GFP. Immunocytochemical analysis revealed that 30-60% of the p53-expressing neurons showed diffuse cytochrome c immunoreactivity, indicative of mitochondrial cytochrome c release (Fig. 4E). In contrast, the vast majority of neurons infected with the GFP adenovirus exhibited punctate cytochrome c immunoreactivity, with none or few showing a diffuse pattern (Fig. 4E), a pattern similar to that seen for uninfected neurons (Figs. 4C, 6A). Thus, p53 overexpression is sufficient to cause cytochrome c release and neuronal apoptosis accompanied by increases in p21 and Apaf-1 but is not sufficient to induce either JNK activation or BimEL upregulation, two of the most proximal events seen after NGF withdrawal.
Figure 6.

ΔNp73 prevents cytochrome c release from mitochondria and BimEL upregulation in sympathetic neurons after NGF withdrawal. A, Quantitation of cytochrome c distribution at 24 hr post-NGF withdrawal in established cultures of neonatal rat sympathetic neurons that were infected with either LacZ or ΔNp73β adenovirus, the latter of which coexpresses GFP, and withdrawn from NGF on DIV5. After 24 hr, cells were fixed and immunostained for β-galactosidase and cytochrome c (for Ad 5 LacZ) or for GFP and cytochrome c (for Ad5ΔNp73β). Nuclei were visualized by Hoechst dye, and the percentage of infected neurons showing punctate (black bars) versus diffuse (white bars) cytochrome c immunostaining was determined. Error bars indicate the mean ± SEM (n = 3 independent experiments). B, Western blot analysis for BimEL in cultured neonatal rat sympathetic neurons that were established for 3 DIV, infected with Ad5LacZ, Ad5ΔNp73α, or Ad5ΔNp73β, withdrawn from NGF on DIV5, and then analyzed 20 hr later. As a control, neurons were maintained in NGF for the entirety of the experiment. The positive controls are lysates of cultured human embryonic kidney 293 cells or HeLa cells. The blots were reprobed for the ERK proteins to ensure equal loading of protein (p44 ERK), and adenoviral gene transfer was confirmed by probing the same lysates with antibodies specific for β-galactosidase and p73. These blots are representative of those obtained in three independent experiments. The fold-induction of BimEL from two independent experiments is also graphed, demonstrating the consistency of the rescue with ΔNp73. The asterisk (*) in the p73 reprobe denotes a nonspecific band. A background band (**) is present because of incomplete stripping of the ERK blot.
ΔNp73 blocks the induction of the p53 target genes p21WAF1 and Apaf-1 as it inhibits neuronal apoptosis after NGF withdrawal
Having shown that p53 induces only a subset of the alterations associated with NGF withdrawal-induced apoptosis, we then asked which aspects of this apoptotic cascade were inhibited by ΔNp73. We have shown previously that when sympathetic neurons are cultured in NGF to promote their survival, ΔNp73 levels are increased. Withdrawal of NGF dramatically decreases ΔNp73 levels coincident with neuronal apoptosis, and this apoptosis can be inhibited by rescuing the decrease in ΔNp73 (Pozniak et al., 2000). We therefore overexpressed ΔNp73α or ΔNp73β in cultured sympathetic neurons, withdrew them from NGF, and asked how this inhibited apoptosis. Cells rescued by either ΔNp73 isoform remained healthy in the absence of NGF for at least 4 d, as demonstrated by phase microscopy and MTT staining (data not shown). For all of the ensuing biochemical studies, similar results were obtained with both isoforms.
We initially examined expression of the direct p53 target genes p21WAF1 and Apaf-1. In this regard, although we have shown previously that p21WAF1 is increased after NGF withdrawal and p53 overexpression (Aloyz et al., 1998; Deshmukh et al., 2000), Apaf-1 has not been examined. We therefore first asked whether Apaf-1 levels were increased after NGF withdrawal, as they were after p53 overexpression (Fig. 4A). Western blot and densitometric analyses of mouse sympathetic neurons established in NGF for 5 d and then switched into media with or without NGF for 24 hr revealed that Apaf-1 levels were consistently but modestly increased during NGF withdrawal-induced apoptosis (Fig. 5A), as were levels of p21WAF1 (Fig. 5B).
Figure 5.
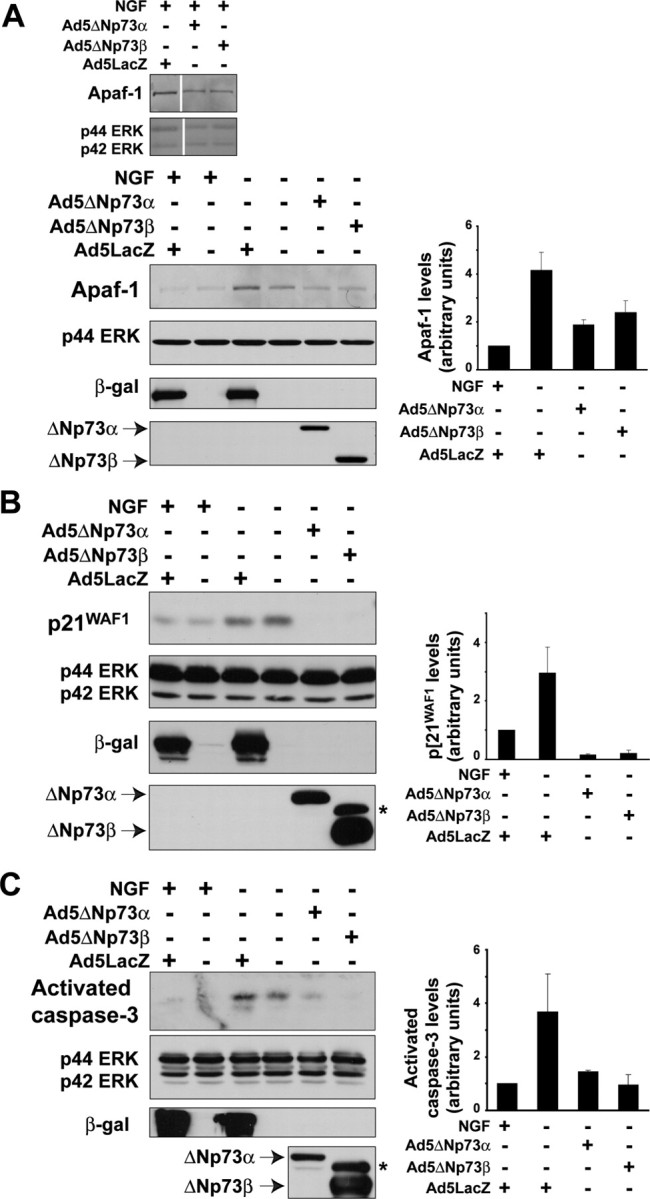
ΔNp73 blocks the increases in p21WAF1 and Apaf-1 and cleavage of caspase-3 in sympathetic neurons withdrawn from NGF. A-C, Western blot analysis of cultured neonatal mouse (A) or rat (B, C) sympathetic neurons that were established in the presence of NGF, transduced with the recombinant adenoviruses Ad5ΔNp73α, Ad5ΔNp73β, or Ad5LacZ as indicated, withdrawn from NGF on DIV5, and lysed 24 hr later. Controls were maintained in NGF for the entirety of the experiment. Western blots were then probed with antibodies specific for Apaf-1 (A), for p21WAF1 (B), or for cleaved activated caspase-3 (C). All blots were then reprobed for the ERKs to demonstrate equal loading (p42/p44 ERK). Adenoviral gene transfer was confirmed by performing Western blots on the same lysates with antibodies specific for β-galactosidase and for p73. The average fold-induction ± SEM of the relevant protein from two independent experiments is also graphed, demonstrating the consistency of the rescue with ΔNp73. The asterisk (*) in the p73 reprobe denotes a nonspecific band.
We next asked whether ΔNp73 antagonized these p53-dependent alterations; sympathetic neurons were established in NGF and then transduced with adenoviruses expressing either ΔNp73 (Ad5ΔNp73) or β-galactosidase (Ad5LacZ). Neurons were then withdrawn from NGF and analyzed 24 hr later. Western blot and densitometry analyses revealed that overexpression of ΔNp73 but not β-galactosidase prevented the increased levels of Apaf1 seen after NGF withdrawal (Fig. 5A). Similar results were obtained for p21WAF1, except that p21 levels were even lower than baseline control levels in the ΔNp73-expressing neurons (Fig. 5B). ΔNp73 also inhibited the cleavage and activation of the effector caspase, caspase-3 (Fig. 5C), consistent with the observed rescue of neuronal survival (Pozniak et al., 2000). Thus, ΔNp73 inhibits neuronal apoptosis upstream of p21, Apaf-1, and caspase-3, possibly by antagonizing the transcriptional actions of p53.
ΔNp73 blocks mitochondrial activation after NGF withdrawal
Because both NGF withdrawal and p53 overexpression (Fig. 4E) directly induce a mitochondrial apoptotic transition, we asked whether ΔNp73 would inhibit cytochrome c release. Neurons were established in NGF, infected with adenoviruses expressing either ΔNp73 or β-galactosidase, and were then withdrawn from NGF for 24 hr. Control neurons were maintained in NGF for the entirety of the experiment. Immunocytochemistry and quantitative analysis revealed that in neurons expressing control β-galactosidase and maintained in NGF, cytochrome c staining was punctate and nuclei were intact, as was expected (punctuate, 86 ± 9.8%; diffuse, 13 ± 9.8%) (Fig. 6A). Similar staining patterns were observed in neurons expressing ΔNp73β and maintained in NGF (punctuate, 80 ± 3.2%; diffuse, 20 ± 3.2%) (Fig. 6A). Conversely, in neurons withdrawn from NGF and expressing β-galactosidase, most cells exhibited faint, diffuse, cytochrome c staining and had pyknotic nuclei, indicating that these apoptotic cells had indeed released cytochrome c (punctuate, 27 ± 4.3%; diffuse, 72.8 ± 4.3) (Fig. 6A). Interestingly, when neurons expressing ΔNp73 were withdrawn from NGF, cytochrome c staining in the transduced neurons was primarily bright and punctate (punctate, 74 ± 4.5%; diffuse, 26 ± 4.5%) (Fig. 6A), a similar pattern to that observed in NGF-maintained neurons. Thus, ΔNp73 acts to inhibit the release of cytochrome c after NGF withdrawal.
Proapoptotic and anti-apoptotic Bcl-2 family members such as Bim participate in maintaining mitochondrial integrity. Because ΔNp73 was acting upstream of mitochondrial cytochrome c release, we asked whether ΔNp73 modulated the levels of BimEL. Western blot analysis demonstrated that the upregulation of BimEL seen after NGF withdrawal was completely prevented by overexpression of ΔNp73 isoforms (Fig. 6B). Because p53 is not sufficient to induce BimEL, we conclude that ΔNp73 acts potentially by inhibiting p53-dependent actions at the mitochondrion as well as p53-independent upregulation of BimEL.
ΔNp73 binds to JNK and inhibits JNK activation after NGF withdrawal
The induction of BimEL is thought to be downstream of JNK activation after NGF withdrawal (Putcha et al., 2001; Whitfield et al., 2001). Because ΔNp73 inhibited this induction, we examined the activation of JNK, one of the most proximal apoptotic events to occur after NGF withdrawal (Estus et al., 1994; Ham et al., 1995; Eilers et al., 1998). To assess JNK activation, we used two approaches; Western blot analysis with a phospho-specific JNK antibody and an in vitro immune complex kinase assay using GST-c-jun (1-169) as a substrate. For both, sympathetic neurons were established in NGF, infected with either Ad5ΔNp73 or Ad5LacZ, and then withdrawn from NGF. Eight hours later, lysates were collected for biochemical analysis. Western blot analysis with the phospho-specific JNK antibody revealed that, as previously reported, JNK activation was increased 8 hr after NGF withdrawal, and this activation was unaffected by infection with Ad5LacZ (Fig. 7A). In contrast, overexpression of ΔNp73 inhibited the increased phosphorylation of JNK seen using this assay (Fig. 7A). The immune complex kinase assay confirmed this result; JNK activity was increased after NGF withdrawal, as evidenced by increased in vitro phosphorylation of the GST-c-jun substrate, an increase that was inhibited by ΔNp73 but not β-galactosidase (Fig. 7B).
Figure 7.

A, B, ΔNp73 binds to JNK and inhibits activation of JNK, an early apoptotic signaling event after NGF withdrawal of sympathetic neurons. A, B, Established cultures of neonatal rat sympathetic neurons were infected with Ad5LacZ, Ad5ΔNp73α, or Ad5ΔNp73β adenovirus, withdrawn from NGF after 5 DIV and then analyzed 8 hr later. A, Western blot analysis of neurons treated as indicated and then probed with an antibody specific to the phosphorylated, activated form of JNKs 1 and 2 (P-p46/p54 JNK). The blot was then reprobed first for total JNK protein (p46/p54 JNK) and then for ERK protein (p42/p44 ERK) to ensure that equal amounts of protein were loaded in each lane. Adenoviral gene transfer was confirmed by probing the same lysates with antibodies specific for β-galactosidase and p73. The fold induction of phosphorylated JNK from a second independent experiment is also graphed, demonstrating the consistency of the rescue with ΔNp73. The asterisk (*) in the p73 reprobe denotes a nonspecific band. B, In vitro JNK activity assay of neurons treated as indicated, using GST-c-jun (1-169) as a substrate (arrow). Autophosphorylated JNK is marked with an asterisk (*). The blot was then probed for JNK to visualize the amounts of immunoprecipitated JNK (p46 JNK). The IgG heavy chain (IgG) is also present. C-E, ΔNp73 binds directly to JNK. C, Lysates of cultured cortical neurons were incubated with a GST-ΔNp73β fusion protein or as a control with GST alone, and the GST fusion protein was then precipitated with glutathione Sepharose beads. Precipitates were analyzed on Western blots, and the blots were probed with an antibody specific to JNK. The input cortical neuron lysate is also shown for comparison. Note that JNK is present in the GST-ΔNp73β precipitate but not in the GST-alone precipitates. D, Cortical neurons were infected with recombinant adenoviruses expressing either ΔNp73α orβ, and nuclear fractions were immunoprecipitated with an anti-p73 antibody. Immunoprecipitates were analyzed by Western blot analysis with an antibody specific to JNK (p46 JNK). The blots were reprobed with an antibody specific for p73 (bottom). The input cortical neuron lysate is also shown for comparison. Note that JNK associates with both ΔNp73 isoforms. E, Purified, active, phosphorylated JNKα1 or inactive, nonphosphorylated JNKα1 was mixed with GST-ΔNp73β bound to glutathione Sepharose beads. The beads were then precipitated and analyzed on Western blots. As a positive control, aliquots of the mixture containing active or inactive JNKα1 were run on the same blot. Note that JNKα1 is present in the GST-ΔNp73β precipitates but not in the GST-alone precipitates.
We next asked whether the prevention of JNK activation by ΔNp73 was attributable to a direct interaction between the two proteins. To answer this question, we performed GST pull-down assays by incubating GST-ΔNp73β (Pozniak et al., 2000) with nuclear extracts of cortical neurons, which express abundant JNK. This approach revealed that GST-ΔNp73β was able to interact with JNK in cortical neuron lysates (Fig. 7C). GST alone did not interact with JNK, demonstrating the specificity of this interaction (Fig. 7C). We then confirmed that ΔNp73 could interact with endogenously expressed JNK in cortical neurons. Cortical neurons were infected with Ad5ΔNp73α or Ad5ΔNp73β, and ΔNp73 was immunoprecipitated from an enriched nuclear lysate with an antibody specific to p73. Western blotting of the immunoprecipitated proteins with a JNK antibody showed that JNK associated with immunoprecipitated ΔNp73 in cortical neurons (Fig. 7D).
Because all of these assays involved neuronal lysates, it was possible that the interaction between ΔNp73 and JNK was mediated indirectly by another associated protein. To address this possibility, we directly mixed GST-ΔNp73β with purified JNK1 that was either phosphorylated (active) or not (inactive). We then pulled down the GST-ΔNp73β and asked whether it was associated with JNK. This analysis revealed that both active and inactive JNKα1 directly associated with GST-ΔNp73β but not with GST alone (Fig. 7E). Thus, ΔNp73 inhibits JNK activation after NGF withdrawal, likely by directly interacting with JNK itself.
Discussion
The data presented here support the hypothesis that ΔNp73 mediates its neuronal survival effects via p53-dependent and -independent mechanisms, and that it does so upstream of the mitochondrial apoptotic transition. Specifically, our findings support four major conclusions. First, our genetic study shows that loss of p53 is sufficient to partially rescue the enhanced sympathetic neuron death seen in p73-/- mice, demonstrating that these two proteins interact in a functionally antagonistic manner during naturally occurring neuronal death. However, this rescue is only partial, indicating that p73 must mediate neuronal survival via other targets as well. Interestingly, our genetic study also suggests that p73 plays a role in cell size regulation, and that it might do so in a p53-independent manner. Second, the studies we report here with p53-/- neurons, together with our previous studies showing that ΔNp73 can directly rescue p53-mediated neuronal apoptosis (Pozniak et al., 2000), strongly argue that ΔNp73 mediates neuronal survival by p53-dependent and -independent pathways. Third, our biochemical and cell biological studies indicate that ΔNp73 inhibits NGF withdrawal-induced neuronal apoptosis at multiple points, including some that are upstream of the mitochondrial apoptotic transition. Some of these effects are consistent with inhibition of the proapoptotic actions of p53, whereas others, such as the inhibition of JNK activation, likely represent p53-independent actions. Fourth, our studies demonstrate that ΔNp73 can directly bind to and inhibit JNK, thereby identifying a novel target for this prosurvival protein. Together, these findings support a model where ΔNp73 acts as a potent and early inhibitor of neuronal apoptotic signaling cascades, mediating its effects both via p53 and via at least one other target protein, JNK.
We demonstrated previously that the ΔNp73 isoforms are highly potent survival proteins, inhibiting the death of cultured sympathetic and cortical neurons as mediated by a variety of stimuli, including NGF withdrawal and DNA damage (Pozniak et al., 2000, 2002). Moreover, we previously demonstrated enhanced death of p73-/- postnatal sympathetic and cortical neurons in vivo, indicating that this gene is essential for the survival of at least some populations of developing neurons. How then does ΔNp73 inhibit neuronal apoptosis? Data presented here argue that ΔNp73 functions partially by antagonizing p53. Although p53 is best known for its proapoptotic actions in mature neurons after insults such as excitotoxicity and axonal injury (Miller et al., 2000), we have shown previously that it is also important for death of developing sympathetic neurons (Aloyz et al., 1998). Our data suggest that one way ΔNp73 antagonizes p53 is at the transcriptional level, as described previously in cell lines (Kartasheva et al., 2002; Stiewe et al., 2002). Specifically, ΔNp73 blocked the upregulation of two direct p53 transcriptional targets, p21WAF1 (el-Deiry et al., 1993) and Apaf-1 (Fortin et al., 2001; Robles et al., 2001) after NGF withdrawal. ΔNp73 coincidentally prevented the upregulation of BimEL, a proapoptotic Bcl-2 family member. However, because Bim expression is not induced by p53 expression in sympathetic neurons, and because Bim is not known to be a direct transcriptional target of p53, we propose that this latter response is likely mediated indirectly via inhibition of JNK.
In addition to blocking increases in p53 target gene products, our data indicate that ΔNp73 inhibits apoptosis outside of the nucleus. In particular, we show that ΔNp73 inhibited the mitochondrial apoptotic transition. In light of recent reports that suggest that p53 can act directly at the mitochondrion to cause cytochrome c release as well as associate with proteins that regulate mitochondrial integrity (Schuler et al., 2000; Mihara et al., 2003; Chipuk et al., 2004), one possible role for ΔNp73 is to directly inhibit the actions of p53 at the mitochondrion. However, because ΔNp73 also inhibited the increase in BimEL, an increase that is involved in cytochrome c release after NGF withdrawal, and because p53 transcriptionally upregulates other proapoptotic Bcl-2 family members such as Puma and Noxa (Oda et al., 2000; Nakano and Vousden, 2001; Yu et al., 2001), these mitochondrial effects might also be the indirect consequence of transcriptional inhibition.
Our data showing only a partial rescue of the p73-/- phenotype with the deletion of one or two p53 alleles argues that ΔNp73 has prosurvival targets other than p53. What are these other targets? As discussed above, one additional target is JNK. We show here that ΔNp73 inhibits JNK activation, directly associates with JNK1, and can be coimmunoprecipitated with JNK directly from neuronal nuclei. Such a direct inhibition of JNK activation has broad implications, because this kinase is a key mediator of cellular apoptosis in response to many cues, including osmotic shock (Galcheva-Gargova et al., 1994), trophic factor withdrawal (Xia et al., 1995; Eilers et al., 1998), UV irradiation (Derijard et al., 1994; Tournier et al., 2000), and excitotoxicity (Yang et al., 1997). Such an interaction is not without precedent, because one study suggested that p53 binds directly to JNK (Fuchs et al., 1998). Thus, multiple members of the p53 family may be able to bind to and regulate JNK, making this an unexpected integration point for cell survival.
In addition to JNK, we propose that the third family member, p63 (Yang et al., 1998, 1999), is another p53-independent target of ΔNp73. This speculation derives from the finding that, in vivo, p53-/- neurons show a deficit in, but not an absence of, apoptosis and that cultured p53-/- sympathetic neurons apparently lose much of their resistance to apoptosis during late embryogenesis (Vogel and Parada, 1998) so that by birth, they display only a modest decrease in the rate of apoptosis after NGF withdrawal (Besirli et al., 2003; our data). In addition, as confirmed in this study, our in vivo data show that p53-/- animals do not have a further deficit in apoptosis compared with p53+/- animals (Aloyz et al., 1998), suggesting the existence of a compensatory mechanism in the p53 knock-out background. One potential explanation for all of these findings is that the family member TAp63 collaborates with p53 to promote developmental sympathetic neuron apoptosis and might actually compensate for the loss of p53 in a p53-/- background. In this regard, our recent studies (W. B. Jacobs, A. Mills, F.D.M., and D.R.K., unpublished data) support this hypothesis and show that cultured p63-/- sympathetic neurons display a deficit in apoptosis after NGF withdrawal that is more dramatic than cultured p53-/- sympathetic neurons. On the basis of these findings, we therefore propose a model where it is ultimately the balance of full-length, proapoptotic p53/TAp73/TAp63 family members relative to the prosurvival N-terminal truncated ΔNp73/ΔNp63 isoforms that determines the life versus death of any given neuron. This type of rheostat model is reminiscent of models proposed for more downstream apoptotic checkpoints such as that involving the Bcl-2 family (for review, see Chao and Korsmeyer, 1998) and implies that even relatively modest alterations in levels of these proteins could have profound implications for neuronal survival.
A somewhat unexpected finding reported here is the decreased neuronal size in the p73-/- animals, suggesting that p73 may have effects on cell growth as well as survival in the sympathetic nervous system. Because the p73-/- animals we studied lack all p73 isoforms in all tissues, we cannot conclude that this is a cell-autonomous effect. However, reduced cell size has been reported in organisms lacking other prosurvival proteins, such as Akt in Drosophila and mouse models (Verdu et al., 1999; Tuttle et al., 2001). Furthermore, our finding that p53 deletion can rescue the survival of p73-/- neurons, but not the cell size, suggests that the cell size phenotype is not attributable to an altered microenvironment. Other tumor suppressor genes, notably phosphatase and tensin homolog deleted on chromosome 10 (PTEN), are important in controlling cell size in neurons (Backman et al., 2001; Kwon et al., 2001), and p53 itself can transcriptionally regulate PTEN expression (Stambolic et al., 2001). It will be interesting to determine whether the other p53 family members play any role in regulating PTEN.
Together, these data indicate that p73 comprises one of the earliest checkpoints in the neuronal apoptotic cascade, acting as far upstream as JNK activation. It will therefore be important to elucidate the intracellular mechanisms that regulate the levels and stability of ΔNp73. In this regard, although we have previously shown that withdrawal of NGF leads to a rapid decrease in the levels of ΔNp73 (Pozniak et al., 2000), the signaling pathways responsible for the NGF-induced upregulation and/or stabilization of ΔNp73 have not yet been defined. A likely candidate for this regulation is the PI3-kinase-Akt pathway, which comprises the major NGF-mediated survival pathway in these neurons (Kaplan and Miller, 2000). This pathway has been previously shown to directly regulate the level and activation state of the Bcl-2 family (Datta et al., 1997) and to regulate levels of the IAPs (inhibitor of apoptosis) (Wiese et al., 1999; Jordan et al., 2001), thereby providing multiple checkpoints at different levels to ensure that neurons are only eliminated when they have failed to compete successfully for this trophic factor during the period of naturally occurring neuronal death.
Footnotes
This work was supported by research grants from the Canadian Institutes of Health Research (CIHR) and the Canadian Stroke Network. F.D.M. is a CIHR Senior Investigator, D.R.K. and F.D.M. are Canada Research Chairs, A.F.L. was supported by a CIHR M.D./Ph.D. studentship, D.K.H. was supported by a Natural Sciences and Engineering Research Council of Canada studentship, and G.S.W. was supported by a Hospital for Sick Children Restracomp Award.
Correspondence should be addressed to Dr. Freda Miller, Senior Scientist and Professor, Developmental Biology, Hospital for Sick Children, Room 3203 Black Wing, 555 University Avenue, Toronto, Ontario, M5G 1X8 Canada. E-mail: fredam@sickkids.ca.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249174-11$15.00/0
References
- Aloyz RS, Bamji SX, Pozniak CD, Toma JG, Atwal J, Kaplan DR, Miller FD (1998) p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J Cell Biol 143: 1691-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR (1995) High-frequency developmental abnormalities in p53-deficient mice. Curr Biol 5: 931-936. [DOI] [PubMed] [Google Scholar]
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW (2001) Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29: 396-403. [DOI] [PubMed] [Google Scholar]
- Bamji SX, Majdan M, Pozniak CD, Belliveau DJ, Aloyz R, Kohn J, Causing CG, Miller FD (1998) The p75 neurotrophin receptor mediates neuronal apoptosis and is essential for naturally occurring sympathetic neuron death. J Cell Biol 140: 911-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besirli CG, Deckwerth TL, Crowder RJ, Freeman RS, Johnson Jr EM (2003) Cytosine arabinoside rapidly activates Bax-dependent apoptosis and a delayed Bax-independent death pathway in sympathetic neurons. Cell Death Differ 10: 1045-1058. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ (1998) BCL-2 family: regulators of cell death. Annu Rev Immunol 16: 395-419. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010-1014. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Chung K, Greenwood D, Hulsebosch CE (1984) An empirical method for converting nucleolar counts to neuronal numbers. J Neurosci Methods 12: 125-132. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231-241. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson Jr EM (1993) Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol 123: 1207-1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ (1994) JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 76: 1025-1037. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson Jr EM (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol 150: 131-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers A, Whitfield J, Babij C, Rubin LL, Ham J (1998) Role of the Jun kinase pathway in the regulation of c-Jun expression and apoptosis in sympathetic neurons. J Neurosci 18: 1713-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817-825. [DOI] [PubMed] [Google Scholar]
- Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson Jr EM (1994) Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol 127: 1717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin A, Cregan SP, MacLaurin JG, Kushwaha N, Hickman ES, Thompson CS, Hakim A, Albert PR, Cecconi F, Helin K, Park DS, Slack RS (2001) APAF1 is a key transcriptional target for p53 in the regulation of neuronal cell death. J Cell Biol 155: 207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson Jr EM (1994) Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron 12: 343-355. [DOI] [PubMed] [Google Scholar]
- Fuchs SY, Adler V, Pincus MR, Ronai Z (1998) MEKK1/JNK signaling stabilizes and activates p53. Proc Natl Acad Sci USA 95: 10541-10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galcheva-Gargova Z, Derijard B, Wu IH, Davis RJ (1994) An osmosensing signal transduction pathway in mammalian cells. Science 265: 806-808. [DOI] [PubMed] [Google Scholar]
- Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL (1995) A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron 14: 927-939. [DOI] [PubMed] [Google Scholar]
- Jordan BW, Dinev D, LeMellay V, Troppmair J, Gotz R, Wixler L, Sendtner M, Ludwig S, Rapp UR (2001) Neurotrophin receptor-interacting mage homologue is an inducible inhibitor of apoptosis protein-interacting protein that augments cell death. J Biol Chem 276: 39985-39989. [DOI] [PubMed] [Google Scholar]
- Jost CA, Marin MC, Kaelin Jr WG (1997) p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 389: 191-194. [DOI] [PubMed] [Google Scholar]
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D (1997) Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 90: 809-819. [DOI] [PubMed] [Google Scholar]
- Kao CC, Yew PR, Berk AJ (1990) Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179: 806-814. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- Kartasheva NN, Contente A, Lenz-Stoppler C, Roth J, Dobbelstein M (2002) p53 induces the expression of its antagonist p73 Delta N, establishing an autoregulatory feedback loop. Oncogene 21: 4715-4727. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29: 404-411. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479-489. [DOI] [PubMed] [Google Scholar]
- Ma Y, Campenot RB, Miller FD (1992) Concentration-dependent regulation of neuronal gene expression by nerve growth factor. J Cell Biol 117: 135-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Walsh GS, Aloyz R, Miller FD (2001) TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J Cell Biol 155: 1275-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM (2003) p53 has a direct apoptogenic role at the mitochondria. Mol Cell 11: 577-590. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR (2001) On Trk for retrograde signaling. Neuron 32: 767-770. [DOI] [PubMed] [Google Scholar]
- Miller FD, Pozniak CD, Walsh GS (2000) Neuronal life and death: an essential role for the p53 family. Cell Death Differ 7: 880-888. [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683-694. [DOI] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL (1998) Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol 142: 1583-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053-1058. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453-501. [DOI] [PubMed] [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, Carter BD (2002) c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol 158: 453-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DS, Farinelli SE, Greene LA (1996) Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem 271: 8161-8169. [DOI] [PubMed] [Google Scholar]
- Park DS, Levine B, Ferrari G, Greene LA (1997) Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci 17: 8975-8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD (2000) An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 289: 304-306. [DOI] [PubMed] [Google Scholar]
- Pozniak CD, Barnabe-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD (2002) p73 is required for survival and maintenance of CNS neurons. J Neurosci 22: 9800-9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson Jr EM (1999) BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci 19: 7476-7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615-628. [DOI] [PubMed] [Google Scholar]
- Robles AI, Bemmels NA, Foraker AB, Harris CC (2001) APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res 61: 6660-6664. [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T (1995) A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 10: 175-180. [DOI] [PubMed] [Google Scholar]
- Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P, Green DR (2000) p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. J Biol Chem 275: 7337-7342. [DOI] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD (1996) Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol 135: 1085-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M (1994) Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368: 246-249. [DOI] [PubMed] [Google Scholar]
- Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, Mak TW (2001) Regulation of PTEN transcription by p53. Mol Cell 8: 317-325. [DOI] [PubMed] [Google Scholar]
- Stiewe T, Theseling CC, Putzer BM (2002) Transactivation-deficient Delta TA-p73 inhibits p53 by direct competition for DNA binding: implications for tumorigenesis. J Biol Chem 277: 14177-14185. [DOI] [PubMed] [Google Scholar]
- Toma JG, El-Bizri H, Barnabe-Heider F, Aloyz R, Miller FD (2000) Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci 20: 7648-7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870-874. [DOI] [PubMed] [Google Scholar]
- Tuttle RL, Gill NS, Pugh W, Lee JP, Koeberlein B, Furth EE, Polonsky KS, Naji A, Birnbaum MJ (2001) Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 7: 1133-1137. [DOI] [PubMed] [Google Scholar]
- Vaillant AR, Mazzoni I, Tudan C, Boudreau M, Kaplan DR, Miller FD (1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase-Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146: 955-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu J, Buratovich MA, Wilder EL, Birnbaum MJ (1999) Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol 1: 500-506. [DOI] [PubMed] [Google Scholar]
- Vogel KS, Parada LF (1998) Sympathetic neuron survival and proliferation are prolonged by loss of p53 and neurofibromin. Mol Cell Neurosci 11: 19-28. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J (2001) Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 29: 629-643. [DOI] [PubMed] [Google Scholar]
- Wiese S, Digby MR, Gunnersen JM, Gotz R, Pei G, Holtmann B, Lowenthal J, Sendtner M (1999) The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat Neurosci 2: 978-983. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326-1331. [DOI] [PubMed] [Google Scholar]
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, Andrews NC, Caput D, McKeon F (1998) p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell 2: 305-316. [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, Tabin C, Sharpe A, Caput D, Crum C, McKeon F (1999) p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398: 714-718. [DOI] [PubMed] [Google Scholar]
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, McKeon F, Caput D (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404: 99-103. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389: 865-870. [DOI] [PubMed] [Google Scholar]
- Yew PR, Berk AJ (1992) Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357: 82-85. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell 7: 673-682. [DOI] [PubMed] [Google Scholar]