

Schwann cells (SCs) cover most of the surface of all axons in peripheral nerves. Axons and these glial cells are not only in intimate physical contact but also in constant and dynamic communication, each one influencing and regulating the development, function, and maintenance of the other. In recent years, there has been significant progress in the understanding of the molecular mechanisms of axon-Schwann cell interactions, particularly those relevant for postnatal development and maintenance of nerve function and structure. In this review, we discuss recent progress in four aspects of axon-Schwann cell interactions, including the roles of the neuregulin1 (NRG1)-erbB signaling pathway, the mechanisms underlying the formation and function of the node of Ranvier, the role of perisynaptic Schwann cells at the neuromuscular junction, and the mechanisms that generate Schwann cell tumors.
Along the entire length of mammalian peripheral nerves, axons of motor, sensory, and autonomic neurons are in close association with SCs. The intimate contact between SCs and peripheral axons provided a first indication that these cells interact in important ways. In the mature nervous system, Schwann cells can be divided into four classes: myelinating cells (MSCs), nonmyelinating cells (NMSCs), perisynaptic Schwann cells (PSCs) (also known as terminal Schwann cells), and satellite cells of peripheral ganglia. These classes are based on their morphology, biochemical makeup, and the neuronal types (or area of their axons) with which they associate. MSCs, the best characterized SC, wrap around all large-diameter axons, including all motor neurons and some sensory neurons. Each MSC associates with a single axon and creates the myelin sheath necessary for saltatory nerve conduction (Fig. 1A). NMSCs associate with small-diameter axons of C-fibers emanating from many sensory and all postganglionic sympathetic neurons. Each NMSC wraps around several sensory axons to form a Remak bundle, keeping individual axons separated by thin extensions of the Schwann cell body (Fig. 1B). Finally, located more peripherally, PSCs reside at the neuromuscular junctions (NMJ), where they cover, without completely wrapping around, the presynaptic terminal of motor axons (Fig. 1C). Satellite cells, which will not be discussed here, associate with neuronal cell bodies in peripheral ganglia.
Figure 1.

Myelinated, unmyelinated, and perisynaptic Schwann cells as seen with the electron microscope. A, Cross section of a myelinated axon of an adult mouse sciatic nerve. The myelin sheath (MS) surrounding the axon (Ax) and the Schwann cell nucleus (S) are clearly visible. B, Cross section of a bundle of unmyelinated axons of an adult mouse sciatic nerve. The Schwann cell forms the Remak bundle, a bouquet-like bundle of thin axons, each separated from its neighbor by thin cytoplasmic extensions of the Schwann cell. C, Cross section of a frog neuromuscular junction reveals three juxtaposed cellular elements: the perisynaptic Schwann cell, nerve terminal (N), and muscle fiber (M). The perisynaptic Schwann cell body (S indicates nucleus) and its processes cap the nerve terminal, but the processes do not wrap around the nerve terminal region facing acetylcholine receptors on muscle.
Schwann cells, their origins, and their adult characteristics
The diverse types of SCs found in the adult nerve are primarily derived from a single precursor cell type, the neural crest cell. Some SCs may derive from placodes and ventral neural tube. The multipotent and actively migratory neural crest cells migrate into the peripheral nerves during embryonic development, in which they mature in a stepwise process, giving rise to all SCs (Fig. 2). By embryonic day 12 (E12) to E13 in mice, Schwann cell precursors begin to express three differentiation markers: P0 (myelin protein 0), GAP43 (growth-associated protein 43), and F-spondin, (Jessen and Mirsky, 1999). From E15 to the time of birth, these precursors give rise to immature Schwann cells, which express S100β and low levels of the myelin protein P0 (Jessen et al., 1994). Finally, after birth, the immature SCs differentiate into myelinating, nonmyelinating, and perisynaptic phenotypes, a process that continues over several weeks. Axons provide signals that regulate the choice between the different SC phenotypes, but molecular identity of these signals remains unknown. Undoubtedly, signals from the different axonal types and regions are important.
Figure 2.
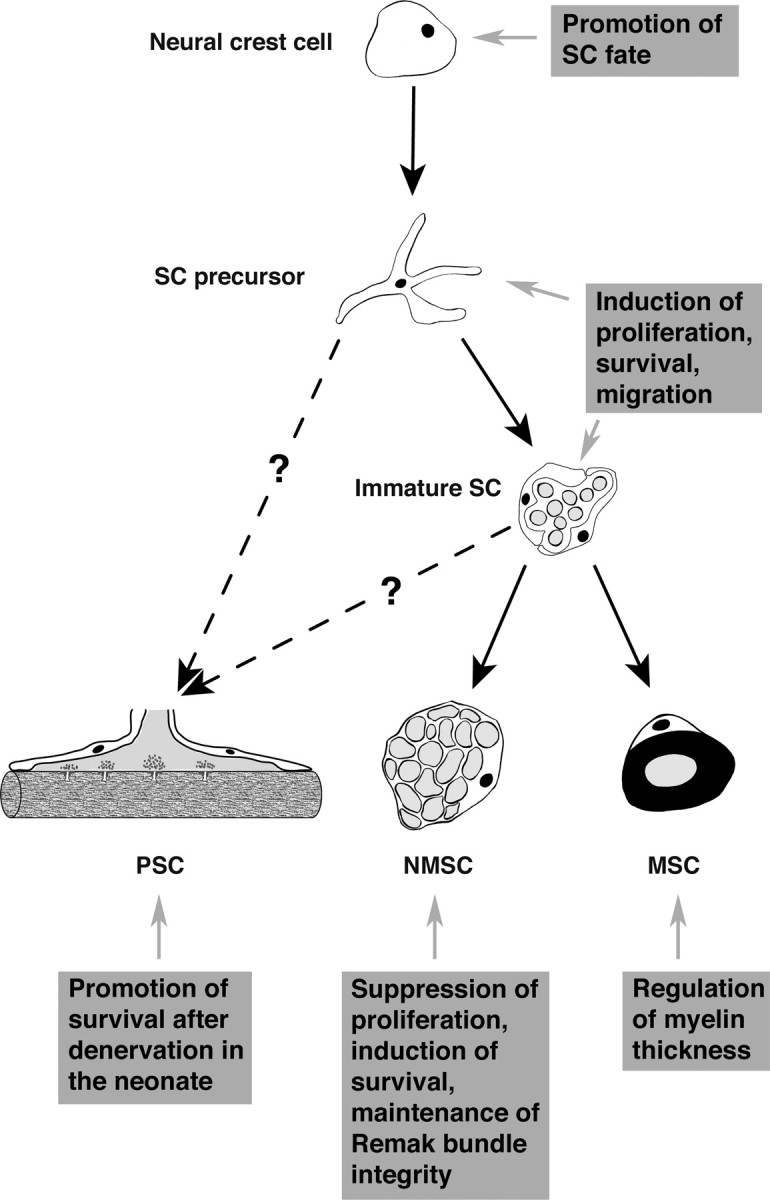
NRG1-erbB signaling and Schwann cell development. During development, neural crest cells give rise to Schwann cell precursors, which then develop into the three adult phenotypes: PSCs, NMSCs, or MSCs. Whereas, during their differentiation into MSCs and NMSCs, the precursors proceed through a stage called immature Schwann cell, the direct precursor of PSCs remains unknown. NRG1-erbB signaling regulates important aspects of Schwann cell biology at each step of their development (see boxed text).
The three types of SCs not only differ in the type of axons with which they associate, their location along axons, and their morphology, but they also differ dramatically in their biochemical composition. MSCs, the best characterized biochemically, express myelin proteins such as MBP (myelin basic protein), PMP22 (peripheral myelin protein 22), P0, MAG (myelin-associated glycoprotein), and MAL (myelin and lymphocyte protein). These proteins are critical for the formation and function of myelin sheaths. The NMSCs and PSCs have been characterized to a much lesser extent. NMSCs can be distinguished from their neighboring MSCs by their high levels of glial fibrillary acidic protein (GFAP) (Jessen et al., 1990), the low-affinity neurotrophin receptor p75 (Jessen et al., 1990) and the cell adhesion molecule L1 (Faissner et al., 1984). No specific markers exist for PSCs, but, within the NMJ, these cells can be easily visualized and studied using S100β as a marker (Woolf et al., 1992) (see Fig. 4).
Figure 4.
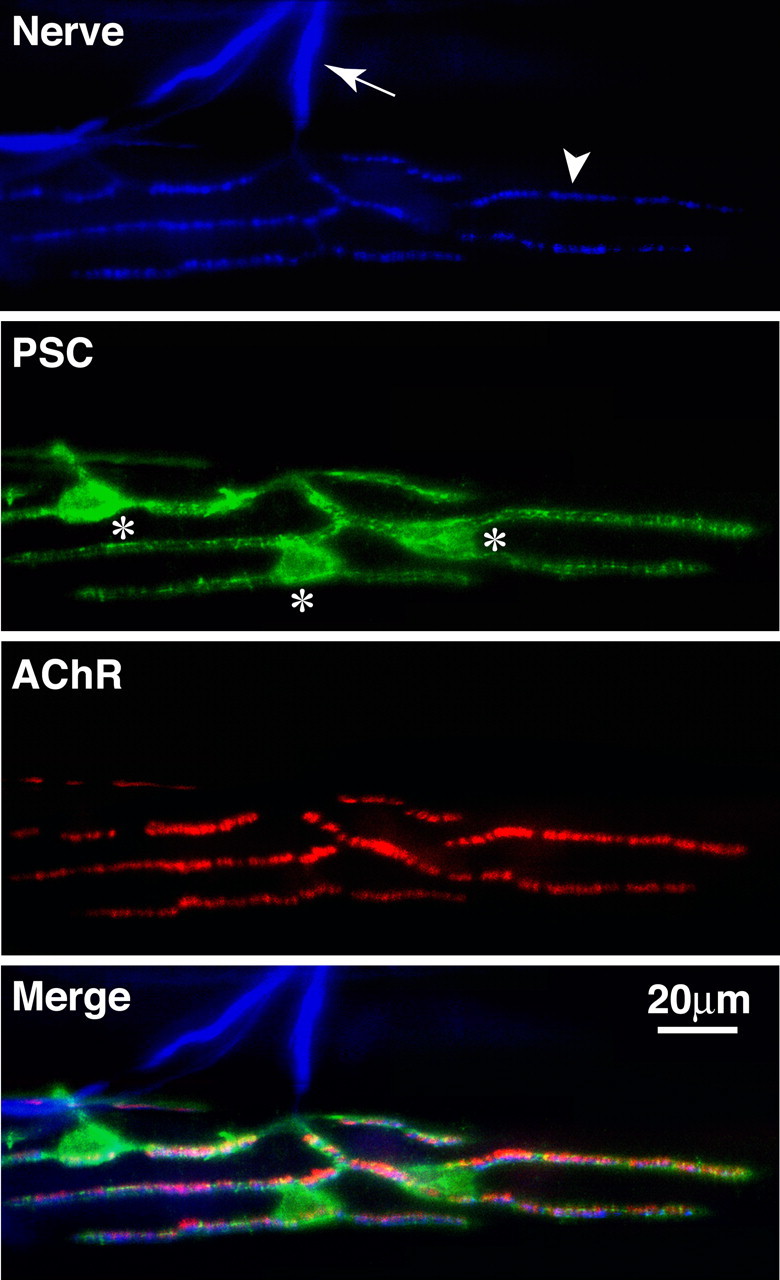
Perisynaptic Schwann cells at the neuromuscular junction. Frog skeletal muscle triple labeled with anti-neurofilament for axons (first panel, arrow) and synapsin I antibodies for nerve terminals (first panel, arrowhead), a monoclonal antibody, 2A12, for perisynaptic Schwann cells (second panel, the cell bodies are marked with *), and α-bungarotoxin for postsynaptic acetylcholine receptors (third panel). The merged image is shown in the fourth panel.
Myelinating, nonmyelinating, and perisynaptic SCs also differ in their function. As with their other aspects, MSCs are the best understood. These cells form the myelin sheath critical for rapid saltatory nerve conduction. They also regulate the formation and contribute to the structure of the nodes of Ranvier (see below). MSCs influence several aspects of axonal structure, including axonal diameter, axonal neurofilament spacing, and phosphorylation (Hsieh et al., 1994). In contrast, the functional roles of NMSCs have been very poorly investigated, but recent studies indicate that they play critical roles in the maintenance and function of unmyelinated axons and nociception in the adult (see below). PSCs in amphibians and mammals appear to play important roles in the maturation and function of the NMJ, as well as in its regeneration after injury (see below).
NRG1-erbB signaling in axon-Schwann cell interactions
Numerous molecules mediate specific aspects of the interactions between peripheral axons and SCs, including MAG (Yin et al., 1998), p75 (Cosgaya et al., 2002), IGF1 (Syroid et al., 1999), integrins (Feltri et al., 2002), and TGF-β (Guenard et al., 1995). The growth factor NRG1 and its receptors, the erbB receptors tyrosine kinases, have emerged as key regulators of axon-SC interactions at every stage and for all SC types. NRG1 is also known as glial growth factor, a protein purified as a mitogen for Schwann cells in culture (Marchionni et al., 1993). Early studies showed that spinal cord motoneurons, DRG sensory neurons, and autonomic neurons express NRG1 (Corfas et al., 1995) and that SCs express erbB2 and erbB3 receptors (Meyer and Birchmeier, 1995). Thus, it seemed reasonable that this ligand-receptor system would mediate interactions between these neurons and SCs. Indeed, many in vitro studies provided compelling evidence that NRG1 regulates many aspects of SC development. NRG1 can induce the differentiation of neural crest cells into the SC phenotype (Shah et al., 1994), the maturation of SC precursors isolated from embryonic nerves (Dong et al., 1995), and the survival of SCs isolated from neonate nerves (Syroid et al., 1996). NRG1 also promotes SC motility and migration (Mahanthappa et al., 1996) and induces SC proliferation in culture (Marchionni et al., 1993). NRG1 appears to also regulate physiological properties of SCs because it induces expression of Na+ channels (Wilson and Chiu, 1993) and increases gap junction communication between SCs (Chandross et al., 1996).
The importance of NRG1-erbB signaling in SC development has also been demonstrated in vivo with genetically modified mice. The first series of mice lacking NRG1, erbB2, or erbB3 established the essential role of these molecules in Schwann cell precursor development (Lee et al., 1995; Meyer and Birchmeier, 1995; Erickson et al., 1997; Riethmacher et al., 1997). These mice have cranial ganglia defects, as well as reduction of Schwann cells, enteric ganglia, and adrenal chromaffin cells, suggesting that the primary site of action for NRG1-erbB signaling is neural crest cells (Erickson et al., 1997; Riethmacher et al., 1997; Lin et al., 2000). Unfortunately, these mutant mice die as embryos because of heart defects; thus, these initial studies were limited to early stages of development.
Subsequently, NRG1-erbB signaling in axon-SC interactions were examined using more elaborated animal models, including knock-outs of specific NRG1 isoforms (Kramer et al., 1996; Meyer et al., 1997; Wolpowitz et al., 2000) and rescue of early lethality by expression of erbB receptors in the heart (Morris et al., 1999; Woldeyesus et al., 1999). Altogether, these studies showed that (1) different NRG1 isoforms have similar yet somewhat different roles in SC development, (2) defects in SC development attributable to lack of NRG1-erbB signaling in embryogenesis result in the degeneration of both sensory and motor neurons in mice and (3) defects in NRG1-erbB signaling during embryonic development lead to NMJ defects. These studies highlighted the importance of SCs in the development and maintenance of peripheral nerves.
More recently, several studies have explored NRG1-erbB signaling in axon-SC interactions with conditional knock-outs (Garratt et al., 2000), mice heterozygous for NRG, erbB2, and erbB3 (Michailov et al., 2004), and cell-specific blockade or erbB receptors in transgenic mice (Chen et al., 2003). These studies showed that the roles of erbB signaling in SCs differ between postnatal and embryonic cells, as well as between myelinating and nonmyelinating Schwann cells.
To interfere with NRG1-erbB signaling between unmyelinated axons and NMSCs, Chen et al. (2003) generated transgenic mice expressing a dominant-negative erbB receptor in NMSCs. These mice had normal peripheral nerves until the third postnatal week but then developed a small fiber neuropathy with loss of thermal nociception, consistent with dysfunction of unmyelinated C-fiber axons. The loss of erbB signaling in NMSCs resulted in the loss of Remak bundles and extensive proliferation of NMSCs. These results were surprising, because they indicated that, in adult NMSCs, erbB signaling prevents rather than induces proliferation, opposite to its role during development. The rate of SC apoptosis was also increased in these mice, demonstrating that erbB signaling is a pro-survival stimulus for adult NMSCs. The mutant mice had significant loss of unmyelinated axons by postnatal day 30, indicating that NMSC are involved in maintenance of unmyelinated axon integrity throughout life. Interestingly, even at stages in which mice were unable to respond to thermal stimuli, they still retained 50% of their C-fiber axons, suggesting that NMSCs may play a role in electrical conduction of unmyelinated axons as MSCs do for myelinated axons. At later stages, the number of unmyelinated DRG neurons was dramatically reduced, indicating that NMSCs are involved in the promotion of long-term sensory neurons survival. This DRG loss occurred in conjunction with a dramatic reduction in the levels of expression of glial cell line-derived neurotrophic factor, a trophic factor expressed by SCs and known to be important for the survival of a population of C-fiber neurons. These results show that, as part of a set of reciprocal axon-SC interactions, adult NMSCs are important in the maintenance of C-fiber sensory neurons as a source of essential trophic support.
The roles of NRG1-erbB signaling in MSCs were first studied by Garratt et al. (2000). They generated mice in which the erbB2 gene was deleted by expression of Cre recombinase under the control of the promoter of Krox20, a transcription factor expressed by MSCs. These mice showed hypomyelination of peripheral nerves, revealing that erbB signaling is necessary for normal myelination. Moreover, at early postnatal stages, there was a significant reduction in the number of developing SCs in the ventral and dorsal roots, suggesting that erbB signaling also regulates the SC precursor pool. Interestingly, adult nerves contained normal numbers of SCs, leading the authors to suggest that other mechanisms compensated for the lack of erbB signaling. This line of inquiry was taken a step further by Michailov et al. (2004). Their study showed that mice with only one copy of the NRG1 gene have thinner myelin, whereas mice overexpressing one of the NRG1 isoforms (type III NRG1) in neurons under the control of the Thy1.2 promoter have thicker myelin. Interestingly, type I NRG1 did not have the same effect. Because NRG1 heterozygotes showed the same phenotype as observed in nrg1: erbb2:erbb3 triple heterozygotes, the authors concluded that axonal NRG is the rate-limiting factor for myelination. This study also found normal internodal length in the mutant mice, suggesting that NRG1-erbB signaling is not involved in the regulation of SC length. This surprising result suggested that the regulation of myelin thickness and internode length might be regulated by different mechanisms. However, this may have resulted from an incomplete blockade of erbB signaling in these mice.
NRG1-erbB signaling appears also to regulate important aspects of PSC biology. Trachtenberg and Thompson (1996) found that, in the neonate, PSCs die after muscle denervation and that this apoptosis could be prevented by application of NRG1. Furthermore, exogenous NRG1 applied to normal neonatal muscles resulted in retraction of nerve terminals and alterations in PSC morphology (Trachtenberg and Thompson, 1997). Because both SCs and muscle have erbB receptors, it is not clear which cell is the primary target of NRG1, but the investigators speculated that NRG1 acted on PSCs.
These studies provide irrefutable support for the essential roles of NRG1-erbB signaling in axon-SC interactions during development and in the adult. Nevertheless, many important questions remain to be answered regarding the mechanisms by which NRG1-erbB signaling regulates SC biology, including how signaling through the same erbB receptors can result in different biological outcomes in SCs depending on the NRG1 isoform used or the state of differentiation of the glia.
Axoglial interactions at the node of Ranvier
As discussed above, myelin is essential for efficient and rapid propagation of action potentials. However, this function also depends on the molecular specialization of the nodes of Ranvier, the short periodical interruptions in the myelin sheath that are regularly spaced at intervals of ∼100 times the axonal diameter. The nodal region is organized into several distinct domains, nodes, paranodes, and juxtaparanodal region (Fig. 3). Each domain contains a unique set of ion channels, cell adhesion molecules, and cytoplasmic adaptor proteins (Poliak and Peles, 2003; Salzer, 2003). Disruption in nodal organization results in pathophysiological changes often seen in demyelinating diseases. The local differentiation of myelinated axons is tightly regulated by MSCs through contact-dependent mechanisms.
Figure 3.

Molecular structure of the node of Ranvier. Longitudinal section through the nodal region of a peripheral myelinated axon showing the organization and composition of axonal and glial domains. The axon is covered by an MSC, which in turn is surrounded by a basal lamina. In the paranodal region, the myelin sheath forms a series of paranodal loops (PNL) that invaginate and appose the axon creating a septate-like structure. At the node, the outermost cytoplasmic extension of MSC contains numerous microvilli that contact the axolemma. Specific sets of proteins are enriched in each domain of both axon (red text) and Schwann cells (blue text).
The nodal axolemma is characterized by high density (>1200/mm2) of Na+ channels essential for the generation of the action potential during saltatory conduction. Na+ channels colocalize with KCNQ2 K+ channels (Devaux et al., 2004) and several other transmembrane and cytoskeletal proteins. These include the cell adhesion molecules of the Ig superfamily (Ig-CAMs) NrCAM and neurofascin 186 (NF186) (Davis et al., 1996), the cytoskeletal adaptor ankyrin G (Kordeli et al., 1995), and the actin-binding protein spectrin βIV (Berghs et al., 2000). Axoglial contact at the nodes can be mediated by both the nodal CAMs and the β subunit of Na+ channel (Isom, 2002). Ankyrin G, a membrane-cytoskeleton adaptor that links integral membrane proteins to the spectrin cytoskeleton, interacts with Na+ channels (Srinivasan et al., 1988), KCNQ2 (Devaux et al., 2004), NF186 and NrCAM (Garver et al., 1997), as well as to βIV spectrin, a spectrin isoform that is enriched at the nodes of Ranvier and axon initial segments (Berghs et al., 2000). The interaction between ankyrin G and βIV spectrin may provide further anchorage of the nodal Na+ channel and Ig-CAMs to the axonal cytoskeleton. The nodal axolemma is contacted by microvilli emanating from the outer aspect of MSCs. Three ERM proteins ezrin, radixin, and moesin, as well as ezrin-binding protein EBP50 and Rho-A GTPase, are localized at the microvilli (Melendez-Vasquez et al., 2001; Scherer et al., 2001). Several extracellular matrix proteins are present in the nodal gap under the basal lamina, including the hyaluronan-binding proteoglycan versican (Apostolski et al., 1994), tenascin C (Rieger et al., 1986; Martini et al., 1990), and syndecans (Goutebroze et al., 2003). Dystroglycan is also located at the nodes (Saito et al., 2003). SC-specific ablation of dystroglycan results in disorganization of the microvilli, marked reduction in nodal Na+ channels, and consequently impaired nerve conduction (Saito et al., 2003).
The paranodal junctions (PNJ) that are formed between the axon and the myelinating cell flank the nodes of Ranvier. In this region, the compact myelin lamellae open up into a series of cytoplasmic loops that spiral around the axon, forming a series of septate-like junctions with the axolemma. These junctions appear relatively late during myelination, first generated closer to the nodes by the most outer paranodal loop and then forming gradually as additional loops are attached to the axon. As a result, they are composed of a number of rings that represent each turn of the myelin wrap. The PNJ was proposed to provide attachment of the myelin sheath to the axon, to separate the electrical activity at the node of Ranvier from the internodal region under the compact myelin sheath, and to serve as a boundary that limits the lateral diffusion of membrane components. The axonal membrane at the paranodes contains a complex of cell adhesion molecules that include Caspr (Einheber et al., 1997; Peles et al., 1997) [contactin-associated protein, also known as paranodin (Menegoz et al., 1997)] and the glycosylphosphatidylinositol (GPI)-linked cell adhesion molecule contactin (Rios et al., 2000). This complex is connected to the axonal cytoskeleton through protein 4.1B (Gollan et al., 2002). The glial membrane at the PNJ contains neurofascin 155 (NF155), a spliced isoform of the cell adhesion molecule neurofascin that is specifically found at the glial loops (Tait et al., 2000). Although NF155 binds directly to contactin, it is presently unclear whether the Caspr-contactin complex interacts directly with NF155 (Charles et al., 2002; Gollan et al., 2003). Both Caspr and contactin are essential for the formation of the paranodal junction, and, in their absence, the ultrastructure of the paranodes is severely altered. Specifically, the gap between glial and axonal membranes is increased, and the electron-dense material forming the septa that are the hallmark of the PNJ in wild-type mice is absent (Bhat et al., 2001; Boyle et al., 2001; Gollan et al., 2003). Similar to other so-called paranodal mutants (Poliak and Peles, 2003), such as the galactolipids (Dupree et al., 1999; Poliak et al., 2001) and sulfatide-deficient mice (Ishibashi et al., 2002), the absence of a normal paranodal junction results in only a minor expansion of the nodes but causes a shift in the accumulation of K+ channels from the juxtaparanodal region to the paranodes (Bhat et al., 2001; Boyle et al., 2001; Gollan et al., 2003). Thus, an important function of these junctions is to form a barrier between the nodal Na+ channels and the juxtaparanodal K+ channels.
The juxtaparanodal region is located beneath the compact myelin at both sides of each internodal interval. It is characterized by the presence of delayed rectifier K+ channels of the Shaker family, Kv1.1, Kv1.2, and their Kvβ2 subunit, that may stabilize conduction and help to maintain the internodal resting potential (Wang et al., 1993; Vabnick et al., 1999). At this site, the channels colocalize and create a complex with Caspr2, the second member of a family of putative cell recognition molecules (Poliak et al., 1999). Two other proteins that are found at the juxtaparanodal region are TAG-1 (transient axonal glycoprotein 1), a GPI-anchored cell adhesion molecule related to contactin (Traka et al., 2002), and connexin 29, which is found at the glial membrane (Altevogt et al., 2002). Axonal Caspr2 creates a cis-complex with TAG-1 that associates with a glial-derived TAG-1. In the absence of Caspr2 or TAG-1, K+ channels do not accumulate at the juxtaparanodal region, and, instead, they are distributed along the internodes, suggesting that Caspr2 and TAG-1 form a scaffold that maintains K+ channels at their correct juxtaparanodal position (Poliak et al., 2003; Traka et al., 2003).
During the development of myelinated nerves, the different nodal domains are formed gradually. Na+ channels are first clustered at the nodes, followed by the generation of the paranodal junction, and only then by the clustering of K+ channels at the juxtaparanodal region (Vabnick et al., 1996, 1999). Na+ channels cluster initially at sites adjacent to the edges of processes extended by MSCs (Vabnick et al., 1996; Ching et al., 1999). Additional longitudinal growth of these glial cell processes is associated with the movement of Na+ channel clusters until ultimately two neighboring clusters appear to fuse, indicating that these channels are positioned by direct glial cell contact (Pedraza et al., 2001). In sciatic nerve, NrCAM, NF186, and ankyrin G precede Na+ channels, suggesting that these cell adhesion molecules bind ankyrin G, which in turns recruits Na+ channels (Lambert et al., 1997; Jenkins and Bennett, 2002). In support of this model, the addition of a soluble NrCAM to myelinating dorsal root ganglia cultures was shown to inhibit Na+ clustering (Lustig et al., 2001). Moreover, the appearance of ankyrin G and Na+ channels at the nodes is delayed in NrCAM null mice (Custer et al., 2003), indicating that this adhesion molecule participates in the clustering of these channels. Cell-cell contact controls the two other domains near the nodes; the formation of the paranodes requires Caspr and contactin (Bhat et al., 2001; Boyle et al., 2001; Gollan et al., 2003), whereas the formation of the juxtaparanodal region depends on the presence of Caspr2 and the contactin-related molecule TAG-1 (Poliak et al., 2003; Traka et al., 2003).
In summary, several proteins that may mediate the interactions between myelinating Schwann cells and their underlying axons have been identified, and their role in the local differentiation of the nodal region is being explored. However, despite this progress, the exact molecular mechanisms by which myelinating glial cells control the formation of the nodes and, particularly, the clustering of Na+ channels should probably await the identification of novel components in the future.
Perisynaptic Schwann cells in neuromuscular junction development and maintenance
PSCs cap the presynaptic motor nerve terminal, which in turn aligns with postsynaptic acetylcholine receptors (AChRs) at vertebrate NMJs (Fig. 1). The arrangement of these three intimately juxtaposed cellular elements was demonstrated by electron microscopy more than four decades ago (Birks et al., 1960), but it was not until the 1990s that it became clear that PSCs play active roles in synaptic function, growth, and maintenance. One key finding was the profuse sprouting of PSC processes observed after nerve injury at the mammalian NMJs (Reynolds and Woolf, 1992). The significance of this observation was soon recognized and extended by Thompson and colleagues, who demonstrated that PSC sprouts may lead nerve terminal growth during synaptic regeneration and sprouting (Son and Thompson, 1995). Using repeated in vivo imaging, Ko and colleagues have shown that PSC processes are dynamic and may also lead nerve terminal sprouts during synaptic remodeling at frog NMJs (Chen et al., 1991; Chen and Ko, 1994; Ko and Chen, 1996). PSCs have also been shown to sense synaptic activity by raising their intracellular calcium (Jahromi et al., 1992; Reist and Smith, 1992) and to be capable of modulating synaptic transmission at the NMJ in response to manipulation of G-protein pathways or internal calcium levels (Robitaille, 1998; Castonguay and Robitaille, 2001). Coincidentally, the role of glial cells in synaptic function and development in the CNS also began to unfold in the 1990s (Newman and Volterra, 2004). The importance of glial cells as active and integral components of chemical synapse has now been well acknowledged and has led to the concept of the tripartite synapse (Araque et al., 1999).
The frog NMJ has served as an excellent model to investigate the roles of PSCs. Two vital probes, peanut agglutinin (PNA), which labels the extracellular matrix associated with frog PSCs (Ko, 1987), and a monoclonal antibody (mAb), 2A12, which labels the external membrane surface of frog PSCs (Astrow et al., 1998), provide powerful tools for the study of PSC behavior at adult and developing NMJs in vivo (Fig. 4). In addition, mAb 2A12 can be used with complement-mediated lysis to selectively ablate PSCs en masse from frog muscles (Reddy et al., 2003).
Nerve-muscle contacts can be formed in the absence of Schwann cells in tissue culture (Kullberg et al., 1977), and transient nerve-muscle contacts have also been seen in mouse mutants that lack of Schwann cells (Riethmacher et al., 1997; Morris et al., 1999; Lin et al., 2000; Wolpowitz et al., 2000). Thus, it appears that the initial formation of nerve-muscle contact does not require PSCs. However, PSCs are present and dynamic soon after the initial NMJ formation. During tadpole development, PSCs partially colocalize with developing nerve terminals at the earliest discernible NMJs but quickly cover the full extent of NMJs and often extend profuse processes beyond the nerve terminals by tens or hundreds of micrometers. Repeated, in vivo observations of identified tadpole NMJs show that developing nerve terminals grow along these PSC sprouts in tadpoles (Reddy et al., 2003). When PSC were specifically ablated, very few NMJs showed nerve terminal growth and nearly one-half underwent retraction, in contrast to control muscles, which showed robust NMJ growth. Furthermore, whereas ∼10% of NMJs observed in controls were new synapses, no new synapses were seen in PSC-ablated muscles. These results demonstrate that PSCs promote synaptic growth and are vital for the stability of developing NMJs in vivo.
The mechanisms by which SCs contribute to neuromuscular synaptogenesis are not completely understood. In cultures containing embryonic Xenopus spinal neurons and muscle, neurotrophic factors and elevated cAMP induce a dramatic increase in neuron survival and neurite outgrowth (Peng et al., 2003). However, the number of nerve-muscle contacts that showed AChR clusters was greatly inhibited. Interestingly, addition of medium conditioned by SCs or TGF-β1 increased AChR clusters at neuron-muscle contacts (Peng et al., 2003; Feng and Ko, 2004). These findings suggested that Schwann cell-derived factors allow neurons to switch from a “growth phase” to a “synaptogenic phase.” In addition, SC-conditioned medium modulates transmitter release at the nerve-muscle contacts in Xenopus culture, by increasing the frequency but not the amplitude of spontaneous synaptic current. The identity of the factors that promote these effects is not known, but they appear to be small molecules of <5 kDa (Cao and Ko, 2001).
PSC ablation has provided an effective tool to selectively “knock-out” PSCs and thus investigate the roles of these cells in the maintenance and function of adult NMJs in vivo (Reddy et al., 2003). After acute PSC ablation (up to 5 hr after ablation), no abnormality of nerve terminal morphology or clusters of AChRs was reported at the light microscopic level. Despite the damage of PSCs, the ultrastructure of nerve terminals and muscles was unchanged, suggesting that acute ablation of PSCs does not alter synaptic structure. Moreover, miniature endplate potentials (mepps), evoked endplate potentials (epps), quantal contents, and paired-pulse facilitation and synaptic depression in response to high-frequency stimulation before and after acute PSC ablation were unchanged. Furthermore, acute ablation of PSCs did not affect muscle twitch tension in response to nerve stimulation. All in all, these findings suggest that PSCs, despite their capability to modulate transmitter release in response to pharmacological manipulations (Auld et al., 2003), do not seem to play a significant role in acute synaptic modulation and the overall functioning of adult muscles.
In contrast, PSCs are essential for the long-term maintenance of synaptic structure and function (Reddy et al., 2003). One week after PSC ablation, the frequency of mepps, the amplitude of epps, and the mean quantal content were reduced approximately by half, whereas mepp size was not altered. Nerve-induced muscle twitch tension was significantly reduced 1 week after PSC removal. These changes in presynaptic function were accompanied by partial or total retraction of nerve terminals, as seen with light microscopy. Electron microscopy showed “empty” synaptic gutters resulting from the retraction of nerve terminals. However, there were no signs of either nerve damage typically seen after axotomy or detachment of the remaining length of the retracting nerve terminals. Thus, it seems that, rather than acting as a “glue” to mechanically attach nerve terminals, the PSCs and their derived factors maintain synaptic structure and function. The identity of these factors remains to be investigated.
Repeated in vivo imaging of double-labeled NMJs has shown that, in contrast to mammals, frog PSC sprouting after axotomy occurs with the arrival of regenerating nerve terminals. PSCs extend processes ahead of regenerating nerve terminals, which grow along the preceding PSC processes (Koirala et al., 2000). These observations suggest that regenerating nerve terminals induce PSCs sprouting, and PSC sprouts, in turn, lead and guide nerve terminal sprouts. Similar roles for PSCs in leading growing nerve terminals during synaptic remodeling (Chen et al., 1991; Macleod et al., 2001) and during sprouting induced by nerve implants (Chen and Ko, 1994; Ko and Chen, 1996) have been reported. In addition to a guiding role, in the frog, SCs may also play a role in the aggregation of AChRs at extrajunctional sites during nerve reinnervation and sprouting by expressing active isoforms of agrin (Yang et al., 2001).
Evidence for PSCs sprouts leading regenerating nerve terminals in vivo has also been made in mammalian muscles (O'Malley et al., 1999; Kang et al., 2003). These in vivo observations have confirmed previous suggestions inferred from static images that regenerating axons indeed grow along PSC sprouts at mammalian NMJs. Together, in vivo imaging studies suggest that PSCs guide and lead the growth of regenerating and sprouting nerve terminals at adult vertebrate NMJs. Whether PSCs are absolutely required for guiding nerve terminal sprouting at adult NMJs is not clear. This question could be answered by ablating PSCs at regenerating NMJs.
In summary, studies from the past decade have revealed many active and essential roles of PSCs in synaptic function, formation, and maintenance throughout the life of vertebrate NMJs (for review, see Auld et al., 2003; Kang et al., 2003; Koirala et al., 2003). These studies suggest that PSCs, similar to CNS glial cells (for review, see Ullian et al., 2004), may instruct neurons to make larger, stronger, and more stable synapses. However, little is know about the identity of the molecules that PSCs and nerve terminals communicate with each other at the NMJ. To have a complete understanding of the tripartite neuromuscular synapse, the future challenge is to unravel the molecular mechanisms of the reciprocal interactions among PSCs, nerve terminals, and muscles.
Schwann cell tumors
As described above, communication between axons and SCs is critical for normal nerve function, and disruption of the intimate interactions of SCs with axons results in debilitating peripheral neuropathies, including those caused by peripheral nerve tumors. These tumors can be classified as neurofibromas and schwannomas. Neurofibromas are unencapsulated benign tumors that contain Schwann cells, mast cells, fibroblasts, and perineurial cells comingled with axons in an abundance of collagen-rich matrix. Schwann cells account for 60-80% of the cells in neurofibromas, as defined by staining for the S100β protein (Stefansson et al., 1982; Peltonen et al., 1988). In contrast, schwannomas are encapsulated benign tumors almost entirely composed of Schwann cells that are perched on, but not comingled with, normal nerve bundles (Fig. 5). Neurofibromas occasionally progress to malignant peripheral nerve sheath tumors (MPNST) (Evans et al., 2002), whereas schwannomas apparently do so only after radiation exposure. Schwannomas consist almost entirely of S100β-positive cells (Johnson et al., 1988). At the electron microscopic level, interdigitating SC processes are frequent, and schwannoma cells have characteristic SC basal lamina (Cravioto, 1969; Erlandson and Woodruff, 1982). Schwann cells within the tumor are surrounded by a collagen-poor, laminin-rich matrix.
Figure 5.

Schwann cell tumors. In neurofibromas, Schwann cells fibroblasts and perineurial cells are with in collagen-rich tumor matrix; axons and mast cells are common. Neurofibromas are not encapsulated, although the plexiform neurofibroma develops inside the perineurium (not shown). Schwannomas are encapsulated by a collagenous sheath and are made up almost entirely of S100β-positive Schwann cells, with little or no fibroblast involvement; axons are present only at the boundary of the tumor with the associated nerve trunk.
Clues to tumor formation come from inherited human diseases in which affected individuals are predisposed to develop SC tumors. Neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2) arise from mutations in different genes, each of which plays a key role in regulating SC function. The Nf1 gene on human chromosome 17 encodes an intracellular signaling molecule that functions as a GTPase activating protein for Ras proteins, whereas the Nf2 gene on human chromosome 22 encodes a cytoskeletal-membrane linking protein. Patients with NF1 and NF2 typically present with different types of benign SC tumors in which most SCs lose contact with axons. Neurofibromas are the hallmark of NF1 disease, whereas schwannomas are the hallmark of NF2 disease (Fig. 5). There is great interest in neurofibromatosis type 1 because it is one of the most common of all inherited human diseases, affecting 1 in 2500-3500 individuals of all races worldwide (Rasmussen and Friedman, 2000). The prevalence of NF1 is similar to all the Charcot-Marie-Tooth peripheral neuropathies together (1:2000).
NF1 is inherited as a dominant trait. It is a complex disease with an unpredictable clinical course. Diagnostic criteria, two or more of which define a diagnosis of NF1, include the following: cafe-au-lait macules, freckling, optic glioma, Lisch nodules, a bony lesion, a first degree relative with NF1, and two or more benign nerve sheath tumors (neurofibromas) (Gutmann et al., 1997). Numerous abnormalities can be observed in individual NF1 patients. At least one-half of NF1 children have learning disabilities (for review, see Kayl and Moore, 2000). Short stature, macrocephaly, benign pilocytic astrocytomas, and scoliosis are also frequent (Friedman and Birch, 1997).
The 350 kb Nf1 gene on human chromosome 17q11.2 is a tumor suppressor gene, and complete loss is correlated with tumor formation (Legius et al., 1993). NF1 disease is thought to be caused by loss of NF1 protein because defined Nf1 mutations predict deletions and rearrangements with no evidence for dominant-negative effects; mutations are found scattered throughout the gene (Fahsold et al., 2000). No relationship between genotype and phenotype has been defined except for patients with gross gene deletions who may have deletions in gene(s) contiguous to Nf1. These individuals have a severe NF1 phenotype, with large neurofibroma burden (Lopez-Correa et al., 2001).
Neurofibromas develop in most or all NF1 patients (Friedman and Birch, 1997). NF1 patients can have a few or thousands of these benign peripheral nerve tumors. Discrete neurofibromas can be associated with any peripheral nerve or with small nerve endings under the skin, typically appear during or after puberty, and are prone to growth during pregnancy (Dugoff and Sujansky, 1996). In contrast, plexiform neurofibromas expand within the perineurium and displace surrounding tissue. Plexiform neurofibromas are associated with major nerve trunks and appear in young children (for review, see Wiestler et al., 1994). Currently, the only therapy for neurofibromas is surgical removal.
Recent data strongly support the view that Schwann cells are a crucial pathogenic cell type in neurofibroma formation. Neurofibroma SCs aberrantly stimulate angiogenesis and are invasive (Sheela et al., 1990), and mutations in both Nf1 alleles are present in neurofibroma SCs but not neurofibroma fibroblasts (Kluwe et al., 1999; Serra et al., 2001). A conditional (cre/lox) allele of Nf1 was used to ablate Nf1 in the SC lineage. These mice developed peripheral nerve tumors, but only when all the cells in the mice were also Nf1+/- (Zhu et al., 2002), suggesting a role for a permissive haploinsufficient environment in conjunction with mutant SCs.
NF1 encodes a GTPase activating protein for Ras proteins called neurofibromin (for review, see Donovan et al., 2002). Thus, loss of neurofibromin through mutation at the Nf1 locus might be expected to result in a failure to terminate Ras signals. Indeed, increased levels of Ras-GTP are detected in SCs isolated from neurofibromas (Sherman et al., 2000). Ras-GTP is also elevated in SCs hemizygous or null at Nf1 using a biochemical assay (Kim et al., 1995). Some Nf1 phenotypes are likely caused by increased Ras-GTP because morphological changes and decreased cell division in Nf1 mutant SCs are mimicked in SCs expressing a constitutively activated Ha-Ras allele (Ridley et al., 1988; Kim et al., 1995). In addition, farnesyl protein transferase inhibitor, a drug that inhibits H-Ras activity, reverses proliferation abnormalities in mouse SCs lacking Nf1 (Kim et al., 1997).
Neurofibromin also has ill-defined, non-Ras-related functions. Mutation in Drosophila Nf1 results in small sluggish flies with a block in PACAP (pituitary adenylate cyclase-activating polypeptide)-stimulated enhancement of inwardly rectifying K+ channels at the larval body wall NMJ (Guo et al., 1997). This is not an effect of the classical Ras-MAPK (mitogen-activated protein kinase) pathway, but rather results from abnormalities in the cAMP-mediated opening of the channel. Enlarged peripheral nerves in NF1 mutant flies result from Ras or non-Ras pathway defects (Yager et al., 2001). Neurofibromin is also a substrate of PKA. Aberrant K+ currents have also been described in SCs from MPNST cell lines and could be induced in normal human SCs by exposure to cAMP analogs (Fieber, 1998). PAKs (p21-activated kinases) have also been implicated in SC transformation (Tang et al., 1998). R-Ras and R-Ras2/TC21 may mediate some effects of Nf1 loss in SCs (Huang et al., 2004).
It seems reasonable that Nf1 mutant SCs would reveal deregulation of molecules necessary for normal neuron-glial interaction and provide therapeutic targets for NF1 patients. To study neurofibroma formation resulting from Nf1 mutation, mice with a targeted mutation in Nf1 were developed (Brannan et al., 1994; Jacks et al., 1994). Nf1-/- mouse embryos die by E14.5, and adult Nf1+/- mice do not spontaneously develop neurofibromas. However, neurofibromas formed when Nf1-/- embryonic stem cells were added to wild-type blastocysts (Cichowski et al., 1999) or after wounding of Nf1+/- peripheral nerves (Rizvi et al., 2002). Moreover, when dorsal root ganglion cells are purified from Nf1 mutant embryos at E12.5, the Nf1+/- and Nf1-/- Schwann cells from these cultures do show some abnormalities. Notably, an additional morphologically transformed, fast-growing, p75NGFR, S100β, and GFAP-expressing subpopulation of cells, denoted Nf1-/- TXF, is also present in these cultures (Kim et al., 1997; Rizvi et al., 2002). These cells have been used as a model of neurofibroma formation.
Recently, studies using the candidate gene approach and more global gene expression profiling have provided new insights into neurofibroma formation. As an example of the candidate gene approach, Ratner and colleagues found that Nf1-/- TXF cells aberrantly express the epidermal growth factor receptor (EGFR) (DeClue et al., 2000). Supporting use of the Nf1-/- TXF cells as a model for tumorigenesis, EGFR is also aberrantly expressed in a subset of S100β-expressing cells in neurofibromas and in mouse and human MPNST cell lines, in which EGFR antagonists inhibit cell growth (DeClue et al., 2000; Li et al., 2002). This has led to an ongoing therapeutic trial of EGFR antagonists in MPNST.
Applying global gene expression profiling to the Nf1 mutant mice has revealed numerous new candidate genes. For example, cDNA microarray analysis showed that expression of Blbp (brain lipid binding protein)/Fabp7 (fatty acid binding protein) was increased in Nf1-/- TXF cells (Miller et al., 2003). The correlation between the higher expression levels of BLBP, which has been shown to mediate adhesion between neurons and glia (Feng et al., 1994), and the defects in the ability of Nf1-/- TXF Schwann cells to extend processes along axons suggested that these events could be related (Miller et al., 2003). Strikingly, BLBP blocking antibodies enabled process outgrowth from Nf1-/- TXF cells and restored interaction with axons (Miller et al., 2003). BLBP expression was also detected in EGFR-positive cell lines derived from human MPNST and Nf1:p53 double-mutant mice (Li et al., 2002). Thus, these results not only validate the utility of the Nf1-/- TXF system to identify genes relevant to tumorigenesis but implicate BLBP in the disease phenotype.
In summary, peripheral nerve tumors that form in neurofibromatosis type 1 disrupt axon-SC interactions and peripheral nerve structure. Currently available knock-out mice and SC culture models are available to study peripheral nerve tumorigenesis, and insights into the involvement of Ras signaling, proteins including BLBP, and tyrosine kinase pathways driven by EGFR in these tumors are providing targets for possible therapeutic interventions in NF1. Many important questions remain, including the role of non-SCs in neurofibromas, identification of additional proteins that are biomarkers or therapeutic targets in disease, and identification of the pathway by which loss of NF1 disrupts axon-glial interactions.
Footnotes
This work was supported by National Institutes of Health Grants NS35884 (G.C.), DC04820 (G.C.), NS017954 (C.-P.K.), NS28840 (N.R.), and DAMD-17-02-1-0679 (N.R.). Support was also provided by the National Multiple Sclerosis Society (N.R. and E.P.), the United States-Israel Binational Science Foundation (E.P.), and the Israel Science Foundation (E.P.). We thank Samir Koirala and Kristine Roy for their critical reading of this manuscript, Alex Rosenbaum for the illustrations, and Carlos Rio, Yoshie Sugiura, and Zhihua Feng for micrographs.
Correspondence should be addressed to Dr. Gabriel Corfas, Division of Neuroscience, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115. E-mail: gabriel.corfas@childrens.harvard.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249250-11$15.00/0
References
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL (2002) Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22: 6458-6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolski S, Sadiq SA, Hays A, Corbo M, Suturkova-Milosevic L, Chaliff P, Stefansson K, LeBaron RG, Ruoslahti E, Hays AP, Latov N (1994) Identification of Gal(beta 1-3)GalNAc bearing glycoproteins at the nodes of Ranvier in peripheral nerve. J Neurosci Res 38: 134-141. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22: 208-215. [DOI] [PubMed] [Google Scholar]
- Astrow SH, Qiang H, Ko CP (1998) Perisynaptic Schwann cells at neuromuscular junctions revealed by a novel monoclonal antibody. J Neurocytol 27: 667-681. [DOI] [PubMed] [Google Scholar]
- Auld DS, Colomar A, Belair EL, Castonguay A, Pinard A, Rousse I, Thomas S, Robitaille R (2003) Modulation of neurotransmission by reciprocal synapse-glial interactions at the neuromuscular junction. J Neurocytol 32: 1003-1015. [DOI] [PubMed] [Google Scholar]
- Berghs S, Aggujaro D, Dirkx Jr R, Maksimova E, Stabach P, Hermel JM, Zhang JP, Philbrick W, Slepnev V, Ort T, Solimena M (2000) betaIV spectrin, a new spectrin localized at axon initial segments and nodes of ranvier in the central and peripheral nervous system. J Cell Biol 151: 985-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, St. Martin M, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30: 369-383. [DOI] [PubMed] [Google Scholar]
- Birks R, Katz B, Miledi R (1960) Physiological and structural changes at the amphibian myoneural junction, in the course of nerve degeneration. J Physiol (Lond) 150: 145-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG (1994) Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 8: 1019-1029. [DOI] [PubMed] [Google Scholar]
- Cao G, Ko CP (2001) Schwann cell-conditioned medium modulates synaptic activities at Xenopus neuromuscular junctions in vitro Soc Neurosci Abstr 27: 711.12. [Google Scholar]
- Castonguay A, Robitaille R (2001) Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci 21: 1911-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandross KJ, Spray DC, Cohen RI, Kumar NM, Kremer M, Dermietzel R, Kessler JA (1996) TNF alpha inhibits Schwann cell proliferation, connexin46 expression, and gap junctional communication. Mol Cell Neurosci 7: 479-500. [DOI] [PubMed] [Google Scholar]
- Charles P, Tait S, Faivre-Sarrailh C, Barbin G, Gunn-Moore F, Denisenko-Nehrbass N, Guennoc AM, Girault JA, Brophy PJ, Lubetzki C (2002) Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol 12: 217-220. [DOI] [PubMed] [Google Scholar]
- Chen L, Ko CP (1994) Extension of synaptic extracellular matrix during nerve terminal sprouting in living frog neuromuscular junctions. J Neurosci 14: 796-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LL, Folsom DB, Ko CP (1991) The remodeling of synaptic extracellular matrix and its dynamic relationship with nerve terminals at living frog neuromuscular junctions. J Neurosci 11: 2920-2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Rio C, Ji RR, Dikkes P, Coggeshall RE, Woolf CJ, Corfas G (2003) Disruption of ErbB receptor signaling in adult nonmyelinating Schwann cells causes progressive sensory loss. Nat Neurosci 6: 1186-1193. [DOI] [PubMed] [Google Scholar]
- Ching W, Zanazzi G, Levinson SR, Salzer JL (1999) Clustering of neuronal sodium channels requires contact with myelinating Schwann cells. J Neurocytol 28: 295-301. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T (1999) Mouse models of tumor development in neurofibromatosis type 1. Science 286: 2172-2176. [DOI] [PubMed] [Google Scholar]
- Corfas G, Rosen KM, Aratake H, Krauss R, Fischbach GD (1995) Differential expression of ARIA isoforms in the rat brain. Neuron 14: 103-115. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM (2002) The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298: 1245-1248. [DOI] [PubMed] [Google Scholar]
- Cravioto H (1969) The ultrastructure of acoustic nerve tumors. Acta Neuropathol (Berl) 12: 116-140. [DOI] [PubMed] [Google Scholar]
- Custer AW, Kazarinova-Noyes K, Sakurai T, Xu X, Simon W, Grumet M, Shrager P (2003) The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J Neurosci 23: 10032-10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JQ, Lambert S, Bennett V (1996) Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J Cell Biol 135: 1355-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N (2000) Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest 105: 1233-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS (2004) KCNQ2 is a nodal K+ channel. J Neurosci 24: 1236-1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Brennan A, Liu N, Yarden Y, Lefkowitz G, Mirsky R, Jessen KR (1995) Neu differentiation factor is a neuron-glia signal and regulates survival, proliferation, and maturation of rat Schwann cell precursors. Neuron 15: 585-596. [DOI] [PubMed] [Google Scholar]
- Donovan S, Shannon KM, Bollag G (2002) GTPase activating proteins: critical regulators of intracellular signaling. Biochim Biophys Acta 1602: 23-45. [DOI] [PubMed] [Google Scholar]
- Dugoff L, Sujansky E (1996) Neurofibromatosis type 1 and pregnancy. Am J Med Genet 66: 7-10. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Girault JA, Popko B (1999) Axo-glial interactions regulate the localization of axonal paranodal proteins. J Cell Biol 147: 1145-1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Zanazzi G, Ching W, Scherer S, Milner TA, Peles E, Salzer JL (1997) The axonal membrane protein Caspr, a homologue of neurexin IV, is a component of the septate-like paranodal junctions that assemble during myelination. J Cell Biol 139: 1495-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson SL, O'Shea KS, Ghaboosi N, Loverro L, Frantz G, Bauer M, Lu LH, Moore MW (1997) ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development 124: 4999-5011. [DOI] [PubMed] [Google Scholar]
- Erlandson RA, Woodruff JM (1982) Peripheral nerve sheath tumors: an electron microscopic study of 43 cases. Cancer 49: 273-287. [DOI] [PubMed] [Google Scholar]
- Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A (2002) Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 39: 311-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, Abdel-Nour M, Gewies A, Peters H, Kaufmann D, Buske A, Tinschert S, Nurnberg P (2000) Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet 66: 790-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faissner A, Kruse J, Nieke J, Schachner M (1984) Expression of neural cell adhesion molecule L1 during development, in neurological mutants and in the peripheral nervous system. Brain Res 317: 69-82. [DOI] [PubMed] [Google Scholar]
- Feltri ML, Graus Porta D, Previtali SC, Nodari A, Migliavacca B, Cassetti A, Littlewood-Evans A, Reichardt LF, Messing A, Quattrini A, Mueller U, Wrabetz L (2002) Conditional disruption of beta 1 integrin in Schwann cells impedes interactions with axons. J Cell Biol 156: 199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Ko CP (2004) Transforming growth factor (TGF)-beta 1 mediates Schwann cell-induced synaptogenesis at the neuromuscular junction in vitro Soc Neurosci Abstr 30: 385:18. [Google Scholar]
- Feng L, Hatten ME, Heintz N (1994) Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron 12: 895-908. [DOI] [PubMed] [Google Scholar]
- Fieber LA (1998) Ionic currents in normal and neurofibromatosis type 1-affected human Schwann cells: induction of tumor cell K current in normal Schwann cells by cyclic AMP. J Neurosci Res 54: 495-506. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Birch PH (1997) Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet 70: 138-143. [DOI] [PubMed] [Google Scholar]
- Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C (2000) A dual role of erbB2 in myelination and in expansion of the schwann cell precursor pool. J Cell Biol 148: 1035-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver TD, Ren Q, Tuvia S, Bennett V (1997) Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol 137: 703-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollan L, Sabanay H, Poliak S, Berglund EO, Ranscht B, Peles E (2002) Retention of a cell adhesion complex at the paranodal junction requires the cytoplasmic region of Caspr. J Cell Biol 157: 1247-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollan L, Salomon D, Salzer JL, Peles E (2003) Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol 163: 1213-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutebroze L, Carnaud M, Denisenko N, Boutterin MC, Girault JA (2003) Syndecan-3 and syndecan-4 are enriched in Schwann cell perinodal processes. BMC Neurosci 4: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenard V, Gwynn LA, Wood PM (1995) Transforming growth factor-beta blocks myelination but not ensheathment of axons by Schwann cells in vitro J Neurosci 15: 419-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo HF, The I, Hannan F, Bernards A, Zhong Y (1997) Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276: 795-798. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D (1997) The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278: 51-57. [PubMed] [Google Scholar]
- Hsieh ST, Kidd GJ, Crawford TO, Xu Z, Lin WM, Trapp BD, Cleveland DW, Griffin JW (1994) Regional modulation of neurofilament organization by myelination in normal axons. J Neurosci 14: 6392-6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Rangwala F, Fulkerson PC, Ling B, Reed E, Cox AD, Kamholz J, Ratner N (2004) Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene 23: 368-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, Baba H (2002) A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci 22: 6507-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL (2002) The role of sodium channels in cell adhesion. Front Biosci 7: 12-23. [DOI] [PubMed] [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA (1994) Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet 7: 353-361. [DOI] [PubMed] [Google Scholar]
- Jahromi BS, Robitaille R, Charlton MP (1992) Transmitter release increases intracellular calcium in perisynaptic Schwann cells in situ. Neuron 8: 1069-1077. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci USA 99: 2303-2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R (1999) Schwann cells and their precursors emerge as major regulators of nerve development. Trends Neurosci 22: 402-410. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Morgan L, Stewart HJ, Mirsky R (1990) Three markers of adult non-myelin-forming Schwann cells, 217c(Ran-1), A5E3 and GFAP: development and regulation by neuron-Schwann cell interactions. Development 109: 91-103. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Brennan A, Morgan L, Mirsky R, Kent A, Hashimoto Y, Gavrilovic J (1994) The Schwann cell precursor and its fate: a study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron 12: 509-527. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Glick AD, Davis BW (1988) Immunohistochemical evaluation of Leu-7, myelin basic-protein, S100-protein, glial-fibrillary acidic-protein, and LN3 immunoreactivity in nerve sheath tumors and sarcomas. Arch Pathol Lab Med 112: 155-160. [PubMed] [Google Scholar]
- Kang H, Tian L, Thompson W (2003) Terminal Schwann cells guide the reinnervation of muscle after nerve injury. J Neurocytol 32: 975-985. [DOI] [PubMed] [Google Scholar]
- Kayl AE, Moore III BD (2000) Behavioral phenotype of neurofibromatosis, type 1. Ment Retard Dev Disabil Res Rev 6: 117-124. [DOI] [PubMed] [Google Scholar]
- Kim HA, Rosenbaum T, Marchionni MA, Ratner N, DeClue JE (1995) Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene 11: 325-335. [PubMed] [Google Scholar]
- Kim HA, Ling B, Ratner N (1997) Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol 17: 862-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe L, Friedrich R, Mautner VF (1999) Loss of NF1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer 24: 283-285. [DOI] [PubMed] [Google Scholar]
- Ko CP (1987) A lectin, peanut agglutinin, as a probe for the extracellular matrix in living neuromuscular junctions. J Neurocytol 16: 567-576. [DOI] [PubMed] [Google Scholar]
- Ko CP, Chen L (1996) Synaptic remodeling revealed by repeated in vivo observations and electron microscopy of identified frog neuromuscular junctions. J Neurosci 16: 1780-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Qiang H, Ko CP (2000) Reciprocal interactions between perisynaptic Schwann cells and regenerating nerve terminals at the frog neuromuscular junction. J Neurobiol 44: 343-360. [DOI] [PubMed] [Google Scholar]
- Koirala S, Reddy LV, Ko CP (2003) Roles of glial cells in the formation, function, and maintenance of the neuromuscular junction. J Neurocytol 32: 987-1002. [DOI] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V (1995) AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270: 2352-2359. [DOI] [PubMed] [Google Scholar]
- Kramer R, Bucay N, Kane DJ, Martin LE, Tarpley JE, Theill LE (1996) Neuregulins with an Ig-like domain are essential for mouse myocardial and neuronal development. Proc Natl Acad Sci USA 93: 4833-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullberg RW, Lentz TL, Cohen MW (1977) Development of the myotomal neuromuscular junction in Xenopus laevis: an electrophysiological and fine-structural study. Dev Biol 60: 101-129. [DOI] [PubMed] [Google Scholar]
- Lambert S, Davis JQ, Bennett V (1997) Morphogenesis of the node of Ranvier: co-clusters of ankyrin and ankyrin-binding integral proteins define early developmental intermediates. J Neurosci 17: 7025-7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C (1995) Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378: 394-398. [DOI] [PubMed] [Google Scholar]
- Legius E, Marchuk DA, Collins FS, Glover TW (1993) Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet 3: 122-126. [DOI] [PubMed] [Google Scholar]
- Li H, Velasco-Miguel S, Vass WC, Parada LF, DeClue JE (2002) Epidermal growth factor receptor signaling pathways are associated with tumorigenesis in the Nf1:p53 mouse tumor model. Cancer Res 62: 4507-4513. [PubMed] [Google Scholar]
- Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF (2000) Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA 97: 1299-1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Correa C, Dorschner M, Brems H, Lazaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E (2001) Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet 10: 1387-1392. [DOI] [PubMed] [Google Scholar]
- Lustig M, Zanazzi G, Sakurai T, Blanco C, Levinson SR, Lambert S, Grumet M, Salzer JL (2001) Nr-CAM and neurofascin interactions regulate ankyrin G and sodium channel clustering at the node of Ranvier. Curr Biol 11: 1864-1869. [DOI] [PubMed] [Google Scholar]
- Macleod GT, Dickens PA, Bennett MR (2001) Formation and function of synapses with respect to Schwann cells at the end of motor nerve terminal branches on mature amphibian (Bufo marinus) muscle. J Neurosci 21: 2380-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanthappa NK, Anton ES, Matthew WD (1996) Glial growth factor 2, a soluble neuregulin, directly increases Schwann cell motility and indirectly promotes neurite outgrowth. J Neurosci 16: 4673-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni MA, Goodearl AD, Chen MS, Bermingham-McDonogh O, Kirk C, Hendricks M, Danehy F, Misumi D, Sudhalter J, Kobayashi K, Wroblewski D, Lynch C, Baldassare M, Hiles I, Davis JB, Hsuan J, Totty MF, Otsu M, McBurney RN, Waterfield MD, et al. (1993) Glial growth factors are alternatively spliced erbB2 ligands expressed in the nervous system. Nature 362: 312-318. [DOI] [PubMed] [Google Scholar]
- Martini R, Schachner M, Faissner A (1990) Enhanced expression of the extracellular matrix molecule J1/tenascin in the regenerating adult mouse sciatic nerve. J Neurocytol 19: 601-616. [DOI] [PubMed] [Google Scholar]
- Melendez-Vasquez CV, Rios JC, Zanazzi G, Lambert S, Bretscher A, Salzer JL (2001) Nodes of Ranvier form in association with ezrin-radixin-moesin (ERM)-positive Schwann cell processes. Proc Natl Acad Sci USA 98: 1235-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegoz M, Gaspar P, Le Bert M, Galvez T, Burgaya F, Palfrey C, Ezan P, Arnos F, Girault JA (1997) Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron 19: 319-331. [DOI] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C (1995) Multiple essential functions of neuregulin in development. Nature 378: 386-390. [DOI] [PubMed] [Google Scholar]
- Meyer D, Yamaai T, Garratt A, Riethmacher-Sonnenberg E, Kane D, Theill LE, Birchmeier C (1997) Isoform-specific expression and function of neuregulin. Development 124: 3575-3586. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA (2004) Axonal neuregulin-1 regulates myelin sheath thickness. Science 304: 700-703. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Li H, Rizvi TA, Huang Y, Johansson G, Bowersock J, Sidani A, Vitullo J, Vogel K, Parysek LM, DeClue JE, Ratner N (2003) Brain lipid binding protein in axon-Schwann cell interactions and peripheral nerve tumorigenesis. Mol Cell Biol 23: 2213-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF (1999) Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23: 273-283. [DOI] [PubMed] [Google Scholar]
- Newman EA, Volterra A (2004) Glial control of synaptic function. Glia 47: 207-208. [DOI] [PubMed] [Google Scholar]
- O'Malley JP, Waran MT, Balice-Gordon RJ (1999) In vivo observations of terminal Schwann cells at normal, denervated, and reinnervated mouse neuromuscular junctions. J Neurobiol 38: 270-286. [PubMed] [Google Scholar]
- Pedraza L, Huang JK, Colman DR (2001) Organizing principles of the axoglial apparatus. Neuron 30: 335-344. [DOI] [PubMed] [Google Scholar]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, Schlessinger J (1997) Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J 16: 978-988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen J, Jaakkola S, Lebwohl M, Renvall S, Risteli L, Virtanen I, Uitto J (1988) Cellular differentiation and expression of matrix genes in type 1 neurofibromatosis. Lab Invest 59: 760-771. [PubMed] [Google Scholar]
- Peng HB, Yang JF, Dai Z, Lee CW, Hung HW, Feng ZH, Ko CP (2003) Differential effects of neurotrophins and schwann cell-derived signals on neuronal survival/growth and synaptogenesis. J Neurosci 23: 5050-5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Peles E (2003) The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4: 968-980. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E (1999) Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associates with K+ channels. Neuron 24: 1037-1047. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Salomon D, Berglund EO, Ohara R, Ranscht B, Peles E (2001) Localization of Caspr2 in myelinated nerves depends on axon-glia interactions and the generation of barriers along the axon. J Neurosci 21: 7568-7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E (2003) Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol 162: 1149-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SA, Friedman JM (2000) NF1 gene and neurofibromatosis 1. Am J Epidemiol 151: 33-40. [DOI] [PubMed] [Google Scholar]
- Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP (2003) Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron 40: 563-580. [DOI] [PubMed] [Google Scholar]
- Reist NE, Smith SJ (1992) Neurally evoked calcium transients in terminal Schwann cells at the neuromuscular junction. Proc Natl Acad Sci USA 89: 7625-7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ML, Woolf CJ (1992) Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol 21: 50-66. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Noble M, Land H (1988) Ras-mediated cell cycle arrest is altered by nuclear oncogenes to induce Schwann cell transformation. EMBO J 7: 1635-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger F, Daniloff JK, Pincon-Raymond M, Crossin KL, Grumet M, Edelman GM (1986) Neuronal cell adhesion molecules and cytotactin are colocalized at the node of Ranvier. J Cell Biol 103: 379-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C (1997) Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389: 725-730. [DOI] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, Salzer JL (2000) Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci 20: 8354-8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi TA, Huang Y, Sidani A, Atit R, Largaespada DA, Boissy RE, Ratner N (2002) A novel cytokine pathway suppresses glial cell melanogenesis after injury to adult nerve. J Neurosci 22: 9831-9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R (1998) Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron 21: 847-855. [DOI] [PubMed] [Google Scholar]
- Saito F, Moore SA, Barresi R, Henry MD, Messing A, Ross-Barta SE, Cohn RD, Williamson RA, Sluka KA, Sherman DL, Brophy PJ, Schmelzer JD, Low PA, Wrabetz L, Feltri ML, Campbell KP (2003) Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron 38: 747-758. [DOI] [PubMed] [Google Scholar]
- Salzer JL (2003) Polarized domains of myelinated axons. Neuron 40: 297-318. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Xu T, Crino P, Arroyo EJ, Gutmann DH (2001) Ezrin, radixin, and moesin are components of Schwann cell microvilli. J Neurosci Res 65: 150-164. [DOI] [PubMed] [Google Scholar]
- Serra E, Rosenbaum T, Nadal M, Winner U, Ars E, Estivill X, Lazaro C (2001) Mitotic recombination effects homozygosity for NF1 germline mutations in neurofibromas. Nat Genet 28: 294-296. [DOI] [PubMed] [Google Scholar]
- Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ (1994) Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell 77: 349-360. [DOI] [PubMed] [Google Scholar]
- Sheela S, Riccardi VM, Ratner N (1990) Angiogenic and invasive properties of neurofibroma Schwann cells. J Cell Biol 111: 645-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N (2000) Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem 275: 30740-30745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995) Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron 14: 133-141. [DOI] [PubMed] [Google Scholar]
- Srinivasan Y, Elmer L, Davis J, Bennett V, Angelides K (1988) Ankyrin and spectrin associate with voltage-dependent sodium channels in brain. Nature 333: 177-180. [DOI] [PubMed] [Google Scholar]
- Stefansson K, Wollmann R, Jerkovic M (1982) S-100 protein in soft-tissue tumors derived from Schwann cells and melanocytes. Am J Pathol 106: 261-268. [PMC free article] [PubMed] [Google Scholar]
- Syroid DE, Maycox PR, Burrola PG, Liu N, Wen D, Lee KF, Lemke G, Kilpatrick TJ (1996) Cell death in the Schwann cell lineage and its regulation by neuregulin. Proc Natl Acad Sci USA 93: 9229-9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syroid DE, Zorick TS, Arbet-Engels C, Kilpatrick TJ, Eckhart W, Lemke G (1999) A role for insulin-like growth factor-I in the regulation of Schwann cell survival. J Neurosci 19: 2059-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait S, Gunn-Moore F, Collinson JM, Huang J, Lubetzki C, Pedraza L, Sherman DL, Colman DR, Brophy PJ (2000) An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol 150: 657-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Marwaha S, Rutkowski JL, Tennekoon GI, Phillips PC, Field J (1998) A role for Pak protein kinases in Schwann cell transformation. Proc Natl Acad Sci USA 95: 5139-5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Thompson WJ (1996) Schwann cell apoptosis at developing neuromuscular junctions is regulated by glial growth factor. Nature 379: 174-177. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Thompson WJ (1997) Nerve terminal withdrawal from rat neuromuscular junctions induced by neuregulin and Schwann cells. J Neurosci 17: 6243-6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Dupree JL, Popko B, Karagogeos D (2002) The neuronal adhesion protein TAG-1 is expressed by Schwann cells and oligodendrocytes and is localized to the juxtaparanodal region of myelinated fibers. J Neurosci 22: 3016-3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Goutebroze L, Denisenko N, Bessa M, Nifli A, Havaki S, Iwakura Y, Fukamauchi F, Watanabe K, Soliven B, Girault JA, Karagogeos D (2003) Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol 162: 1161-1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA (2004) Role for glia in synaptogenesis. Glia 47: 209-216. [DOI] [PubMed] [Google Scholar]
- Vabnick I, Novakovic SD, Levinson SR, Schachner M, Shrager P (1996) The clustering of axonal sodium channels during development of the peripheral nervous system. J Neurosci 16: 4914-4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Trimmer JS, Schwarz TL, Levinson SR, Risal D, Shrager P (1999) Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci 19: 747-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Tempel BL (1993) Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 365: 75-79. [DOI] [PubMed] [Google Scholar]
- Wiestler OD, Brustle O, Eibl RH, Radner H, Aguzzi A, Kleihues P (1994) Oncogene transfer into the brain. Recent Results Cancer Res 135: 55-66. [DOI] [PubMed] [Google Scholar]
- Wilson GF, Chiu SY (1993) Mitogenic factors regulate ion channels in Schwann cells cultured from newborn rat sciatic nerve. J Physiol (Lond) 470: 501-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldeyesus MT, Britsch S, Riethmacher D, Xu L, Sonnenberg-Riethmacher E, Abou-Rebyeh F, Harvey R, Caroni P, Birchmeier C (1999) Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev 13: 2538-2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW (2000) Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron 25: 79-91. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Reynolds ML, Chong MS, Emson P, Irwin N, Benowitz LI (1992) Denervation of the motor endplate results in the rapid expression by terminal Schwann cells of the growth-associated protein GAP-43. J Neurosci 12: 3999-4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager J, Richards S, Hekmat-Scafe DS, Hurd DD, Sundaresan V, Caprette DR, Saxton WM, Carlson JR, Stern M (2001) Control of Drosophila perineurial glial growth by interacting neurotransmitter-mediated signaling pathways. Proc Natl Acad Sci USA 98: 10445-10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JF, Cao G, Koirala S, Reddy LV, Ko CP (2001) Schwann cells express active agrin and enhance aggregation of acetylcholine receptors on muscle fibers. J Neurosci 21: 9572-9584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P, Lee VM, Li C, Roder J, Trapp BD (1998) Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 18: 1953-1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF (2002) Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science 296: 920-922. [DOI] [PMC free article] [PubMed] [Google Scholar]