

For over a decade, evidence has mounted that nerve cell death in the CNS is often intimately linked to a process of cell division. Mitotic markers appear in neurons at risk for death in a variety of neurodegenerative conditions, in mouse and in humans. Beyond correlation, studies have shown that experimentally driving the cell cycle in a mature neuron leads to cell death rather than cell division, and blocking cell-cycle initiation can prevent many types of neuronal cell death. This unlikely linkage of cell cycle and cell death pathways is little appreciated among neuroscientists. As only one example, bromodeoxyuridine (BrdU) labeling is often uncritically accepted as proof of neurogenesis when it may well be attributable to a cell cycle-related cell death. This review is meant to enhance appreciation for the relevance of this phenomenon to development and neurodegenerative diseases, in particular the neurodegeneration found in Alzheimer's disease (AD). A brief overview of the participation of mitotic events in human Alzheimer's disease and its mouse models is presented. Against this background, we consider evidence that links various APP (amyloid precursor protein) binding proteins with the cell cycle in Alzheimer's disease. We also examine the role played by oxidative stress as a trigger for cell cycle-related neuronal death. Finally, we discuss the biochemical details of the lethal neuronal cell cycle events and present evidence that non-canonical pathways of DNA replication are probably involved.
Background
Since the earliest days of neurobiology, the study of neuronal cell death has been a field full of surprises. For example, the very contention that the death of neurons might be a normal part of the developmental program of the brain was greeted initially with great skepticism, and its formal demonstration by Hamburger (1975) and others came as quite a surprise to most workers in the field. Later, after the existence of target-related cell death and its dependence on trophic support had been firmly established, Oppenheim and colleagues uncovered yet another surprise. They showed that blocking neuronal activity during the critical period of a neuron, rather than mimicking the effects of target deprivation as expected, actually spared the doomed neurons from the fate of death (Pittman and Oppenheim, 1979). It is in this long tradition of surprises that we review a series of discoveries relating instances of neuronal cell death to a process normally linked to birth and regeneration: the cell cycle. By this time, however, the existence of a paradox such as this should come as no surprise.
The textbooks are clear: after they leave the ventricular zone (VZ) or subventricular zone (SVZ), neurons are aggressively postmitotic. The nature of this permanent exit from the cell cycle is still poorly understood, but the fact that no known cancer originates from an adult neuron is strong evidence that the restriction knows absolutely no work around. It is true that a small number of neurons are generated in the adult, but their origin is believed to be in the remnants of the VZ and SVZ (and their final fate is still debated).
The strange case of the T-antigen transgenics
The fact that a differentiated neuron never divides is an enormous impediment to tissue culture studies of nerve cells. Primary cultures of neurons cannot be propagated; they must be continually reestablished (with apologies to aficionados of neurosphere culture). In 1992, Feddersen et al. (1992) decided that they would rectify this situation using genetic engineering of transgenic mice. They were determined to force cell division in a neuron, so they took a powerful oncogene, the SV40 (simian virus 40) T-antigen, and drove its expression in transgenic mice using a Purkinje cell-specific promoter from the pcp2 gene. Legend has it they were eagerly awaiting culture dishes full of dividing Purkinje cells; what they got instead was ataxic mice. The founder transgenics did indeed express T-antigen exclusively in Purkinje cells and, through its sequestration of the retinoblastoma protein, released what appeared to be an initial attempt at a cell cycle. Purkinje cell nuclei incorporated BrdU and swelled in size, but rather than dividing, the Purkinje cells died. In this same year, al-Ubaidi et al. (1992) attempted an identical feat with a rhodopsin promoter and achieved the same surprising results: blind mice attributable to dead photoreceptors. Other authors were investigating the effect of elimination of the entire retinoblastoma gene. Three different laboratories reported essentially the same phenotype: in the absence of RB, neurons exiting the ventricular zone continue to proliferate, fail to differentiate, and subsequently die (Clarke et al., 1992; Jacks et al., 1992; Lee et al., 1992). From these early experiments, the concept arose that a cell division forced on a mature neuron is a lethal event.
Over the next several years, a body of correlative data began to accumulate to further develop this theme and move it from the realm of transgenic oddity into the mainstream of the neurodegeneration literature. Herrup and Busser showed that two models of CNS target-related cell death were accompanied by the reexpression of cell cycle proteins and incorporation of BrdU into the DNA of the target-deprived neurons hours before their death (Herrup and Busser, 1995). Tissue culture studies from the laboratories of Greene, Park, and others provided more detailed mechanistic data on the process. These groups were also the first to show that blocking the cell cycle with cyclin-dependent kinase inhibition could block neuronal cell death in culture (Park et al., 1996, 1997). Finally, a substantial number of laboratories used immunocytochemistry to show the ectopic neuronal reexpression of cell cycle proteins in regions of human brain in which degeneration occurs. Alzheimer's disease is reviewed in greater detail below, but cell cycle-associated neurodegeneration is also found in amyotrophic lateral sclerosis, stroke, and some forms of encephalitis (Jordan-Sciutto et al., 2002; Love, 2003; Ranganathan and Bowser, 2003). The fact that cell cycle events are bedfellows, however strange, of neuronal cell death in many human neurodegenerative diseases compels us to pose several difficult but important questions.
The questions
The prevalence of cell cycle events in the process of neuronal cell death means that we are facing a challenge of some magnitude. Clearly the therapeutic dimension of the discoveries urges thorough attention. However, how does one study cell cycle in a cell that by definition does not divide? We have addressed a series of questions in this review in an effort to put limits on the extent of our current ignorance. First, we consider the phenomenology of cell cycle reactivation in the context of a single human disease, Alzheimer's disease, and its mouse models. This normative work serves as a platform to ask more mechanistic questions. What kind of events can trigger a lethal mitosis in a neuron? What is the nature of the cellular signaling events involved? Just what is a cell cycle in an adult neuron, and what is the relationship of the chemistry of this attempt at rejuvenation to that seen in a population of normally dividing cells? We close with a set of predictions for the future of this exciting field.
A statement of the problem: cell cycle events in Alzheimer's disease
AD is the most common form of late-life dementia. It presents with a range of problems that include the inability to form recent memories, executive dysfunction, and other behavioral symptoms. Instances of AD are divided into two basic types: familial and sporadic. Familial forms (FAD) are relatively rare and are nearly always caused by a single autosomal dominant mutation with near 100% penetrance. They typically strike the victim before the age of 65 and thus are known as early-onset AD. Sporadic AD, in contrast, strikes after 65. Although genetic risk factors are known for sporadic AD, none is of sufficient predictive value to make it a cost-effective screening tool. On autopsy, the pathological signature of AD is a collection of abnormal deposits: neurofibrillary tangles consisting of aggregates of hyperphosphorylated forms of the microtubule-associated protein, tau, and waxy protein plaques consisting of fragments of the membrane protein APP. Neuronal death is regionally variable with heavy losses of neurons (up to 80%) found in hippocampus and other limbic regions of cortex, as well as in basal nucleus, locus ceruleus, and the dorsal raphe. Although familial forms account for <10% of all AD, much of our knowledge of the biological basis of the disease comes from these forms. Most FAD can be accounted for by mutations in one of three major disease genes: APP itself or the subunits of the APP-cleaving γ-secretases, PS1 (presenilin 1) or PS2. A full articulation of the amyloid hypothesis is well outside the focus of this article (for information and a taste of the ongoing debates in the field, see Neve and Robakis, 1998; Selkoe, 2002; Lee et al., 2004). The puzzle in the field has always been that, although dementia in AD is likely caused by the neuronal atrophy and loss, the path from plaques and tangles to regionally variable cell loss is not clear. What is becoming clear, however, is that the path passes directly through the cell cycle.
Vincent and Davies were perhaps the first to show definitively that active cell cycle components are present in neurons in the AD brain (Vincent et al., 1996). In close succession, several other laboratories reported that a wide range of cell cycle proteins, including cell cycle inhibitors, are present in neurons in autopsy specimens from AD patients (Fig. 1A) but not in age matched controls (Arendt et al., 1996; McShea et al., 1997; Nagy et al., 1997; Busser et al., 1998). These studies all rely primarily on protein-based evidence (e.g., immunocytochemistry) to show the reappearance of cell cycle kinases, their activators, and inhibitors; the data offer only indirect support of the idea that neurons are entering a real cell cycle. Yang et al. (2001) made an important additional observation. The authors used fluorescent in situ hybridization (FISH) to show that actual DNA replication has occurred in at-risk neurons in the AD brain (Fig. 1B). The same laboratory also showed that the involvement of cell cycle processes occurs in early stages of the disease process, not just during end-stage disease (Y. Yang et al., 2003). In both immunocytochemical and FISH preparations, 4-10% of the neurons in susceptible regions are positive for cell cycle evidence, whereas in a disease with a 10 year course, <0.05% would be expected if death after cycling were a rapid process. To account for the relatively high percentage of cell cycle-positive neurons, Yang et al. speculated that the neurons in the AD brain must take a long time to die after they begin their “cycle.”
Figure 1.

Cell cycle events in hippocampal pyramidal neurons of human and mouse. A, A neuron in the CA1 hippocampal region of a patient who died with Alzheimer's disease. The neuron indicated by the black arrow is immunopositive for the PCNA protein, an S-phase marker (from Busser et al., 1998). B, A CA3 area pyramidal cell from an Alzheimer's patient hybridized with a unique genomic probe from chromosome 11. Note the four spots of hybridization (white arrows) indicating the full replication of this portion of the genome (from Yang et al., 2001). C, The CA1 region of an R1.40 transgenic mouse stained with an antibody to cyclin A (green) and counterstained with propidium iodide (red). Note that the three neurons are immunopositive for this S-phase marker (Y. Yang, B. T. Lamb, and K. Herrup, unpublished observations).
When one is dealing with postmortem human brain tissue as source of material, one has little “experimental” control. DNA replication, easily shown with BrdU incorporation in mouse, must be shown by other means in humans, and although longitudinal pathological studies may be straightforward in mice (in which genotype and environmental variation can be held to a minimum) in humans, nearly all pathological tissue is from individuals at the end of the disease course. The ability to examine earlier stages depends heavily on good fortune. Thus, there was strong impetus to develop a number of different transgenic mouse models of AD that overexpress the human FAD genes in the context of the mouse (Hock and Lamb, 2001). Plaques are found in many of these models, especially when APP and PS1 transgenes are combined, but none develops any neurofibrillary tangles. In a recent example of a triple transgenic mouse (APP/PS1/tau), tangles were observed in the neuropil, documenting that such deposits are possible in mice (Oddo et al., 2003). This would seem to be a good start on mimicking the neuropathology of AD, yet the neurons just do not seem to get the message that there is a problem. Neuritic atrophy is found in some transgenics (German et al., 2003), but only one of the nearly one dozen mouse models has any reported loss of neurons (Calhoun et al., 1998). Furthermore, in this line, the loss amounts to less than one-quarter of the hippocampal CA1 pyramidal neurons. Other laboratories have searched for cell loss and found none (Irizarry et al., 1997). Abnormal behavioral phenotypes are found, including memory deficits, suggesting that neuronal dysfunction is going on at some level. The source of the dysfunction is unclear, however, because clearance of Aβ plaque peptide by immunization seems to result in a behavioral “cure” within days, although the plaques themselves linger for weeks (Dodart et al., 2002; Kotilinek et al., 2002).
But what does all this have to do with the cell cycle? The absence of neurodegeneration in model after model in which the other features of the AD pathology are captured so well is a bit odd. The quantification of DNA replication in human AD brain, however, suggests that cycling neurons might take a long time to die, perhaps as much as 1 year (Busser et al., 1998; Yang et al., 2001; Y. Yang et al., 2003). This prompted Yang and Herrup (2004) to determine whether the reexpression of cell cycle markers, the presumed beginning of the neuronal cell loss, were observed in any of the mouse models. It is. In four different plaque-bearing models, both immunocytochemistry and FISH successfully show that neurons in the most vulnerable areas have begun a true cell cycle event (Fig. 1C), although neuronal cell loss has not been reported in any of these models. The timing of these events is also informative. In the best-studied model, cell cycle events are already present by 6 months, many months before the first evidence of plaques at over 1 year.
This view of AD from the vantage point of the cell cycle suggests several conclusions, but also raises many questions. The human studies make it plain that cell cycle events are commonly associated with the death of neurons in the AD brain. Furthermore, their presence in the APP transgenic mice imparts a certain unity to the mouse genetic models and the human pathology that had been missing. However, although the concordance of mouse and man is gratifying in some ways, it is troubling in others. The relative well-being of the “cycling” mouse and human neurons leads to the following question: if a cell cycle is such a bad thing, how is it that many neurons can “cycle” yet not die? We consider this and other questions further below.
The signaling events linking the amyloid precursor protein to cell cycle and death
One of the strongest pieces of evidence in support of the hypothesis that the Aβ plaque-forming peptide is the true culprit in Alzheimer's comes from human genetics. The three major FAD genes all encode proteins that enhance the processing of APP into the highly amyloidogenic forms of Aβ. The logic is compelling, yet, in a truly rigorous sense, the genetics only points to APP and the γ-secretase as disease-causing entities. The Aβ connection is inferential. This is relevant because of the possibility that APP itself may act as a signaling receptor and influence neuronal survival in different ways. This idea was first proposed on the basis of its predicted amino acid sequence, which suggested that APP was a type 1 intrinsic membrane protein consistent with the structure of a “cell surface receptor” (Kang et al., 1987). That APP might actually function this way was supported when it was found that its intracellular domain interacts with the heterotrimeric G-protein Go (Nishimoto et al., 1993) and further that a percentage of APP is found on the cell surface in neurons (Jung et al., 1996; Storey et al., 1996; Perez et al., 1997).
As noted above, a failure of regulation of the cell cycle, which can push neurons into apoptotic death, occurs in neurons in AD brain. However, the signaling mechanisms that trigger these events have not been defined. The discovery that at least one of the signaling pathways mediated by APP overexpression leads to neuronal DNA synthesis and apoptosis (Chen et al., 2000; McPhie et al., 2003) suggests that this signaling pathway may play a role in the coexistence of both the cell cycle events and the neurodegeneration seen in AD. A search of cDNA libraries uncovered genes encoding proteins that interact with the cytodomain of APP. One of these, APP-BP1 (binding protein 1) (Chow et al., 1996), drives the cell cycle through the S-M checkpoint in dividing cells, and overexpression of APP-BP1 in neurons causes them to die (Chen et al., 2000). Notably, expression of FAD mutants of APP in neurons results in (1) an increase in expression of APPBP1, (2) entry of the neurons into the S-phase of the cell cycle, and (3) neuronal apoptosis. Furthermore, APP-BP1 is overexpressed in at-risk regions of human AD brain relative to cognitively intact controls. APP-BP1 is thus a candidate for participation in the events leading to the loss of cell cycle regulation and DNA replication that appears to occur in AD neurons.
A second binding protein for APP, PAK3 (p21-activated kinase 3), is a serine/threonine kinase (McPhie et al., 2003). This molecule also is implicated in the DNA synthesis and neuronal apoptosis caused by FAD mutants of APP. A dominant-negative kinase mutant of PAK3 inhibits the neuronal apoptosis and DNA synthesis; this effect is abolished by deletion of the PAK3 APP-binding domain or by coexpression of a peptide representing this binding domain. The involvement of PAK3 specifically in FAD APP-mediated apoptosis rather than in general apoptotic pathways is suggested by the facts that a dominant-positive mutant of PAK3 does not alone cause neuronal apoptosis and that the dominant-negative mutant of PAK3 does not inhibit chemically induced apoptosis. Pertussis toxin, which inactivates the heterotrimeric G-proteins Go and Gi, inhibits the apoptosis and DNA synthesis caused by FAD APP mutants; the apoptosis and DNA synthesis are rescued by coexpression of a pertussis toxin-insensitive Go. FAD APP-mediated DNA synthesis precedes FAD APP-mediated apoptosis in neurons, and inhibition of neuronal entry into the cell cycle inhibits the apoptosis.
It is hypothesized that APP and its C-terminal binding proteins, G0 PAK3 and APP-BP1, have a regulated interaction that activates a signaling pathway important for normal brain function, perhaps regulating synaptic remodeling or neurogenesis during learning. Dysfunction of this interaction by FAD mutations in APP causes sustained disruption of the pathway in neurons, leading to entry into the cell cycle and subsequent apoptosis. Thus, this signaling pathway may play a role in AD neurodegeneration.
Oxidative stress, cell cycle, and neurodegeneration
The focus on APP and its metabolites is a strong tradition in the Alzheimer's field. As mentioned above, the logic for this emphasis in understanding FAD is clear. However, >90% of the current cases of AD are believed to be sporadic, in which the largest risk factor by far is age. The biological meaning of age is a difficult concept, one that has seen more and more attention in recent years. Many variables change slowly during the course of an organism's lifetime, but one of the most reliable is the widespread accumulation of oxidative damage. Whereas oxygen is essential for life, its metabolites, known as reactive oxygen species (ROS), can cause extensive cellular damage. Up to 1% of the mitochondrial electron flow can generate ROS, which are detoxified by cellular antioxidants. However, a deleterious condition known as oxidative stress occurs when ROS levels exceed the antioxidant capacity of the cell. Unchecked, excessive ROS can lead to nucleic acid, lipid, and protein damage and ultimately cell death via apoptosis or necrosis.
Given their high metabolic rate, limited regeneration potential, and relatively low baseline levels of antioxidants, neurons are thought to be particularly vulnerable to ROS. Studies of human postmortem brain tissue have repeatedly implicated oxidative stress in neuronal death, particularly in later-onset neurodegenerative disorders. For example, DNA and protein oxidation, and lipid peroxidation are observed in the substantia nigra of patients with Parkinson's disease (PD) (Jenner and Olanow, 1996). The increase in ROS correlates with multiple findings of mitochondrial complex 1 deficiencies in PD patients, studies promoted by the finding that MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), paraquat, and rotenone compounds known to inhibit complex 1, also induce PD pathophysiology (Parker and Swerdlow, 1998; Dawson and Dawson, 2003). Similarly, increases in various biomarkers of oxidative stress have also been observed in diseased regions of postmortem brain tissue from patients with Alzheimer's disease, amyotrophic lateral sclerosis, prion disorders, Huntington's disease, and progressive supranuclear palsy (Andersen, 2004; Barnham et al., 2004).
Because cell cycle misregulation, like oxidative stress, is associated with human late-onset neurodegenerative disorders, the suggestion arises that these processes may be molecularly intertwined. At first, this idea of a connection seems to run counter to the many reports that ROS-induced DNA damage correlates with cell cycle arrest, not reentry. The effects of ROS on cell cycle progression, however, appear to be dose dependent. For example, whereas high doses of H2O2 induce cell cycle arrest and cell death, low doses stimulate proliferation or survival. This suggests that below toxic concentrations, H2O2, like nitric oxide, can serve as a signaling molecule (D'Souza et al., 1993; Burdon, 1995; Burdon et al., 1996; Herbert et al., 1996; Ohguro et al., 1999; Clerk, 2003). A better example is the harlequin (Hq) mutant mouse.
Harlequin is an X-linked mutation; Hq/Hq and Hq/Y mice develop progressive ataxia associated with the death of both granule and Purkinje cells in cerebellum, as well as ganglion, amacrine, horizontal, and photoreceptor cells of the retina (Klein et al., 2002). The Hq mutation is caused by a proviral insertion in the first intron of the apoptosis-inducing factor (Aif) gene, resulting in an 80-90% reduction of both transcripts and protein. AIF is a ubiquitously expressed flavoprotein with structural homology to gluthione reductase, an important recycling component of the glutathione H2O2-scavenging system (Susin et al., 1999; Maté et al., 2002). Downregulation of AIF in Hq mutant mice is associated with increases in oxidative damage: lipid peroxidation, catalase, and total glutathione. Significantly, these increases are observed months before the onset of neurodegeneration. In keeping with these findings, mutant cerebellar granule cells are more sensitive to H2O2-mediated cell death, a sensitivity that is rescued by expression of full-length Aif cDNA. Furthermore, there is an increase in the levels of DNA oxidation, as measured by the presence of 8-hydroxyguanine (8-OHdG), a DNA lesion resulting from unrepaired oxidative damage. Together, these studies suggest that AIF regulates the amount of ROS in cerebellar and retinal neurons by unidentified mechanisms (Lipton and Bossy-Wetzel, 2002; Bonni, 2003).
In addition to the changes in oxidation damage repair, the neurons in the Hq mouse also suffer from a failure of cell cycle control. PCNA (proliferating cell nuclear antigen) and CDC47 (cell division cycle 47) immunostaining are found in susceptible Hq neurons as is BrdU-labeling, demonstrating that Hq retinal neurons reenter a cell cycle (Klein et al., 2002). The number of granule cells demonstrating cell cycle markers increases with age, peaking at 7 months when most cell death occurs. That cell cycle reentry is tightly associated with cell death in these cells is indicated by the finding that all apoptotic neurons express S-phase markers. Furthermore, all neurons that had re-entered into S-phase had oxidized DNA, as evidenced by their immunoreactivity with antibodies to 8-OHdG. Not all 8-OHdG-positive neurons, however, were cell cycle positive, suggesting that the oxidative damage precedes cell cycle reentry.
The mechanisms by which oxidative stress induces cell cycle abnormalities in postmitotic neurons is not known (Klein and Ackerman, 2003). ROS might alter the components of a mitogenic signaling pathway by ectopically activating either a growth factor receptor (Holbrook and Ikeyama, 2002) or its signaling molecules such MAP (mitogen-activated protein) kinase and NF-κB (nuclear factor κB) that are farther downstream (Martindale and Holbrook, 2002). Another intriguing possibility is that the ROS-induced DNA damage itself might initiate cell cycle reentry in postmitotic neurons. The relative timing of the appearance of oxidized DNA (8-OHdG immunoreactivity) and cell cycle events (BrdU, PCNA, etc.) in Hq neurons is one piece of evidence, but there are others. Abortive cell cycle reentry and subsequent apoptosis is known to occur in cultured postmitotic neurons treated with compounds that induce chromosomal breaks and base mis-incorporation (Kruman et al., 2004). Also, DNA repair proteins have been shown to physically interact with proteins involved in DNA replication (Nagelhus et al., 1997; Otterlei et al., 1999), further suggesting a linkage. Finally, expression and/or activity of several DNA repair enzymes appear to be regulated by the cell cycle and are highest in proliferating cells (Nilsen et al., 2000; Choi et al., 2002). Perhaps the DNA strand breaks and oxidized DNA bases that have been observed in postmortem tissue from patients with neurodegenerative disorders (Cotman and Su, 1996; Davydov et al., 2003) are not efficiently repaired. If not, it may be that they introduce mutations and thus interfere with neuronal transcription and other DNA-dependent processes. If the damage is too great, a critical component of the cell cycle control pathway might be lost in cells, leading to apoptosis: divide and die.
What is the nature of the cell cycle events observed in Alzheimer's disease?
This naturally returns the discussion to the question is just what is a cell cycle in a nondividing cell besides a classic oxymoron. The studies of AIF carry a clear message: there are more ways than one to engage a neuronal cell cycle, but whenever this happens the net effect is usually bad. The examples of APP signaling and oxidative damage are but two examples (and the Aβ peptide itself is another). However, if all of these stimuli lead to the same common pathway, then surely we would be well advised to learn as much as we can about the events that are triggered. With the war on cancer now nearly 35 years old, the depth of our knowledge of the normal mechanics of the eukaryotic cell cycle is considerable. Classically, the reentry of a mammalian cell into the cell cycle depends on extracellular proliferative signals, which exert their effect by the sequential activation of CDK (cyclin-dependent kinase) complexes (for review, see Grana and Reddy, 1995). The cyclin D/CDK4-6 complexes control the transition of cells from G0- to G1-phase. The G1/S transition and the S-phase progression are regulated in part by the activation of the cyclin E/CDK2 complex and of the cyclin A/CDK2 complex, respectively. The cyclin B/CDK1 complex is the regulator of the G2/M transition. After an appropriate stimulus to a differentiated neuron (the Aβ peptide, for example), there is a rise in cyclin D1 and an increase in the levels of phosphorylation of the retinoblastoma protein. This is followed by the induction of cyclins E and A. The treated neurons start DNA replication but then appear to undergo apoptosis instead of completing a full mitotic cycle (Copani et al., 1999). This abortive attempt at cell division has true biological meaning, however, because analysis by fluorescent-activated cell-sorting techniques has shown that blockade of the G1/S transition using a cyclin D1 antisense or a dominant-negative mutant of CDK2 prevented both Aβ-induced DNA replication and apoptosis (Copani et al., 1999). These experiments, as well as the studies cited above, show that highly differentiated neurons can be induced to express significant amounts of cell cycle proteins (e.g., cyclins) and enter a process of DNA replication that leads to death and not division (Freeman et al., 1994; Park et al., 1997; Giovanni et al., 1999). The reactivation of the cell cycle is an obligatory step in this pathway (not just a by product) because blocking the cycle blocks the death. The question that remains is how does the ectopic S-phase trigger the death of neurons. This understanding requires a more detailed analysis of the biochemistry of eukaryotic DNA replication.
The DNA replication machinery is activated by Aβ in adult neurons
In proliferating cells, replication involves three distinct DNA polymerases (pols), polα, polδ, and polϵ (for review, see Hubscher et al., 2000). Polα, with its associated primase activity, is unique among DNA pols because of its ability to initiate de novo DNA replication by synthesizing short RNA-DNA primers. These are extended by either polδ or polϵ, which are endowed with a proofreading exonuclease activity that is suited to minimizing errors during replication. The complete polα consists of four polypeptides with molecular mass of 180, 70, 58, and 49 kDa. The p180 subunit carries the DNA polymerase activity, whereas the p49 subunit harbors the catalytic primase activity. Polα helps to hold the cell cycle in check (for review, see Arezi and Kuchta, 2000), whereas polδ and polϵ can function as DNA repair enzymes, correcting any mismatch errors that have escaped the editing process during replication. Thus, the coordinated regulation of the activity of multiple DNA polymerases is a fundamental aspect of DNA replication in normally dividing cells.
There are other DNA polymerases that are specialized to specifically ignore DNA damage during replication. The tradeoff is that these enzymes introduce errors at a high frequency when they operate on undamaged DNA (Friedberg et al., 2002). One of these, polβ, participates in base excision repair, a pathway that is used to replace any oxidized bases that may appear over time (Sobol et al., 1996). The precise repertoire of DNA polymerases that are expressed by neurons is unknown. Most of our current information comes from studies of the differential sensitivity of the polymerases to different inhibitors. In isolated neuronal fractions, polβ activity tends to predominate, although some polδ/ϵ and a little polα activity are also present (Raji et al., 2002).
In dissociated culture, it appears that neurons constitutively express polδ and its ancillary protein PCNA, whereas polϵ is undetectable (Copani et al., 2002). After exposure to Aβ, neurons express the p49 and the p58 subunits of the primase/polα complex, whereas the expression of replicative polymerases (polα, polδ, and polϵ) remains unaffected. Unexpectedly, the repair enzyme polβ is induced in response to Aβ in a cell cycle-dependent manner that is unusual in proliferating cells. This increase in protein appears to have functional consequences because the knockdown of polβ prevented Aβ-induced DNA synthesis and the ensuing apoptosis. Similar effects are observed by knocking down the p49 primase subunit. These results suggest the existence of a primase-directed noncanonical pathway of DNA replication, which is mediated by polβ in Aβ-treated neurons (Fig. 2).
Figure 2.
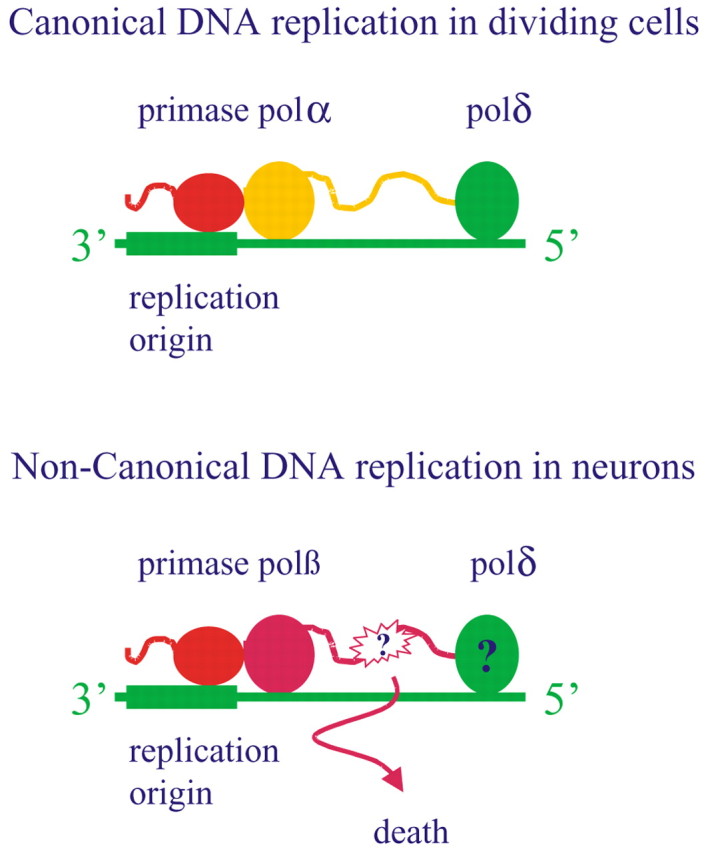
A noncanonical pathway of DNA replication is mediated by polβ and is causally related to neuronal death. In dividing cells (top), DNA replication (yellow strand) is accomplished by DNA polα and polδ/ϵ, which follow one another in the elongation of the short RNA primer synthesized by the primase (red). In neurons challenged with Aβ (bottom), DNA replication (purple strand) is performed by polβ that substitutes for polα. The error-prone activity of polβ might generate DNA damage, eventually leading to cell death. Some DNA replication is possibly mediated by polδ but does not contribute to neuronal death (Copani et al., 2002).
These results are as important as they are unexpected. Nonetheless, they leave unanswered the question of how this unorthodox pathway of DNA replication leads to neuronal apoptosis. Given that polβ is an error-prone enzyme, it is possible that its use contributes to a level of DNA damage that proves toxic to the neuron. Such damage is reported in AD neurons (Su et al., 1997), along with an increased expression of p53, a sensor of DNA damage (de la Monte et al., 1997). When polβ induction is prevented, the increase in p53 is not seen (Copani et al., 2002). The presence of hyperploid neurons in AD brain (Yang et al., 2001) suggests that this error-prone replication mechanism somehow proceeds without signaling to cell cycle checkpoints. If this entire process were driven by polβ, the prediction would be that the abnormal neurons are loaded with unsustainable amounts of DNA damage. In other words, de novo DNA synthesis might be a potential source of replication errors that are sufficient to trigger the activation of a p53-dependent death pathway in neurons. This process need not be all-or-none; the outcome might depend on the severity of the insult and/or on the vulnerability of the neurons (for examples of the different phenotypes of neuronal death in the harlequin mutant mice, see Klein and Ackerman, 2003) (Fig. 3).
Figure 3.

The involvement of DNA replication in neuronal death depends on the type of insult and the susceptibility of neurons. A, A susceptible neuron (smaller circle) in the presence of a severe insult (thick vertical arrow), which possibly causes extensive DNA damage, dies without the contribution of de novo DNA synthesis. B, In a less vulnerable neuron (bigger circle) exposed to other types of insults, including Aβ or mild oxidative stress, de novo DNA synthesis becomes essential for neuronal death.
Conclusions and cautions
Cell cycling, it seems, is incompatible with the complex differentiated state of a neuron. Forcing the former in the presence of the latter results in neuronal death by an as yet unknown mechanism. In other words, if you are a neuron, “you divide, you die.” In an era of information overload, one might wish to avoid the implications of this deceptively simple relationship, but evidence is mounting that there is new biology under the surface. Although there are significant technical hurdles to overcome in the study of cell cycle regulation in adult neurons, these difficulties must be addressed. This is true in part because of the growing number of situations in which the behavior of nerve cells is governed by the divide-and-die dictum. In neurodegenerative disease as well as in normal development, we are becoming aware that the state of the genome is not always static. Major genomic changes involving extensive amounts of new DNA synthesis can occur. These changes are visible either by monitoring the incorporation of BrdU or through the direct query of the genome using FISH techniques. That genome-level changes occur is certain. It is the nature of these events that is unclear at present.
The questions that remain are large but clear. What is the state of the genome in a CNS neuron? The prohibition against cell division can be overcome in most other cells (hence, the large number of different types of cancer), but there are hints that things are not straightforward in the brain (Geller and Potter, 1999; Rehen et al., 2001; Kaushal et al., 2003; A. H. Yang et al., 2003). So what is different about a nerve cell? What is it about the reengagement of a cell cycle process that kills a neuron? It would help the discovery process if we could determine whether the events described in this article represent a true mitotic cell cycle or something different. Unraveling the biochemistry and cell biology of the process we have called a neuronal cell cycle is an important first step. Along the way, we need to discover whether the decision to divide is destructive (as suggested by studies such as the T-antigen transgenics) or protective (as hinted at by the apparent long survival of the hyperploid neurons in mouse and man). Other questions have importance for therapeutic intervention in specific disease situations. What drives a neuron to divide? Is it activity, heightened trophic factor levels, other cytokines, or something novel? What are the signaling pathways that relay this impulse to the cell “division” machinery?
Cell cycling is a as old as life itself. In most situations, it is a generative force that creates new cells from old. However, in the brain, there are persistent associations between cell cycling and the loss of neurons to cell death. This is a paradox to be sure, but, in a field that is known for its surprises, we should probably expect nothing less.
Footnotes
We acknowledge the sources of funding that helped to support the writing of this article and the conduct of the much of the work described. These sources are as follows: National Institutes of Health (NIH) Grants P50 AG08012 and R01 NS20591, the Blanchette Hooker Rockefeller Fund, and the Coins for Alzheimer's Research Trust Fund Foundation (K.H.); the Alzheimer's Association and Ministry of Instruction, University, and Research cofin 2003 (A.C.); NIH Grant AG12954 (R.N.); and NIH Grant AG19358 (S.L.A.).
Correspondence should be addressed to Karl Herrup, Department of Neurosciences, Case School of Medicine (E504), Department of Neurology, University Hospitals of Cleveland, 10900 Euclid Avenue, Cleveland, OH 44120. E-mail: kxh26@case.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249232-08$15.00/0
References
- al-Ubaidi MR, Hollyfield JG, Overbeek PA, Baehr W (1992) Photoreceptor degeneration induced by the expression of simian virus 40 large tumor antigen in the retina of transgenic mice. Proc Natl Acad Sci USA 89: 1194-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Rev Neurosci [Suppl] 10: S18-S25. [DOI] [PubMed] [Google Scholar]
- Arendt T, Rodel L, Gartner U, Holzer M (1996) Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport 7: 3047-3049. [DOI] [PubMed] [Google Scholar]
- Arezi B, Kuchta RD (2000) Eukaryotic DNA primase. Trends Biochem Sci 25: 572-576. [DOI] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3: 205-214. [DOI] [PubMed] [Google Scholar]
- Bonni A (2003) Neurodegeneration: a non-apoptotic role for AIF in the brain. Curr Biol 13: R19-R21. [DOI] [PubMed] [Google Scholar]
- Burdon RH (1995) Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic Biol Med 18: 775-794. [DOI] [PubMed] [Google Scholar]
- Burdon RH, Gill V, Alliangana D (1996) Hydrogen peroxide in relation to proliferation and apoptosis in BHK-21 hamster fibroblasts. Free Radic Res 24: 81-93. [DOI] [PubMed] [Google Scholar]
- Busser J, Geldmacher DS, Herrup K (1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci 18: 2801-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M (1998) Neuron loss in APP transgenic mice. Nature 395: 755-756. [DOI] [PubMed] [Google Scholar]
- Chen Y, McPhie DL, Hirschberg J, Neve RL (2000) The amyloid precursor protein-binding protein APP-BP1 drives the cell cycle through the S-M checkpoint and causes apoptosis in neurons. J Biol Chem 275: 8929-8935. [DOI] [PubMed] [Google Scholar]
- Choi EK, Lee YH, Choi YS, Kwon HM, Choi MS, Ro JY, Park SK, Yu E (2002) Heterogeneous expression of Ku70 in human tissues is associated with morphological and functional alterations of the nucleus. J Pathol 198: 121-130. [DOI] [PubMed] [Google Scholar]
- Chow N, Korenberg JR, Chen XN, Neve RL (1996) APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem 271: 11339-11346. [DOI] [PubMed] [Google Scholar]
- Clarke A, Maandag E, van Roon M, van der Lugt N, van der Valk M, Hooper M, Berns A, te Riele H (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359: 328-330. [DOI] [PubMed] [Google Scholar]
- Clerk A (2003) The radical balance between life and death. J Mol Cell Cardiol 35: 599-602. [DOI] [PubMed] [Google Scholar]
- Copani A, Condorelli F, Caruso A, Vancheri C, Sala A, Giuffrida Stella AM, Canonico PL, Nicoletti F, Sortino MA (1999) Mitotic signaling by beta-amyloid causes neuronal death. FASEB J 13: 2225-2234. [PubMed] [Google Scholar]
- Copani A, Sortino MA, Caricasole A, Chiechio S, Chisari M, Battaglia G, Giuffrida-Stella AM, Vancheri C, Nicoletti F (2002) Erratic expression of DNA polymerases by beta-amyloid causes neuronal death. FASEB J 16: 2006-2008. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Su JH (1996) Mechanisms of neuronal death in Alzheimer's disease. Brain Pathol 6: 493-506. [DOI] [PubMed] [Google Scholar]
- D'Souza RJ, Phillips HM, Jones PW, Strange RC, Aber GM (1993) Interactions of hydrogen peroxide with interleukin-6 and platelet-derived growth factor in determining mesangial cell growth: effect of repeated oxidant stress. Clin Sci (Lond) 85: 747-751. [DOI] [PubMed] [Google Scholar]
- Davydov V, Hansen LA, Shackelford DA (2003) Is DNA repair compromised in Alzheimer's disease? Neurobiol Aging 24: 953-968. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson's disease. Science 302: 819-822. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Sohn YK, Wands JR (1997) Correlates of p53- and Fas (CD95)-mediated apoptosis in Alzheimer's disease. J Neurol Sci 152: 73-83. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, De-Long CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci 5: 452-457. [DOI] [PubMed] [Google Scholar]
- Feddersen RM, Ehlenfeldt R, Yunis WS, Clark HB, Orr HT (1992) Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron 9: 955-966. [DOI] [PubMed] [Google Scholar]
- Freeman RS, Estus S, Johnson Jr EM (1994) Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron 12: 343-355. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Wagner R, Radman M (2002) Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science 296: 1627-1630. [DOI] [PubMed] [Google Scholar]
- Geller LN, Potter H (1999) Chromosome missegregation and trisomy 21 mosaicism in Alzheimer's disease. Neurobiol Dis 6: 167-179. [DOI] [PubMed] [Google Scholar]
- German DC, Yazdani U, Speciale SG, Pasbakhsh P, Games D, Liang CL (2003) Cholinergic neuropathology in a mouse model of Alzheimer's disease. J Comp Neurol 462: 371-381. [DOI] [PubMed] [Google Scholar]
- Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS (1999) Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F x DP, in B-amyloid-induced neuronal death. J Biol Chem 274: 19011-19016. [DOI] [PubMed] [Google Scholar]
- Grana X, Reddy EP (1995) Cell cycle control in mammalian cells: role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 11: 211-219. [PubMed] [Google Scholar]
- Hamburger V (1975) Cell death in the development of the lateral motor column of the chick embryo. J Comp Neurol 160: 535-546. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Bono F, Savi P (1996) The mitogenic effect of H2O2 for vascular smooth muscle cells is mediated by an increase of the affinity of basic fibroblast growth factor for its receptor. FEBS Lett 395: 43-47. [DOI] [PubMed] [Google Scholar]
- Herrup K, Busser JC (1995) The induction of multiple cell cycle events precedes target-related neuronal death. Development 121: 2385-2395. [DOI] [PubMed] [Google Scholar]
- Hock Jr BJ, Lamb BT (2001) Transgenic mouse models of Alzheimer's disease. Trends Genet 17: S7-S12. [DOI] [PubMed] [Google Scholar]
- Holbrook NJ, Ikeyama S (2002) Age-related decline in cellular response to oxidative stress: links to growth factor signaling pathways with common defects. Biochem Pharmacol 64: 999-1005. [DOI] [PubMed] [Google Scholar]
- Hubscher U, Nasheuer HP, Syvaoja JE (2000) Eukaryotic DNA polymerases, a growing family. Trends Biochem Sci 25: 143-147. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT (1997) Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt E, Bronson R, Goodell M, Weinberg R (1992) Effects of an Rb mutation in the mouse. Nature 359: 295-300. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson's disease. Neurology 47: S161-S170. [DOI] [PubMed] [Google Scholar]
- Jordan-Sciutto KL, Wang G, Murphey-Corb M, Wiley CA (2002) Cell cycle proteins exhibit altered expression patterns in lentiviral-associated encephalitis. J Neurosci 22: 2185-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SS, Nalbantoglu J, Cashman NR (1996) Alzheimer's beta-amyloid precursor protein is expressed on the surface of immediately ex vivo brain cells: a flow cytometric study. J Neurosci Res 46: 336-348. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B (1987) The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 325: 733-736. [DOI] [PubMed] [Google Scholar]
- Kaushal D, Contos JJ, Treuner K, Yang AH, Kingsbury MA, Rehen SK, McConnell MJ, Okabe M, Barlow C, Chun J (2003) Alteration of gene expression by chromosome loss in the postnatal mouse brain. J Neurosci 23: 5599-5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Ackerman SL (2003) Oxidative stress, cell cycle, and neurodegeneration. J Clin Invest 111: 785-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL (2002) The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 419: 367-374. [DOI] [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH (2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci 22: 6331-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae Jr R, Gorospe M, Mattson MP (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41: 549-561. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang C-Y, Hu N, Wang Y-CJ, Lai C-C, Herrup K, Lee W-H, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288-294. [DOI] [PubMed] [Google Scholar]
- Lee HG, Casadesus G, Zhu X, Joseph JA, Perry G, Smith MA (2004) Perspectives on the amyloid-beta cascade hypothesis. J Alzheimers Dis 6: 137-145. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel E (2002) Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell 111: 147-150. [DOI] [PubMed] [Google Scholar]
- Love S (2003) Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci Lett 353: 29-32. [DOI] [PubMed] [Google Scholar]
- Martindale JL, Holbrook NJ (2002) Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 192: 1-15. [DOI] [PubMed] [Google Scholar]
- Maté MJ, Ortiz-Lombardia M, Boitel B, Haouz A, Tello D, Susin SA, Penninger J, Kroemer G, Alzari PM (2002) The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol 9: 442-446. [DOI] [PubMed] [Google Scholar]
- McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL (2003) DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci 23: 6914-6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShea A, Harris PL, Webster KR, Wahl AF, Smith MA (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol 150: 1933-1939. [PMC free article] [PubMed] [Google Scholar]
- Nagelhus TA, Haug T, Singh KK, Keshav KF, Skorpen F, Otterlei M, Bharati S, Lindmo T, Benichou S, Benarous R, Krokan HE (1997) A sequence in the N-terminal region of human uracil-DNA glycosylase with homology to XPA interacts with the C-terminal part of the 34-kDa subunit of replication protein A. J Biol Chem 272: 6561-6566. [DOI] [PubMed] [Google Scholar]
- Nagy Z, Esiri M, Cato A, Smith A (1997) Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 94: 6-15. [DOI] [PubMed] [Google Scholar]
- Neve RL, Robakis NK (1998) Alzheimer's disease: a re-examination of the amyloid hypothesis. Trends Neurosci 21: 15-19. [DOI] [PubMed] [Google Scholar]
- Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, Daly G, Krokan HE, Lindahl T, Barnes DE (2000) Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell 5: 1059-1065. [DOI] [PubMed] [Google Scholar]
- Nishimoto I, Okamoto T, Matsuura Y, Takahashi S, Murayama Y, Ogata E (1993) Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o). Nature 362: 75-79. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409-421. [DOI] [PubMed] [Google Scholar]
- Ohguro N, Fukuda M, Sasabe T, Tano Y (1999) Concentration dependent effects of hydrogen peroxide on lens epithelial cells. Br J Ophthalmol 83: 1064-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE (1999) Post-replicative base excision repair in replication foci. EMBO J 18: 3834-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Farinelli S, Greene L (1996) Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem 271: 8161-8169. [DOI] [PubMed] [Google Scholar]
- Park D, Levine B, Ferrari G, Greene L (1997) Cyclin-dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci 17: 8975-8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker Jr WD, Swerdlow RH (1998) Mitochondrial dysfunction in idiopathic Parkinson disease. Am J Hum Genet 62: 758-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH (1997) The beta-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J Neurosci 17: 9407-9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman R, Oppenheim RW (1979) Cell death of motoneurons in the chick embryo spinal cord. IV. Evidence that a functional neuromuscular interaction is involved in the regulation of naturally occurring cell death and the stabilization of synapses. J Comp Neurol 187: 425-446. [DOI] [PubMed] [Google Scholar]
- Raji NS, Krishna TH, Rao KS (2002) DNA-polymerase alpha, beta, delta and epsilon activities in isolated neuronal and astroglial cell fractions from developing and aging rat cerebral cortex. Int J Dev Neurosci 20: 491-496. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Bowser R (2003) Alterations in G(1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol 162: 823-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J (2001) Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci USA 98: 13361-13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest 110: 1375-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH (1996) Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 379: 183-186. [DOI] [PubMed] [Google Scholar]
- Storey E, Beyreuther K, Masters CL (1996) Alzheimer's disease amyloid precursor protein on the surface of cortical neurons in primary culture colocalizes with adhesion patch components. Brain Res 735: 217-231. [DOI] [PubMed] [Google Scholar]
- Su JH, Deng G, Cotman CW (1997) Neuronal DNA damage precedes tangle formation and is associated with up-regulation of nitrotyrosine in Alzheimer's disease brain. Brain Res 774: 193-199. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441-446. [DOI] [PubMed] [Google Scholar]
- Vincent I, Rosado M, Davies P (1996) Mitotic mechanisms in Alzheimer's disease? J Cell Biol 132: 413-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AH, Kaushal D, Rehen SK, Kriedt K, Kingsbury MA, McConnell MJ, Chun J (2003) Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J Neurosci 23: 10454-10462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Herrup K (2004) Cell cycle markers as potential outcome measures in preclinical trials: evidence from four mouse models. Paper presented at 56th Annual Meeting of the American Academy of Neurology, San Francisco, CA, April.
- Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci 21: 2661-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Mufson E, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer disease. J Neurosci 23: 2557-2563. [DOI] [PMC free article] [PubMed] [Google Scholar]