

Abstract
Long-term synaptic plasticity is both protein synthesis-dependent and synapse-specific. Therefore, the identity of the newly synthesized proteins, their localization, and mechanism of regulation are critical to our understanding of this process. Tissue plasminogen activator (tPA) is a secreted protease required for some forms of long-term synaptic plasticity. Here, we show tPA activity is rapidly increased in hippocampal neurons after glutamate stimulation. This increase in tPA activity corresponds to an increase in tPA protein synthesis that results from the translational activation of mRNA present at the time of stimulation. Furthermore, the mRNA encoding tPA is present in dendrites and is rapidly polyadenylated after glutamate stimulation. Both the polyadenylation of tPA mRNA and the subsequent increase in tPA protein is dependent on metabotropic glutamate receptor (mGluR) activation. A similar mGluR-dependent increase in tPA activity was detected after stimulation of a synaptic fraction isolated from the hippocampus, suggesting tPA synthesis is occurring in the synaptodendritic region. Finally, we demonstrate that tPA mRNA is bound by the mRNA-binding protein CPEB (cytoplasmic polyadenylation element binding protein-1), a protein known to regulate mRNA translation via polyadenylation. These results indicate that neurons are capable of synthesizing a secreted protein in the synaptic region, that mGluR activation induces mRNA polyadenylation and translation of specific mRNA, and suggest a model for synaptic plasticity whereby translational regulation of an immediate early gene precedes the increase in gene transcription.
Keywords: CPEB, dendrite, tPA, hippocampus, synaptoneurosome, mGluR
Introduction
The serine protease tissue plasminogen activator (tPA) is released by both neurons and glia in the brain (Teesalu et al., 2001; Tsirka, 2001). tPA expression can be found in many brain regions, including the hippocampus, amygdala, and cerebellum, where it plays a role in experience-driven synaptic plasticity (Baranes et al., 1998; Pawlak et al., 2003; Seeds et al., 2003). Overwhelming evidence suggests that tPA plays a critical role in synaptic changes associated with learning and memory. In the hippocampus, tPA synthesis is induced during stimuli that produce a long-lasting increase in synaptic strength [long-term potentiation (LTP)], and tPA-deficient mice have impaired synaptic plasticity and memory, whereas mice overexpressing tPA have enhanced LTP and learning (Qian et al., 1993; Huang et al., 1996; Baranes et al., 1998; Madani et al., 1999). In addition to hippocampal-dependent learning, tPA has been implicated in visual cortical plasticity, stress-induced anxiety-like behavior in the amygdala, and cerebellar motor learning (Mataga et al., 2002; Pawlak et al., 2003; Seeds et al., 2003).
How tPA release contributes to synaptic plasticity is unclear. However, several mechanisms have been proposed, including digestion of extracellular matrix proteins and partial proteolysis of the NMDA receptor (Nicole et al., 2001; Matys and Strickland, 2003), as well as nonproteolytic mechanisms, including the activation of protein kinase A via a cell surface receptor (Zhuo et al., 2000). Despite the clear evidence that tPA is involved in synaptic plasticity, little is known regarding the regulation of its synthesis and the site of release.
Identified as an immediate-early gene, transcription of tPA mRNA increases in the hippocampus after strong synaptic activation (Qian et al., 1993). Although there is little doubt that neural stimulation increases transcription of tPA, an increase of released tPA can be detected as early as 5 min after stimulation, although the increase in mRNA isn't detected until ∼30 min later (Baranes et al., 1998). This raises the possibility of translational regulation of tPA mRNA, at early times after stimulation, contributing to the LTP mechanism. Taken together with the finding that excess tPA is associated with neural cell death (Tsirka and Strickland, 1996; Siao et al., 2003), we felt that it was an ideal candidate to be regulated at the translational level.
In this study, we examined the possibility that mRNA encoding tPA was present in neurons before stimulation and that early synthesis of tPA is regulated by mRNA translational activation at stimulated synapses. This would provide the neuron with a rapid and spatially localized release of tPA. We found that glutamate stimulation of cultured neurons or isolated synaptoneurosomes leads to the polyadenylation and translation of tPA mRNA. Importantly, glutamate-induced tPA protein synthesis is dependent on the activation of metabotropic glutamate receptor (mGluRs) and independent of NMDA receptor (NMDAR) activation. However, glutamate also induces the polyadenylation and translation of the α-subunit of calcium-calmodulin-dependent protein kinase II (CaMKII) mRNA, a process that is NMDAR-dependent (Wells et al., 2001; Huang et al., 2002). This suggests that neurons are able to activate translation of specific mRNAs through NMDA and mGluR activation.
Materials and Methods
Cell cultures. Cultures of rat hippocampal neurons were made as described previously (Goslin and Banker, 1991). Briefly, the hippocampus was removed from embryonic day 18 (E18) rat embryos, trypsinized (0.25%), dissociated by trituration, and plated onto poly-l-lysine-coated (1 mg/ml) glass coverslips (100,000 cells/ml) for 3 hr. The coverslips were then transferred to dishes containing a monolayer of glial cells in growth medium (N2.1 media). Cultures were maintained by feeding with N2.1 twice per week.
Glial cells were isolated from postnatal day 1 rat pups (Goslin and Banker, 1991) and plated into T75 flasks at a concentration of 250,000 cells/ml. These cells were grown for 1-2 weeks, trypsinized, and replated into uncoated 12 well dishes at 100,000 cells/ml. When prepared in this way, the cultures contain no neurons as assayed by Western blot analysis using the neural specific anti-NeuN antibody (Chemicon, Temecula, CA) (data not shown).
Immunohistochemistry. Western blot analysis of rodent brain homogenate using several commercial anti-human tPA antibodies revealed multiple immunoreactive bands; therefore, we affinity purified a commercial antibody (American Diagnostica, Greenwich, CT) using the full-length recombinant mouse tPA protein (Calbiochem, La Jolla, CA; mouse and rat tPA proteins are >90% identical). The resultant antibody yielded a single band at ∼68 kDa when brain homogenate was probed by polyacrylamide gel electrophoresis followed by Western blot analysis. Immunohistochemistry on hippocampal neurons 14-16 d in vitro (DIV) was performed as described previously (Wu et al., 1998; Wells et al., 2001). Synapses were detected with anti-synaptophysin (1:2000; Chemicon), and tPA was localized using the antibody described above (1:500). Fluorescent and phase images were taken on a Nikon (Tokyo, Japan) Eclipse E800 microscope and processed using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA).
Stimulation protocol. Individual coverslips containing hippocampal neurons 14-16 DIV were transferred to 12 well plates and stimulated with glutamate for 1-2 min. The cultures were then washed with fresh media that lacked glutamate for an additional 20 min. Cells were harvested with buffer, and tPA levels were analyzed by zymography. Where appropriate, drugs were applied 5 min before stimulation and remained in the incubation media for the duration of the experiment. Poly(A)-tail length was also determined after the same stimulation protocol. Samples from two or three coverslips per treatment group were combined before running the analysis [zymography or poly(A)-tail assay].
tPA zymography. tPA activity was assayed by direct zymography (Heussen and Dowdle, 1980). Briefly, samples were mixed with sample buffer without reducing reagent at room temperature. The samples were then run on a 10% polyacrylamide gel containing casein (1 mg/ml; Sigma, St. Louis, MO) and plasminogen (13 μg/ml; American Diagnostica). The gel was washed for 30 min with 2.5% Triton X-100 to remove SDS and further washed for 10 min in 0.1 m Tris buffer, pH 8.0. The gel was incubated in the Tris buffer for 12-24 hr at 37°C to allow caseinolysis to occur. The gel was then stained with Coomassie Brilliant Blue (G-250) and destained with 20% methanol and 10% acetic acid. On the darkly stained casein background, PA activity was visualized as light bands resulting from casein degradation. The caseinolysis band detected at 68 kDa is specific for tPA (Heussen and Dowdle, 1980) and corresponds to the band of purified tPA run in the same gel (data not shown). The zymogram was analyzed using Epi Chemi II Darkroom (UVP, Upland, CA) equipped with the image analysis program LabWorks. All pictures of zymography are presented as inverted images prepared using Photoshop 7.0.
Probe preparation. Four different oligonucleotide probes (45-53 nucleotides in length) complementary to tPA mRNA and directed at regions in the 3′-untranslated region (UTR), 5′-UTR, and coding region, were used for in situ hybridization. Oligonucleotide probes were end-labeled with digoxigenin (DIG) according to manufacturer specifications (DIG oligonucleotide tailing kit second generation; Roche Applied Science, Indianapolis, IN). Two oligonucleotide probes complementary to the enhanced green fluorescent protein (eGFP) mRNA coding region were also prepared. Probe sequences: tPA1: tctgggtttaaggagctgggctttctttgtcattgcttcatgttgtctt; tPA2: gtgggaggatggggaatatttgatttcaagggttttagggggctgtggt; tPA3: agcaggacgcacaacagctctcccttcattttgctccccgtttcttccgtgtc; tPA4: cagtgccgacagaactgctctcctgggcacactgactggaatctgacctc; eGFP1: atggcggacttgaagaagtcgtgctgcttcatgtggtcggggtag; and eGFP2: cgagatctgagtccggacttgtacagctcgtccatgccgagagtga.
Hybridization. In situ hybridization was performed according to Bassell et al. (1998) with slight modifications. Briefly, 12-16 DIV hippocampal neurons were fixed for 15 min at room temperature with 4% paraformaldehyde in 1× PBS and 5 mm MgCl2. Cells were washed three times in 1× PBS with 5 mm MgCl2 and permeabilized for 5 min at room temperature with 0.1% Triton X-100. Cells were equilibrated for 20 min at room temperature in 1× SSC, 50% formamide, and 10 mm sodium phosphate, pH 7.0. Coverslips were hybridized face down for 5 hr at 37°C on a 40 μl drop of probe-hybridization buffer mixture consisting of 40 ng of tPA probes in 50% formamide, 2× SSC, 10% dextran sulfate, 40 μg tRNA, 0.2% BSA, 10 μg salmon sperm DNA, 20 μm vanadyl ribonucleoside complex, and 10 mm sodium phosphate. After hybridization, cells were washed for 20 min at 37°C in 50% formamide with 1× SSC. Cells were then washed at room temperature in 1× SSC on a rocker. Neurons were incubated for 1 hr at room temperature in 0.1 m Tris, pH 7.5, 0.15 m NaCl, and 1% BSA before incubation overnight at 4°C with a 1:300 dilution of mouse anti-DIG-Cy3 in block solution. Cy3-labeled probes were visualized on Nikon Eclipse E800 microscope, and controls and tPA images were identically processed using Adobe Photoshop 7.0.
The specificity of the tPA mRNA probes was demonstrated by using probes specific for GFP mRNA, an mRNA not expressed in hippocampal neurons. The concentration of probe was identical to that of the tPA probes, and the coverslips were processed in parallel with coverslips treated with tPA probes. In addition, the hybridization signal detected with DIG-labeled tPA probes could be eliminated by hybridizing labeled probe with a 100-fold higher concentration of unlabeled tPA probes (data not shown).
Preparation of synaptoneurosome. Synaptoneurosome (SN) fractions were isolated as previously reported with slight modification (Quinlan et al., 1999; Wells et al., 2001). Briefly, the tissues were homogenized in ice-cold buffer (50 mm HEPES, 100 mm NaCl, 3 mm KAc, and 1 mm MgSO4, pH 7.4) and were centrifuged at 2000 × g for 1 min. Supernatants were passed through two 100 μm nylon mesh filters, followed by a 5 μm pore filter. The filtrate was then centrifuged at 1000 × g for 10 min and then was gently resuspended with same buffer at a protein concentration of 2 mg/ml.
For Western blot, equal amounts of total protein (20 μg) from the SN fractions were resolved on a 8-10% polyacrylamide gel, blotted, and probed with antibodies against several synaptic marker proteins including α-CaMKII (Chemicon), postsynaptic density-95 kDa (PSD-95) (Upstate Biotechnology, Charlottesville, VA), and NMDAR1 (PharMingen, San Diego, CA), followed by horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA). The immunoreactivity was revealed using ECL reagents (Amersham, Buckinghampshire, UK).
For in vitro stimulation analysis, the hippocampus was removed from 4- to 6-week-old Sprague Dawley rats, bisected, and a SN fraction was prepared from each half. These samples were then combined and separated into equal aliquots for the stimulation assay. The stimulated SNs were treated with 100 μm glutamate for 30 min at 37°C. Where indicated, drugs were added 5 min before glutamate addition and kept in throughout the duration of the stimulation (30 min). After stimulation, an equal volume of 2× zymography buffer (4% SDS, 0.25 M Tris, 20% glycerol) was added to each sample, and tPA activity was measured by zymography.
Polyadenylation of tPA mRNA. To test the poly(A)-tail length of tPA mRNA, the poly(A)-tail test (PAT) assay was performed according to Salles et al. (1992) with minor modifications. Briefly, total RNA was isolated from brain (Rneasy; Qiagen, Valencia, CA) or cultured hippocampal neurons and SN preparations (Cell to cDNA II; Ambion, Houston, TX), and 1 μg of total RNA was used for the reverse transcription with an oligo(dT)-anchor primer (5′ GCGAGCTCCGCGGCCGCGT(12)). Subsequent PCR was performed using the oligo(dT)-anchor and a specific upstream primer for the test RNA. The upstream primers were: tPA 5′ TCCAAGAGAAGGAAGGGGTT; neurofilament-1 (NFM1) 5′ GAGATGTATTACGCAAAGTACC. The PCR products were visualized with ethidium bromide and resolved in a 2% agarose gel.
Distribution of tPA mRNA in synaptoneurosome. To determine relative distribution of tPA mRNA between crude homogenate and SN, we used an end-point RT-PCR analysis (Havik et al., 2003). Briefly, total RNA was prepared from rat hippocampus or SN, and 0.5-4 μg was reverse-transcribed for 1 hr at 37°C. An aliquot (2-4 μl) of cDNA was used for PCR amplification of several mRNAs using Hot start Taq polymerase (Qiagen). Crude homogenate samples were amplified for 30 cycles, and SN samples were amplified for 45 cycles. Each cycle consisted of denaturation at 95°C for 30 sec, annealing at 55°C for 1 min, and extension at 72°C for 1 min. After amplification, equal volumes of crude hippocampus and SN PCR products were loaded on 1.5% agarose gel side by side, and band intensity was determined using Epi Chemi II Darkroom (UVP) equipped with UV transilluminator and CCD camera. The integrated density values obtained from SN sample was divided by those obtained from crude hippocampus sample to obtain a ratio indicating the relative distribution in the SN for each particular mRNA species. The primers used in this analysis are: tPA, 5′TCAGATGAGATGACAGGGAAATGCC (forward) and 5′ATCATACAGTTCTCCCAGCC (reverse); Histone H1, 5′GGTGGCTTTCAAGAAGACCAA (forward) and 5′TGAGGTCTGTTTGCTGTCCTT (reverse); glial fibrillary acidic protein (GFAP), 5′GCTAATGACTATCGCCGCCAACT (forward) and 5′ATCCCGCATCTCCACCGTCTTTAC (reverse); β-Actin, 5′GGTCAGGTCATCACTATCGGC (forward) and 5′CGCAGCTCAGTAACAGTCCGCC (reverse); and α-CAMKII, 5′TTCAGGGGCAATAGCAAGCAACAC (forward) and 5′ATGGGGAGGGAGAGCACGAAGATT (reverse).
Cytoplasmic polyadenylation element binding protein-1 binding of tPA mRNA. Glutathione S-transferase (GST) fusion proteins were constructed using a pCDNA4 expression plasmid (Invitrogen, Carlsbad, CA) and the sequence coding for full-length human cytoplasmic polyadenylation element binding protein-1 (CPEB) (a generous gift from Dr. Angus MacNicol). The resultant fusion protein (GST-CPEB) was expressed in COS7 cells by transfecting the plasmid using Lipofectamine 2000 (Invitrogen). The full-length 3′-UTR of tPA was amplified from rat brain using RT-PCR and cloned into pCR2.1 vector (Invitrogen). The 3′-UTR was then in vitro transcribed using T7 RNA polymerase (New England Biolabs, Beverly, MA). COS7 cells expressing the GST-CPEB fusion protein or mock transfected were lysed, and the soluble fraction was mixed with in vitro transcribed tPA 3′-UTR. This mixture was added to glutathione-Sepharose, and the bound protein was separated and washed three times. Bound RNA was recovered by phenol:chloroform extraction and analyzed by RT-PCR for the presence of tPA 3′-UTR. To minimize RNA degradation, all the reagents were prepared in RNase-free buffer, and RNase inhibitor was added to RNA-GST CPEB fusion protein incubation mixture at a final concentration of 200 U/ml (Roche, Mannheim, Germany).
Immunoprecipitation of CPEB from rat brain homogenate was performed as previously described (Brown et al., 2001), with slight modifications. Briefly, both hemispheres of the hippocampus were removed from two Sprague Dawley rats (4 weeks old) and homogenized in 2 ml of ice-cold buffer (10 mm HEPES, pH 7.4, 200 mm NaCl, 30 mm EDTA, and 0.5% Triton X-100) with 200 U/ml RNase Inhibitor (Roche). After centrifugation for 5 min at 2000 × g, the supernatants were raised to 400 mm NaCl and spun at 16,000 × g for 30 min. An aliquot of the resulting supernatant was mixed with 10 μg of rabbit anti-CPEB for 20 min and immunoprecipitated with 50 μl protein A for 30 min. The immunoprecipitates were washed three times for 10 min each and bound RNA was recovered by phenol:chloroform and ethanol precipitation. After resuspension in Rnase-free water, the precipitates were treated with DNase I (50 U/ml; Ambion) for 15 min at 37°C and reverse-transcribed for RT-PCR analysis.
Statistical analysis. Data are expressed as the mean ± SEM and analyzed for statistical significance by using one-way ANOVA followed by Neuman-Keuls test as a post hoc test, and a p value <0.05 was considered significant.
Results
tPA localization to the synapse
To determine the cellular localization of tPA, cultured hippocampal neurons (14 DIV) were immunostained with an affinity-purified anti-tPA antibody (Fig. 1). Synapses were identified using an antibody to the synaptic vesicle protein synaptophysin. tPA immunoreactivity was observed in the cell body and in large puncta that colocalized with synaptophysin, as well as in more discrete puncta within dendrites that were not localized to synapses (Fig. 1B,C).
Figure 1.

Expression of tPA protein localizes to dendrites and synapses. A, Western blot of rat hippocampal homogenate probed with an affinity-purified anti-tPA antibody recognizes a single band of ∼68 kDa (arrow). Molecular weight markers shown on the left. B, tPA immunoreactivity (red) is detected in the cell body and distributed in puncta on hippocampal neurons in culture. This distribution is similar to that of synaptophysin (green). C, Images in B overlaid with each other and on the bright-field phase image showing dendritic distribution of tPA. Yellow puncta indicate a colocalization of tPA with synaptophysin (arrows). Box indicates an area of the cell shown at higher magnification in the right panel showing tPA protein also distributed in small puncta throughout the dendrite (arrowheads). Scale bars: B, C, left panel, 20 μm; C, right panel, 5 μm.
Glutamate stimulates neuronal tPA synthesis
The presence of tPA at synapses raises the possibility that tPA might be released after synaptic activation. To examine this possibility, we stimulated cultured hippocampal neurons with glutamate and measured the levels of tPA activity in the released and cell-associated fractions by zymography. To our surprise we saw a rapid increase in the tPA activity in both fractions, suggesting that the total level of tPA had increased (Fig. 2A). To determine if this were the case, a discrete glutamate stimulation (2 min) was applied, followed by a wash-out of the glutamate-containing media (including any immediately released tPA) with fresh media. To ensure glutamate receptor inactivation, in some experiments fresh media containing a glutamate receptor antagonist mixture (50 μm AP-5 plus 10 μm CNQX) was washed in. The results were the same with both protocols (data not shown). The cells were harvested 20 min after the washout of glutamate and processed for zymography. A dose-dependent increase in total tPA activity was detected (Fig. 2A). This significant increase in both released and cell-associated tPA levels indicated that an increase in neural activity was driving a rapid increase in total tPA activity.
Figure 2.

Stimulation of tPA synthesis by glutamate in hippocampal neurons in culture. A, Hippocampal neurons in culture (14-16 DIV) were treated with increasing concentrations of glutamate and the level of tPA activity was examined by zymography. Shown is a dose-dependent increase in both released tPA and cell associated tPA (n = 4). B, Astrocytes in culture (14 DIV) were treated with glutamate as in A. Zymography of total tPA activity shows glutamate does not stimulate tPA in glial cells (n = 4). C, Hippocampal neurons in culture were stimulated with 20 μm glutamate for 20 min and then processed for Western blot analysis using antibodies specific for tPA and PAI-1. Glutamate stimulation resulted in a significant increase in tPA protein (121.5 ± 9.2% of unstimulated; p < 0.05; n = 8) but no change in the tPA inhibitor PAI-1 (101.9 ± 2.2% of unstimulated; p < 0.4; n = 5). For comparison to the zymography data shown in A, the size of the increase in tPA protein stimulated by 20 μm glutamate was indistinguishable from that obtained by 100 μm glutamate (data not shown). A representative zymogram is shown above A and B, whereas a representative Western blot is shown in C. *Statistically significant difference compared with unstimulated group (p < 0.05; mean ± SEM; n = 4).
The hippocampal cultures consist of ∼95% neurons and 5% glia. Therefore, to determine if this increase in tPA activity was coming from neurons or glia, we stimulated a pure glial cell culture (see Materials and Methods) with glutamate and found no change in the total tPA activity (Fig. 2B). Under the conditions in which we perform the zymography, all tPA protein is activated; therefore, the tPA activity is representing the total level of tPA protein present. To confirm this, we processed glutamate-stimulated hippocampal neurons for tPA protein levels by Western blot analysis (Fig. 2C) and show a significant increase in tPA protein (121.5% ± 9.2% of unstimulated; p < 0.05; n = 8). However, the level of plasminogen activator inhibitor 1 (PAI-1), the primary inhibitor of tPA activity in the brain, remains constant (Fig. 2C). Taken together, this indicates that changes in tPA activity assessed by zymography are reflective of a change in total tPA protein.
To determine if the increase in tPA was mediated by new protein synthesis, we pretreated cultured neurons with various protein synthesis inhibitors 5 min before glutamate stimulation. Translation inhibitors cycloheximide (100 μm) and anisomycin (20 μm) significantly inhibited the glutamate-induced increase in tPA (Fig. 3A). In addition, the polyadenylation inhibitor cordycepin (20 μm) similarly prevented the increase in tPA. Cordycepin (3′-deoxyadenosine) can activate adenosine receptors; therefore, we treated neurons with adenosine (20 μm) before glutamate stimulation. Adenosine did not inhibit glutamate-induced tPA synthesis (Glu: 157.97 ± 22.45% and Glu plus adenosine: 153.31 ± 17.75% compared with control). The transcription inhibitor actinomycin D (5 μm) had no effect on the glutamate-induced increase in tPA levels (Fig. 3A). To assure that we were completely blocking new transcription (Hyman and Davidson, 1970), we pretreated the neurons with actinomycin D for 30 min before glutamate stimulation, yet tPA was still induced (Glu: 159.15 ± 22.32% and Glu + act. D: 155.36 ± 27.11% compared with control), indicating that the increase in tPA was derived from an activation of mRNA translation, and furthermore, that this mRNA was already present at the time of the stimulation.
Figure 3.

Glutamate-induced tPA increase is dependent on protein synthesis and mediated by mGluR activation. A, Glutamate-induced tPA increase is mediated by translation of tPA mRNA present at the time of stimulation. Hippocampal neurons in culture were treated as in Figure 2 and processed for zymography. Translation inhibitors cycloheximide (CX) and anisomycin (AN) as well as the mRNA polyadenylation inhibitor cordycepin (CD) inhibited the glutamate-induced increase in tPA. However, the transcription inhibitor actinomycin D (AD) had no effect on the tPA increase. B, Activation of the mGluR is required for glutamate-induced tPA synthesis. The mGluR antagonist MCPG inhibited the tPA increase by glutamate, whereas the NMDAR antagonist AP-5 and the AMPA receptor antagonist CNQX had no effect. C, The group I mGluR-selective agonist DHPG alone increased tPA level to the same extent as glutamate. *Statistically significant difference compared with unstimulated (CON) cells; #statistically significant difference compared with glutamate stimulated group (p < 0.05; mean ± SEM; n = 4; representative zymogram shown above each graph).
To determine the glutamate receptor subtype responsible for inducing the increase in tPA, glutamate stimulations were performed in the presence of various glutamate receptor antagonists. In cultured neurons pretreated with 200 μm α-methyl-4-carboxyphenylglycine (MCPG), a group I mGluR antagonist, glutamate failed to produce an increase in tPA levels (Fig. 3B). However, neither the NMDA-type glutamate receptor antagonist AP-5 (50 μm) nor the non-NMDAR antagonist CNQX (10 μm) inhibited the tPA increase (Fig. 3B). In addition, the specific group I mGluR agonist dihydroxyphenylglycine (DHPG) (25 μm) resulted in an increase in tPA levels that were similar to that induced by glutamate (Fig. 3C).
Site of tPA synthesis
To determine if the mRNA for tPA is present in dendrites, we performed fluorescent in situ hybridization (FISH) on hippocampal neurons using probes specific for tPA (tPA-FISH) (Fig. 4). To control for nonspecific binding of the tPA probes, tPA-FISH was compared with fluorescent probes directed against green fluorescent protein, an mRNA not found in hippocampal neurons (GFP-FISH) (Fig. 4) or tPA-FISH performed in the presence of 100-fold excess of unlabeled tPA probes (data not shown). Results were similar using either control. Images obtained using the same exposure time, and processed identically revealed tPA mRNA within the cell body and in small puncta that extended into dendrites (Fig. 4).
Figure 4.

Expression of tPA mRNA in dendrites. A, Hippocampal neurons were processed for tPA fluorescent in situ hybridization (tPA-FISH) or green fluorescent protein-FISH (GFP-FISH). tPA mRNA is detected in the cell body and in small puncta expressed along the dendritic arbors (arrows). GFP mRNA is not expressed in these neurons; therefore, GFP-FISH demonstrates the specificity and strength of tPA signal. B, A higher magnification of the boxed areas in A reveals tPA mRNA is often found at branch points along the dendrites as well as in structures resembling synaptic spines (arrowheads). Similar results were obtained in three experiments from three separate cultures ranging from 12-14 DIV. Scale bars: A, 20 μm; B, 5 μm.
To investigate the presence and possible release of tPA from the synaptic compartment, we generated SNs, a biochemical preparation that enriches for synaptic components (Hollingsworth et al., 1985). Electron microscopy of our hippocampal SN fraction reveals profiles consisting of adjacent resealed membranes, with one compartment containing presumptive synaptic vesicles opposite a membrane resembling a postsynaptic density (Fig. 5A). Furthermore, the SN fraction enriches for synaptic proteins, including PSD-95, NMDAR1, synaptophysin, and α-CaMKII, whereas the cytoskeletal protein α-tubulin 1, the nuclear protein histone H1, and the glia-expressed GFAP are not SN-enriched (Fig. 5B). Consistent with the immunohistochemical localization of tPA to synapses, zymographic analysis shows tPA enriched in the SN fraction at least threefold compared with crude homogenates (Fig. 5B).
Figure 5.
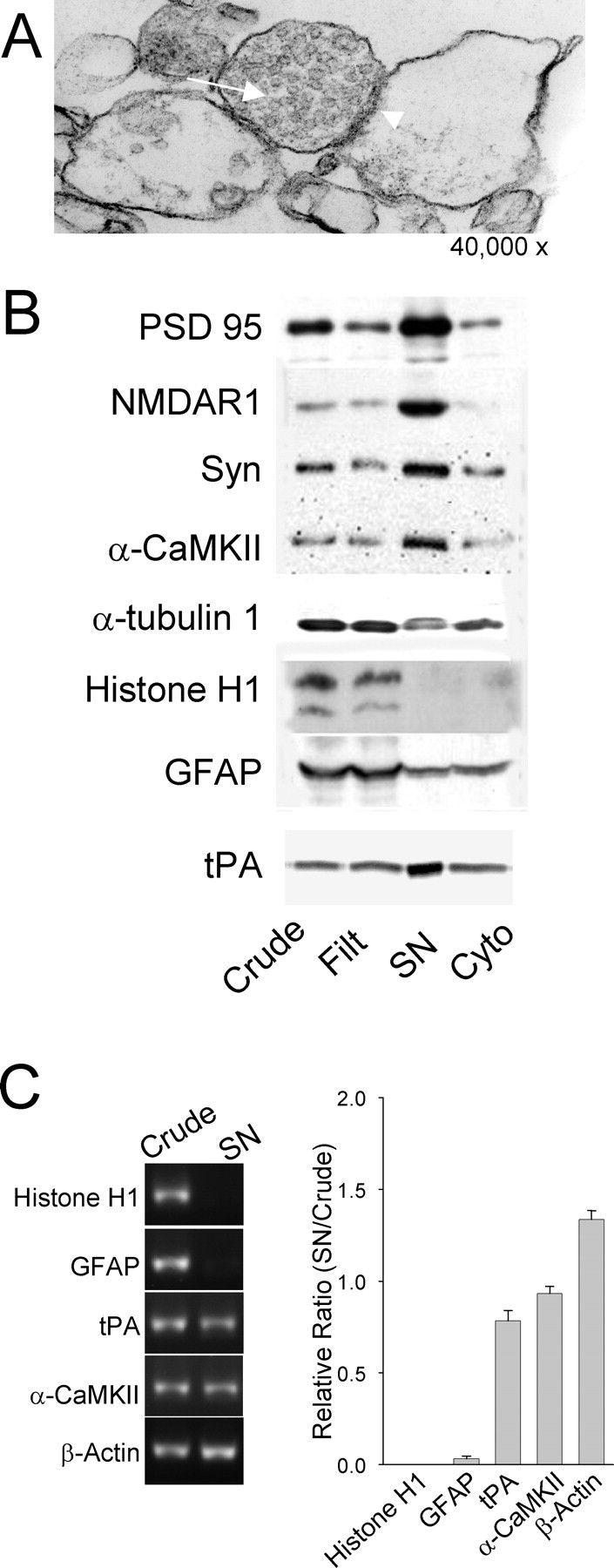
Expression of tPA protein and mRNA in the synaptic fraction isolated from the hippocampus. A, Electron micrograph of SN fraction isolated from hippocampus showing a typical profile consisting of adjacent resealed membranes with one side containing synaptic vesicles (arrow) and the adjacent membrane resembling a postsynaptic density (arrowhead). B, Characterization of protein enrichment of SN fraction showing tPA in synaptic fraction. Crude homogenate (Crude), filtrate, synaptoneurosome pellet (SN), and supernatant (Cyto) fractions were analyzed by Western blot to determine the distribution pattern of synaptic and nonsynaptic proteins or by zymography for tPA. Synaptic proteins PSD-95, NMDAR1, synaptophysin, α-CaMKII, and tPA were enriched in the SN compared with crude. The glial protein GFAP, the nuclear protein Histone H1, and the cytoskeletal protein α-tubulin were either diminished or absent in the SN fraction. C, Presence of tPA mRNA in the synaptic fraction. RT-PCR analysis using mRNA-specific primers for the indicated mRNA was performed from the same crude homogenate and SN preparations. The mRNAs for tPA, α-CaMKII, and β-actin are relatively enriched in the SN fraction compared with nondendritically localized mRNAs for Histone H1 and GFAP. A ratio of the SN band intensity divided by the level in the crude homogenate is plotted on the right (n = 3).
The amount of tPA mRNA expressed in hippocampal neurons is very low (Baranes et al., 1998). Therefore, to assess the relative distribution of tPA mRNA we amplified the mRNA by RT-PCR from both the crude homogenate and SN fraction and compared its distribution with that of other mRNAs (Fig. 5C). This endpoint analysis allows for the comparison of the relative amount of mRNA in the synaptic fraction compared with the total expressed for that particular mRNA. It is not a quantitative analysis, so absolute amounts cannot be determined. However, this analysis did reveal that the mRNA encoding tPA is present in our hippocampal SN preparation and that its relative distribution is similar to known dendritically targeted mRNAs encoding α-CaMKII and β-actin (Mayford et al., 1996; Eom et al., 2003) (Fig. 5C). As controls for the sensitivity of the assay and as a further indication that the SN fraction is indeed a synaptic fraction, mRNAs not thought to be dendritically targeted (encoding histone H1 and GFAP) are not detected in the SN fraction.
To determine if tPA mRNA localized to the synaptic compartment could be under the same translational control we detected in intact neurons, we developed an in vitro assay using the SN fraction. Glutamate stimulation of the SN fraction isolated from the hippocampus yields a dose-dependent increase in tPA levels detected by zymography (Fig. 6A). No changes in the levels of GFAP or PSD-95 were detected in Western blot analysis, however, an increase in α-CaMKII protein was observed (data not shown). Consistent with results obtained with cultured hippocampal neurons, protein translation inhibitor cycloheximide prevented the increase in tPA, whereas the transcription inhibitor actinomycin D had no effect on glutamate stimulated tPA synthesis (Fig. 6B). SN fractions treated with MCPG and to a lesser extent CNQX, but not AP-5, showed an inhibition of the glutamate-induced increase in tPA (Fig. 6C). Furthermore, DHPG but not the group II mGluR-selective agonist 4-aminopyrrolidine-2,4-dicarboxylate (APDC) (20 μm) increased SN tPA levels (Fig. 6D). This mGluR-mediated increase in tPA translation is dependent on polyadenylation of tPA mRNA because cordycepin blocks the increase (Fig. 6B).
Figure 6.
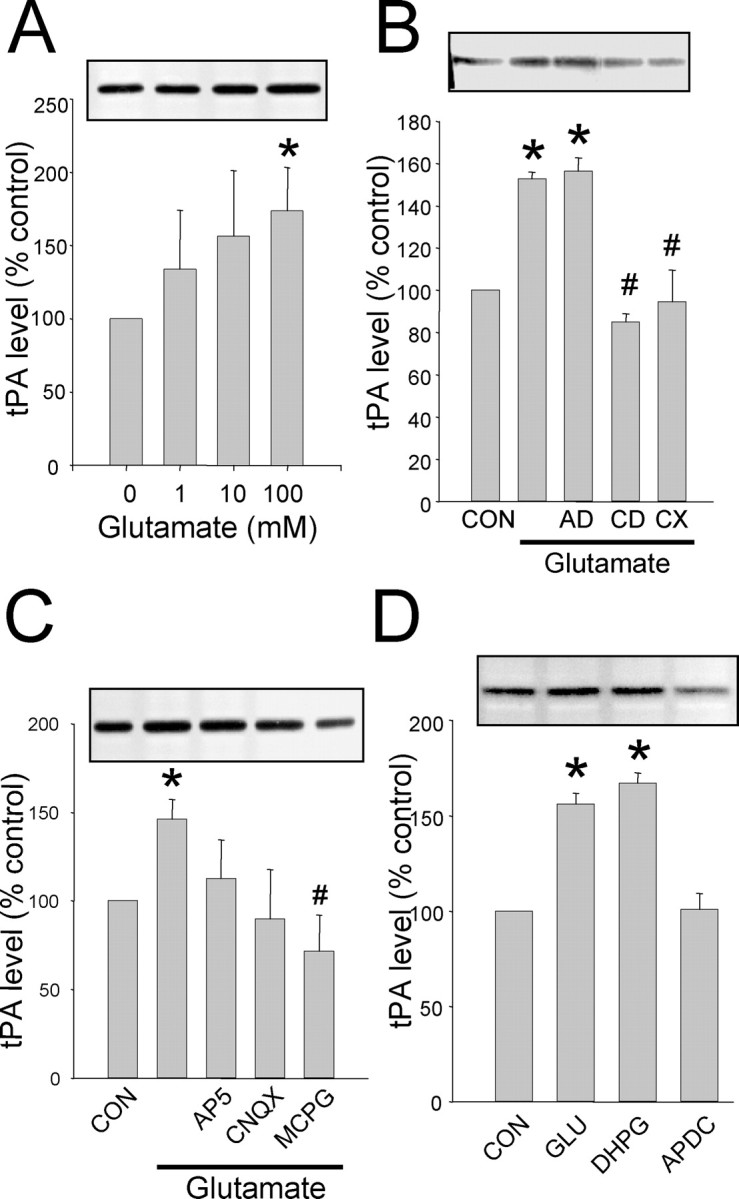
Synaptic protein synthesis of tPA induced by glutamate. A, The SN fraction was isolated from the hippocampus and stimulated with increasing concentrations of glutamate for 30 min, then tPA was analyzed by zymography. B, SNs were pretreated with AD, CD, and CX before and during glutamate (100 μm) stimulation as in A. C, tPA synthesis is dependent on the activation of mGluR. All glutamate receptor antagonists were added 5 min before glutamate stimulation as in B. D, The type I mGluR agonist DHPG mimics glutamate stimulation; however, the group II mGluR agonist APDC does not increase tPA levels. *Statistically significant difference from control (CON); #statistically significant difference from glutamate-stimulated group (p < 0.05; mean ± SEM; n = 4 representative zymograms shown above each graph).
Regulation of tPA mRNA translation
These results suggested that glutamate, via the activation of mGluR, was inducing a rapid translation of tPA mRNA into protein. Because of its sensitivity to cordycepin, we determined the status of the poly(A)-tail length of tPA mRNA by using a PAT assay (Wu et al., 1998; Huang et al., 2002). As early as 2 min after glutamate stimulation, an increase in poly(A)-tail length for tPA mRNA was observed (Fig. 7A). Similar kinetics were observed for α-CaMKII mRNA, the only other dendritic mRNA thus far shown to be regulated by polyadenylation after synaptic activation (Wu et al., 1998; Wells et al., 2001; Huang et al., 2002). Poly(A)-tail length of NFM-1 mRNA was not changed by glutamate stimulation (Fig. 7). Furthermore, the polyadenylation inhibitor cordycepin and the mGluR antagonist MCPG prevented the glutamate-induced increase in the poly(A)-tail length of tPA mRNA (Fig. 7B). Glutamate stimulation leads to the polyadenylation and translation of α-CaMKII mRNA that is completely blocked by the NMDAR antagonist AP-5 (Wells et al., 2001; Huang et al., 2002). Taken together, these data support a model whereby hippocampal neurons can activate translation of distinct mRNAs downstream of either NMDAR or mGluR activation.
Figure 7.

tPA mRNA is rapidly polyadenylated after glutamate stimulation. A, Cultured hippocampal neurons (14-16 DIV) were stimulated with glutamate for 2, 5, and 10 min, and the poly(A)-tail length was determined by PAT assay. An increase in poly(A)-tail length was evident even at 2 min after glutamate stimulation. B, Inhibition of polyadenylation by cordycepin (CD) and MCPG. Cultured neurons as in A were treated with CD, MCPG, or AP-5 before and during glutamate stimulation. Poly(A)-tail length was determined 10 min after glutamate stimulation. CD and MCPG both inhibited the glutamate-induced polyadenylation of tPA mRNA. Neurofilament mRNA (NFM1) polyadenylation was not altered under any condition. Representative experiment shown; similar results were obtained in n = 3 experiments for both A and B. Scale bar shown on side of tPA gels represents 250 nucleotides.
The polyadenylation of tPA mRNA at activated synapses led us to investigate whether tPA mRNA isolated from the brain could bind CPEB. To determine if tPA mRNA is bound by CPEB, we immunoprecipitated (IP) endogenous CPEB from rat brain homogenates, isolated the mRNA that co-IP, and used mRNA-specific primers to detect the presence of tPA mRNA (Fig. 8). The CPE-containing mRNAs encoding tPA and α-CaMKII co-IP with CPEB, whereas the non-CPE containing mRNA encoding GFAP did not. Because CPEB is thought to bind mRNA in the 3′-UTR, we expressed a GST-CPEB fusion protein in COS7 cells, harvested the cells, and combined the soluble fraction with in vitro transcribed tPA 3′-UTR. Figure 8B shows that the CPEB fusion protein was capable of binding to the 3′-UTR of tPA mRNA.
Figure 8.

CPEB binds tPA mRNA through an interaction with the 3′-UTR. A, Native CPEB binds to endogenous tPA mRNA. CPEB was immunoprecipitated (IP) from rat brain homogenate using either an anti-CPEB antibody (α-CPEB) or a nonspecific IgG (CON) and Western blotted to determine the specificity of the IP (left panel). RT-PCR was then performed after the IPs to determine what mRNAs were bound by CPEB. The CPE-containing mRNAs encoding α-CaMKII and tPA were both found to associate with CPEB, whereas the non-CPE-containing mRNA encoding GFAP was not (right panel). B, Full-length human CPEB was expressed as a GST-CPEB fusion protein in COS7 cells and combined with in vitro transcribed RNA encoding the entire 3′-UTR of rat tPA mRNA. The top panel is the RT-PCR product of the input RNA indicating equivalent loading. The bottom panel shows the RT-PCR product if GST alone (CON) or GST-CPEB was used to pull down tPA 3′-UTR.
Discussion
There is mounting evidence that tPA is involved in synaptic plasticity throughout the brain. Mice deficient in tPA have impaired LTP in the hippocampus and impaired hippocampal and cerebellar-dependent learning (Baranes et al., 1998; Calabresi et al., 2000; Seeds et al., 2003). Our results demonstrate that tPA expression in the hippocampus is under translational control and argues for a mechanism by which strong glutamate stimulation elicits an immediate tPA response that precedes an increase in mRNA synthesis. This activity-induced tPA synthesis is mediated through the activation of a type I mGluR and leads to the polyadenylation of tPA mRNA, possibly through its interaction with the mRNA binding protein CPEB.
Glutamate-induced tPA mRNA polyadenylation and translation is dependent on mGluR activation. This is consistent with earlier reports demonstrating mGluR-mediated dendritic protein synthesis (Weiler and Greenough, 1993; Weiler et al., 1997; Kacharmina et al., 2000; Ju et al., 2004). In addition, DHPG treatment induces a form of hippocampal LTD that is dependent on dendritic protein synthesis (Huber et al., 2000). Furthermore, tPA induction has also been implicated in hippocampal LTD (Napolitano et al., 1999; Calabresi et al., 2000) as well as hippocampal LTP (Qian et al., 1993; Baranes et al., 1998). This suggests tPA may facilitate structural changes during many forms of synaptic plasticity. Located in a narrow zone surrounding the postsynaptic specialization, mGluRs may be specially situated to respond to glutamate only during synaptic stimulation that will lead to changes in synaptic strength (Lujan et al., 1997; Takumi et al., 1999). Taken together, this suggest that mGluR activation of tPA synthesis may have a role in establishing structural changes involved in both the strengthening and weakening of synaptic connections.
The mechanism by which tPA mRNA is polyadenylated may be regulated by the binding of CPEB. The binding site for CPEB is located in the 3′-UTR of responsive mRNAs and is called the cytoplasmic polyadenylation element (CPE) (Mendez and Richter, 2001). The CPE consensus sequence (UUUUUAAU) is usually located within 100 nucleotides of the hexanucleotide sequence AAUAAA, which signifies the end of the 3′-UTR (Mendez and Richter, 2001). The 96 nucleotides upstream of the hexanucleotide sequence in the tPA 3′-UTR sequence have two of these consensus sites (UUUUAAU-27nt-UAUUUUAAU-53nt-AAUAAA-; GenBank accession number J03520). CPEB-mediated polyadenylation is activated after CPEB phosphorylation (Richter, 2000; Atkins et al., 2004). Here, we show that tPA mRNA binds to CPEB through a sequence in the 3′-UTR and that tPA mRNA is polyadenylated before the increase in tPA activity. Although these results indicate that CPEB could be mediating tPA mRNA polyadenylation, Huang et al. (2002) demonstrated that DHPG stimulation in the hippocampus does not phosphorylate CPEB. The mechanism of CPEB action on tPA mRNA is still unclear, for example binding alone may influence deadenylation (see below) or a nonphosphorylatable CPEB protein family member may be responsible for tPA mRNA translational regulation (Theis et al., 2003). We are currently investigating if CPEB binding and/or phosphorylation is required for tPA mRNA polyadenylation. However, the current findings clearly show that mGluR activation can lead to tPA mRNA polyadenylation and translation, and suggest that the different glutamate receptors can activate distinct mRNAs.
Cytoplasmic polyadenylation of mRNA could lead to an increase in translation by at least two mechanisms. In the oocyte, CPEB-mediated polyadenylation acts to promote binding sites for poly(A)-binding protein, which in turn relieves translational inhibition and recruits the initiation factor eIF4G to the 5′-cap, thus promoting translation initiation (Cao and Richter, 2002). Alternatively, the elongation of the poly(A)-tail and/or the binding of CPEB could lead to the stabilization of tPA mRNA. Specifically, tPA mRNA contains AU-rich elements (ARE) in the 3′-UTR that are known to promote destabilization of mRNA through deadenylation (Chen et al., 1995; Chen and Shyu, 1995). The polyadenylation of tPA mRNA, or CPEB binding and “masking” the AREs could prolong the half-life of tPA mRNA and promote greater translation.
We present several lines of evidence suggesting that the immediate tPA response to synaptic stimulation is mediated by mRNA translation at the synapse. First, the mRNA encoding tPA is present in dendrites and is localized to the synaptic fraction in a similar proportion to that of known dendritic mRNAs encoding α-CaMKII and β-actin. Second, both intact hippocampal neurons and a biochemical fraction isolated from the hippocampus containing intact synapses respond to glutamate by synthesizing new tPA that does not require new mRNA transcription. Finally, tPA mRNA can interact with CPEB (Fig. 8), a postsynaptic density protein (Wu et al., 1998). Although this is the first report of a secreted protein being synthesized in the dendritic region, others have demonstrated the synthesis and membrane insertion of a transmembrane protein using a dendrite in culture isolated from its cell body (Kacharmina et al., 2000; Ju et al., 2004). In addition, the presence of protein synthetic machinery within postsynaptic spines, including membranes analogous to rough endoplasmic reticulum and the trans-golgi network, was recently demonstrated by immunoelectron microscopy (Pierce et al., 2001). Furthermore, a redistribution of polyribosomes into the dendritic spine is seen after LTP-inducing stimulation in the hippocampus (Ostroff et al., 2002). Taken together, we feel this argues strongly for the postsynaptic synthesis of tPA.
Transcription of tPA mRNA is also induced after synaptic activation, peeking at ∼4 hr (Qian et al., 1993; Fiumelli et al., 1999; Seeds et al., 2003). Consistent with these reports, we observed that actinomycin D significantly inhibited tPA increase 60 min after glutamate stimulation in cultured neurons. This suggests a model of temporal regulation of tPA expression after synaptic activation: first, an immediate and synapse-specific synthesis and release of tPA, mediated by the activation of mGluR, followed by an NMDAR-dependent transcriptional activation and the upregulation of tPA mRNA (Qian et al., 1993). This newly synthesized tPA mRNA might be transported out of the cell body to the dendrites to consolidate and maintain the changes induced by the immediate response. Indeed, a recent study has demonstrated that CPEB may be involved in the transport of CPE-containing mRNA into the dendritic region (Huang et al., 2003). Finally, in Aplysia, a form of CPEB protein has been proposed to act as a synaptic tag, marking the activated synapses (Si et al., 2003). If this model holds true in the mammalian nervous system, this would allow for new protein synthesis of tPA to be directed to only the previously activated synapses, localizing any structural changes induced by tPA.
Footnotes
This work was supported by the Korean Science and Engineering Foundation (C.Y.S.), the Ellison Medical Foundation (D.G.W.), and the National Institute of Mental Health (D.G.W.). We thank Dr. Angus MacNicol, University of Arkansas Medical School, for the hCPEB clone.
Correspondence should be addressed to David Wells, Department of Molecular, Cellular, and Developmental Biology, Yale University, 219 Prospect Street, P. O. Box 208103, New Haven, CT 06520-8103. E-mail: david.wells@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249425-09$15.00/0
References
- Atkins CM, Nozaki N, Shigeri Y, Soderling TR (2004) Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II. J Neurosci 24: 5193-5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranes D, Lederfein D, Huang Y-Y, Chen M, Bailey CH, Kandel ER (1998) Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 21: 813-825. [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang HL, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS (1998) Sorting of b-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18: 251-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477-487. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Napolitano M, Centonze D, Marfia GA, Gubellini P, Teule MA, Berretta N, Bernardi G, Frati L, Tolu M, Gulino A (2000) Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur J Neurosci 12: 1002-1012. [DOI] [PubMed] [Google Scholar]
- Cao Q, Richter JD (2002) Dissolution of the maskin-eIF4E complex by cytoplasmic polyadenylation and poly(A)-binding protein controls cyclin B1 mRNA translation and oocyte maturation. EMBO J 21: 3852-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Xu N, Shyu A (1995) mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol Cell Biol 15: 5777-5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Shyu AB (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci 20: 465-470. [DOI] [PubMed] [Google Scholar]
- Eom T, Antar LN, Singer RH, Bassell GJ (2003) Localization of a {beta}actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J Neurosci 23: 10433-10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiumelli H, Jabaudon D, Magistretti PJ, Martin J-L (1999) BDNF stimulates expression, activity and release of tissue-type plasminogen activator in mouse cortical neurons. Eur J Neurosci 11: 1639-1646. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G (1991) Rat hippocampal neurons in low-density culture. In: Culturing nerve cells (Banker G, Goslin K, eds), pp 251-282. Cambridge: MIT.
- Havik B, Rokke H, Bardsen K, Davanger S, Bramham CR (2003) Bursts of high-frequency stimulation trigger rapid delivery of pre-existing a-CaMKII mRNA to synapses: a mechanism in dendritic protein synthesis during long-term potentiation in adult awake rats. Eur J Neurosci 17: 2679-2689. [DOI] [PubMed] [Google Scholar]
- Heussen C, Dowdle EB (1980) Electrophoretic analysis of plasminogen activators in polyacrylamide gels containing sodium dodecyl sulfate and co-polymerized substrates. Anal Biochem 102: 196-202. [DOI] [PubMed] [Google Scholar]
- Hollingsworth EB, McNeal ET, Burton JL, Williams RJ, Daly JW, Creveling CR (1985) Biochemical characterization of filtered synaptoneurosome preparation from guinea pig cerebral cortex: cyclic adenosine 3′:5′-monophosphate-generating systems, receptors, and enzymes. J Neurosci 5: 2240-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-S, Jung M-Y, Sarkissian M, Richter JD (2002) N-methyl-D-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J 21: 2139-2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-S, Carson JH, Barbarese E, Richter JD (2003) Facilitation of dendritic mRNA transport by CPEB. Genes Dev 17: 638-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-Y, Bach ME, Lipp H-P, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind J-M, Mulligan R, Collen D, Carmeliet P (1996) Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci USA 93: 8699-8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254-1256. [DOI] [PubMed] [Google Scholar]
- Hyman RW, Davidson N (1970) Kinetics of the in vitro inhibition of transcription by actinomycin. J Mol Biol 50: 421-438. [DOI] [PubMed] [Google Scholar]
- Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC (2004) Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci 7: 244-253. [DOI] [PubMed] [Google Scholar]
- Kacharmina JE, Job C, Crino P, Eberwine J (2000) Stimulation of glutamate receptor protein synthesis and membrane insertion within isolated neuronal dendrites. Proc Natl Acad Sci USA 97: 11545-11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P (1997) Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat 13: 219-241. [DOI] [PubMed] [Google Scholar]
- Madani R, Hulo S, Toni N, Madani H, Steimer T, Muller D, Vassalli J-D (1999) Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J 18: 3007-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataga N, Nagai N, Hensch TK (2002) Permissive proteolytic activity for visual cortical plasticity. Proc Natl Acad Sci USA 99: 7717-7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys T, Strickland S (2003) Tissue plasminogen activator and NMDA receptor cleavage. Nat Med 9: 371-372. [DOI] [PubMed] [Google Scholar]
- Mayford M, Baranes D, Podsypanina K, Kandel ER (1996) The 3′-untranslated region of CaMKIIa is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci USA 93: 13250-13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Richter JD (2001) Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol 2: 521-529. [DOI] [PubMed] [Google Scholar]
- Napolitano M, Marfia GA, Vacca A, Centonze D, Bellavia D, Di Marcotullio L, Frati L, Bernardi G, Gulino A, Calabresi P (1999) Modulation of gene expression following long-term synaptic depression in the striatum. Brain Res Mol Brain Res 72: 89-96. [DOI] [PubMed] [Google Scholar]
- Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A (2001) The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med 7: 59-64. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM (2002) Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35: 535-545. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S (2003) Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci 6: 168-174. [DOI] [PubMed] [Google Scholar]
- Pierce JP, Mayer T, McCarthy JB (2001) Evidence for a satellite secretory pathway in neuronal dendritic spines. Curr Biol 11: 351-355. [DOI] [PubMed] [Google Scholar]
- Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D (1993) Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 361: 453-457. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF (1999) Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci 2: 352-357. [DOI] [PubMed] [Google Scholar]
- Richter JD (2000) Influence of polyadenylation-induced translation on metazoan development and neuronal synaptic function. In: Translational control of gene expression (Sonenberg N, Hershey J, Mathews M, eds), pp 785-805. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Salles FJ, Darrow FJ, O'Connell ML, Strickland S (1992) Isolation of novel murine maternal mRNAs regulated by cytoplasmic polyadenylation. Genes Dev 6: 1202-1212. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Ferguson JE (2003) Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci 23: 7368-7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si K, Giustetto M, Etkin A, Hsu R, Janisiewicz AM, Miniaci MC, Kim J-H, Zhu H, Kandel ER (2003) A neuronal isoform of CPEB regulates local protein synthesis and stabilizes synapse-specific long-term facilitation in Aplysia Cell 115: 893-904. [DOI] [PubMed] [Google Scholar]
- Siao C-J, Fernandez SR, Tsirka SE (2003) Cell type-specific roles for tissue plasminogen activator released by neurons or microglia after excitotoxic injury. J Neurosci 23: 3234-3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, Matsubara A, Rinvik E, Ottersen OP (1999) The arrangement of glutamate receptors in excitatory synapses. Ann NY Acad Sci 868: 474-482. [DOI] [PubMed] [Google Scholar]
- Teesalu T, Kulla A, Asser T, Koskiniemi M, Vaheri A (2001) Tissue plasminogen activator as a key effector in neurobiology and neuropathology. Biochem Soc Trans 30: 183-189. [DOI] [PubMed] [Google Scholar]
- Theis M, Si K, Kandel ER (2003) Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc Natl Acad Sci USA 100: 9602-9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka SE (2001) Tissue plasminogen activator as a modulator of neuronal survival and function. Biochem Soc Trans 30: 222-225. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Strickland S (1996) Neuronal cell death and tPA. Nature 384: 123-124. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT (1993) Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA 90: 7168-7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci USA 94: 5395-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DG, Dong X, Quinlan EM, Huang YS, Bear MF, Richter JD, Fallon JR (2001) A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J Neurosci 21: 9541-9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD (1998) CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron 21: 1129-1139. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Holtzman DM, Li Y, Osaka H, DeMaro J, Jacquin M, Bu G (2000) Role of tissue plasminogen activator receptor LRP in hippocampal long-term potentiation. J Neurosci 20: 542-549. [DOI] [PMC free article] [PubMed] [Google Scholar]