

Abstract
Huntington's disease (HD) and eight additional inherited neurological disorders are caused by CAG triplet-repeat expansions leading to expanded polyglutamine-sequences in their respective proteins. These triplet-CAG repeat disorders have in common the formation of aberrant intraneuronal proteinaceous inclusions containing the expanded polyglutamine sequences. These aggregates have been postulated to contribute to pathogenesis caused by conformational toxicity, sequestration of other polyglutamine-containing proteins, or by interfering with certain enzymatic activities. Testing these hypotheses has been hampered by the difficulty to isolate these aggregates from brain. Here we report that polyglutamine aggregates can be isolated from the brain of the Tet/HD94 conditional mouse model of HD, by following a method based on high salt buffer homogenization, nonionic detergent extraction, and gradient fractionation. We then verified that the method can be successfully applied to postmortem HD brains. Immunoelectron microscopy, both in human and mouse samples, revealed that the stable component of the inclusions are mutant huntingtin-containing and ubiquitin-containing fibrils. Atomic-force microscopy revealed that these fibrils have a “beads on a string” morphology. Thus, they resemble the in vitro assembled filaments made of recombinant mutant-huntingtin, as well as the Aβ and α-synuclein amyloid protofibrils. Finally, by shutting down transgene expression in the Tet/HD94 conditional mouse model of HD, we were able to demonstrate that these filaments, although stable in vitro, are susceptible to revert in vivo, thus demonstrating that the previously reported reversal of ubiquitin-immunoreactive inclusions does not simply reflect disassembling of the inclusions into their constituent fibrils and suggesting that any associated conformational or protein-sequestration toxicity is also likely to revert.
Keywords: Huntington's disease, aggregate purification, immunoelectron microscopy, atomic force microscopy, reversal, conditional mouse model
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by a CAG triplet-repeat expansion coding for a poly-glutamine (polyQ) sequence in the N-terminal region of the huntingtin (htt) protein (HDCRG, 1993). Patients suffer from motor dysfunction, cognitive decline, and psychological disturbances over 10-15 years until death, caused by brain atrophy mainly in striatum and cortex (Vonsattel and DiFiglia, 1998).
At least eight other autosomal dominant neurological diseases are also caused by a polyQ expansion mutation in their respective proteins. These CAG triplet-repeat disorders include spinal and bulbar muscular atrophy, several forms of spinocerebellar ataxia, and dentatorubral and pallidolusyan atrophy (Zoghbi and Orr, 2000). All these diseases share an interesting commonality; the presence of intraneuronal aggregates containing the expanded polyQ in the affected areas of the brain (Ross, 1997; Nakamura et al., 2001).
Despite the presence of intraneuronal aggregates in all CAG triplet-repeat disorders, the relevance of aggregate formation to the etiology of HD is unclear. Aggregates have been implicated as the trigger for neurodegeneration, because both aggregates and pathogenicity are caused by the same polyQ length threshold (Scherzinger et al., 1997). Furthermore, in a transgenic mouse model, these aggregates precede the onset of symptoms (Davies et al., 1997). Nevertheless, although some studies using transfected cells support aggregate-induced toxicity (Waelter et al., 2001), others dissociate inclusion formation and cell death (Kim et al., 1999; Martin-Aparicio et al., 2001), and even suggest that intranuclear inclusions may reflect a protective mechanism against soluble htt-induced toxicity (Saudou et al., 1998; Kopito, 2000).
Although the role of polyQ-containing aggregates in HD pathogenesis is still a matter of debate, several mechanisms by which aggregates could trigger toxicity have been postulated. These include conformational toxicity, as it has been proposed for other neurodegenerative diseases with intraneuronal aberrant aggregates of amyloidogenic proteins like Aβ in Alzheimer's disease and α-synuclein in Parkinson's disease (Bucciantini et al., 2002). Another proposed pathogenic mechanism is the sequestration of other proteins containing nonpathogenic PolyQ sequences like CBP (Nucifora et al., 2001) or htt transcribed from the nonmutated allele (Zuccato et al., 2001). Finally, because intraneuronal aggregates are stained with anti-ubiquitin and anti-proteasome antibodies, an impairment of the ubiquitin proteasome system by the aggregates has been proposed as the pathogenic mechanism. Interestingly, work on transfected cell models support this hypothesis (Bence et al., 2001; Jana et al., 2001), although no evidence has been found so far in tissue from patients or mouse models (Diaz-Hernandez et al., 2003; Hernández et al., 2004).
PolyQ aggregates have never been purified from the brain of patients of any of the CAG triplet-repeat disorders or from the brain of any of the many available mouse models (Rubinsztein, 2002). The ability to purify these aggregates from the brain of HD patients or from animal models would, by allowing their biochemical and ultrastructural characterization, offer key information about the possible contribution of the aggregates to the pathogenesis of HD and other CAG triplet-repeat disorders. Similarly to what has been done with isolated proteinaceous deposits from Alzheimer's disease (Selkoe, 1991) and Parkinson's disease brains (Pollanen et al., 1993).
Here we report the purification of huntingtin aggregates from the Tet/HD94 conditional mouse model of HD (Yamamoto et al., 2000; Martin-Aparicio et al., 2001) and from postmortem HD brains. Biochemical studies, together with immunoelectron microscopy and atomic-force microscopy ultrastructural analysis, revealed that the stable components of HD inclusions are amyloid-like huntingtin filaments. Finally, by shutting down transgene expression in the Tet/HD94 mice, we were able to demonstrate that these filaments, although stable in vitro, are susceptible to revert in vivo.
Materials and Methods
Animals
Tet/HD94 mice were generated as previously described (Yamamoto et al., 2000). Mice were bred at the Centro de Biología Molecular “Severo Ochoa” (Madrid, Spain). Four or five mice were housed per cage with food and water available ad libitum. Mice were maintained in a temperature-controlled environment on a 12 hr light/dark cycle with light onset at 7:00 A.M.
Antibodies
The following antibodies were used: anti-N-terminal mutant huntingtin CAG53b (amino acids 1-118 with a 51 polyQ stretch, a kind gift from Dr. Wanker, Berlin, Germany), anti-α-tubulin (Sigma, St. Louis, MO), anti-β-galactosidase (Promega, Madison, WI), anti-ubiquitin (Dako, Carpinteria, CA), anti-poly-ubiquitin conjugates (FK-2; Affinity Bioreagents, Golden, CO), anti-N-terminal htt (N-18; Santa Cruz Biotechnology, Santa Cruz, CA), and anti-expanded polyQ (1C2; Chemicon, Temecula, CA).
Human samples
Brain specimens used in this study were removed at autopsy from three HD (one woman aged 65 years and two men aged 71 and 68 years) and three age-matched controls (one woman aged 80 years and two men aged 63 and 79 years) following the protocols of nervous tissue donation approved by the local Ethical Committees of the Barcelona and Bellvitge brain banks. The postmortem delay in tissue processing was between 4 and 15 hr in both groups. The neuropathological examination in HD cases revealed a diagnosis of HD grade 4 following the criteria of Vonsattel et al. (1985), revised in Vonsattel and DiFiglia (1998).
Immunohistochemistry
Mouse brain. Mice were anesthetized with a xylazine-ketamine solution and transcardially perfused with 4% paraformaldehyde (PFA) in Sorensen's buffer for 10 min. Brains were postfixed in 4% PFA for 2 hr at 4°C and cryoprotected in 30% sucrose solution. Thirty micrometer sagittal sections were cut on a freezing microtome (Leica, Nussloch, Germany) and collected in 0.1% azide-PBS solution. Next, brain sections were pretreated for 1 hr with 1% BSA, 5% FBS, and 0.2% Triton X-100 and then incubated with primary antibodies at the following dilutions: anti-polyubiquitin FK2 (1:500) and anti-N-terminal htt CAG53b (1:5000). Finally, brain sections were incubated in avidin-biotin complex using the Elite Vectastain kit (Vector Laboratories, Burlingame, CA). Chromogen reactions were performed with 0.05% diaminobenzidine (DAB; Sigma) and 0.01% H2O2 for 10 min. Sections were coverslipped with Fluorosave.
Human brain. Samples (2-mm-thick) of the frontal cortex and striatum from control and HD subjects were fixed at the time of the autopsy in 4% paraformaldehyde for 24-48 hr and then immersed in 30% buffered sucrose for 48 hr. Once cryoprotected, the samples were frozen and stored at -80°C until use. Then, 30 μm sections were obtained with a cryostat and processed for free-floating immunohistochemistry in the same conditions as described for the mouse samples. The primary antibodies assayed were used at the same concentration previously described for mouse samples. Immunoreaction was visualized with 0.05% DAB and 0.01% H2O2 (brown precipitate).
Tissue processing for electron microscopy
For electron microscopy, immunostained vibratome sections were processed as previously described (Lucas et al., 2001). Briefly, the sections were postfixed in 2% OsO4 for 1 hr, dehydrated, embedded in Araldite, and flat-mounted in Formvar-coated slides, using plastic coverslips. After polymerization, selected areas were photographed, trimmed, reembedded in Araldite, and resectioned at 1 μm. These semithin sections were rephotographed and resectioned in ultrathin sections. The ultrathin sections were observed in a Jeol electron microscope, without heavy metal staining to avoid artifactual precipitates.
Aggregate purification procedure
For the isolation of huntingtin- and ubiquitin-containing aggregates from brain tissue, we used protocol similar to the one previously used to isolate aberrant intraneuronal proteinaceous deposits from the brains of Alzheimer's disease (Greenberg and Davies, 1990) and Parkinson's disease patients (Pollanen et al., 1992). Briefly, the entire forebrain of Tet/HD94 mice or cortical dissects from HD grade 4 cases (1-2 gm) was homogenized in 6 ml of cold buffer 1 (10 mm Tris, 1 mm EGTA, 0.8 m NaCl, 10% sucrose, 0.1% Triton X-100, and protease inhibitors) (2 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin) in a glass homogenizer. After centrifugation at 27,000 × g for 20 min at 4°C, the supernatant S1 was saved, and pellet P1 was homogenized again in buffer 1 and then was centrifuged at 27,000 × g for 20 min. The supernatants S1 and S2 were combined, adjusted to 1% (w/v) N-lauroylsarcosine (Sarkosyl) and 1% (v/v) 2 mercaptoethanol, and incubated at room temperature for 2 hr in agitation on an orbital shaker. After centrifugation at 100,000 × g for 45 min the huntingtin filaments pellets were homogenized in 2 ml of buffer 3 (10 mm Tris, 1 mm EGTA, 0.8 m NaCl, 10% sucrose, and protease inhibitors) (2 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin) and layered over a discontinuous sucrose gradient consisting of 4 ml of 50% sucrose and 4 ml of 20% sucrose in 10 mm Tris, 1 mm EGTA, 0.8 m NaCl. After centrifugation for 2 hr at 150,000 × g, the htt filaments were collected from 20-50% interface and were stored at -70°C until used for further analysis.
Western blot analysis
Mouse samples. Extracts for Western blot analysis were obtained from different steps of Huntingtin filament-enriched fraction preparation and treated with protease inhibitors (2 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml pepstatin). The samples were homogenized at 4°C, and protein content was determined by Bradford. Total protein (20 μg) was electrophoresed on 10% SDS-PAGE gel and transferred to a nitrocellulose membrane (Schleicher & Schuell, Dassell, Germany). The experiments were performed using the following primary antibodies: polyclonal anti-polyglutamine expansion 1C2 (1:500), monoclonal anti-poly-ubiquitin FK2 clone (1:2000), polyclonal anti-huntingtin N-terminal CAG53b (1:5000), polyclonal anti-ubiquitin (1:1000), and polyclonal anti-N-terminal htt N-18 (1:100). The filters were incubated with the antibody at 4°C overnight in 5% nonfat dried milk. A secondary goat anti-mouse (monoclonal antibodies) or goat anti-rabbit (polyclonal antibodies) antibody (both 1:5000; Invitrogen, San Diego, CA) was used followed by ECL detection (Amersham Biosciences, Arlington Heights, IL).
Human samples. Fresh samples of the frontal cortex and striatum were obtained at the time of the autopsy, and they were immediately frozen on dry ice and stored at -80°C for biochemical studies. Control and HD brain samples were processed following the same protocol as described for mouse samples.
Quantification of immunoreactivity was performed by using a GS-710 Calibrated Imaging densitometer scanner controller by Quantity One PC software from Bio-Rad (Hercules, CA). In all cases, the average intensity value of the pixels in a background-selected region was calculated and was subtracted from each pixel in the samples. The densitometry values obtained in the linear range of detection with these antibodies were normalized with respect to the values obtained with an anti-α-tubulin antibody to correct for any deviation in loaded amounts of protein. Statistical analysis was performed using one-way ANOVA followed by Bonferroni test.
Immunoelectron microscopy of purified aggregates
Single immunoelectron microscopy was performed after adsorption of the samples to electron microscopy carbon-coated grids and incubation with the first antibody (1:100) for 1 hr at room temperature. After extensive washing, the grids were incubated with the secondary antibody (1: 40) conjugated with 10-nm-diameter gold particles. Finally, the samples were stained with 2% uranyl acetate for 1 min. Transmission electron microscopy was performed in a Jeol (Peabody, MA) model 1200EX electron microscope operated at 100 kV.
Double immunoelectron microscopy with the polyclonal antibody CAG53b and the monoclonal antibody FK2 was developed as described before for single immunoelectron microscopy but mixing both antibodies (1:300 CAG53b and 1:500 FK2). After extensive washing, the grids were incubated with secondary antibodies (anti-mouse conjugated with 10-nm-diameter gold particles and anti-rabbit conjugated with 5-nm-diameter gold particles, both 1:40). The samples were processed as described before for single-immunolabeling.
Atomic force microscopy of purified aggregates
A 5 μl aliquot of the different fractions of the sucrose gradient was deposited onto freshly cleaved mica (Electron Microscopy Sciences, Fort Washington, PA) and allowed to adsorb for 1 min. The sample was washed with deionized and double-distilled water, dried with nitrogen gas, and imaged in air with a commercial microscope (Nanotec Electronica, Madrid, Spain) operating in noncontact dynamic mode as previously described (Moreno-Herrero et al., 2001, 2004). Images were processed using WSxM freeware. To enhance surface details, the Laplacian operator was applied to the topographic image, thus generating the images in the inserts.
Immunofluorescence studies
After pelleting and washing, aliquots from different steps of Huntingtin filament-enriched fraction preparation were deposited onto coverslips pretreated with poly-l-lysine and maintained for 45 min to allow setting and sticking to the substrate. After treated with NH4Cl 50 mm, glycine 1 m, 1% BSA, and 1% Triton X-100 in PBS buffer, the samples were incubated with primary antibodies at the following dilutions: polyclonal anti-huntingtin N-terminal CAG53b (1:5000) and monoclonal anti-poly-ubiquitin conjugates (FK2) (1:10,000). Subsequently, samples were washed with PBS buffer and incubated with secondary antibodies at the following dilutions: goat anti-rabbit IgG labeled with Texas Red from Molecular Probes (Eugene, OR) (1:400) and goat anti-mouse IgG labeled with Oregon green 488 from Molecular Probes (1:200). Finally, samples were washed with PBS and mounted following the standard procedures. Controls were performed by following the same procedure but substituting the primary antibodies with preimmune serum (in the case of polyclonal primary antibodies) or with an unrelated monoclonal antibody, or just omitting any primary antibody.
Colocalization of two markers was analyzed by taking successive Oregon green 488 and Texas Red fluorescent images using an Axioskop 2 plus microscope and a CCD camera (Coolsnap FX color). Positive signal was considered for the different antibodies if the mean intensity value was >140 on a 0-255 scale with 0 = white and 255 = black. The cutoff value of 140 was determined from visual analysis of immunolabeling and by comparison with control (maximal level obtained with pre-absorbed antibodies).
Flow cytometry
After incubation with blocking buffer (PBS, 1% BSA, and 1% FCS) for 20 min at 4°C, samples were incubated with primary antibodies at the following dilutions: polyclonal anti-huntingtin N-terminal CAG53b (1:300) and monoclonal anti-poly-ubiquitin conjugates (FK2) (1:300) for 20 min at 4°C. Subsequently, samples were washed with PBS buffer and incubated with goat anti-rabbit IgG labeled with Alexa 488 and goat anti-mouse IgG labeled with Alexa 647 for 20 min at 4°C. To check staining specificity, controls were performed by following the same procedure but substituting the primary antibodies with PBS in presence of 1% BSA. Samples were analyzed on a flow cytometer FACsCalibur (BD Biosciences, Erembodegem, Belgium) using CellQuest software (BD Biosciences).
Thioflavine S fluorescence spectroscopy
The 20-50 fraction of the purification procedure was incubated with PBS containing 10 μm thioflavine S (ThS) at room temperature for 15 min. Fluorescence was then measured with an Aminco Bowman Series 2 Luminescence Spectrometer (Thermo Spectronic) with an excitation filter of 440 nm and an emission filter of 510 nm. Measurements were performed at room temperature either in plain PBS media or in PBS supplemented with 10 μm ThS. Background fluorescence and light scattering of the sample without ThS was subtracted.
Results
Isolation of mutant htt aggregates from the brain of Tet/HD94 mice
Tet/HD94 mice develop neuropil and nuclear inclusions that can be detected with antibodies raised against N-terminal htt and against ubiquitin (Yamamoto et al., 2000; Martin-Aparicio et al., 2001; Diaz-Hernandez et al., 2003) (Fig. 1A,B). Because exon 1 mutant htt expression in Tet/HD94 mice is driven by the CamKIIα promoter, inclusions are abundant in the whole forebrain. Interestingly, similarly to what happens in humans (DiFiglia et al., 1997; Gutekunst et al., 1999), inclusions are more abundant in cortex (Fig. 1A) than in striatum (Fig. 1B) of Tet/HD94 mice. Also in good agreement with what has been described in other HD mouse models (Davies et al., 1997; Yu et al., 2003) and in humans (DiFiglia et al., 1997; Gutekunst et al., 1999), ultrastructural analysis of Tet/HD94 brain sections immunostained with anti-N-terminal htt or anti-ubiquitin antibodies revealed that inclusions are composed of fibrillar structures (Fig. 1C,D).
Figure 1.
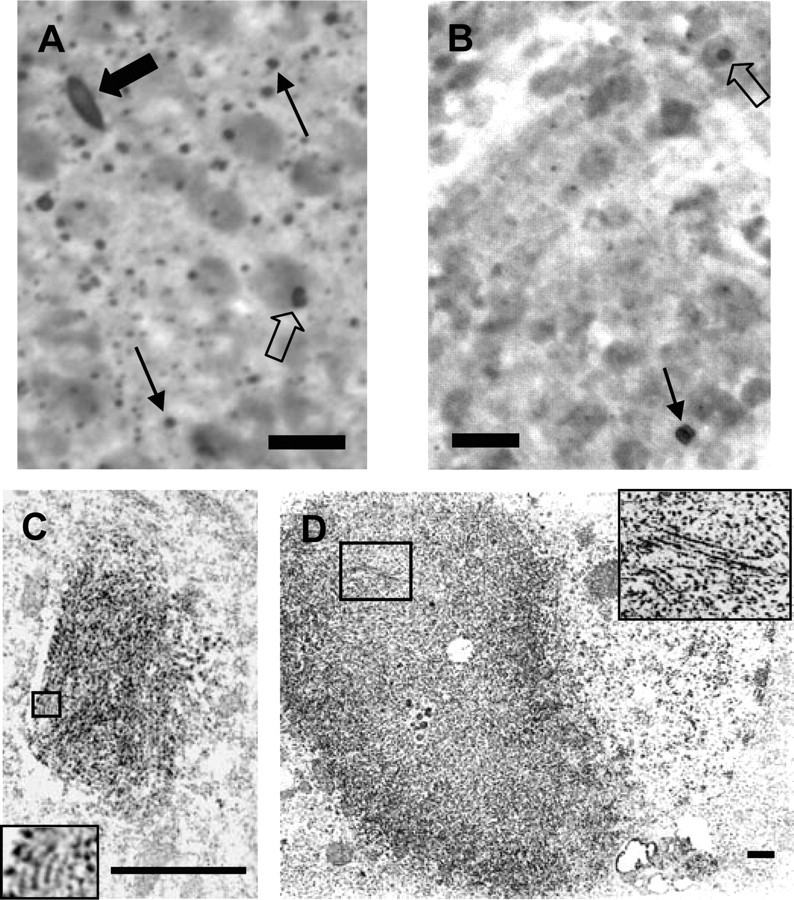
Neuropil and nuclear inclusions in the cortex and striatum of Tet/HD94 mice: immunohistochemical detection and ultrastructural analysis. A, B, Immunohistochemistry with anti-N-terminal mutant htt (CAG53b) antibody revealed the presence of aggregates in the cortex (A) and striatum (B) of Tet/HD94 mice. Both nuclear (empty arrows) and neuropil (black thin arrows) inclusions, as well as dystrophic neurites (black solid arrows) can be detected. Scale bar, 20 μm. C, D, Ultrastructural transmission electron microscopy analysis of CAG53b stained neuropil inclusion (C) or dystrophic neurite (D) from the cortex of a Tet/HD94 mouse. Inset in C shows a 3.5× magnification of the indicated area. Inset in D shows a 2.5× magnification of the indicated area. Scale bars, 500 nm.
For the biochemical isolation of aggregates, we thus excised the entire forebrain of Tet/HD94 and control littermates and subjected them to a procedure similar to the one used to isolate the aberrant intraneuronal proteinaceous deposits from the brain of Alzheimer's disease (Greenberg and Davies, 1990) and Parkinson's disease patients (Pollanen et al., 1992). Essentially, tissue is homogenized in a high salt buffer, followed by extraction with the nonionic detergent Sarkosyl, and subsequent sucrose gradient fractionation (see Materials and Methods) (Fig. 2A).
Figure 2.
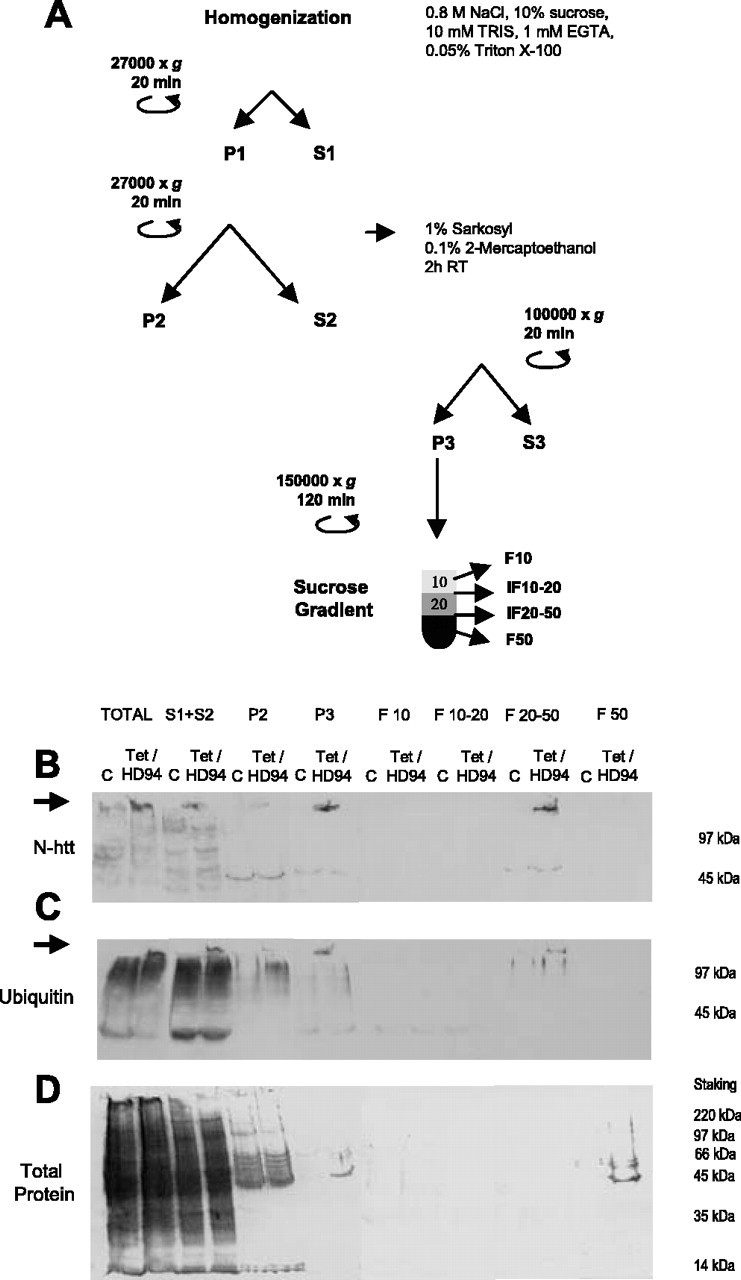
Isolation of N-terminal htt-containing and ubiquitin-containing aggregates from the brain of Tet/HD94 mice. A, Protocol for isolation of aggregates from the forebrain of Tet/HD94 mice. B-D, The progressive enrichment in aggregated material in the different fractions along the procedure is monitored by Western blot with anti-N-terminal mutant htt (CAG53b) antibody (B) or anti-poly-ubiquitin antibody (C). Arrows indicate the accumulation of aggregated material in the stacking gel. D shows total protein content by Ponceau red staining of the nitrocellulose membrane.
To verify that the subsequent steps of the procedure led to progressive isolation of polyQ-containing aggregates, we took advantage of previously published work. More precisely, it had been shown that polyQ inclusions are SDS and Triton X-100-resistant in cell transfection experiments (Kazantsev et al., 1999) and that they can be monitored in cell transfection and in in vitro assembly experiments as the material retained in a filter trap or in the stacking gel of SDS-PAGE that is immunoreactive with anti-N-terminal htt or anti-polyQ antibodies (Scherzinger et al., 1997, 1999). Therefore, during the optimization of the purification procedure we were able to track the polyQ-containing aggregates by Western blot as the material retained in the stacking gel and that is recognized by N-terminal mutant htt (Fig. 2B), polyubiquitin (Fig. 2C), or polyQ (1C2; data not shown) antibodies. The subsequent enrichment in polyQ-containing aggregates was made evident by Ponceau red or Coomasie blue staining of the total protein content in the different steps of the purification procedure (Fig. 2D) (data not shown). In parallel, we also analyzed the effect of the different steps of the purification procedure on the number and size of N-terminal htt- and ubiquitin-containing particles by double immunofluorescence in suspensions of the different fractions and subsequent analysis either by fluorescence microscopy on poly-lysine-coated coverslips or by flow cytometry (see Fig. 5).
Figure 5.

The size and number of spheroid inclusions decrease progressively during the purification procedure. A, B, Flow cytometry analysis of the total number of CAG53b and ubiquitin-positive aggregates after homogenization of Tet/HD94 forebrain tissue at different NaCl concentrations (A) and percentage of these inclusions with diameter ≥2.5 μm (B). C, Semiautomated counting of double-labeled particles according to their diameter. D, Comparison of the effect of homogenization of tissue in isotonic (0.15 m NaCl) or high salt (0.8 m NaCl) buffer on the distribution of the huntingtin-containing aggregated material between P2 and P3 fractions. E, Solubilization of monomeric exon 1 mutant htt band (44 kDa) by subjecting the P3 fraction to boiling during 15 min in 10% SDS and 10% βME containing buffer (+); when this solubilization step was omitted (-), all htt immunoreactivity remained in the stacking gel. F, Flow cytometry analysis of the total number of CAG53b-positive and ubiquitin-positive aggregates remaining when high salt homogenates (Control) are subjected to 1% Sarkosyl extraction for 2 hr at room temperature (Sarkosyl). G, Thioflavine S fluorescence in the 20-50 fraction of Tet/HD94 mice with respect to the same fraction of control mice. *p < 0.05.
As it can be seen in Figure 2, B and C, total homogenates in high salt buffer from Tet/HD94 forebrain contain N-terminal htt-positive and ubiquitin-positive aggregated material that is not present in the homogenates from control littermates. Repeated low field centrifugation to get rid of myelin sheets as well as non-lysed membrane organelles and nuclei resulted in a soluble fraction (S1 plus S2) containing most of the N-terminal htt-containing and ubiquitin-containing aggregated material, but lower total protein content (Fig. 2B-D). Extraction of the S1 plus S2 fraction with the nonionic detergent Sarkosyl in the presence of β-mercaptho-ethanol, followed by high field centrifugation resulted in a pellet fraction (P3) highly enriched in htt-containing and ubiquitin-containing aggregated material (Fig. 2B-D). Finally, we were able to separate the aggregated material from the little residual soluble protein present in P3 (Fig. 2A-D) by a sucrose gradient centrifugation. After this, the htt-containing and ubiquitin-containing aggregated material was retained in the 20-30% sucrose interphase, whereas the residual soluble protein was in the pellet and/or in the 50% sucrose phase. The gradient was accordingly simplified to 10, 20, and 50% sucrose phases in subsequent experiments, as shown in Figure 2A.
N-terminal huntingtin-containing and ubiquitin-containing filaments are the high salt-resistant and detergent-resistant component of Huntington's disease inclusions
We next analyzed by immuno-EM, the morphology of the htt-containing and ubiquitin-containing aggregated material present in P3 and in the 20-50% interphase. Immunogold labeling of the P3 fractions revealed that the N-terminal htt (Fig. 3A-C) and ubiquitin-containing (Fig. 3D,E) aggregates isolated from Tet/HD94 mouse brain were filamentous structures similar to those seen in the inclusions by ultrastructural analysis of Tet/HD94 brain tissue (Fig. 1C,D). Thus suggesting that, during the purification procedure, spheroid inclusions disassemble into their constituent filamentous material. In good agreement with the Western blot data, no immunoreactive material was found in P3 fractions from control littermates (data not shown). The length of the purified filaments varied considerably from 150 nm to >1 μm. They were essentially straight fibrils, especially those shorter than 200 nm. However, longer ones showed multiple mild turns (15-20°) without any apparent periodicity (Fig. 3A-E) and, occasionally, they also showed more dramatic turns (up to 60-70°) (data not shown) (Fig. 4B,C). The width also varied considerably from one filament to another, ranging from 15 to 40 nm. The thinnest filaments (Fig. 3B,D) were quite uniform in width (15-20 nm). Thicker ones, on the contrary, were less homogeneous, and they seemed to be composed of two or more aligned thin filaments (Fig. 3C,E).
Figure 3.
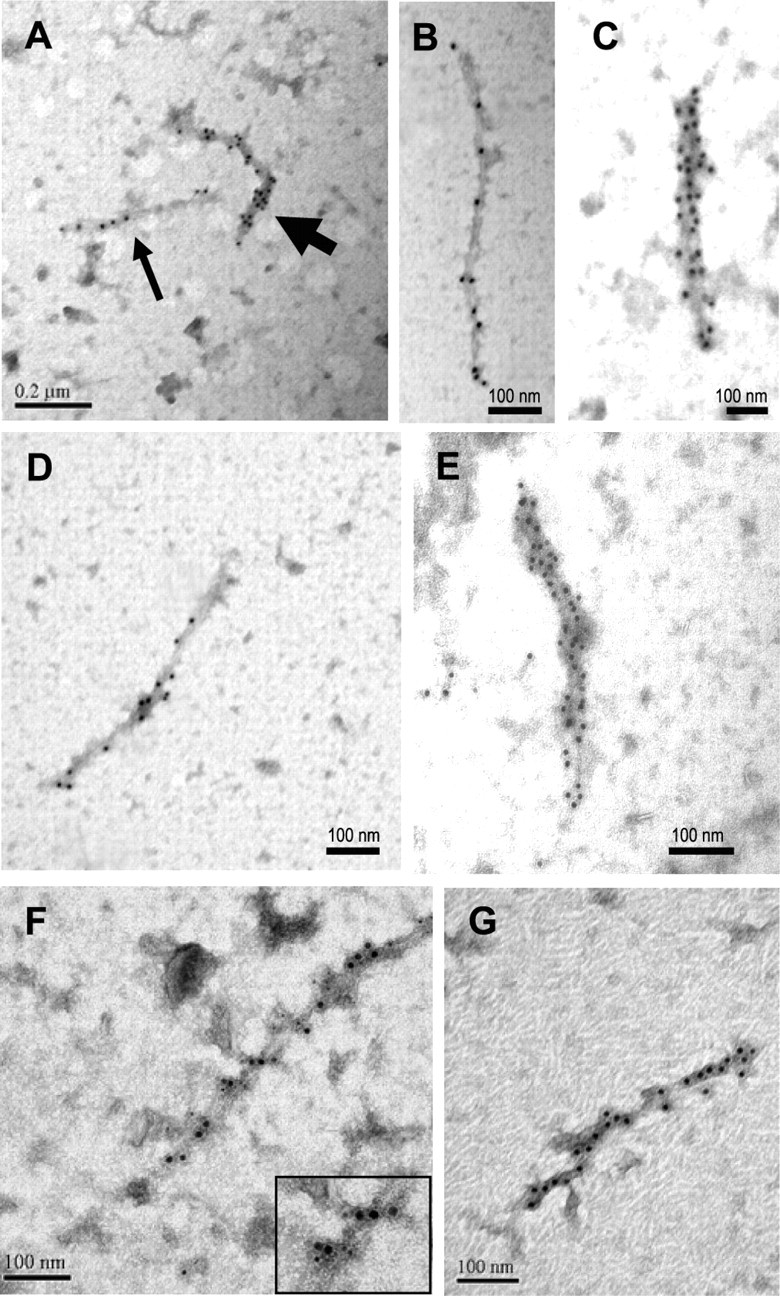
Immunoelectron microscopy analysis of the aggregates isolated from the brain of Tet/HD94 mice. P3 (A-F) or 20-50% (G) fractions of the purification procedure were subjected to immunodecoration with anti-N-terminal mutant htt (CAG53b) antibody (A-C, G), or with anti-poly-ubiquitin antibody (D, E) or with both antibodies (F) and secondary antibodies conjugated with gold particles. Thin arrow in A indicates a thin filament like those shown at higher magnification in B and D. Thick arrow in A indicates a thicker filament like those shown at higher magnification in C and E. For the double immunodetection in F, CAG53b staining is detected with a secondary antibody conjugated with 10 nm gold particles, and poly-ubiquitin staining is detected with a secondary antibody conjugated with 5 nm gold particles. Inset in bottom right corner of F shows a higher magnification for a better discrimination of 10 and 5 nm gold particles.
Figure 4.

AFM analysis of filaments purified from Tet/HD94 mice. P3 (A) and 20-50% fractions (B, C) of the purification procedure were analyzed by AFM. C shows a higher magnification of the filament depicted in B. To enhance the details of the structure, the Laplacian operator was applied to the topographic images (inserts). A bead on a string morphology is clearly visible from the data. In C are depicted the trajectories of the height measurements (1 and 2) shown in the bottom right quadrant.
Because N-terminal htt-immunodecorated fibrils (Fig. 3A-C) had similar morphologies to those decorated with anti-ubiquitin antibodies (Fig. 3D,E), it was plausible that both molecules coexist in the same filaments. To test this, we performed double-labeling immuno-EM experiments with secondary antibodies conjugated with gold particles of different sizes. As shown in Figure 3F, 5 and 10 nm gold particles codecorate the same filaments, thus demonstrating that N-terminal htt and ubiquitin coexist in the same fibrils.
Immuno-EM experiments in the 20-50% fraction revealed that the N-terminal htt-containing and ubiquitin-containing material consisted of the same type of filaments as described for the P3 fraction but, in good agreement with what was shown in Figure 2D-F, virtually devoid of unlabeled contaminant material (Fig. 3G) (data not shown).
Atomic force microscopy analysis of mutant N-terminal htt filaments from Tet/HD94 mice
For a more precise analysis of the morphology of the aggregated material purified from Tet/HD94 mouse brain, we performed atomic force microscopy (AFM) studies on the P3 (Fig. 4A) and 20-50% (Fig. 4B,C) fractions. In good agreement with the immuno-EM and Western blot data, the 20-50% samples show a cleaner appearance and a higher density of filaments. Similar to what has been described regarding the immuno-EM experiments, individual filaments were straight with occasional turns and were several hundred nanometer long. Interestingly, AFM revealed that the filaments were not uniform in shape but presented a beads on a string morphology, thus resembling the in vitro assembled filaments made of recombinant N-terminal mutant huntingtin (Poirier et al., 2002), as well as the Aβ and α-synuclein amyloid protofibrils (Dobson, 2003). AFM analysis also allowed us to measure the height of these filaments. Because Tet/HD94 filaments had a beads on a string morphology, their maximal height of the Tet/HD94 filaments was, therefore, that of the central part of the beads, which varied from 6 to 7.5 nm (Fig. 4C, measurement 1 as an example of a given bead, profile 2 to see the variable height of the peaks). The minimal height, on the opposite, was measured in the border between adjacent beads and was ≥5 nm (Fig. 4C, height profile 2). Regarding the filament width, although this measurement is affected by the finite size of the tip, we were able to estimate widths of 13 and 18 nm for the areas of low and high height, respectively (Fig. 4C, measurement 1). As mentioned above, the morphology of the filaments is similar to a succession of bead-shape features. The length of each of these beads was quite variable. We found a bead length as small as 5 nm and a maximum length of 20 nm, with a mean value of 14 ± 4 nm (Fig. 4C, height profile 2).
The size and number of spheroid inclusions decrease progressively during the purification procedure
As mentioned above, during the optimization of the purification procedure we analyzed the effect of the different steps on the number and size of N-terminal htt-immunolabeled and ubiquitin-immunolabeled particles by double immunofluorescence labeling in suspensions of the different fractions and subsequent analysis either by fluorescence microscopy on polylysine-coated coverslips or by flow cytometry (Fig. 5). In good agreement with the immunohistochemistry data shown in Figure 1, A and B, after isotonic homogenization, N-terminal htt and ubiquitin double-labeled particles corresponded to spheroids with diameters ranging between 0.5 and 5 μm (data not shown). Interestingly, 95.2% of the N-terminal htt-labeled particles were also ubiquitin-positive. We then analyzed the effect of homogenizing the tissue in high salt buffer (0.8 or 1.2 m NaCl) in the number and size of these particles. Flow cytometry analysis revealed that the total number of N-terminal mutant htt-labeled particles did not change significantly (Fig. 5A). However, the percentage of particles larger than the reference 2.5-μm-diameter beads decreased significantly by the raise in salt concentration to 0.8 or 1.2 m NaCl by 48 and 53%, respectively (p < 0.1) (Fig. 5B).
To further explore the effect of high salt on those particles <2.5 μm, we performed semiautomated counting of double-labeled particles (Fig. 5C). By this analysis, a dramatic decrease in the percentage of particles with diameters between 2 and 4 μm or >4 μm was also observed. Interestingly, we found that 0.8 m NaCl resulted in a 110% increase (p < 0.05) in the percentage of particles between 1 and 2 μm, and 180% (p < 0.05) increase in those detected particles <1 μm. Very similar results were obtained with 1.2 m NaCl. Therefore, diminishing protein-protein interactions by exposure to high salt buffer seems to be enough for partial disassembling of the inclusions into their constituent filaments (that cannot be detected by immunofluorescence based techniques).
Interestingly, the decrease in the size of the inclusions caused by high salt exposure is key for their separation from non-lysed membrane organelles and myelin sheets by low field centrifugation. As shown in Figure 5D, if the purification procedure is performed after homogenization in isotonic buffer, 100% of the aggregated material goes to P2 instead of P3.
Next, we analyzed the effect of Sarkosyl and β-mercaptoethanol (βME), either individually or combined, on the number and size of the particles previously exposed to 0.8 m NaCl. As shown in Figure 5F, Sarkosyl incubation by itself produced a dramatic decrease in the number of particles, as revealed by flow cytometry analysis. Similar results were obtained by semiautomated counting of double-labeled particles. Interestingly, βME by itself had no effect, but potentiated the effect of Sarkosyl in decreasing both the number and size of double-labeled particles (data not shown).
In summary, these data suggest that, along the biochemical procedure, inclusions disassemble into smaller aggregated material that can be detected by immunobiochemical procedures but not by optical microscopy. To confirm that the remaining aggregated material detected by Western blot at the top of the gel corresponds to aggregates made of monomeric exon 1 mutant huntingtin, we subjected the P3 and 20-50 fractions to extraction with a solution containing 10% each of SDS and βME at 100°C for 15 min, in a similar way to what has been done to solubilize tau from purified paired helical filaments isolated from Alzheimer's disease brains (Iqbal et al., 1984). As shown in Figure 5E, this resulted in detection of a 44 kDa N-terminal httimmunoreactive band that corresponds to the monomeric transgenic exon 1 mutant huntingtin produced by neurons of Tet/HD94 mice (Martin-Aparicio et al., 2001) together with a reduction in the htt-immunoreactive material at the top of the gel.
Finally, because AFM analysis of P3 and 20-50 fractions revealed filaments with a beads on string morphology similar to that found in amyloid fibrils made of Aβ or α-synuclein, we decided to incubate the 20-50 fraction with the amyloid-labeling dye thioflavine S. As shown in Figure 5G, there is a remarkable 205.8 ± 16.2% increase (p < 0.05) in thioflavine S fluorescence in the 20-50 fraction from Tet/HD94 mice with respect to the same fraction from control mice, thus strongly suggesting that the purified material corresponds, indeed, to amyloid aggregates.
Tet/HD94 filaments revert in vivo after silencing mutant N-terminal htt transgene expression
We have previously reported that HD inclusions are dynamic structures in vivo. More precisely, we showed that abolishing transgene expression in symptomatic Tet/HD94 mice (by administering the tetracycline analog doxycycline in the drinking water) leads to disappearance of the inclusions detected by immunohistochemistry (Yamamoto et al., 2000), with proteasome activity being involved in this phenomenon (Martin-Aparicio et al., 2001). However, in view of the here reported observation that along the biochemical procedure, inclusions disassemble into smaller filamentous aggregated material that can be detected by immunobiochemical procedures but not by optical microscopy, we wondered if these high salt-resistant and detergent-resistant filaments persist after silencing of mutant N-terminal htt transgene expression in Tet/HD94 mice.
To test this, we performed the above described purification procedure in parallel with forebrain tissue from untreated Tet/HD94 mice, Tet/HD94 mice that were given 2 mg/ml doxycycline in the drinking water for 4 months (as in Yamamoto et al., 2000), and untreated control littermates. As shown in Figure 6A, silencing mutant huntingtin expression in Tet/HD94 mice by doxycycline treatment for 4 months resulted in a dramatic decrease in aggregated material immunodetected with anti-N-terminal htt and anti-ubiquitin antibodies (Fig. 6A, total homogenates), thus ruling out that the decreased inclusion load seen by immunohistochemistry was a consequence of mere disassembling into their constituent filamentous material. The actual load of filaments was also dramatically decreased as a consequence of transgene expression shutdown, as it can be seen in the P3 and 20-50 fractions of the biochemical purification procedure (Fig. 6A). More precisely, the aggregated material detected with anti-N-terminal mutant htt showed a 89.88 ± 14.9% decrease in the P3 fraction and a 93.75 ± 12.30% decrease in the 20-50 fraction. Similarly, the aggregated material detected with the anti-polyubiquitin antibody showed a 70.82 ± 14.6% decrease in the P3 fraction and a 90.74 ± 15.85% decrease in the 20-50 fraction. The decrease in the number of filaments after mutant-htt transgene shutdown was also evident by immuno-EM analysis of these two fractions (Fig. 6B-D). As shown in Figure 6D, transgene shutdown results in a 75% decrease (p < 0.05) in the number of mutant htt-labeled filaments. The remaining filaments in doxycycline-treated Tet/HD94 mice seemed shorter than the average seen in untreated Tet/HD94 mice (Fig. 6C). However, when we quantified the number of gold particles per decorated structure as an indirect indication of filament length, such decrease did not reach statistical significance (data not shown).
Figure 6.

N-terminal mutant htt-containing and polyubiquitin-containing filaments revert in vivo after silencing mutant N-terminal htt expression in Tet/HD94 mice. A, Western blot analysis of the N-terminal mutant htt-containing and polyubiquitin-containing aggregated material during the different steps of the purification procedure from the forebrain of Tet/HD94 or control littermates (C) either treated (+) or untreated (-) with 5 mg/ml doxycycline in the drinking water for the last 4 months. Arrows indicate the accumulation of aggregated material in the stacking gel. B—D, Immuno-EM analysis of the filaments isolated from the forebrain of Tet/HD94 mice: without pharmacological treatment (B) or after 4 months of transgene expression shutdown by the administration of 5 mg/ml doxycycline in the drinking water (C). D shows a quantification of the total number of filaments isolated from either treated (+) or untreated (-) Tet/HD94 mice. *p < 0.05.
Mutant htt-containing and ubiquitin-containing aggregated material can be purified from HD postmortem brains, and it also corresponds to filaments
The antibodies used for immunohistochemical detection of inclusions in the brain of Tet/HD94 mice (anti-N-terminal mutant htt and anti-polyubiquitin) also detect inclusions in the cortex of human HD brains (Fig. 7A,B). We thus decided to subject cortical samples from grade 4 HD brains to the same biochemical purification procedure described for mouse tissue and monitored the isolation of aggregated material with these antibodies. The P3 fraction from HD brains, but not from control human brain contained aggregated material that is immunoreactive with N-terminal mutant htt antibody (Fig. 7C) and with ubiquitin antibody (data not shown). Similar to what has been described for mouse tissue; sucrose gradient centrifugation allowed further purification of the mutant htt-containing and ubiquitin-containing aggregated material that is retained in the 20-50% interphase (Fig. 7C).
Figure 7.
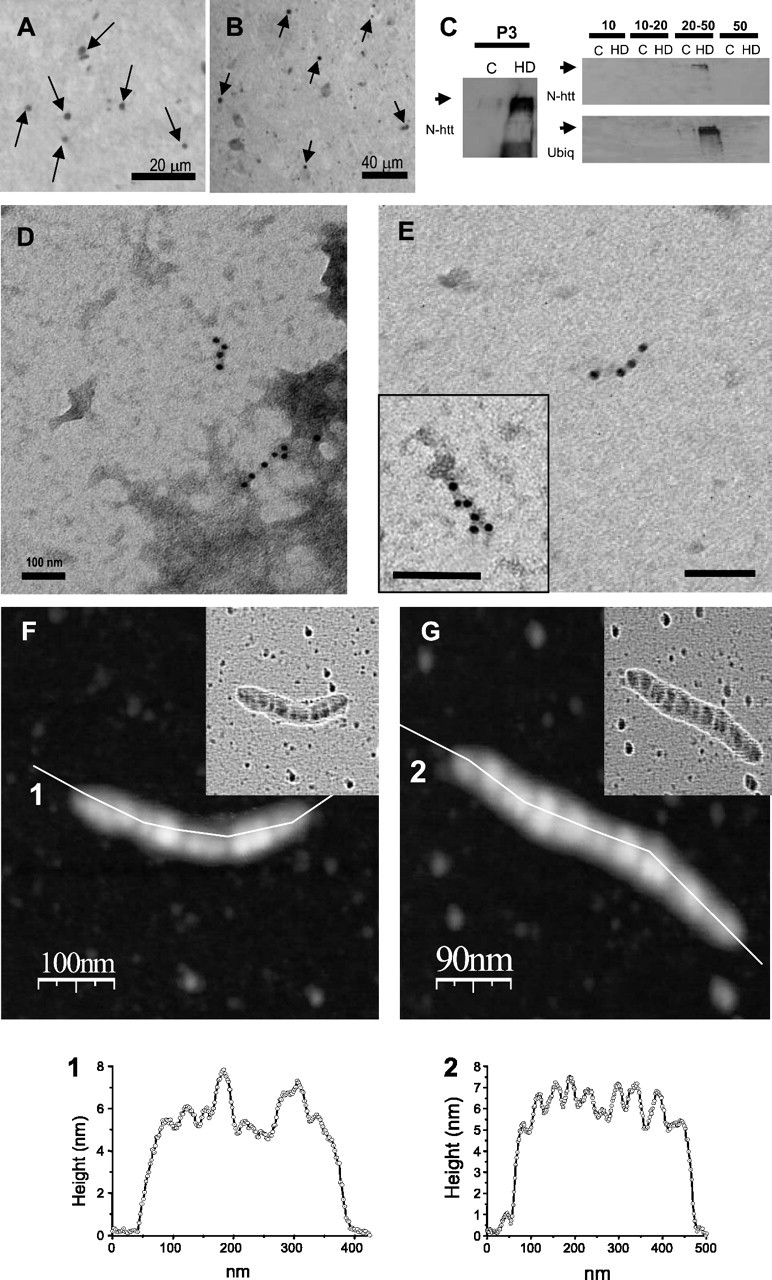
Mutant htt-containing and polyubiquitin-containing aggregated material can be purified from HD postmortem brains, and it also corresponds to filaments. A, B, Immunohistochemistry of cortical sections from a grade 4 HD brain with anti-polyubiquitin (A) and anti N-terminal mutant htt (B) antibodies. Arrows indicate some of the many stained neuropil inclusions. C, Western blot analysis of the mutant htt-containing and polyubiquitin-containing aggregated material present in the P3 fraction of the purification procedure and in its sucrose fractionation, from the cortex of either a control subject (C) or from a grade 4 HD brain (HD). D, E, Immuno-EM analysis of the P3 (D) or 20-50% (E) fractions with two different anti-N-terminal htt antibodies: CAG53b (D, E, inset) or N-18 (full image in E). F-G, AFM analysis of filaments isolated from the cortex of an HD brain with the corresponding height profiles (1 and 2). Laplacian operator filtered images are shown as inserts in the topographic images.
Immunoelectron microscopy analysis with CAG53b antibody of the P3 fraction again demonstrated that the immunoreactive aggregated material corresponded to filamentous structures (Fig. 7D). As shown in Figure 7E, the 20-50 fraction had less contaminant material and we were able to decorate the purified filamentous structures with two different antibodies against N-terminal htt (N-18 and CAG53b antibodies) (Fig. 7E, full image and insert, respectively) and with anti-polyubiquitin and anti-polyQ antibodies (data not shown). The filaments from the human HD samples were much shorter that those previously shown from the brain of the mouse model, with lengths ranging from 100 up to 250 nm, and they often showed mild turns, thus resulting in a short bent filament morphology (Fig 7D-G). In comparison to the filaments isolated from Tet/HD94 mice, AFM analysis (Fig 7F,G) revealed that human HD filaments are shorter (as above described in the immuno-EM analysis) and thicker (the measured width was 40 ± 4 nm). The human HD filaments also presented a beads on a string morphology similar to that found in Tet/HD94 mice. As for the mouse filaments, the height of the human filaments varied between 5.5 and 7.5 nm (Fig. 7F,G, height profiles 1 and 2). However, the length of each bead was larger and more variable (between 27 and 44 nm) in the human filaments, with an average value of 38 ± 9 nm.
Discussion
Here we report, for the first time, a protocol to isolate the aberrant proteinaceous aggregates found in the brain of HD patients. This protocol, based on high salt homogenization, nonionic detergent extraction, and sucrose gradient, was optimized in our conditional mouse model of HD and revealed that the stable aggregated material corresponds to mutant huntingtin-containing and ubiquitin-containing filaments. Furthermore, AFM revealed that these filaments had a beads on a string morphology very similar to the one described for in vitro assembled filaments made of recombinant mutant htt and of other amyloidogenic proteins. These fibrils, although very stable in vitro, are susceptible to revert in vivo as here demonstrated by shutting down transgene expression in the conditional mouse model. These studies will also allow further conformational studies, enzymatic assays, and proteomic analysis that will be crucial to clarify the contribution of aggregates to HD pathogenesis.
Previous reports of HD aggregate isolation were restricted to transfected cells and were based on the use of GFP fusion forms of mutant exon 1 htt (to allow separation in a cell sorter) and/or CsCl gradient fractionation (Scherzinger et al., 1999; Suhr et al., 2001; Hazeki et al., 2002; Mitsui et al., 2002). Studies on transfected cells very often do not reflect the situation in the tissue of an HD patient mainly because of the massive overexpression achieved by transfection. In contrast, in the mouse models, the levels of toxic proteins are adjusted to a maximum compatible with the normal development of the animals but leading to progression to a relevant phenotype during adult life. As an example, transfection of mutant huntingtin into primary cultured neurons results in cell death (Saudou et al., 1998), whereas primary neuronal cultures from mouse models do not show decreased viability (Martin-Aparicio et al., 2001). Accordingly, no loss of striatal neurons is detected at early stages of phenotype progression in mouse models, despite obvious striatal atrophy and/or motor deficits (Hansson et al., 1999; Guidetti et al., 2001; Martin-Aparicio et al., 2001) or in grade 0 HD postmortem brains (Vonsattel et al., 1985). Similarly, proteasome enzymatic activity assays with small fluorogenic substrates in extracts from transfected and transduced cells revealed decreased 20S proteasome activity (Bence et al., 2001; Jana et al., 2001; Nishitoh et al., 2002), whereas no inhibition was detected in extracts from adult mice with overt phenotype and an extensive load of polyQ aggregates (Diaz-Hernandez et al., 2003; Hernández et al., 2004). For all these reasons it was crucial to develop a method to isolate htt aggregates from mouse models and eventually from HD postmortem brains.
The enrichment in N-terminal htt-positive and ubiquitin-positive aggregated material in P3 and 20-50 fractions despite the progressive disruption of the particles detected by double immunofluorescence suggests that, along the biochemical procedure, inclusions disassemble into smaller aggregated material that can be detected by immunobiochemical procedures but not by optical microscopy. Because the EM and AFM studies revealed that this aggregated material corresponds to filaments similar to those detected inside the intact inclusion by ultrastructural analysis of brain sections, it is conceivable that along the purification procedure, inclusions disassemble into their constituent filaments.
The beads on a string morphology of the filaments revealed by AFM is in contrast with the morphology of other filament-like structures found in other neurodegenerative diseases like the paired helical filaments isolated from Alzheimer's disease patients, which present a clear helical and ribbon-like morphology (Pollanen et al., 1994; Moreno-Herrero et al., 2001, 2004). However, the beads on a string morphology of the here purified filaments is identical to that reported after in vitro assembling of recombinant exon 1 mutant htt (Poirier et al., 2002) and very similar to those generated by other amyloidogenic proteins such as Aβ, α-synuclein, and yeast (prion) Sup35 NM peptide (Harper et al., 1999; Rochet et al., 2000). The in vitro generation of these amyloid fibrillar deposits is characterized by sequential formation of globular assemblies (like the beads of the filaments) that can associate linearly to form single protofibrils (like the here reported individual 15-20 nm filaments). Finally, mature fiber formation results from the lateral association of two or more protofibrils (like the here reported 40 nm fibers). However, because the thin fibrils are larger in diameter than previously described protofibrils, and these are usually thought of as being transient intermediates (Dobson, 2003), the possibility also exists that the thin fibrils could be mature fibrils and that the thicker ones could result from longitudinal association of those thin fibrils.
The filaments obtained after in vitro assembling of recombinant mutant exon 1 htt (Poirier et al., 2002) and the here reported after purification from the mouse model and from HD postmortem brains have identical beads on a string morphology. They also have the same height as measured by AFM. However, as mentioned above, they differ in the diameter of the beads and therefore, in the width resulting filaments (5 nm in the in vitro assembled filaments, 13-18 nm in the filaments isolated from Tet/HD94 mouse brains, and 36-44 nm in the filaments isolated from postmortem HD brains). This increasing diameter of the globular beads most likely reflects the increasing complexity of the protein entities forming the aggregates. In the in vitro assembled filaments, the only constituent protein is recombinant exon 1 htt with a 44 polyQ repeat. In the mouse model, the mutant htt also corresponds to exon 1 htt (in this case, with a 94 polyQ repeat), but it is not the only constituent because, at least, polyubiquitin chains are also present in the filament. In the case of the human filaments, the mutant htt molecules forming the filaments are expected to be heterogeneous depending on their degree of endoproteolytic cleavage from the full length form. Some of the described endoproteolytic events lead to htt fragments very similar to those generated from exon 1 constructs or transgenes (Lunkes et al., 2002), whereas others result in significantly longer forms of N-terminal mutant htt (Goldberg et al., 1996; Wellington et al., 2000).
The N-terminal mutant htt in vitro assembled filaments have been shown to be amyloid aggregates (Scherzinger et al., 1997, 1999; Poirier et al., 2002). In view of the morphological similarity and the thioflavine S fluorescence data, it is reasonable to think that filaments isolated from brain tissue are also amyloid aggregates. More precisely, they resemble the protofibril stage of amyloid polymerization (Dobson, 2003). Interestingly, protofibrils have been suggested to be more toxic than mature amyloid aggregates (Conway et al., 2001; Klein et al., 2001). In such case, accumulation of protofibrilar aggregates into mature inclusions would represent a protective mechanism for the cell. We have previously shown that inclusions are dynamic structures in vivo and that they revert in parallel with behavioral phenotype improvement (Yamamoto et al., 2000; Martin-Aparicio et al., 2001). If inclusion reversal were attributable simply to disassembling of the inclusions into filaments, they should have been accompanied by a detriment of the cellular physiology and behavioral phenotype. In good agreement with the behavioral improvement, here we found that the filaments, despite being very stable in vitro, are susceptible to revert in vivo.
In summary, the here described work demonstrates that mutant htt aggregated material can be isolated from the brain of HD mouse models and from postmortem HD brains. The biochemical and ultrastructural characterization of the isolated material has revealed that it is formed by htt-containing and ubiquitin-containing filamentous aggregates with a bead on string morphology, and, more importantly, that these filamentous aggregates are susceptible to revert in vivo, thus suggesting that any associated toxicity is also likely to revert.
Footnotes
This work was supported by grants from Comunidad Autónoma de Madrid, Fundación “La Caixa,” Spanish Comisión Interministerial de Ciencia y Tecnología, and by institutional grants from Fundación Ramón Areces and from Fondo de Investigaciones Sanitarias. Miguel Díaz-Hernández is recipient of a postdoctoral fellowship from Comunidad Autónoma de Madrid. We thank Drs. Javier Díaz-Nido and Francisco Wandosell for helpful discussion and comments and Dr. Erich E. Wanker for kindly providing CAG53b antibody. We are also grateful to Javier Palacín, Carlos Sánchez, Virginia García de Yébenes, Raquel Cuadros, Cristina Plata, and Elena Langa for technical assistance.
Correspondence should be addressed to José J. Lucas, Centro de Biología Molecular “Severo Ochoa,” Consejo Superior de Investigaciones Científicas, Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain. E-mail: jjlucas@cbm.uam.es.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249361-11$15.00/0
References
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: 1552-1555. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416: 507-511. [DOI] [PubMed] [Google Scholar]
- Conway KA, Rochet JC, Bieganski RM, Lansbury Jr PT (2001) Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alphasynuclein adduct. Science 294: 1346-1349. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90: 537-548. [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez M, Hernandez F, Martin-Aparicio E, Gomez-Ramos P, Moran MA, Castano JG, Ferrer I, Avila J, Lucas JJ (2003) Neuronal induction of the immunoproteasome in Huntington's disease. J Neurosci 23: 11653-11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Davies S, Bates G, Vonsattel J, Aronin N (1997) Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277: 1990-1993. [DOI] [PubMed] [Google Scholar]
- Dobson CM (2003) Protein folding and misfolding. Nature 426: 884-890. [DOI] [PubMed] [Google Scholar]
- Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR (1996) Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet 13: 442-449. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827-5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell Jr WO, Schwarcz R, Tagle DA (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol 169: 340-350. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ (1999) Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci 19: 2522-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Peters A, Leist M, Nicotera P, Castilho RF, Brundin P (1999) Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA 96: 8727-8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury Jr PT (1999) Assembly of A beta amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry 38: 8972-8980. [DOI] [PubMed] [Google Scholar]
- Hazeki N, Tsukamoto T, Yazawa I, Koyama M, Hattori S, Someki I, Iwatsubo T, Nakamura K, Goto J, Kanazawa I (2002) Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine. Biochem Biophys Res Commun 294: 429-440. [DOI] [PubMed] [Google Scholar]
- Hernández F, Díaz-Hernández M, Avila J, Lucas JJ (2004) Testing the ubiquitin-proteasome hypothesis of neurodegeneration in vivo. Trends Neurosci 27: 66-70. [DOI] [PubMed] [Google Scholar]
- HDCRG: Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971-983. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Zaidi T, Thompson CH, Merz PA, Wisniewski HM (1984) Alzheimer paired helical filaments: bulk isolation, solubility, and protein composition. Acta Neuropathol (Berl) 62: 167-177. [DOI] [PubMed] [Google Scholar]
- Jana NR, Zemskov EA, Wang G, Nukina N (2001) Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet 10: 1049-1059. [DOI] [PubMed] [Google Scholar]
- Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D (1999) Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA 96: 11404-11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M (1999) Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci 19: 964-973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE (2001) Targeting small Abeta oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci 24: 219-224. [DOI] [PubMed] [Google Scholar]
- Kopito RR (2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10: 524-530. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J (2001) Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J 20: 27-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y (2002) Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell 10: 259-269. [DOI] [PubMed] [Google Scholar]
- Martin-Aparicio E, Yamamoto A, Hernandez F, Hen R, Avila J, Lucas JJ (2001) Proteasomal-dependent aggregate reversal and absence of cell death in a conditional mouse model of Huntington's disease. J Neurosci 21: 8772-8781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, Nakayama H, Akagi T, Nekooki M, Ohtawa K, Takio K, Hashikawa T, Nukina N (2002) Purification of polyglutamine aggregates and identification of elongation factor-1alpha and heat shock protein 84 as aggregate-interacting proteins. J Neurosci 22: 9267-9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Herrero F, Valpuesta JM, Perez M, Colchero J, Baro AM, Avila J, Montejo De Garcini E (2001) Characterization by atomic force microscopy and cryoelectron microscopy of tau polymers assembled in Alzheimer's disease. J Alzheimers Dis 3: 443-451. [DOI] [PubMed] [Google Scholar]
- Moreno-Herrero F, Perez M, Baro AM, Avila J (2004) Characterization by atomic force microscopy of Alzheimer paired helical filaments under physiological conditions. Biophys J 86: 517-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441-1448. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora Jr FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA (2001) Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 291: 2423-2428. [DOI] [PubMed] [Google Scholar]
- Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem 277: 41032-41037. [DOI] [PubMed] [Google Scholar]
- Pollanen MS, Bergeron C, Weyer L (1992) Detergent-insoluble cortical Lewy body fibrils share epitopes with neurofilament and tau. J Neurochem 58: 1953-1956. [DOI] [PubMed] [Google Scholar]
- Pollanen MS, Dickson DW, Bergeron C (1993) Pathology and biology of the Lewy body. J Neuropathol Exp Neurol 52: 183-191. [DOI] [PubMed] [Google Scholar]
- Pollanen MS, Markiewicz P, Bergeron C, Goh MC (1994) Twisted ribbon structure of paired helical filaments revealed by atomic force microscopy. Am J Pathol 144: 869-873. [PMC free article] [PubMed] [Google Scholar]
- Rochet JC, Conway KA, Lansbury Jr PT (2000) Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse alpha-synuclein. Biochemistry 39: 10619-10626. [DOI] [PubMed] [Google Scholar]
- Ross CA (1997) Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19: 1147-1150. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC (2002) Lessons from animal models of Huntington's disease. Trends Genet 18: 202-209. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55-66. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90: 549-558. [DOI] [PubMed] [Google Scholar]
- Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE (1999) Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington's disease pathology. Proc Natl Acad Sci USA 96: 4604-4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6: 487-498. [DOI] [PubMed] [Google Scholar]
- Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH (2001) Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 153: 283-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M (1998) Huntington disease. J Neuropathol Exp Neurol 57: 369-384. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson Jr EP (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559-577. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE (2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12: 1393-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR (2000) Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem 275: 19831-19838. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57-66. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ (2003) Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci 23: 2193-2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217-247. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of Huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493-498. [DOI] [PubMed] [Google Scholar]